

Abstract
The phlACBD genes responsible for the biosynthesis of the antifungal metabolite 2,4-diacetylphloroglucinol (PHL) by the biocontrol strain Pseudomonas fluorescens F113 are regulated at the transcriptional level by the pathway-specific repressor PhlF. Strong evidence suggests that this regulation occurs mainly in the early logarithmic phase of growth. First, the expression of the phlF gene is relatively high between 3 and 13 h of growth and relatively low thereafter, with the phlACBD operon following an opposite expression profile. Second, the kinetics of PHL biosynthesis are specifically altered in the logarithmic phase in a P. fluorescens F113 phlF mutant. The phlA-phlF intergenic region presents a complex organization in that phlACBD is transcribed from a σ70 RNA polymerase-dependent promoter that is likely to overlap the promoter of the divergently transcribed phlF gene. The repression by PhlF is due to its interaction with an inverted repeated sequence, phO, located downstream of the phlA transcriptional start site. Cross-linking experiments indicate that PhlF can dimerize in solution, and thus PhlF may bind phO as a dimer or higher-order complex. Furthermore, it is now demonstrated that certain regulators of PHL synthesis act by modulating PhlF binding to phO. PHL, which has previously been shown to be an autoinducer of PHL biosynthesis, interacts with PhlF to destabilize the PhlF-phO complex. Conversely, the PhlF-phO complex is stabilized by the presence of salicylate, which has been shown to be an inhibitor of phlA expression.
2,4-Diacetylphloroglucinol (PHL) is a phenolic molecule produced by many fluorescent pseudomonad bacteria (3, 23, 42, 47, 55, 56, 61, 63). It has antifungal (28, 55, 58), antibacterial (28), antihelminthic (7, 21), and phytotoxic (23, 48) activities. PHL is a polyketide synthesized by condensation of three molecules of acetyl coenzyme A with one molecule of malonyl coenzyme A to produce the precursor monoacetylphloroglucinol, which is subsequently transacetylated to generate PHL (37, 55).
The genes involved in the biosynthesis of PHL have been cloned and sequenced from a number of Pseudomonas strains (3, 4, 11, 17, 37, 55, 61). Genetic and sequence data show that the phl locus is comprised of a number of transcriptional units. The structural genes, phlA, phlC, phlB, and phlD, are transcribed as a single operon (phlACBD), with the putative permease gene, phlE, probably transcribed from its own promoter further downstream. The transcriptional repressor, phlF, is located upstream of the phlACBD operon and is transcribed in the opposite direction (4, 11, 54). This organization is common to many polyketide biosynthetic loci (14, 41, 43).
There is evidence that PHL production is influenced by environmental and nutritional signals as well as by transcriptional and posttranscriptional regulatory systems. The environmentally responsive GacS/GacA two-component regulatory system is required for production of PHL, with mutation of either component completely abolishing PHL biosynthesis (10, 11, 15). Transcriptional studies have demonstrated that the GacS/GacA system positively regulates expression of both phlACBD and phlF (11, 20). At another level, sigma factors may also play a role in regulation at the phl locus. It has been shown that overexpression of the housekeeping sigma factor rpoD, encoding σ70, or mutation of the stationary-phase sigma factor rpoS increased PHL production (38, 53, 64).
Pathway-specific regulators are also involved in regulation of PHL biosynthesis. The phl biosynthetic gene cluster is negatively regulated by the repressor PhlF and positively modulated by the genetically linked phlH gene, which encodes a putative regulator (11, 54). Posttranscriptional control via homologues of the Escherichia coli CsrA and CsrB molecules also plays a role in controlling PHL production. This system, which comprises a translational repressor protein (CsrA/RsmA) and a regulatory RNA (CsrB/RsmB), has been described in a number of gram-negative bacteria (6, 30-33, 36, 50). Recently, we reported the characterization of the prrB gene encoding a regulatory RNA that is a functional homologue of CsrB/RsmB. Overexpression of prrB RNA restores PHL production in gacA and gacS mutants and leads to overproduction of PHL in a wild-type P. fluorescens strain (1).
It has been established that PHL is an autoregulator, positively influencing its own production. Furthermore, salicylate and other secondary metabolites (fusaric acid and pyoluteorin) have a negative effect on PHL production. In both cases, an intact phlF gene is required, suggesting that PHL and salicylate act via PhlF (54). Thus, the interaction of PhlF with coregulators and the phlACBD promoter plays an important role in regulating PHL production. This paper describes the characterization of the promoter of the phlACBD gene cluster and identification of the PhlF binding site (operator). It is also established that the autoinducer (PHL) and the corepressor (salicylate) interact physically with the PhlF repressor to modulate PhlF binding to its operator sequence.
MATERIALS AND METHODS
Bacterial strains, plasmids, and culture conditions.
The bacterial strains and plasmids used in this study are listed in Table 1. P. fluorescens F113 and derivatives were routinely grown at 28°C in Luria-Bertani (LB) broth and in minimal medium (SA) with sucrose (50 mM) and asparagine (17.5 mM), respectively, as sole carbon and nitrogen sources. SA medium was supplemented with 100 μM FeCl3. E. coli strains were grown at 37°C in LB broth (52). Antibiotics were used at the following concentrations: for P. fluorescens, tetracycline, 75 μg/ml, and chloramphenicol, 250 μg/ml; for E. coli, tetracycline, 25 μg/ml, chloramphenicol, 30 μg/ml, and ampicillin, 100 μg/ml.
TABLE 1.
Strains and plasmids used in this study
| Strain or plasmid | Description | Reference |
|---|---|---|
| Pseudomonas fluorescens F113 | Wild-type; HCM+ Prt+ Lac− | 55 |
| Escherichia coli DH5α | φ80 lacZΔM15 Δ(lacZYA-argF)U169 hsdR17 recA1 endA1 thi-1 | 52 |
| Plasmids | ||
| pMP190 | Promoterless lacZ vector, IncQ Cmr Smr | 57 |
| pMP220 | Promoterless lacZ vector, IncP Tcr | 57 |
| pQE-60.27 | 700-bp NcoI-BglII fragment containing PCR-amplified phlF in pQE-60 | 12 |
| pCU107 | phACBD-lacZ, 5-kb EcoRI fragment from pCU203 in pMP220, Tcr | 12 |
| pCU109 | phlF-lacZ, 1.8-kb SalI-KpnI fragment from pCU203 in pMP190, Cmr | 12 |
Recombinant DNA techniques.
Small-scale and large-scale plasmid DNA isolations were performed using Qiagen plasmid mini and maxi kits, respectively, according to the manufacturer's specifications (Qiagen, Inc.). Plasmids were introduced into E. coli and P. fluorescens by electroporation (16) or mobilized into P. fluorescens by triparental matings using the helper plasmid pRK2013 (19).
Construction of transcriptional fusions and β-galactosidase assays.
To characterize the transcriptional activity of the phlACBD and phlF genes, we used pCU107 and pCU109 constructs bearing lacZ transcriptionally fused with phlACBD and phlF, respectively (11, 12). The kinetics of expression of biosynthetic fusions in different backgrounds were monitored by performing β-galactosidase assays (40). P. fluorescens F113 cells were grown with shaking (150 rpm) in 100-ml flasks containing 25 ml of medium. All measurements were performed in triplicate. For time course experiments, samples were taken from cultures at the indicated times.
RNA techniques.
Total RNA was isolated from 7 × 109 cells of wild-type P. fluorescens F113 grown for 18 h on LB broth with the RNeasy total RNA isolation kit according to the manufacturer's specifications (Qiagen, Inc.). RNA integrity was checked by electrophoresis in 1.2% (wt/vol) agarose gels containing 0.66% (vol/vol) formaldehyde. Primer extension was performed by the method of Pujic et al. (44) with the following modifications: 250 pmol of primer PE-A, which hybridized 71 nucleotides downstream of the ATG translation start codon of phlA, was end labeled using T4 polynucleotide kinase and [γ32P]ATP. Labeled primers and total RNA were hybridized at 65°C for 5 min and allowed to cool at room temperature for 1.5 h. Reverse transcription was performed at 42°C for 1 h using avian myeloblastosis virus reverse transcriptase. Sequence reactions were performed with the same primers using P. fluorescens F113 plasmid DNA as a template.
Purification of PhlF.
We have previously reported the purification of a histidine-tagged PhlF repressor (23.6 kDa) (12). A protein of smaller size (18 kDa) invariably copurified with the histidine-tagged PhlF protein. This 18-kDa protein is thought to be a derivative of PhlF truncated by approximately 50 amino acids at its N terminus. Pure PhlF protein was needed in this study for two reasons: first, to avoid any possible competition between the two proteins in the binding assays, especially when analyzing the influence of PHL on PhlF binding activity, and second, to avoid any interference between the functional full-sized PhlF and the truncated form in cross-linking experiments. For these reasons we optimized the purification conditions to purify only the full-length PhlF. A number of parameters were modified relative to the previous protocol (12). The procedure is detailed below, the major modifications being the use of a different strain of E. coli and different inducing conditions.
The PhlF-overexpressing plasmid pQE-60.27 was introduced into E. coli DH5α by electroporation. Then 300 ml of LB broth containing ampicillin (100 mg/ml) was inoculated at an absorbance at 600 nm of 0.1 by an overnight culture of DH5α/pQE-60.27 and grown at 37°C with shaking (200 rpm) to an optical density at 600 nm (OD600) of 0.5. Overexpression of phlF was induced by addition of 0.2 mM isopropylthiogalactopyranoside (IPTG). After incubation at 30°C for 4.5 h, the cells were harvested by centrifugation (5,000 × g) for 15 min at 4°C, washed twice in LB broth, and resuspended in 4 ml of lysis buffer (50 mM Na2PO3, 300 mM NaCl, 10 mM imidazole, 40 mg of lysozyme). Cells were then incubated on ice for 15 min and passed twice though a French press. Cell debris was removed from the crude extract by centrifugation (10,000 × g) for 40 min at 4°C, and the 6xHis-tagged PhlF protein was purified under nondenaturing conditions using Ni-nitrilotriacetic acid (Ni-NTA) spin columns, following the manufacturer's protocol (Qiagen, Inc.). The purified PhlF-6xHis was then dialyzed overnight at 4°C against 1× TE (Tris-EDTA) buffer.
Gel retardation.
DNA fragment 7EH of 145 bp covers the common sequence between 2.9 and 7.U used by Delany et al. (12). This fragment of the intergenic region was amplified using specific primers 7A5E and 7A3H with pCU109 as the DNA template. Four double-stranded oligonucleotides, p12d, p11d, p10d, and p9d, of 35 bp each were obtained by annealing oligonucleotides p12 and p12c; p11 and p11c; p10 and p10c, and p9 and p9c, respectively. Each double-stranded oligonucleotide overlaps the oligonucleotides adjacent to it by 4 bp. The four oligonucleotides cover the full length of the overlap between probes 2.9 and 7.U. The probes were 5′ end labeled with [γ32P]CTP using polynucleotide kinase (NEB). The labeled probes were extracted once with phenol-chloroform and ethanol precipitated. The sequences of the oligonucleotides used for PCR amplification and gel retardation will be provided upon request.
Gel shift assays were performed as described by Liu et al. (34). The binding reaction was performed in 40 μl of binding buffer (12 mM HEPES-NaOH [pH 7.9], 4 mM Tris-HCl [pH 7.9], 75 mM KCl, 10 mM MgCl2, 5 mM CaCl2, 1.0 mM dithiothreitol) containing 1 μg of salmon sperm DNA, 50 ng of poly(dI-dC), 2 μg of bovine serum albumin, 1 μl of probe, and 3 μg of purified PhlF-6xHis protein. The reaction mixture was incubated for 20 min at room temperature and directly subjected to polyacrylamide gel electrophoresis (PAGE) in a 5% (wt/vol) polyacrylamide gel containing 2.5% (vol/vol) glycerol in TAE (Tris-acetate-EDTA) buffer (40 mM Tris-acetate [pH 7.5], 2 mM EDTA). In general, electrophoresis was performed at 110 V for 2.5 h. When probes p12d, p11d, p10d, and p9d were used, electrophoresis was performed at 80 V for 1.5 h. Gels were dried and examined by autoradiography with X-ray film (Biomax; Kodak).
To analyze the effects of PHL and salicylate on PhlF binding activity, a set of solutions with different concentrations of the two metabolites were prepared in ethanol (70%). To investigate whether ethanol interferes with PhlF activity, a mobility shift assay was monitored in the presence of different concentrations of ethanol. We found that PhlF binding activity was not altered by ethanol concentrations up to 6% (data not shown). Note that the highest concentration of ethanol routinely used in binding assays with PHL and salicylate was less than 4%. As a control to determine the specificity of the effect of PHL on PhlF binding, a control band shift was performed. This used the cI2009 repressor of lactococcal bacteriophage TUC2009, which represses transcription from the early lytic promoter PR located in the cI2009-CRO2009 intergenic region (27, 59).
Footprinting.
Chemical footprinting of the PhlF-DNA complex was performed using a combined gel retardation-1,10-phenanthroline-copper ion footprinting procedure (24). Briefly, labeled probe 7EH was digested with EcoRI, extracted once with phenol-chloroform, and ethanol precipitated. Digested probe was resuspended in water, and gel retardation was then performed as described above. The full retardation gel was soaked in 200 ml of 50 mM Tris-HCl (pH 8.0) and treated for 10 min at room temperature with 10 ml of solution A (2 mM 1,10-phenanthroline, 0.45 mM CuSO4) and 10 ml of solution B (58 mM 3-mercaptopropionic acid). The reaction was then quenched by addition of 20 ml of 28 mM 2,9-dimethylphenanthroline for 2 min. The gel was rinsed with distilled water and exposed to X-ray film for 30 min at room temperature. Bands of interest were cut and eluted overnight at 37°C in 500 mM ammonium acetate-1 mM EDTA. Eluted DNA was ethanol precipitated and resuspended in 80% (vol/vol) formaldehyde-10 mM NaOH-1.0 mM EDTA-0.1% (wt/vol) bromophenol blue. The DNA sample was run on an 8% (wt/vol) polyacrylamide sequencing gel.
Glutaraldehyde cross-linking of PhlF.
The glutaraldehyde cross-linking procedure was modified from Derré et al. (13). PhlF (3 μg) was incubated with 10 mM cross-linking reagent glutaraldehyde in 40 μl of a reaction mixture containing 50 mM NaH2PO4, 50 mM NaCl, and 10% (vol/vol) glycerol. After incubation for 1 h at 30°C, the reaction was stopped by the addition sodium dodecyl sulfate (SDS) loading buffer. The samples were boiled and analyzed by SDS-PAGE on 10% (wt/vol) polyacrylamide gels (25). The gel was stained with Coomassie blue.
RESULTS
Kinetics of phlA and phlF gene expression.
Delany et al. (12) established that wild-type P. fluorescens F113 does not produce PHL during early stages of growth. In a phlF mutant, however, the kinetics of PHL production were altered, such that PHL was produced earlier in the growth cycle. From these data, it was concluded that PhlF acts as a repressor of PHL production during the early log phase (12).
To further investigate the mechanism of regulation of PHL biosynthesis by PhlF, we performed a time course experiment monitoring the expression of phlF and phlACBD using transcriptional fusions. P. fluorescens F113 strains containing the phlACBD-lacZ transcriptional fusion carried by pCU107 and the phlF-lacZ transcriptional fusion carried by pCU109 were grown independently on SA medium and analyzed by Miller assays (Fig. 1). Expression of the phlACBD-lacZ construct was low between 0 and 12 h and reached its maximum at 13 to 20 h, when the culture was in stationary phase. This correlates with the peak of PHL production (12). In contrast, expression of the phlF-lacZ construct was highest earlier in the growth cycle, with maximal expression during logarithmic growth (3 to 13 h). These results are consistent with the hypothesis that the low transcriptional activity of phlACBD in the early logarithmic phase is a consequence of high levels of the PhlF repressor.
FIG. 1.

Time course of expression of the phl operon. The transcriptional fusions phlACBD-lacZ (pCU107) and phlF-lacZ (pCU109) were introduced into P. fluorescens F113. Cultures were grown on minimal medium, and absorbance was measured at 600 nm (solid circles, phlF-lacZ; open circles, phlACBD-lacZ). Expression of the fusions was assessed by measuring levels of β-galactosidase. Grey shading represents phlF-lacZ expression, and dark shading represents phlACBD-lacZ expression. Triplicate cultures were assayed. The standard deviations are represented with error bars.
Characterization of the phlA promoter and analysis of the organization of the phlA-phlF intergenic region.
To better understand how PhlF influences the transcriptional activity from the phlA promoter, we decided to characterize the transcriptional start of the phlACBD operon. Total RNA isolated from wild-type P. fluorescens F113 grown for 18 h was subjected to primer extension analysis. This analysis revealed only one specific transcript starting 363 bp upstream of the translational start (Fig. 2). A putative σ70 −10 element (TAGGAT) and a perfectly conserved −35 element (TTGACA) were identified (Fig. 3). The sequences of the two promoter elements are perfectly conserved in two other strains of P. fluorescens, Q2-87 (U41818) and CHA0 (AF207529). Similar promoter organization has already been reported for a number of genes in Pseudomonas aeruginosa (35, 39).
FIG. 2.

Primer extension analysis mapping the phlA transcriptional start site. Primer extension (PE) on total RNA from P. fluorescens F113 was performed using primer PE-A, which maps inside the phlA coding region. Plasmid DNA was sequenced using the same primer. The gel shows the only primer extension stop detected by this analysis.
FIG. 3.

Sequence organization of the phlA-phlF intergenic region. phlA and phlF indicate the start of the phlA and phlF open reading frames, respectively. Start indicates the transcriptional start of the phlACBD operon. The putative σ70 −10 and −35 elements for phlA are underlined. The PhlF binding site, phO, is indicated, with its two inverted repeated sequences shown by arrows. The three regions protected by the PhlF footprint are boxed. From 5′ to 3′, these are box 1, box 2, and box 3, as referred to in the text. The CCAAT (ATTGG) motif of low-temperature-induced genes is underlined. The inverted repeated sequences flanking the −10 element are indicated with arrows. The accession number for this sequence is AF497760.
Detailed analysis of the P. fluorescens phlA-phlF intergenic region revealed the presence of many inverted repeat sequences. Interestingly, in both strain F113 and strain Q2-87, the −10 element is located between inverted repeat sequences (Fig. 3). This could indicate that access of RNA polymerase to the −10 box is regulated. The sequence CCAAT, which represents a common motif of low-temperature-induced genes in E. coli and other bacteria (26, 45, 51), is perfectly conserved at position −40 from the transcriptional start in strains F113 and Q2-87 but not in strain CHA0. The sequence data suggest that the phlA gene is transcribed from a σ70-dependent promoter in all three Pseudomonas strains and indicate that the phl loci in strains F113 and Q2-87 are structurally more similar to each other than to the phl locus in strain CHA0. This is consistent with a recent phylogenetic analysis which established that P. fluorescens strains F113 and Q2-87 are more closely related to each other than to P. fluorescens CHA0 (46). Previously, it has been reported that PHL accumulates to higher levels at reduced temperatures, but it remains to be determined whether the CCAAT motif is involved in this phenomenon (55).
Identification of the PhlF binding site.
Previously, we established that PhlF binds to the phlA-phlF intergenic region and used gel mobility shift analyses to localize the binding site to a 100-bp sequence (12). In order to define the precise PhlF binding site, a more comprehensive gel shift analysis was undertaken using shorter DNA probes (Fig. 4). Fragments 2.9 and 7.U were previously determined to contain the PhlF binding site. A DNA fragment, 7EH, that spanned the overlap between these probes was constructed and analyzed by gel retardation (Fig. 5). PhlF-6xHis, used in the mobility shift assays, was purified according to the modified protocol described in Materials and Methods. It is clearly seen that the migration of fragment 7EH was retarded upon addition of PhlF. No retarded band was seen when a large excess of unlabeled 7EH DNA fragment was added along with PhlF protein to the 7EH labeled fragment.
FIG. 4.

Map of probes used in the mobility shift assays in this study. The names of probes are presented on the left of the figure. Binding of PhlF protein to the probes is indicated as + or −. IGR, intergenic region. The 5′ ends of the phlA and phlF ORFs are indicated
FIG. 5.

Identification of the PhlF binding site. Probe 7EH (2 ng) was analyzed for reduced mobility in a native gel in the presence of 3 μg of purified PhlF. Lane 1 contains labeled 7EH probe; lane 2 contains labeled 7EH probe and 3 μg of PhlF-6xHis; lane 3 contains labeled 7EH probe, 3 μg of PhlF-6xHis, and an excess of unlabeled 7EH probe.
To more precisely localize the PhlF binding site, we designed internal double-stranded primers, p12d, p11d, p10d, and p9d, spanning most of the 7EH region (Fig. 4). Mobility shift experiments established that only p12d was able to bind PhlF (data not shown). Probe p12d contains an inverted repeated sequence of eight bases separated by eight bases that we designate the phl operator, phO (Fig. 3). It was noted that the strength of binding of PhlF to probe 7EH appeared to be stronger than to probe p12d (data not shown).
To address the possibility that weaker interactions between nucleotides within probe 7EH and PhlF stabilize or enhance binding of PhlF to the operator sequence phO, the PhlF footprint on probe 7EH was identified (Fig. 6). It was found that binding of PhlF to 7EH protected the three regions of the probe, designated boxes 1, 2, and 3. Box 1 is a 30-bp stretch of DNA that contains the inverted repeat sequence that has been designated the phO operator (Fig. 3). Boxes 2 and 3, 9 and 7 bp, respectively, are shorter stretches of DNA (Fig. 3). Box 2 is contained within probe p10d, and box 3 is contained within probe p9d, and since neither of these probes can bind PhlF, it is concluded that boxes 2 and 3 do not constitute primary PhlF binding sites. Interaction of PhlF with these boxes may, however, contribute to the stabilization of PhlF bound to the phO operator (box 1). Further support for the premise that phO constitutes the PhlF binding site came from analysis of mutations between the inverted repeat regions. Any sequence changes in this region abolished the ability of PhlF to bind the phO operator (data not shown).
FIG. 6.
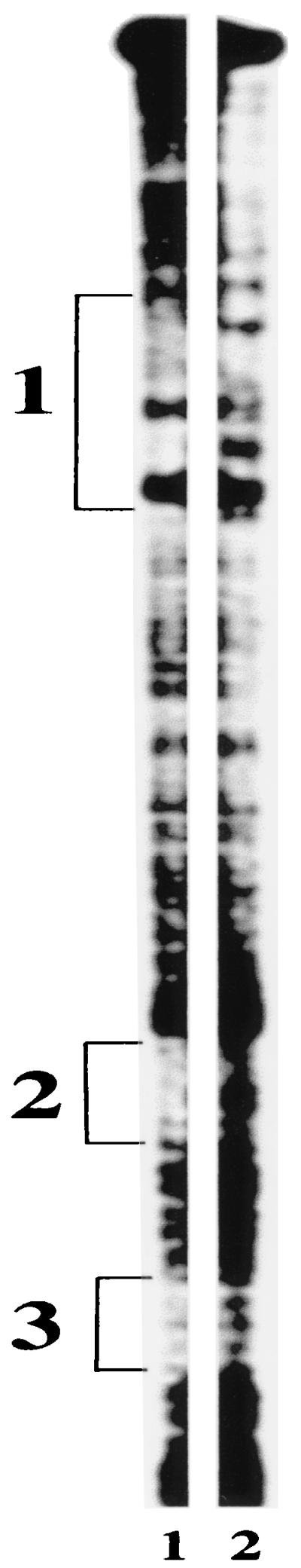
Footprint of PhlF. DNA probe 7EH that was bound by PhlF (lane 1) or unbound (lane 2) was treated with a chemical footprinting agent to locate the PhlF footprint. Three protected regions of the probe were identified and labeled 1, 2, and 3. The positions of these protected regions are shown in Fig. 3.
By analogy to other DNA-binding proteins (13, 29), the structure of phO suggests that the PhlF repressor may be active as a dimer or even a multimer. Dimerization of the PhlF was investigated in vitro by chemical cross-linking with a bifunctional reagent, glutaraldehyde (13). It was found that un-cross-linked PhlF migrates at 30 kDa (theoretical mass of PhlF is 23.6 kDa) and that incubation of a PhlF monomer solution with glutaraldehyde generated an altered form of PhlF with reduced electrophoretic mobility (≈63 kDa) (Fig. 7). These results suggest that PhlF can dimerize in solution and may bind to the phO operator as a dimer. Although in vitro evidence of higher-order protein complexes was not obtained, it remains a possibility that the protein may bind DNA as a larger complex, for example, a tetramer.
FIG. 7.

PhlF forms a dimer in vitro. SDS-PAGE analysis of purified PhlF-6xHis samples (3 μg) without (lane 1) and with (lane 2) treatment with 10 mM glutaraldehyde. Midrange molecular size standards were loaded the left lane; sizes are shown in kilodaltons.
Effect of PHL and salicylate on PhlF binding activity.
Recently, it was reported that PHL induced and salicylate repressed phlA expression via PhlF (54). It was not established how these metabolites influenced PhlF activity. To determine whether PHL could modulate binding of PhlF to the phO operator, PHL was added in increasing concentrations to a binding mixture containing the radiolabeled probe 7EH and PhlF (Fig. 8A). It was found that PHL reduced PhlF binding in a concentration-dependent fashion. This suggests that there is a physical interaction between PHL and PhlF. This effect was independent of the order in which the components were added, indicating that PHL may dissociate preformed PhlF-phO complexes. An alternative explanation is that there may be an equilibrium between free and bound PhlF protein. These data demonstrate that autoinduction of phlACBD transcription by PHL operates by reducing binding of the repressor PhlF to its operator. The specificity of PHL for PhlF was demonstrated by performing a gel shift assay using the repressor protein cI2009 of phage Tuc2009 and a DNA fragment containing its binding site (59). cI2009 binding activity was not affected by 3 mM PHL (Fig. 8B).
FIG. 8.

(A) Impact of PHL on the electrophoretic mobility of the PhlF-7EH probe complex. Increasing amounts of PHL were added to the binding mixture containing 3 μg of PhlF-6xHis and 2 ng of radiolabeled 7EH probe. Pro, free 7EH probe. Samples 1 to 11 contain 0 mM, 0.05 mM, 0.1 mM, 0.2 mM, 0.25 mM, 0.3 mM, 0.4 mM, 0.75 mM, 1 mM, 2 mM, and 3 mM PHL, respectively. (B) Mobility shift assay using an unrelated system composed of repressor protein cI2009 of phage Tuc2009 and a DNA fragment (2880 to 3030) from the genetic switch of Tuc2009. Lanes: Pro, probe without repressor; 0, probe with repressor cI2009; 3 mM, probe with repressor cI2009 and PHL (3 mM).
Salicylate enhances the repression of phlA expression by PhlF in vivo (54). To investigate whether this is due to a direct interaction with the repressor, binding of PhlF to the phO operator in the presence and absence of salicylate (4 mM) was investigated. Band shift assays were performed over a range of PHL concentrations, and the relative concentrations of bound and unbound probe were determined (Fig. 9). It was found that the presence of salicylate results in longer preservation of the PhlF-probe complex even at high concentrations of PHL. The same effect of salicylate was observed independent of the sequence in which the components of the binding reaction were added. These data indicate that salicylate interacts physically with PhlF.
FIG. 9.

Salicylate enhances PhlF binding to phO. Binding reaction mixtures containing 2 ng of radiolabeled 7EH probe, 3 μg of PhlF-6xHis, and increasing amounts of PHL were prepared with or without 4 mM salicylate. Binding was measured by densitometry, and the relative PhlF binding activity in the presence and absence of salicylate was calculated for each concentration of PHL. This is presented as the natural log of the intensity of bound probe divided by the total intensity of the probe. Sal, salicylate.
DISCUSSION
Control of phlACBD expression by PhlF.
Previous studies reported that P. fluorescens does not produce PHL in early log phase (12, 54). In the present study, we have demonstrated that this is likely due to the high expression of the phlF repressor gene during this growth phase. This is supported by our previous finding that in the P. fluorescens F113 phlF mutant, PHL production is derepressed mainly in the early stage of growth (12). It is noteworthy, however, that even in the early stages of growth, when phlF expression is relatively high, phlACBD transcription is not completely absent. Although this indicates that there is not complete repression of phlACBD transcription by PhlF, PHL is not detected during this phase of growth. This is consistent with the view that PHL biosynthesis is regulated at more then one level (1).
To understand the mechanism by which PhlF represses phlA transcription, it was important to determine the transcriptional start of this gene and consequently to characterize its promoter. Our analysis suggests that the phlACBD operon is transcribed from a σ70-dependent promoter. This conclusion is supported by the fact that overexpression of the housekeeping sigma factor rpoD resulted in increased PHL production (53, 64). Many σ70-dependent promoters are regulated by transcriptional repressors that bind upstream of the transcriptional start site and impede binding of RNA polymerase. There are also examples, however, where repressors bind downstream of the transcription start site and function by interfering with promoter clearance (9). This is also the case with PhlF, which has its operator located at +140 to +169 relative to the phlA transcriptional start site.
Consistent with reports that downstream operators are less effective (50), repression by PhlF is not absolute. It is notable, however, that in addition to the phO operator, PhlF also interacts with two DNA regions closer to the transcription start site (Fig. 3 and Fig. 6). The length of these sequences (7 and 9 bp) and the fact that PhlF does not bind to oligonucleotides containing those sequences suggest these are not discrete PhlF binding sites. Rather, it is possible that PhlF bound at the phO operator interacts with these regions to form a more stable interaction. This suggests that binding of PhlF leads to significant structural reorganization of the phlA promoter, and this could also contribute to inhibition of transcription.
Some features of PhlF show similarities to those of the TetR repressor. First, the sequence and structure of the phl operator, phO, are very similar to those of the tetO operator located within the tetR-tetA intergenic region (2). Second, the putative helix-turn-helix motif present at the amino terminus of the PhlF protein (4, 12, 54) shows interesting similarities with the helix-turn-helix of TetR. Third, the active state of TetR is a homodimer (22), and we have shown in this study that PhlF forms a dimer in solution, suggesting that it may be active as a homodimer. Based on these similarities, it can be hypothesized that PhlF and TetR bind DNA in similar ways.
Mechanism of autoinduction.
Many biosynthetic or degradation pathways are regulated by positive or negative feedback (5, 8, 18, 60). Recently, Schnider-Keel and colleagues showed that addition of PHL to the growth medium induced both production of PHL and expression of a phlA′-′lacZ gene fusion (54). Autoinduction by PHL required the phlF gene, but it was not determined whether PHL itself or a breakdown product was required, nor whether PhlF was directly or indirectly involved in the process. Using defined components in vitro, we established that PHL interacts directly with PhlF to reduce its binding to the phO operator. Furthermore, we also determined that the corepressor salicylate also acts directly via PhlF, this time to enhance its binding to phO. Since corepression by salicylate occurs even in the presence of high concentrations of PHL, it is speculated that autoinducer and corepressor recognize different sites on the PhlF protein.
To better characterize the PhlF interaction with the two secondary metabolites, a more detailed analysis is needed. Based on current data, however, we suggest that PHL interacts physically with PhlF and probably induces a conformational change such that the affinity of the repressor for phO drops, resulting in separation of the complex. Salicylate would stabilize the general structure of the repressor and therefore result in longer preservation of the complex. The concentrations of PHL and salicylate required for in vitro assays are higher than the concentrations used in the physiological study reported by Schnider-Keel et al. (54). The reason for that is not known, but it can be speculated that under physiological conditions, the PHL and the salicylate concentrations inside the cells may be significantly higher than those in the medium. Alternatively, it may be that aggregates of PhlF protein, which cannot bind DNA, are still capable of binding PHL or salicylate in the in vitro assays.
Conclusion.
Biosynthesis of PHL is regulated at the transcriptional level by PhlF. The phlA-phlF intergenic region displays a complex organization in that phlA is transcribed from a σ70 RNA polymerase-dependent promoter that is likely to overlap the promoter of the divergently transcribed phlF gene. Repression by PhlF is due to its interaction with a specific sequence, phO, located downstream of the phlA promoter, suggesting that the repression may occur by inhibition of promoter clearance. This repression occurs only in the early log phase, after which the repressor is inefficient because of its interaction with the inducer, PHL. Salicylate can interact with PhlF to stabilize its interaction with the phlA promoter, leading to tighter repression of PHL production. Interaction of these secondary metabolites with PhlF may contribute to the complex regulation of PHL biosynthesis. Further work is required to determine how environmental signals and physiological cues combine to control PHL production.
Acknowledgments
We thank members of the BIOMERIT Research Centre for valuable discussions, Claire Adams and Ultan Walsh for critical reading of the manuscript, Stephen Leach for the gift of the c12009 repressor and its specific probe, and Liam Burgess and Pat Higgins for advice and technical assistance.
This work was supported in part by grants awarded by the Higher Education Authority of Ireland Enterprise Ireland (SC/98/306 and SC/98/261), and the European Commission (BIO4-CT96-0027, BIO4-CT96-0181, FMRX-CT96-0039, and BIO4-CT98-0254).
REFERENCES
- 1.Aarons, S., A. Abbas, C. Adams, A. Fenton, and F. O'Gara. 2000. A regulatory RNA (prrB RNA) modulates expression of secondary metabolite genes in Pseudomonas fluorescens F113. J. Bacteriol. 182:3913-3919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Backes, H., C. Berens, V. Helbl, S. Walter, F. X. Schmid, and W. Hillen. 1997. Combinations of the alpha-helix-turn-alpha-helix motif of TetR with respective residues from LacI or 434Cro: DNA recognition, inducer binding, and urea-dependent denaturation. Biochemistry 36:5311-5322. [DOI] [PubMed] [Google Scholar]
- 3.Bangera, M. G., and L. S. Thomashow. 1996. Characterization of a genomic locus required for synthesis of the antibiotic 2,4-diacetylphloroglucinol by the biological control agent Pseudomonas fluorescens Q2-87. Mol. Plant-Microbe Interact. 9:83-90. [DOI] [PubMed] [Google Scholar]
- 4.Bangera, M. G., and L. S. Thomashow. 1999. Identification and characterization of a gene cluster for synthesis of the polyketide antibiotic 2,4-diacetylphloroglucinol from Pseudomonas fluorescens Q2-87. J. Bacteriol. 181:3155-3163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barthelmebs, L., B. Lecomte, C. Divies, and J.-F. Cavin. 2000. Inducible metabolism of phenolic acids in Pediococcus pentosaceus is encoded by an autoregulated operon which involves a new class of negative transcriptional regulator. J. Bacteriol. 182:6724-6731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blumer, C., S. Heeb, G. Pessi, and D. Haas. 1999. Global GacA-steered control of cyanide and exoprotease production in Pseudomonas fluorescens involves specific ribosome binding sites. Proc. Natl. Acad. Sci. USA 96:14073-14078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bowden, K., J. L. Broadbent, and W. J. Ross. 1965. Some simple antihelmintics. Br. J. Pharmacol 24:714-724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Butler, A. R., S. A. Flint, and E. Cundliffe. 2001. Feedback control of polyketide metabolism during tylosin production. Microbiology 147:795-801. [DOI] [PubMed] [Google Scholar]
- 9.Collado-Vides, J., B. Magasanik, and J. D. Gralla. 1991. Control site location and transcriptional regulation in Escherichia coli. Microbiol. Rev. 55:371-394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Corbell, N., and J. E. Loper. 1995. A global regulator of secondary metabolite production in Pseudomonas fluorescens Pf-5. J. Bacteriol. 177:6230-6236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Delany, I. 1999. Ph.D. thesis. University College, Cork, Ireland.
- 12.Delany, I., M. M. Sheehan, A. Fenton, S. Bardin, S. Aarons, and F. O'Gara. 2000. Regulation of production of the antifungal metabolite 2,4-diacetylphloroglucinol in Pseudomonas fluorescens F113: genetic analysis of phlF as a transcriptional repressor. Microbiology 146:537-543. [DOI] [PubMed] [Google Scholar]
- 13.Derre, I., G. Rapoport, and T. Msadek. 2000. The CtsR regulator of stress response is active as a dimer and specifically degraded in vivo at 37 degrees C. Mol. Microbiol. 38:335-347. [DOI] [PubMed] [Google Scholar]
- 14.Doull, J. L., and L. C. Vining. 1994. Global physiological controls, p. 9-63. In L. C. Vining and C. Stuttard (ed.), Genetics and biochemistry of antibiotic production. Butterworth-Heineman, Newton, Mass.
- 15.Duffy, B. K., and G. Defago. 1999. Environmental factors modulating antibiotic and siderophore biosynthesis by Pseudomonas fluorescens biocontrol strains. Appl. Environ. Microbiol. 65:2429-2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Farinha, M. A., and A. M. Kropinski. 1990. High efficiency electroporation of Pseudomonas aeruginosa using frozen cell suspensions. FEMS Microbiol. Lett. 58:221-225. [DOI] [PubMed] [Google Scholar]
- 17.Fenton, A. M., P. M. Stephens, J. Crowley, M. O'Callaghan, and F. O'Gara. 1992. Exploitation of gene(s) involved in 2,4-diacetylphloroglucinol biosynthesis to confer a new biocontrol capability to a Pseudomonas strain. Appl. Environ. Microbiol. 58:3873-3878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ferrandez, A., J. L. Garcia, and E. Diaz. 2000. Transcriptional regulation of the divergent paa catabolic operons for phenylacetic acid degradation in Escherichia coli. J. Biol. Chem. 275:12214-12222. [DOI] [PubMed] [Google Scholar]
- 19.Figurski, D. H., and D. R. Helinski. 1979. Replication of an origin-containing derivative of plasmid RK2 dependent on a plasmid function provided in trans. Proc. Natl. Acad. Sci. USA 76:1648-1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Haas, D., C. Blumer, and C. Keel. 2000. Biocontrol ability of fluorescent pseudomonads genetically dissected: importance of positive feedback regulation. Curr. Opin. Biotechnol. 11:290-297. [DOI] [PubMed] [Google Scholar]
- 21.Harrison, L. A., L. Letendre, P. Kovacevich, E. Pierson, and D. M. Weller. 1993. Purification of an antibiotic effective against Gaeumannomyces graminis var. tritici produced by biocontrol agent, Pseudomonas auerofaciens. Soil Biol. Biochem. 25:215-221. [Google Scholar]
- 22.Hinrichs, W., C. Kisker, M. Duvel, A. Muller, K. Tovar, W. Hillen, and W. Saenger. 1994. Structure of the Tet repressor-tetracycline complex and regulation of antibiotic resistance. Science 264:418-420. [DOI] [PubMed] [Google Scholar]
- 23.Keel, C., U. Schnider, M. Maurhofer, C. Viossard, J. Laville, U. Burger, P. Wirthner, D. Haas, and G. Defago. 1992. Suppression of root diseases by Pseudomonas fluorescens CHA0: importance of the bacterial secondary metabolite 2,4-diacetylphloroglucinol. Mol. Plant-Microbe Interact. 5:4-13. [Google Scholar]
- 24.Kuwabara, M. D., and D. S. Sigman. 1987. Footprinting DNA-protein complexes in situ following gel retardation assays using 1,10-phenanthroline-copper ion: Escherichia coli RNA polymerase-lac promoter complexes. Biochemistry 26:7234-7238. [DOI] [PubMed] [Google Scholar]
- 25.Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680-685. [DOI] [PubMed] [Google Scholar]
- 26.La Teana, A., A. Brandi, M. Falconi, R. Spurio, C. L. Pon, and C. O. Gualerzi. 1991. Identification of a cold shock transcriptional enhancer of the Escherichia coli gene encoding nucleoid protein H-NS. Proc. Natl. Acad. Sci. USA 88:10907-10911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leach, S. 2001. Ph.D. thesis. University College, Cork, Ireland.
- 28.Levy, E., F. J. Gough, D. K. Berlin, P. W. Guiana, and J. T. Smith. 1992. Inhibition of Septoria tritici and other phytopatogenic fungi and bacteria by Pseudomonas fluorescens and its antibiotics. Plant Pathol. 41:335-341. [Google Scholar]
- 29.Lewis, M., G. Chang, N. C. Horton, M. A. Kercher, H. C. Pace, M. A. Schumacher, R. G. Brennan, and P. Lu. 1996. Crystal structure of the lactose operon repressor and its complexes with DNA and inducer. Science 271:1247-1254. [DOI] [PubMed] [Google Scholar]
- 30.Liu, M. Y., G. Gui, B. Wei, J. F. Preston 3rd, L. Oakford, U. Yuksel, D. P. Giedroc, and T. Romeo. 1997. The RNA molecule CsrB binds to the global regulatory protein CsrA and antagonizes its activity in Escherichia coli. J. Biol. Chem. 272:17502-17510. [DOI] [PubMed] [Google Scholar]
- 31.Liu, M. Y., and T. Romeo. 1997. The global regulator CsrA of Escherichia coli is a specific mRNA-binding protein. J. Bacteriol. 179:4639-4642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu, M. Y., H. Yang, and T. Romeo. 1995. The product of the pleiotropic Escherichia coli gene csrA modulates glycogen biosynthesis via effects on mRNA stability. J. Bacteriol. 177:2663-2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu, Y., Y. Cui, A. Mukherjee, and A. K. Chatterjee. 1998. Characterization of a novel RNA regulator of Erwinia carotovora ssp. carotovora that controls production of extracellular enzymes and secondary metabolites. Mol. Microbiol. 29:219-234. [DOI] [PubMed] [Google Scholar]
- 34.Liu, Y., G. Jiang, Y. Cui, A. Mukherjee, W. L. Ma, and A. K. Chatterjee. 1999. kdgREcc negatively regulates genes for pectinases, cellulase, protease, HarpinEcc, and a global RNA regulator in Erwinia carotovora subsp. carotovora. J. Bacteriol. 181:2411-2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lodge, J., R. Williams, A. Bell, B. Chan, and S. Busby. 1990. Comparison of promoter activities in Escherichia coli and Pseudomonas aeruginosa: use of a new broad-host-range promoter-probe plasmid. FEMS Microbiol. Lett. 55:221-225. [DOI] [PubMed] [Google Scholar]
- 36.Ma, W., Y. Cui, Y. Liu, C. K. Dumenyo, A. Mukherjee, and A. K. Chatterjee. 2001. Molecular characterization of global regulatory RNA species that control pathogenicity factors in Erwinia amylovora and Erwinia herbicola pv. gypsophilae. J. Bacteriol. 183:1870-1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mann, J. 1987. Secondary metabolism. Clarendon Press, Oxford, United Kingdom.
- 38.Maurhofer, M., C. Keel, D. Haas, and G. Defago. 1995. Influence of plant species on disease suppression by Pseudomonas fluorescens strain CHA0 with enhanced antibiotic production. Plant Pathol. 44:40-50. [Google Scholar]
- 39.McLean, B. W., S. L. Wizeman, and M. Kropinski. 1997. Functional analysis of sigma-70 consensus promoters in Pseudomonas aeruginosa and Escherichia coli. Can. J. Microbiol. 43:981-985. [DOI] [PubMed] [Google Scholar]
- 40.Miller, J. H. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 41.Molnar, I., J. F. Aparicio, S. F. Haydock, L. E. Khaw, T. Schwecke, A. Konig, J. Staunton, and P. F. Leadlay. 1996. Organisation of the biosynthetic gene cluster for rapamycin in Streptomyces hygroscopicus: analysis of genes flanking the polyketide synthase. Gene 169:1-7. [DOI] [PubMed] [Google Scholar]
- 42.Nowak-Thompson, B., S. J. Gould, J. Kraus, and L. J. E. 1994. Production of 2,4-diacetylphloroglucinol by the biocontrol agent Pseudomonas fluorescens Pf-5. Can. J. Microbiol. 40:1064-1066. [Google Scholar]
- 43.Otten, S. L., J. Ferguson, and C. R. Hutchinson. 1995. Regulation of daunorubicin production in Streptomyces peucetius by the dnrR2 locus. J. Bacteriol. 177:1216-1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pujic, P., R. Dervyn, A. Sorokin, and S. D. Ehrlich. 1998. The kdgRKAT operon of Bacillus subtilis: detection of the transcript and regulation by the kdgR and ccpA genes. Microbiology 144:3111-3118. [DOI] [PubMed] [Google Scholar]
- 45.Qoronfleh, M. W., C. Debouck, and J. Keller. 1992. Identification and characterization of novel low-temperature-inducible promoters of Escherichia coli. J. Bacteriol. 174:7902-7909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ramette, A., Y. Moenne-Loccoz, and G. Defago. 2001. Polymorphism of the polyketide synthase gene phlD in biocontrol fluorescent pseudomonads producing 2,4-diacetylphloroglucinol and comparison of PhlD with plant polyketide synthases. Mol. Plant-Microbe Interact. 14:639-652. [DOI] [PubMed] [Google Scholar]
- 47.Reddi, T. K., and A. V. Borovkov. 1970. Antibiotic properties of 2,4-diacetylphloroglucinol (2,4-diacetyl-1,3,5-trihydroxybenzene) produced by Pseudomonas fluorescens strain 26-o. Antibiotiki 15:19-21. [PubMed] [Google Scholar]
- 48.Reddi, T. K., Y. P. Khudiakov, and A. V. Borovkov. 1969. Pseudomonas fluorescens strain 26.o, producing phytotoxic substances. Mikrobiologiya 38:909-913. [PubMed] [Google Scholar]
- 49.Rojo, F. 2001. Mechanisms of transcriptional repression. Curr. Opin. Microbiol. 4:145-151. [DOI] [PubMed] [Google Scholar]
- 50.Romeo, T. 1998. Global regulation by the small RNA-binding protein CsrA and the non-coding RNA molecule CsrB. Mol. Microbiol. 29:1321-1330. [DOI] [PubMed] [Google Scholar]
- 51.Sahara, T., M. Suzuki, J. Tsuruha, Y. Takada, and N. Fukunaga. 1999. cis-acting elements responsible for low-temperature-inducible expression of the gene coding for the thermolabile isocitrate dehydrogenase isozyme of a psychrophilic bacterium, Vibrio sp. strain ABE-1. J. Bacteriol. 181:2602-2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 53.Schnider, U., C. Keel, C. Blumer, J. Troxler, G. Defago, and D. Haas. 1995. Amplification of the housekeeping sigma factor in Pseudomonas fluorescens CHA0 enhances antibiotic production and improves biocontrol abilities. J. Bacteriol. 177:5387-5392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schnider-Keel, U., A. Seematter, M. Maurhofer, C. Blumer, B. Duffy, C. Gigot-Bonnefoy, C. Reimmann, R. Notz, G. Defago, D. Haas, and C. Keel. 2000. Autoinduction of 2,4-diacetylphloroglucinol biosynthesis in the biocontrol agent Pseudomonas fluorescens CHA0 and repression by the bacterial metabolites salicylate and pyoluteorin. J. Bacteriol. 182:1215-1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shanahan, P., D. O'Sullivan, P. Simpson, J. Glennon, and F. O'Gara. 1992. Isolation of 2,4-diacetylphloroglucinol from a fluorescent pseudomonad and investigation of physiological parameters influencing its production. Appl. Environ. Microbiol. 58:353-358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sharifi-Tehrani, A., M. Zala, A. Natsch, Y. Moënne-Loccoz, and G. Defago. 1998. Biocontrol of soil-born fungal plant by 2,4-diacetyphloroglucinol-producing fluorescent pseudomonads with different restriction profiles of amplified 16s rDNA. Eur. J. Plant Pathol. 104:631-643. [Google Scholar]
- 57.Spaink, H. P., R. J. Okker, C. A. Wiffelman, E. Pees, and B. J. Lugtenberg. 1987. Promoters in the nodulation region of the Rhizobium leguminosarum Sym plasmid pRL1J1. Plant Mol. Biol. 9:27-39. [DOI] [PubMed] [Google Scholar]
- 58.Tomas-Lorente, F., E. Iniesta-Sanmartin, F. A. Tomas-Barberan, W. Trowitzsch-Kienast, and V. Wray. 1989. Antifungal phloroglucinol derivative and lipophilic flavenoids from Helichrysum decumbens. Phytochemistry 28:1613-1615. [Google Scholar]
- 59.van de Guchte, M., C. Daly, G. F. Fitzgerald, and E. K. Arendt. 1994. Identification of the putative repressor-encoding gene cI of the temperate lactococcal bacteriophage Tuc2009. Gene 144:93-95. [DOI] [PubMed] [Google Scholar]
- 60.Vedler, E., V. Koiv, and A. Heinaru. 2000. TfdR, the LysR-type transcriptional activator, is responsible for the activation of the tfdCB operon of Pseudomonas putida 2,4-dichlorophenoxyacetic acid degradative plasmid pEST4011. Gene 245:161-168. [DOI] [PubMed] [Google Scholar]
- 61.Vincent, M. N., L. A. Harrison, J. M. Brackin, P. A. Kovacevich, P. Mukerji, D. M. Weller, and E. A. Pierson. 1991. Genetic analysis of the antifungal activity of a soilborne Pseudomonas aureofaciens strain. Appl. Environ. Microbiol. 57:2928-2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Weller, D., and L. S. Thomashow. 1993. Use of rhizobacteria for biocontrol. Curr. Opin. Biotechnol. 4:306-311. [Google Scholar]
- 63.Weller, D. M., and R. J. Cook. 1983. Suppression of take-all of wheat by seed treatment with fluorescent pseudomonads. Phytopathology 73:463-469. [Google Scholar]
- 64.Whistler, C. A., N. A. Corbell, A. Sarniguet, W. Ream, and J. E. Loper. 1998. The two-component regulators GacS and GacA influence accumulation of the stationary-phase sigma factor σS and the stress response in Pseudomonas fluorescens Pf-5. J. Bacteriol. 180:6635-6641. [DOI] [PMC free article] [PubMed] [Google Scholar]