

Abstract
The dpr gene is an antioxidant gene which was isolated from the Streptococcus mutans chromosome by its ability to complement an alkyl hydroperoxide reductase-deficient mutant of Escherichia coli, and it was proven to play an indispensable role in oxygen tolerance in S. mutans. Here, we purified the 20-kDa dpr gene product, Dpr, from a crude extract of S. mutans as an iron-binding protein and found that Dpr formed a spherical oligomer about 9 nm in diameter. Molecular weight determinations of Dpr in solution by analytical ultracentrifugation and light-scattering analyses gave values of 223,000 to 292,000, consistent with a subunit composition of 11.5 to 15 subunits per molecule. The purified Dpr contained iron and zinc atoms and had an ability to incorporate up to 480 iron and 11.2 zinc atoms per molecule. Unlike E. coli Dps and two other members of the Dps family, Dpr was unable to bind DNA. One hundred nanomolar Dpr prevented by more than 90% the formation of hydroxyl radical generated by 10 μM iron(II) salt in vitro. The data shown in this study indicate that Dpr may act as a ferritin-like iron-binding protein in S. mutans and may allow this catalase- and heme-peroxidase-deficient bacterium to grow under air by limiting the iron-catalyzed Fenton reaction.
Bacteria living in air rely on defense systems that detoxify reactive oxygen species, such as superoxide, hydrogen peroxide, and hydroxyl radical, which are generated from incomplete reduction of oxygen by enzymatic and nonenzymatic means. These defense systems include (i) enzymes that scavenge reactive oxygen, such as superoxide dismutases (SOD), catalases, and peroxidases (46); (ii) DNA repair enzymes, such as exonuclease III, DNA polymerase, and RecA (46, 50); (iii) protein repair systems, such as thioredoxin and methionine sulfoxide reductase (15a); and (iv) proteins which regulate the cellular metabolism of iron to ameliorate the generation of reactive oxygen species (46, 50).
Lactic acid bacteria, including Streptococcus mutans, cannot synthesize heme and therefore lack catalase and cytochrome oxidases required for energy-linked oxygen metabolism. The growth of lactic acid bacteria, therefore, depends strictly on fermentation. Accordingly, the lactic acid bacteria are considered to have a preference for anaerobiosis. However, many lactic acid bacteria can grow in the presence of oxygen and even consume molecular oxygen through the action of flavoenzymes, such as NADH oxidase, pyruvate oxidase, and α-glycerophosphate oxidase (3, 14, 19, 20, 21, 42). Several antioxidant enzymes, including manganese SOD (13, 33, 38, 41) and none-heme peroxidases, such as manganese-containing catalase (26, 27) and NADH peroxidase (40), which may function as substitutes for catalase, were identified and characterized in lactic acid bacteria. Previously, we identified two components of an NADH-dependent peroxidase (AhpC and Nox-1) from S. mutans (20, 21, 36). While studying an ahpC and nox-1 double-disruption mutant of S. mutans, we found that the mutant still showed the same level of peroxide tolerance as did the wild-type strain, suggesting the existence of one or more other antioxidants in S. mutans (20). In a preceding paper, we identified a dpr (for Dps [DNA binding protein from starved cells] [1]-like peroxide resistance gene) gene as a potential peroxide resistance gene from chromosomal DNA of S. mutans and demonstrated a functional significance for the gene against oxidative stress (55). The dpr disruption mutant of S. mutans could not form colonies on agar plates under air. In addition, although dpr disruption alone did not interfere with growth in liquid cultures, neither the Δdpr ΔahpC Δnox-1 triple mutant nor the Δdpr Δsod double mutant of S. mutans was able to grow aerobically in liquid medium. The synthesis of the 20-kDa dpr gene product, Dpr, was found to be induced by exposure of S. mutans cells to air. Analysis of deduced primary and secondary structures of Dpr suggested that Dpr is a member of the Dps family of proteins (55). Dps is a nonspecific DNA binding protein which accumulates in stationary-phase cells of Escherichia coli (1). Members of the Dps family of proteins form spherical complexes, like ferritin, which are composed of 7 to 12 identical subunits of 16 to 22 kDa, and some of them bind iron (1, 6, 15, 22, 34, 48). To date, three family members, including E. coli Dps, have been shown to bind to DNA for protection from oxidative stress (1, 8, 34). On the other hand, functional divergence of other Dps family proteins was also reported, i.e., the nonheme ferritin of Listeria innocua (6), the fine-tangled-pilus major subunit of Haemophilus ducreyi (7), the neutrophil-activating protein (HP-NAP) of Helicobacter pylori (48), and a cold shock protein from Listeria monocytogenes (18). In this study, we purified Dpr from S. mutans and examined its molecular properties to understand how Dpr confers oxygen tolerance on S. mutans.
MATERIALS AND METHODS
Reagents.
Horse spleen ferritin, apo-horse spleen ferritin (apoferritin), and 3-(2-pyridyl)-5,6-bis(2-[5-furyl sulfonic acid])-1,2,4-triazine (Ferene S) were purchased from Sigma Chemical Co. (St. Louis, Mo.). Deferoxamine methylate (deferoxamine) was from ICN Pharmaceuticals, K. K., Tokyo, Japan. Bovine serum albumin (BSA), bovine liver catalase, ferrous ammonium sulfate, zinc chloride hexahydrate, 2-deoxyribose, xanthine, xanthine oxidase, and cytochrome c were from Wako Pure Chemicals, Osaka, Japan. All other chemicals used were of the best grade commercially available. The stock 100 mM ferrous ammonium sulfate solution in 0.05 N HCl was prepared immediately before use. Apoferritin was further purified by high-performance liquid chromatography (HPLC) with a Mono-Q HR 5/5 column (Amersham Pharmacia Biotech, Tokyo, Japan) using a linear NaCl gradient (0.2 to 0.6 M) in 20 mM Tris-HCl (pH 7.0) because of contamination of SOD activity in the preparation. The purified apoferritin preparation was dialyzed against 0.1 mM MOPS-NaOH (pH 7.0) and concentrated by a CM-50 Centricon ultrafiltration unit (Millipore, Tokyo, Japan) and was used for assays.
Bacterial strains, media, and culture conditions.
S. mutans GS-5 (53) was routinely grown in TYG medium at 37°C as described previously (55).
Preparation and purification of Dpr.
A preculture of 24 ml of S. mutans under atmosphere without shaking was used as the inoculum for 2.4 liters of aerobic culture. The cells were incubated for 13 h with vigorous shaking (120 cycles/min), the culture was centrifuged at 12,000 × g for 10 min, and the pelleted cells were washed with 100 mM potassium phosphate buffer (pH 7.0) containing 0.5 mM EDTA (buffer A). About 10 g (wet weight) of the cells obtained from 2.4 liters of culture was resuspended in 30 ml of buffer A and disrupted by a French pressure cell. Cell debris was removed by centrifugation at 28,000 × g for 30 min, and the supernatant was dialyzed against 20 mM potassium phosphate buffer (pH 7.0) containing 0.5 mM EDTA (buffer B). The dialysate was treated with streptomycin sulfate at a final concentration of 1% (wt/vol) at room temperature for 30 min and centrifuged (28,000 × g). The supernatant obtained (about 30 ml) was dialyzed against 80 mM potassium phosphate buffer (pH 7.0) containing 0.5 mM EDTA (buffer C). Sixteen grams of hydroxylapatite powder (Wako Pure Chemicals, Osaka, Japan) was added to the dialysate and kept for 2 h at 4°C with stirring. The hydroxylapatite was recovered, packed in a glass column, and washed with buffer C. The Dpr fraction, which was eluted with 0.2 M potassium phosphate buffer (pH 7.0) containing 0.5 mM EDTA, was dialyzed against a 10-fold volume of distilled water. The dialysate was then subjected to HPLC on a Mono-Q HR 5/5 anion-exchange column using a linear NaCl gradient (0.15 to 0.45 M) in buffer B. The Dpr fraction, which eluted at 0.3 M NaCl, was then subjected to HPLC on an HA-1000 column (Tosoh, Tokyo, Japan) using a linear potassium phosphate gradient (20 to 180 mM) in buffer B. The Dpr fraction, which eluted at 80 mM potassium phosphate, was subjected to HPLC on a DEAE-5PW column (Tosoh) using a linear NaCl gradient (0.1 to 0.3 M) in buffer B. The purity of the Dpr fraction obtained during various steps of the procedure was checked by SDS-PAGE by the method of Laemmli (28), followed by staining with Coomassie brilliant blue R250. The protein concentration was measured by the method of Lowry et al. (30).
Preparation of apo-Dpr.
Apo-Dpr was prepared by dialyzing the Dpr preparation against 100 volumes of 20 mM MOPS (morpholinepropanesulfonic acid)-NaOH buffer (pH 7.0) containing 1 mM 2,2′-dipyridyl and 0.3% sodium dithionite at 4°C. The dialysis buffer was changed twice over 36 h. The apo-Dpr preparation was dialyzed against 0.1 mM MOPS-NaOH (pH 7.0) and concentrated (14 to 94 μM) by a CM-50 Centricon and was used for all assays. The apo-Dpr preparation contained <1 atom of iron per molecule of Dpr (see below).
Preparation of iron-loaded Dpr.
The iron-loaded Dpr preparation was obtained by incubating 100 μg of the apo-Dpr preparation/ml with 1 mM ferrous ammonium sulfate in 20 mM potassium phosphate buffer (pH 7.0) at room temperature for 1 h. To remove excess iron, the mixture was applied on a DEAE Toyopal 650 M column (Tosoh). The iron-loaded Dpr fraction was eluted with 20 mM potassium phosphate buffer (pH 7.0) containing 0.4 M NaCl. The iron content of the iron-loaded Dpr preparation was determined by atomic absorption spectrometry (170-30; Hitachi, Tokyo, Japan).
The native Dpr and iron-loaded Dpr preparations were also dialyzed against 0.1 mM MOPS-NaOH (pH 7.0), concentrated by a CM-50 Centricon, and used for all assays.
Purification of E. coli Dps.
For expression of Dps, a DNA fragment containing the dps structural gene was amplified from chromosomal DNA of E. coli DH-5α by PCR using the primers AGTACCGCTAAATTAGTTAAATCAAAAG (dps2) and 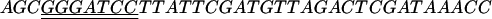 (dps3). In dps3, the double underlining indicates the BamHI site. The amplified fragment was digested with BamHI and inserted into the NcoI-BamHI site of an expression vector, pTrc99A (Amersham Pharmacia Biotech), to construct plasmid pTRCDPS. Then, E. coli DH5α was transformed with pTRCDPS. Purification of E. coli Dps from the crude extract of E. coli DH5α(pTRCDPS) was done as described by Almiron et al. except that an SP-5PW column (Tosoh) was used for the final step of purification. The purified Dps was dialyzed against 50 mM Tris-HCl (pH 8.0) containing 100 mM NaCl and 1 mM EDTA and then concentrated by a CM-50 Centricon.
(dps3). In dps3, the double underlining indicates the BamHI site. The amplified fragment was digested with BamHI and inserted into the NcoI-BamHI site of an expression vector, pTrc99A (Amersham Pharmacia Biotech), to construct plasmid pTRCDPS. Then, E. coli DH5α was transformed with pTRCDPS. Purification of E. coli Dps from the crude extract of E. coli DH5α(pTRCDPS) was done as described by Almiron et al. except that an SP-5PW column (Tosoh) was used for the final step of purification. The purified Dps was dialyzed against 50 mM Tris-HCl (pH 8.0) containing 100 mM NaCl and 1 mM EDTA and then concentrated by a CM-50 Centricon.
Immunoblot analysis.
For immunoblot analysis, proteins on SDS-PAGE gels were electrophoretically transferred to polyvinylidene difluoride membranes (Millipore). The membranes were immunostained with a 10,000-fold dilution of an anti-Dpr antiserum which was prepared from a rabbit immunized with the purified Dpr preparation. The reactive proteins were detected using a goat anti-rabbit antibody conjugated to alkaline phosphatase. The Dpr band was developed with nitroblue tetrazolium and 5-bromo-4-chloro-3-indolyl phosphate as the substrate. The amount of Dpr in the cell extract was determined by quantitative comparisons with the signal generated by amounts of the purified Dpr preparation.
Analytical procedures. (i) N-terminal amino acid sequence.
The protein on the SDS-polyacrylamide gel was blotted onto a polyvinylidene difluoride membrane, and its N-terminal amino acid sequence was determined by a gas phase protein sequencer (model ABI 491; Applied Biosystems) equipped with an on-line amino acid analyzer (model 610A; Applied Biosystems).
(ii) Metal content in Dpr preparations.
The metal contents of native and apo-Dpr preparations were measured by a magnetic sector inductively coupled plasma (ICP) mass spectrometer (ELEMENT; Finnigan MAT) in medium-resolution mode.
(iii) Analytical ultracentrifugation and light-scattering methods.
Sedimentation equilibrium analysis was performed using an Optima XL-A analytical ultracentrifuge (Beckman Instruments, Palo Alto, Calif.) essentially as described previously (37). Purified Dpr was concentrated and rediluted three times into 25 mM potassium phosphate buffer with 1 mM EDTA and 0.15 M NaCl at pH 7.0 using CM-10 Centricon ultrafiltration units. Three concentrations of Dpr (4.4 to 38 μM) were loaded (110 μl each) into three of the six sectors of the cell, and buffer from the filtrate was placed in the reference sectors. Data were collected following equilibration at each speed for 12, 14, and 16 h at 20°C. Global analysis of 10 data sets equilibrated at 5,000, 6,500, 9,000, and 12,000 rpm was performed using the Windows version of NONLIN (24). Only data of less than 1.4 absorbance units were used in the analysis. A value of 0.73466 cm3/g was calculated for the partial specific volume of Dpr from the amino acid composition (29). The buffer density at 1.0077 g/cm3 was measured using a DA-310 precision density meter (Mettler Toledo, Hightstown, N.J.).
Sedimentation velocity experiments were also carried out at 20°C using three samples and the same buffer described above in double-sectored cells at 0.33, 0.68, and 1.05 starting A280 values. Sedimentation data were collected every 4 min at a rotor speed of 35,000 rpm and a radial step size of 0.003 cm. From these data, sedimentation coefficient values at each concentration were calculated using the SVEDBERG program (version 6.23; from J. Philo [http://www.jphilo.mailway.com]) standardized to pure water (corrected for density and viscosity) (29) and were extrapolated to zero concentration to give a value for s020,w of 1.241 × 10−12 s. The molecular weight of Dpr was calculated from the Svedberg equation (51) using this s020,w value and the corrected translational diffusion coefficient (D020,w), which was obtained by dynamic light-scattering analysis (see below).
Light-scattering data were acquired using a BI-2030AT correlator operated together with a BI-200SM light-scattering goniometer-photon-counting detector (Brookhaven Instruments, Holtsville, N.Y.) and a model 127 helium-neon laser (35 mW; equipped with a vertical polarization rotator; Spectra Physics, Mountain View, Conn.). For determination of the molecular weight of Dpr by static light-scattering analyses, measurements were made at an angle of 90° in specially formulated microcuvettes (Hellma Cells, Inc.) maintained at 20°C in a refractive-index matching bath (containing 50% glycerol). Samples in the 25 mM potassium phosphate buffer without NaCl were passed through a 0.2-μm-pore-size filter, and the filtrate was collected in acid-washed, dust-free microcuvettes. Sample scattering intensities were corrected for solvent scattering and were expressed relative to a benzene standard (17). Dpr concentrations were measured using the microbiuret protein assay with BSA as a standard (4). A total of 14 intensity measurements from three concentrations of Dpr (0.38 to 1.72 mg/ml) were averaged and used for molecular weight calculations. Weight average molecular weights were determined from the solvent-corrected relative scattering intensity using Rayleigh-Gans-Debye theory (23) and a protein refractive-index increment (dn/dc) of 0.183 ml/g calculated from the amino acid composition (43). Dynamic light scattering was also carried out with the above-mentioned samples to determine the translational diffusion coefficient using the method of cumulants as described previously (17), and the corrected D020,w (51), at (3.97 ± 0.42) × 10−7 cm2/s, was used with the sedimentation coefficient to compute a third, independent value for the molecular weight of Dpr.
(iv) Iron and zinc incorporation.
For electrophoresis, the apo-Dpr preparation (200 μg/ml) was incubated with 500 μM ferrous ammonium sulfate in 20 mM potassium phosphate (pH 7.0) at room temperature for 1 h. The reaction mixture was subjected to native PAGE, and the protein band containing iron on the gel was detected by Ferene S as described previously (9, 55). To examine the capacity of Dpr to incorporate iron and zinc, 100 μg of the apo-Dpr preparation/ml was incubated with 150 to 500 μM ferrous ammonium sulfate or 50 to 200 μM zinc chloride hexahydrate in 20 mM MOPS-NaOH buffer (pH 7.0) at room temperature for 30 min. Iron- or zinc-loaded Dpr was then purified on a PD-10 column (Amersham Pharmacia Biotech) which was equilibrated with 50 mM MOPS-NaOH buffer (pH 7.0), and the iron and zinc contents of Dpr were determined using an atomic absorption spectrometer. The results shown are means for triplicates.
(v) DNA binding assay.
The DNA binding assay was done as described by Almiron et al., who reported that when Dps bound to DNA, the Dps-DNA complex did not enter an agarose gel during electrophoresis (1). According to the method, 5, 10, and 30 μg of the purified or iron-loaded Dpr preparation was added to 200 ng of pUC118, λ DNA, or S. mutans genomic DNA in TE (10 mM Tris-HCl, 1 mM EDTA, pH 8.0) and incubated at 37°C for 30 min. The reaction mixture was then electrophoresed through a 1% Tris-acetate agarose gel. DNA on the gel was detected by staining it with ethidium bromide. E. coli Dps was used as a positive control.
A DNA-cellulose (Sigma) column, which was equilibrated with 50 mM Tris-HCl (pH 8.0) containing 50 mM NaCl and 1 mM EDTA, was also used to examine the DNA binding ability of Dpr. Both the purified and iron-loaded Dpr preparations were recovered in the flowthrough fraction. In contrast, Dps was eluted at ∼0.2 M NaCl by using a linear gradient of 50 to 500 mM NaCl.
(vi) Measurement of SOD and catalase activities.
The activity of SOD was measured by the xanthine oxidase-cytochrome c method (32). One unit of SOD activity was defined as the amount of protein that gave a 50% decrease in the rate of reduction of cytochrome c. Catalase activity was measured according to the method of Beer and Siezer (5). For both assays, 10 to 100 μg of the purified Dpr preparation/ml was used.
(vii) Assay of iron-dependent hydroxyl radical formation.
Assay of iron-dependent hydroxyl radical formation was done by the method described by Halliwell and Gutteridge (16). Iron(II) salt in solution generates hydroxyl radical by the following nonenzymatic reactions (16):
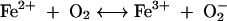 |
(1) |
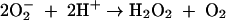 |
(2) |
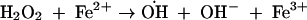 |
(3) |
Hydroxyl radical degrades deoxyribose to a malondialdehyde-like compound which forms a chromogen with thiobarbituric acid. Fluorescence of the chromogen was used as the assay for hydroxyl radical.
The basal reaction mixture contained 10 mM potassium phosphate buffer (pH 7.4), 63 mM NaCl, and 4 mM 2-deoxyribose in a total volume of 0.3 ml. To the basal reaction mixture was added an appropriate amount of Dpr, apoferritin, BSA, deferoxamine, or catalase. The reaction was started by adding ferrous ammonium sulfate to the reaction mixture at a final concentration of 10 μM. After incubation at 37°C for 15 min, 0.25 ml of 1% (wt/vol) thiobarbituric acid and 2.8% (wt/vol) trichloroacetic acid were added to the reaction mixture. After the mixture was boiled for 10 min and rapidly cooled, the amount of chromogen formed in the sample was measured by its fluorescence emission at 553 nm using a Hitachi model 204A fluorescence spectrophotometer (λex = 532 nm).
Electron microscopy.
Approximately 10 μl of iron-loaded Dpr (50 μg/ml) or horse spleen ferritin (50 μg/ml) in 20 mM MOPS-NaOH (pH 7.0) was placed on carbon-coated grids and negatively stained with 1% (wt/vol) sodium phosphotungstic acid (pH 7.4). The samples were examined under an electron microscope (H-8100; Hitachi) at an acceleration voltage of 100 kV.
Homology search.
The amino acid sequences of S. mutans Dpr (AB036428), E. coli nonheme ferritin (S14069), and E. coli bacterioferritin (AF058450.1) were compared with the sequence databases using the BLAST (version 1.49) programs implemented at the EMBL/GenBank/DDBJ, SWISSPROT/NBRF-PIR, and Streptococcus thermophilus Sequencing Group at the Catholic University of Louvain (http://www.biol.ucl.ac.be/gene/genome/) and the Microbial Genome Database available through the National Center for Biotechnology Information server (http://www.ncbi.nlm.nih.gov/Microb_blast/unfinishedgenome.html), including Streptococcus pyogenes, Streptococcus pneumoniae, S. mutans, Streptococcus equi, Streptococcus gordonii, Enterococcus faecalis, and Lactococcus lactis.
RESULTS
Purification of Dpr from S. mutans.
We previously identified the dpr gene product, Dpr, as a 20-kDa protein in the cell extract of S. mutans (55). Dpr was purified to an electrophoretically homogeneous state (Fig. 1A) from crude extracts of S. mutans cells, which were grown aerobically, by successive column chromatography using hydroxylapatite, Mono-Q HR 5/5, HA-1000, and DEAE-5PW as described in Materials and Methods. The purified Dpr preparation gave a single absorption maximum at 280 nm and an A280/260 value of 1.5 ± 0.02, suggesting that the purified Dpr preparation was free from nucleic acid (10). The N-terminal seven residues of the purified 20-kDa protein were identical to the second through the eighth residues of the deduced amino acid sequence of Dpr. The yield and purification of Dpr were 25.4% and 43.5-fold, respectively, and 2.4 mg of the purified Dpr preparation was obtained from 2.4 liters of culture.
FIG. 1.

SDS-PAGE profile for the purified Dpr preparation (A) and its iron-binding activity (B). (A) Molecular weight markers (lane 1) and 2 μg of purified protein (lane 2) were electrophoresed on an SDS-polyacrylamide gel (15.0% [wt/vol]), and the protein bands were stained with Coomassie brilliant blue R250. (B) Native Dpr (lanes 1), apo-Dpr (lanes 2), and iron-loaded Dpr (lanes 3) preparations were electrophoresed on nondenaturing polyacrylamide gel (8.0% [wt/vol]) and stained with Ferene S (i) and Coomassie brilliant blue R250 (ii). Each lane contained 3 μg of Dpr.
Oligomeric structure of Dpr.
Molecular weight determinations were made by three independent methods to determine the subunit composition of Dpr in solution. Sedimentation equilibrium analyses of data sets at a variety of concentrations and speeds yielded molecular weights that exhibited no significant dependence on either protein concentration or rotor speed, consistent with the single-ideal-species model (Fig. 2). Global fits of the data to this model resulted in a weight average molecular weight of 223,000 ± 7,000. Another method for analyzing shape-independent molecular weights of macromolecules, static light scattering, gave an average value of 291,000 ± 22,000 for the molecular weight of Dpr. Finally, using s020,w determined from sedimentation velocity measurements and D020,w from dynamic light-scattering analyses, the molecular weight of Dpr as calculated from the Svedberg equation (51) is 292,000. The theoretical molecular weight of a single subunit of Dpr, given its lack of the initiating methionine residue, is 19,485.8 g/mol. These data, in combination, are consistent with a subunit composition of 11.5 to 15.0 subunits per molecule.
FIG. 2.

Sedimentation equilibrium analysis of Dpr to determine molecular weight. Shown are single data sets for 38 μM Dpr equilibrated at 20°C to 6,500 (circles), 9,000 (triangles), and 12,000 (squares) rpm rotor speeds. The solid lines represent global fits to a single ideal species of 14 data sets. The upper panel depicts the deviations between experimental and calculated data (residuals) for each data set. The weight average molecular weight value for Dpr obtained by this method was 239,000 ± 7,000 (variance = 5.62 × 10−4 with 1,066 degrees of freedom) or 223,000 ± 7,000 (variance = 3.64 × 10−4 with 1,056 degrees of freedom) when offset values were floated (25).
The dimensions of the Dpr oligomer were also studied by electron microscopy. Negatively stained electron microscopy of Dpr showed ferritin-like spherical structures, which are characteristic of Dps family proteins (Fig. 3). The average diameter of Dpr was calculated to be 9.2 nm, using horse spleen ferritin (12.5-nm diameter) as a standard. The value was similar to that of other Dps family proteins (9.0 to 10.14 nm) (1, 6, 8, 48) but smaller than that of eucaryotic and other bacterial ferritins (11.8 to 13.0 nm) (2).
FIG. 3.

Electron micrographs of iron-loaded Dpr (A) and horse spleen ferritin (B) preparations. Bars, 45 nm.
Iron-binding activity of Dpr.
Some Dps family proteins, i.e., ferritin from L. innocua and HP-NAP from H. pylori, were shown to bind iron (6, 48). Previously, we also detected Dpr as an iron-binding protein from the crude extract of S. mutans cells by staining proteins on the polyacrylamide gel with Ferene S (55). In this study, the iron-binding ability of the purified Dpr preparation was examined. As shown in Fig. 1B, both native and iron-loaded Dpr preparations were visualized upon being stained by Ferene S (Fig. 1B, lanes 1 and 3). However, the apo-Dpr preparation was not stained by Ferene S (Fig. 1B, lane 2). The staining intensity of the iron-loaded Dpr preparation was approximately 10 times higher than that of the native one. These data clearly indicate that Dpr can bind to iron and that the native Dpr preparation contains iron.
Metal content in the purified Dpr preparation and maximum incorporation of metals into Dpr.
Metals in the purified Dpr preparation were analyzed by ICP mass spectrometry. It was revealed that native Dpr from five independent preparations contains iron (10.8 to 27.5 atoms per molecule) and zinc (0.1 to 15.9 atoms per molecule). Dpr preparations also contained Cu, Cr, Mn, Co, Ni, and Mo at <1 atom per molecule. One molecule of Dpr is taken here to consist of 12 identical subunits (see below). In contrast to the native Dpr preparation, the apo-Dpr preparation contained <1 atom of metal per molecule examined.
Maximum incorporation of iron into Dpr was examined by adding various concentrations of ferrous ammonium sulfate (150, 200, 250, 300, 400, and 500 μM) to MOPS-NaOH buffer containing 100 μg of the apo-Dpr preparation/ml. At iron concentrations higher than 250 μM, a brown precipitate of iron was formed, indicating that 250 μM iron is a saturation point for the iron-binding capacity of the protein. Apo-Dpr that was loaded with iron at 250 μM was then purified on a PD-10 column, and the iron content of the protein was measured by atomic absorption spectrometry. It was found that the iron-loaded Dpr preparation contained 40.0 ± 2.2 iron atoms per subunit, corresponding to 480 iron atoms per molecule. This value resembles those reported for L. innocua ferritin and H. pylori HP-NAP (500 atoms per molecule) (6, 48).
The zinc binding ability of Dpr was also examined using the method described above. The apo-Dpr preparation incorporated up to 11.2 ± 1.7 zinc atoms per molecule.
DNA binding activity.
E. coli Dps nonspecifically binds DNA to form the stable Dps-DNA complex, which is involved in protection of DNA from hydrogen peroxide by sequestration of the DNA (1, 12, 31, 54). DNA binding activities of other Dps family proteins are also considered to be significant factors in protecting cells from oxidative damage (8, 34). Accordingly, the DNA binding activity of Dpr was assayed. Analyses by agarose gel electrophoresis and DNA-cellulose chromatography were carried out to assess the binding of Dpr to DNA. Addition of the purified E. coli Dps preparation to plasmid DNA resulted in the formation of a Dps-DNA complex that was retained in the well after electrophoresis (Fig. 4, lanes 3 and 4). In contrast, addition of the purified Dpr preparation did not affect the mobility of plasmid DNA (Fig. 4, lanes 5, 6, and 7). Addition of the purified Dpr preparation also did not affect the mobility of λ DNA or S. mutans genomic DNA (data not shown). When an iron-loaded Dpr preparation was used, the same result was obtained (data not shown). In addition, Dpr did not bind to DNA-cellulose, while E. coli Dps bound to DNA-cellulose and eluted at a relatively high salt concentration (∼0.2 M NaCl). These results indicated that Dpr did not associate with DNA under the conditions used.
FIG. 4.

Assay of DNA binding activity of Dpr. Purified Dps or purified Dpr was added to 200 ng of pUC118 DNA and incubated at 37°C for 30 min. The reaction mixture was then electrophoresed through a 1% Tris-acetate agarose gel. DNA on the gel was detected by staining it with ethidium bromide. Marker DNA (lane 1), pUC118 alone (lane 2), pUC118 with Dps (2.5 and 5.0 μg [lanes 3 and 4]), and pUC118 with Dpr (5.0, 10.0, and 30.0 μg [lanes 5, 6, and 7]) are shown.
Enzyme activity in Dpr preparation.
Neither SOD nor catalase activity was detected in the Dpr preparation.
Inhibition of iron-dependent hydroxyl radical formation by Dpr.
The results described above suggest that Dpr functions as a ferritin-like iron-binding protein and that the protection of S. mutans from oxidative stress by Dpr might be due to sequestration of iron which otherwise catalyzes the generation of highly toxic radicals, such as hydroxyl radicals. Therefore, the iron-dependent hydroxyl radical formation in the presence or absence of Dpr was investigated as described in Materials and Methods. The results are shown in Table 1 . The reaction mixture containing 10 μM iron(II) salt generated hydroxyl radical (see Materials and Methods, reactions 1 to 3) to yield a value of 59.4 fluorescence units (Table 1). Addition of either catalase or deferoxamine, which is an iron chelator, to the reaction mixture caused inhibition of the generation of hydroxyl radical. Addition of apo-Dpr also effectively inhibited hydroxyl radical formation. Seventy-five percent inhibition of hydroxyl radical formation resulted from the addition of 20 nM apo-Dpr at a ratio of apo-Dpr to iron of 1:500. In contrast, 1,000 nM apo-ferritin inhibited hydroxyl radical formation by only 12.3%. BSA did not inhibit hydroxyl radical formation.
TABLE 1.
Effects of Dpr on iron(II)-dependent hydroxyl radical formationa
| Addition to reaction mixture containing 10 μM iron(II) | Amt | Extent of hydroxyl radical formation (fluorescence emission [arbitrary units])b | Inhibition (%) |
|---|---|---|---|
| None | 59.4 ± 3.1 | ||
| Deferoxamine | 10 μM | 17.9 ± 4.9 | 69.9 |
| 100 μM | 1.4 ± 0.6 | 97.6 | |
| Catalase | 100 U/ml | 35.7 ± 4.3 | 39.9 |
| 1,000 U/ml | 11.0 ± 0.9 | 81.5 | |
| Apo-Dpr | 20 nM | 14.8 ± 0.8 | 75.1 |
| 100 nM | 4.4 ± 0.5 | 92.6 | |
| BSA | 1,500 nM | 72.8 ± 2.8 | <0 |
| 4,500 nM | 88.7 ± 3.4 | <0 | |
| Apoferritin | 100 nM | 64.0 ± 4.2 | <0 |
| 500 nM | 60.3 ± 1.3 | <0 | |
| 1,000 nM | 52.1 ± 4.6 | 12.3 |
The results are the means ± standard deviations for at least triplicates. Inhibition of hydroxyl radical formation by adding the indicated materials to the reaction mixture is also shown. Data for native and iron-loaded Dpr are shown in Fig. 5 but are not included here for simplicity.
λex = 532 nm, λem = 553 nm.
The effect of the iron content of Dpr on the inhibition of hydroxyl radical formation was examined by using preparations of apo-Dpr, native Dpr with 19.8 iron atoms per molecule, and a preparation of iron-loaded Dpr with 277 iron atoms per molecule. As shown in Fig. 5, the extent of the inhibition of hydroxyl radical formation by native Dpr was similar to that by the apo-Dpr preparation, presumably due to the low iron content of the native Dpr preparation. In contrast, the inhibitory capability of the iron-loaded Dpr preparation was significantly decreased but still remained (Fig. 5). These results indicate that the iron-binding ability of Dpr contributes to the inhibition of hydroxyl radical formation and also suggest that the iron-loaded Dpr preparation is still capable of binding iron. A 50% inhibitory concentration value for the inhibition of hydroxyl radical formation, calculated to be 7.5 nM for apo-Dpr, was 6.9-fold lower than that for iron-loaded Dpr (52 nM).
FIG. 5.

Comparison of the extents of inhibition of hydroxyl radical formation by apo-Dpr, native Dpr, and iron-loaded Dpr. Hydroxyl radical formation generated by 10 μM ferrous iron in the presence of the indicated amounts of apo-Dpr (squares), native Dpr (diamonds), and iron-loaded Dpr (circles) preparations is shown. Fluorescence data (λex = 532 nm; λem = 553 nm) were obtained following the addition of thiobarbituric acid and trichloroacetic acid as described in Materials and Methods. Without Dpr, a fluorescence value of 59.4 (arrow) was obtained. The results shown are the means and standard deviations for triplicates.
DISCUSSION
The dpr gene was originally discovered as a potential peroxide tolerance gene in the S. mutans chromosome by its ability to complement an alkyl hydroperoxide reductase-deficient mutant of E. coli (45) with a tert-butyl hydroperoxide-hypersensitive phenotype, and the gene was shown to play an indispensable role in oxygen tolerance in S. mutans (55). In this study, we purified Dpr from crude extracts of S. mutans and showed, by using electron microscopy and molecular weight determinations of the native Dpr preparation, that Dpr forms spherical oligomers similar to those of other Dps family proteins. The purified Dpr preparation contained a small amount of iron and could incorporate up to 480 iron atoms per molecule. Unlike E. coli Dps and two other members of the Dps family (1, 8, 34), Dpr was unable to bind DNA and did not exhibit catalase activity as reported for Synechococcus sp. DpsA (35). Thus, a role for Dpr in protection against oxidative stress might involve the sequestration of iron, thereby preventing the generation of highly toxic radicals and conferring oxygen tolerance. As shown in our experiments, Dpr prevented hydroxyl radical formation generated by 10 μM iron(II) salt in vitro (Table 1). This result supported our previous suggestion from the in vivo study that the failure of a series of Dpr-defective mutants to grow under aerobic conditions is caused by the excessive generation of hydroxyl radicals (55). The high intracellular concentration of Dpr determined by Western blotting (∼2% of total cellular soluble protein [see Materials and Methods]) might enable cells to be protected from iron-mediated oxidative stress. The contribution of the sequestration of iron by bacterial ferritin to oxidative stress protection was demonstrated in the cases of Campylobacter jejuni and an E. coli fur recA mutant (49, 52). Recently, intracellular “free-iron” concentrations of E. coli and Saccharomyces cerevisiae were reported to be 10.0 and 12.8 μM, respectively, using whole-cell electron paramagnetic resonance analyses (25, 44). Though the amount of free iron in S. mutans is not known, the observed level of Dpr present in S. mutans could prevent hydroxyl radical formation for up to 10.0 μM iron(II). In contrast, apoferritin hardly inhibited hydroxyl radical formation under the conditions shown in Table 1, indicating that under the assay conditions used in this study, there is a competition between nonenzymatic iron oxidation by molecular oxygen (reaction 1) and iron sequestration by Dpr or horse spleen ferritin. Because of this, only an iron-binding protein that can associate with iron faster than it is oxidized could inhibit the reaction. The ability of horse spleen ferritin to incorporate iron [kcat and Km values for iron(II) → iron(III) are reported to be 80 min−1 and 350 μM, respectively (47)] should not be enough to inhibit the reaction, especially for the reaction mixture containing phosphate, which functions as a weak iron chelator and enhances iron oxidation.
Negatively stained protein analyzed by electron microscopy showed ferritin-like spherical structures of Dpr about 9.2 nm in diameter (Fig. 3). Interestingly, the results of dynamic light-scattering analyses to determine the translational diffusion coefficient corresponded to a Stokes radius of 5.5 ± 0.6 nm. This hydrodynamic diameter of about 11.0 nm is in reasonable agreement with the anhydrous diameter from electron microscopy of about 9.2 nm, particularly since the z-average value (23) obtained using the light-scattering method includes bound H2O molecules and is more sensitive to larger particles. Using three independent methods to assess the molecular weight of Dpr and taking into account its subunit molecular weight, our data also indicate the presence of 11.5 to 15.0 subunits per molecule of Dpr in solution. Given this value and the fact that the iron content of the Dpr preparation used for analysis is approximately 20 atoms per molecule, it is likely that Dpr is a dodecamer in solution. Given an expected molecular weight of 233,830, then, one would predict a translational diffusion coefficient of 5.22 × 10−7 cm2/s for a solid spherical molecule rather than the measured value of 3.97 × 10−7 cm2/s, which indicates a larger particle than expected from the molecular weight. This result, together with our electron microscopy data, is consistent with the presence of a cavity in spherical Dpr molecules in which iron atoms can be sequestered, as has been observed previously for ferritins (2).
In bacteria, two types of ferritin molecules, heme-containing bacterioferritin and nonheme ferritin, have been reported (2). Both are composed of 24 identical or similar subunits (2). Recent X-ray crystallographic analyses of two members of the Dps family, E. coli Dps and L. innocua ferritin, revealed them to be spherical dodecamers consisting of four α-helical bundle monomers and also delineated their close structural relationship with other bacterial and eucaryotic ferritin proteins (15, 22). Though the iron-binding ability of E. coli Dps is not certain, it is clear that L. innocua ferritin, H. pylori HP-NAP, and S. mutans Dpr all incorporate iron (6, 48). Therefore, at least some members of the Dps protein family, including Dpr, belong to a new class of small ferritins which appear to exist as dodecamers. Dpr was also able to bind zinc ions. Although a physiological role for the zinc binding ability of Dpr is not known, detoxification of zinc by ferritin during overload has been reported (11, 39). To date, ferritin-like proteins have not been reported in lactic acid bacteria, and neither bacterioferritin nor nonheme ferritin homologues have been found in genomic sequence data of lactic acid bacteria (see Materials and Methods). In contrast, Dpr and Dps family members were widely distributed among the databases, including S. pyogenes (AE006586-6), S. pneumoniae (AF055720-1), Streptococcus thermophilus, Streptococcus suis (AF319974-1), S. gordonii (sgord_bvs_203), E. faecalis (gef_11370), L. lactis (AE006432), Lactobacillus rhamnosus (AF037091-2), and Lactobacillus fermentum (AB049598-1). Taken together with our results from S. mutans, Dps family proteins, as well as Dpr, may act as ferritin in lactic acid bacteria and contribute to the ability of these catalase- and heme-peroxidase-deficient bacteria to grow under air by limiting the iron-catalyzed Fenton reaction.
Acknowledgments
We are grateful to S. Yamasaki and K. Kimura of Tohoku University for their excellent assistance with ICP mass spectrometry and atomic absorption spectrometry. We also thank T. Sato for his help with the electron microscopy.
This work was supported in part by a Grant-in-Aid for Scientific Research (no. 12876016) from the Japan Society for the Promotion of Sciences (JSPS), the Mishima Kaiun Memorial Foundation, and the Casio Science Promotion Foundation. Y. Yamamoto is the recipient of a predoctoral fellowship from JSPS. Research support to L. B. Poole (NIH RO1 GM50389 and an AHA Established Investigatorship) and R. R. Hantgan (NSF MCB-9728122) is also acknowledged.
REFERENCES
- 1.Almiron, M., A. J. Link, D. Furlong, and R. Kolter. 1992. A novel DNA-binding protein with regulatory and protective roles in starved Escherichia coli. Genes Dev. 6:2646-2654. [DOI] [PubMed] [Google Scholar]
- 2.Andrews, S. C. 1998. Iron storage in bacteria. Adv. Microb. Physiol. 40:281-351. [DOI] [PubMed] [Google Scholar]
- 3.Auzat, I., S. Chapuy-Regaud, G. Le Bras, D. Dos Santos, A. D. Ogunniyi, I. Le Thomas, J. R. Garel, J. C. Paton, and M. C. Trombe. 1999. The NADH oxidase of Streptococcus pneumoniae: its involvement in competence and virulence. Mol. Microbiol. 34:1018-1028. [DOI] [PubMed] [Google Scholar]
- 4.Bailey, J. L. 1962. Techniques in protein chemistry, p. 294-295. Elsevier Scientific Publishing Co., Inc., New York, N.Y.
- 5.Beer, R. F. J., and I. W. Siezer. 1952. A spectrophotometric method for measuring the breakdown of hydrogen peroxide by catalase. J. Biol. Chem. 195:276-287. [PubMed] [Google Scholar]
- 6.Bozzi, M., G. Mignogna, S. Stefanini, D. Barra, C. Longhi, P. Valenti, and E. Chiancone. 1997. A novel non-heme iron-binding ferritin related to the DNA-binding proteins of the Dps family in Listeria innocua. J. Biol. Chem. 272:3259-3265. [DOI] [PubMed] [Google Scholar]
- 7.Brentjens, R. J., M. Ketterer, M. A. Apicella, and S. M. Spinola. 1996. Fine tangled pili expressed by Haemophilus ducreyi are a novel class of pili. J. Bacteriol. 178:808-816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen, L., and J. D. Helmann. 1995. Bacillus subtilis MrgA is a Dps (PexB) homologue: evidence for metalloregulation of an oxidative-stress gene. Mol. Microbiol. 18:295-300. [DOI] [PubMed] [Google Scholar]
- 9.Chung, M. C. 1985. A specific iron stain for iron-binding proteins in polyacrylamide gels: application to transferrin and lactoferrin. Anal. Biochem. 148:498-502. [DOI] [PubMed] [Google Scholar]
- 10.Copeland, R. A. 1994. Methods for protein analysis. Chapman and Hall, London, United Kingdom.
- 11.Dundon, W. G., A. Polenghi, G. Del Guidice, R. Rappuoli, and C. Montecucco. 2001. Neutrophil-activating protein (HP-NAP) versus ferritin (Pfr): comparison of synthesis in Helicobacter pylori. FEMS Microbiol. Lett. 199:143-149. [DOI] [PubMed] [Google Scholar]
- 12.Frenkiel-Krispin, D., S. Levin-Zaidman, E. Shimoni, S. G. Wolf, E. J. Wachtel, T. Arad, S. E. Finkel, R. Kolter, and A. Minsky. 2001. Regulated phase transitions of bacterial chromatin: a non-enzymatic pathway for generic DNA protection. EMBO J. 20:1184-1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gibson, C. M., and M. G. Caparon. 1996. Insertional inactivation of Streptococcus pyogenes sod suggests that prtF is regulated in response to a superoxide signal. J. Bacteriol. 178:4688-4695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gibson, C. M., T. C. Mallett, A. Claiborne, and M. G. Caparon. 2000. Contribution of NADH oxidase to aerobic metabolism of Streptococcus pyogenes. J. Bacteriol. 182:448-455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grant, R. A., D. J. Filman, S. E. Finkel, R. Kolter, and J. M. Hogle. 1998. The crystal structure of Dps, a ferritin homolog that binds and protects DNA. Nat. Struct. Biol. 5:294-303. [DOI] [PubMed] [Google Scholar]
- 15a.Grimaud, R., B. Ezraty, J. K. Mitchell, D. Lafitte, C. Briand, P. J. Derrick, and F. Barras. 2001. Repair of oxidized proteins. Identification of a new methionine sulfoxide reductase. J. Biol. Chem. 276:48915-48920. [DOI] [PubMed] [Google Scholar]
- 16.Halliwell, B., and J. M. Gutteridge. 1981. Formation of a thiobarbituric acid reactive substance from deoxyribose in the presence of iron salts. FEBS Lett. 128:347-352. [DOI] [PubMed] [Google Scholar]
- 17.Hantgan, R. R., J. V. Braaten, and M. Rocco. 1993. Dynamic light scattering studies of α2b β3 solution conformation. Biochemistry 32:3935-3941. [DOI] [PubMed] [Google Scholar]
- 18.Hebraud, M., and J. Guzzo. 2000. The main cold shock protein of Listeria monocytogenes belongs to the family of ferritin-like proteins. FEMS Microbiol. Lett. 190:29-34. [DOI] [PubMed] [Google Scholar]
- 19.Higuchi, M., M. Shimada, Y. Yamamoto, T. Hayashi, T. Koga, and Y. Kamio. 1993. Identification of two distinct NADH oxidases corresponding to H2O2-forming oxidase and H2O-forming oxidase induced in Streptococcus mutans. J. Gen. Microbiol. 139:2343-2351. [DOI] [PubMed] [Google Scholar]
- 20.Higuchi, M., Y. Yamamoto, L. B. Poole, M. Shimada, Y. Sato, N. Takahashi, and Y. Kamio. 1999. Functions of two types of NADH oxidases in energy metabolism and oxidative stress of Streptococcus mutans. J. Bacteriol. 181:5940-5947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Higuchi, M., Y. Yamamoto, and Y. Kamio. 2000. Molecular biology of oxygen tolerance in lactic acid bacteria: functions of NADH oxidases and Dpr in oxidative stress. J. Biosci. Bioeng. 90:484-493. [PubMed] [Google Scholar]
- 22.Ilari, A., S. Stefanini, E. Chiancone, and D. Tsernoglou. 2000. The dodecameric ferritin from Listeria innocua contains a novel intersubunit iron-binding site. Nat. Struct. Biol. 7:38-43. [DOI] [PubMed] [Google Scholar]
- 23.Johnson, C. S., and D. A. Gabriel. 1981. Laser light scattering, p. 177-272. In J. E. Bell (ed.), Spectroscopy in biochemistry, vol. II. CRC Press, Boca Raton, Fla. [Google Scholar]
- 24.Johnson, M., J. J. Correia, D. A. Yphantis, and H. Halvorson. 1981. Analysis of data from the analytical ultracentrifuge by nonlinear least-squares techniques. Biophys. J. 36:575-588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Keyer, K., and J. A. Imlay. 1996. Superoxide accelerates DNA damage by elevating free-iron levels. Proc. Natl. Acad. Sci. USA 93:13635-13640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kono, Y., and I. Fridovich. 1983. Isolation and characterization of the pseudocatalase of Lactobacillus plantarum. J. Biol. Chem. 258:6015-6019. [PubMed] [Google Scholar]
- 27.Kono, Y., and I. Fridovich. 1983. Functional significance of manganese catalase in Lactobacillus plantarum. J. Bacteriol. 155:742-746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680-685. [DOI] [PubMed] [Google Scholar]
- 29.Laue, T. M., B. D. Shah, T. M. Ridgeway, and S. L. Pelletier. 1992. Computer-aided interpretation of analytical sedimentation data for proteins, p. 90-125. In S. E. Harding, A. J. Rowe, and J. C. Horton (ed.), Analytical ultracentrifugation in biochemistry and polymer science. The Royal Society of Chemistry, Cambridge, United Kingdom.
- 30.Lowry, O. H., N. J. Rosebrough, A. L. Farr, and R. J. Randall. 1951. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 193:265-275. [PubMed] [Google Scholar]
- 31.Martinez, A., and R. Kolter. 1997. Protection of DNA during oxidative stress by the nonspecific DNA-binding protein Dps. J. Bacteriol. 179:5188-5194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McCord, J. M., and I. Fridovich. 1969. Superoxide dismutase. An enzymatic function for erythrocuprein (hemocuprein). J. Biol. Chem. 224:6049-6055. [PubMed] [Google Scholar]
- 33.Nakayama, K. 1992. Nucleotide sequence of Streptococcus mutans superoxide dismutase gene and isolation of insertion mutants. J. Bacteriol. 174:4928-4934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pena, M. M., W. Burkhart, and G. S. Bullerjahn. 1995. Purification and characterization of a Synechococcus sp. strain PCC 7942 polypeptide structurally similar to the stress-induced Dps/PexB protein of Escherichia coli. Arch. Microbiol. 163:337-344. [DOI] [PubMed] [Google Scholar]
- 35.Pena, M. M., and G. S. Bullerjahn. 1995. The DpsA protein of Synechococcus sp. strain PCC7942 is a DNA-binding hemoprotein. Linkage of the Dps and bacterioferritin protein families. J. Biol. Chem. 270:22478-22482. [DOI] [PubMed] [Google Scholar]
- 36.Poole, L. B., M. Higuchi, M. Shimada, M. L. Calzi, and Y. Kamio. 2000. Streptococcus mutans H2O2-forming NADH oxidase is an alkyl hydroperoxide reductase protein. Free. Radic. Biol. Med. 28:108-120. [DOI] [PubMed] [Google Scholar]
- 37.Poole, L. B., A. Godzik, A. Nayeem, and J. D. Schmitt. 2000. AhpF can be dissected into two functional units: tandem repeats of two thioredoxin-like folds in the N-terminus mediate electron transfer from the thioredoxin reductase-like C-terminus to AhpC. Biochemistry 39:6602-6615. [DOI] [PubMed] [Google Scholar]
- 38.Poyart, C., E. Pellegrini, O. Gaillot, C. Boumaila, M. Baptista, and P. Trieu-Cuot. 2001. Contribution of Mn-cofactored superoxide dismutase (SodA) to the virulence of Streptococcus agalactiae. Infect. Immun. 69:5098-5106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Price, D., and J. G. Joshi. 1982. Ferritin: a zinc detoxicant and a zinc ion donor. Proc. Natl. Acad. Sci. USA 79:3116-3119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ross, R. P., and A. Claiborne. 1991. Cloning, sequence and overexpression of NADH peroxidase from Streptococcus faecalis 10C1. Structural relationship with the flavoprotein disulfide reductases. J. Mol. Biol. 221:857-871. [DOI] [PubMed] [Google Scholar]
- 41.Sanders, J. W., K. J. Leenhouts, A. J. Haandrikman, G. Venema, and J. Kok. 1995. Stress response in Lactococcus lactis: cloning, expression analysis, and mutation of the lactococcal superoxide dismutase gene. J. Bacteriol. 177:5254-5260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schmidt, H. L., W. Stocklein, J. Danzer, P. Kirch, and B. Limbach. 1986. Isolation and properties of an H2O-forming NADH oxidase from Streptococcus faecalis. Eur. J. Biochem. 156:149-155. [DOI] [PubMed] [Google Scholar]
- 43.Spotorno, B., L. Piccinini, G. Tassara, C. Ruggiero, M. Nardini, F. Molina, and M. Rocco. 1997. BEAMS (BEAds Modelling System): a set of computer programs for the generation, the visualization and the computation of the hydrodynamic and conformational properties of bead models of proteins. Eur. Biophys. J. 25:373-384. [Google Scholar]
- 44.Srinivasan, C., A. Liba, J. A. Imlay, J. S. Valentine, and E. B. Gralla. 2000. Yeast lacking superoxide dismutase(s) show elevated levels of “free iron” as measured by whole cell electron paramagnetic resonance. J. Biol. Chem. 275:29187-29192. [DOI] [PubMed] [Google Scholar]
- 45.Storz, G., F. S. Jacobson, L. A. Tartaglia, R. W. Morgan, L. A. Silveira, and B. N. Ames. 1989. An alkyl hydroperoxide reductase induced by oxidative stress in Salmonella typhimurium and Escherichia coli: genetic characterization and cloning of ahp. J. Bacteriol. 171:2049-2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Storz, G., and J. A. Imlay. 1999. Oxidative stress. Curr. Opin. Microbiol. 2:188-194. [DOI] [PubMed] [Google Scholar]
- 47.Sun, S., and N. D. Chasteen. 1992. Ferroxidase kinetics of horse spleen apo-ferritin. J. Biol. Chem. 267:25160-25166. [PubMed] [Google Scholar]
- 48.Tonello, F., W. G. Dundon, B. Satin, M. Molinari, G. Tognon, G. Grandi, G. Del Giudice, R. Rappuoli, and C. Montecucco. 1999. The Helicobacter pylori neutrophil-activating protein is an iron-binding protein with dodecameric structure. Mol. Microbiol. 34:238-246. [DOI] [PubMed] [Google Scholar]
- 49.Touati, D., M. Jacques, B. Tardat, L. Bouchard, and S. Despied. 1995. Lethal oxidative damage and mutagenesis are generated by iron in Δfur mutants of Escherichia coli: protective role of superoxide dismutase. J. Bacteriol. 177:2305-2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Touati, D. 2000. Iron and oxidative stress in bacteria. Arch. Biochem. Biophys. 373:1-6. [DOI] [PubMed] [Google Scholar]
- 51.Van Holde, K. E. 1971. Physical biochemistry, 2nd ed., p. 110-136. Prentice-Hall, Inc., Englewood Cliffs, N.J.
- 52.Wai, S. N., K. Nakayama, K. Umene, T. Moriya, and K. Amako. 1996. Construction of a ferritin-deficient mutant of Campylobacter jejuni: contribution of ferritin to iron storage and protection against oxidative stress. Mol. Microbiol. 20:1127-1134. [DOI] [PubMed] [Google Scholar]
- 53.Wetherell, J. R., Jr., and A. S. Bleiweis. 1975. Antigens of Streptococcus mutans: characterization of a polysaccharide antigen from walls of strain GS-5. Infect. Immun. 12:1341-1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wolf, S. G., D. Frenkiel, T. Arad, S. E. Finkel, R. Kolter, and A. Minsky. 1999. DNA protection by stress-induced biocrystallization. Nature 400:83-85. [DOI] [PubMed] [Google Scholar]
- 55.Yamamoto, Y., M. Higuchi, L. B. Poole, and Y. Kamio. 2000. Role of the dpr product in oxygen tolerance in Streptococcus mutans. J. Bacteriol. 182:3740-3747. [DOI] [PMC free article] [PubMed] [Google Scholar]