

Abstract
The recent years have brought breathtaking advances in the biomedical sciences and biomedical engineering. These advances offer the promise that diseases responsible for most disability and early death may soon be addressed by replacing damaged organs with bio-engineered substitutes. Application of these technologies, however, is impeded by the immune response directed against foreign cells and tissues. Here we consider the potentiality and the limitations of these new technologies and how the technologies might be combined to generate novel approaches to organ replacement that overcome immunological barriers to success.
Keywords: transplantation, stem cells, cloning, graft rejection, organogenesis
The most prevalent causes of death and disability involve failure of the heart, lung, liver and/or kidneys [1]. Organ transplantation remains the preferred approach to treating these diseases; but, application is limited by a severe shortage of organ donors [2] and by the immune response of the recipient against the graft. Recent advances in biomedical science, such as the derivation of stem cells from various sources [3, 4], the reprogramming of somatic cell nuclei to allow therapeutic cloning [5], the manufacturing of matrices that support tissue histogenesis, the construction of mechanical devices [6] and the engineering of pigs to yield organs that evade human immunity [7] would seem to indicate that approaches other than organ transplantation, some pursued for nearly a century [8], may soon become available to treat organ failure. Here we review how the immune system limits the technologies that might be used to replace or augment the function of the heart, lungs, liver and kidneys.
Organ transplantation
Transplanted organs provide immediate and full replacement of organ function. The main limitations to organ transplantation are damage to organ caused by the immune response of the recipient against the graft and short supply of human organs [2, 9]. The immune response to allotransplants can be controlled by administration of immunosuppressive agents. However, while immunosuppression prevents rejection in most cases, it does not prevent chronic rejection of grafts and it heightens susceptibility to infection, cancer and premature atherosclerosis. Accordingly, much effort has been devoted to developing safe and effective ways to induce allogeneic tolerance, i.e. specific immune non-responsiveness. However, non-toxic and reliable means of inducing tolerance are not yet available.
The short supply of organs might be addressed by using lower animals such as pigs as a source, i.e. xenotransplantation [2, 10–12]. Xenotransplants might also resist infection by human viruses and provide the opportunity to plan the transplant in advance, allowing customization of the donor or the recipient according to specific needs [13]. Success of xenotransplantation requires, however, overcoming a number of challenges, including the immune response of the recipient against the graft [10] and the possibility that infectious organisms might be transmitted from the transplant to the recipient and potentially from the recipient to others [14].
We believe that cell and tissue xenografts are much less susceptible to immune-mediated injury than organ xenografts (Figures 1A and 2)[10]. Xenogeneic cell and tissue transplants are mainly susceptible to cell-mediated rejection and have thus been carried out successfully in experimental animals [15, 16] and in human subjects [17, 18] using immunosuppression similar to what would be used to sustain an allograft. Xenogeneic organ xenografts, on the other hand, are subject to cellular rejection and also to humoral rejection, owing to the action of anti-donor antibodies and complement as summarized in Figure 1B. Humoral rejection of organ xenografts is quite severe and resistant to treatment.
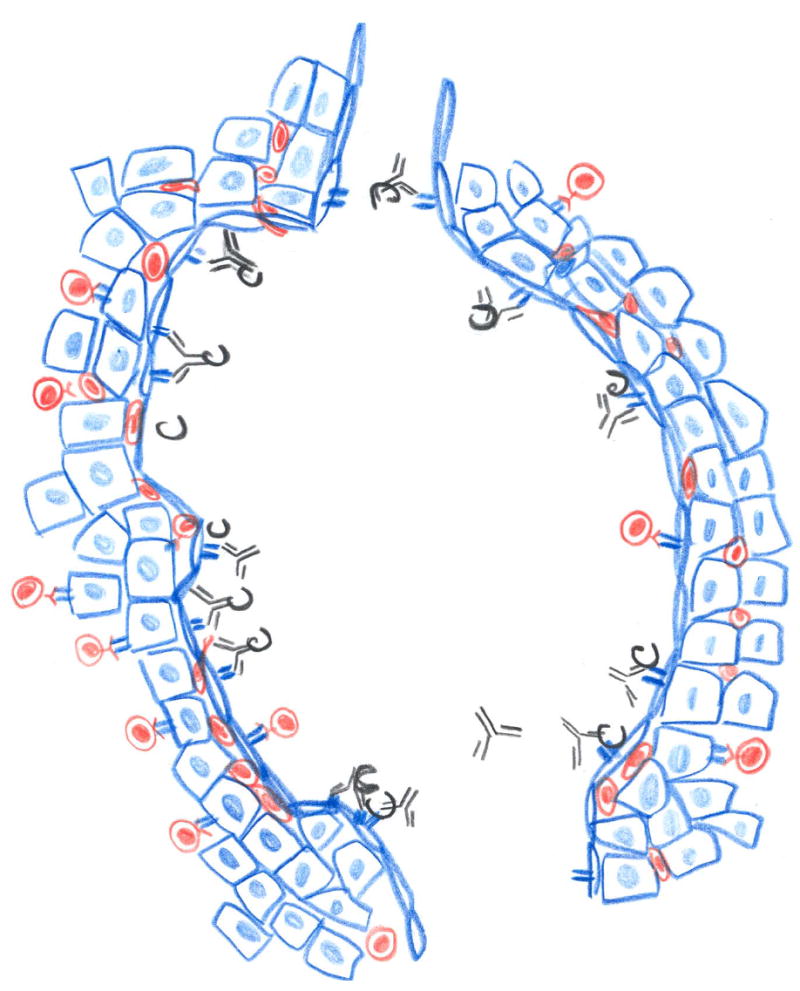
Figure 1. Impact of immunity on outcome of transplants.
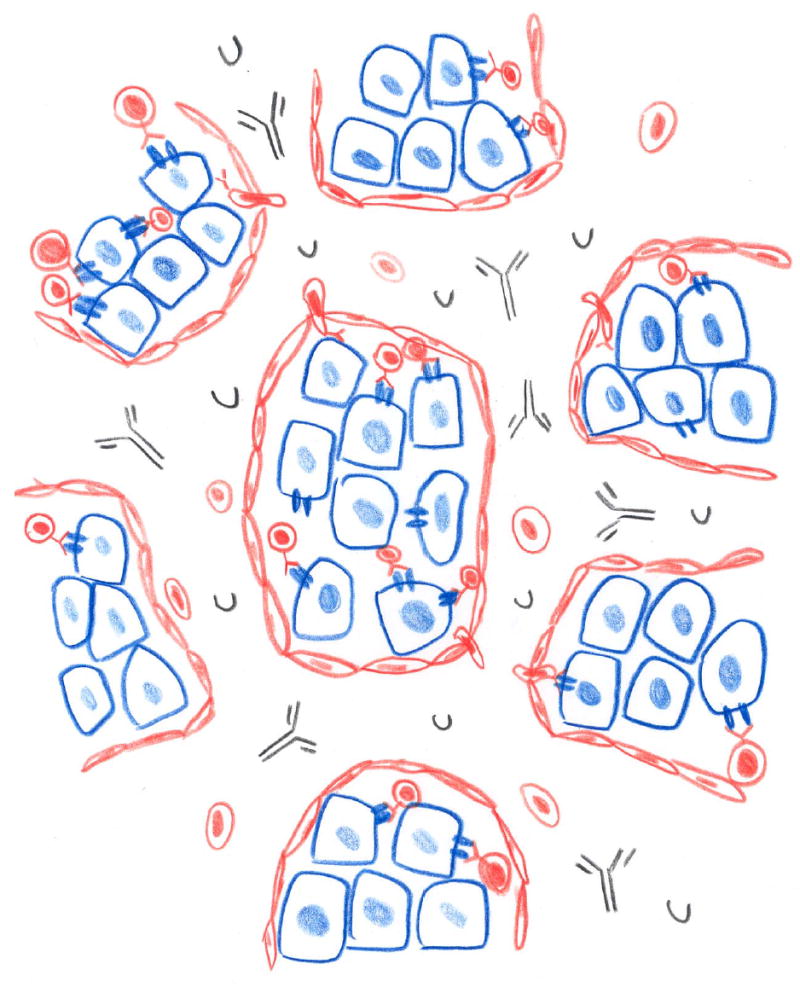
The nature of the immune response to transplantation depends on the genetic difference between the donor and the recipient; the impact of that response on the survival and function of a graft depends to a large extent on the means by which the graft derives its vascular supply, as summarized here. Transplants consisting of cells, such as hepatocytes, or tissues, such as pancreatic islets, engrafted outside of the vasculature are not notably injured by humoral immunity but are subject mainly to cell mediated rejection, whereas organ grafts are attacked by humoral immune responses directed against donor endothelium as well as cell mediated immune responses.
A. Tissue and cell transplants (blue) are vascularized by the in-growth of blood vessels of the recipient (red). The blood vessels are permeated by lymphocytes, but retain antibodies and complement within the lumen and away from the graft (A).
B. Organ grafts (blue) are vascularized by surgical connection of donor (blue) and recipient blood vessels; hence, the vessels in the graft are from the donor. Antibodies and complement can attach to the foreign vascular cells and lymphocytes can permeate the blood vessels (B).
Figure 2. Impact of immunity on various types of grafts.
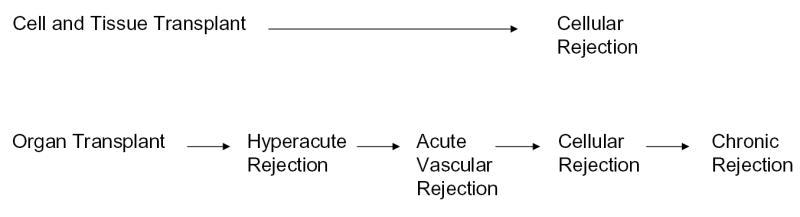
Cell and tissue grafts are mainly susceptible to cellular rejection mediated by T cells. Organ grafts are susceptible to hyperacute and acute vascular and chronic rejection, which may be mediated by antibodies and to cellular rejection mediated by T cells.
More than ten years ago, we suggested that the hurdles to organ xenotransplantation might be overcome by genetic engineering [19]. Efforts toward that end initially focused on expressing human complement regulatory proteins as the product of transgenes in pigs [20, 21]. Organs from pigs expressing human decay accelerating factor or CD46 are not subject to hyperacute rejection [22–24], the most severe immune reaction known. However, these organs still are subject to acute vascular rejection [25] caused by antibodies directed against Galα1-3Gal [26], a blood group-like sugar. To deal with this problem, recent efforts have focused on disrupting α1,3-galactosly transferase (α1,3GT) [7, 27, 28], the enzyme that catalyzes synthesis of Galα1-3Gal, the preeminent target of humoral immunity to organ xenografts [26, 29, 30]. While eliminating expression of Galα1-3Gal and inducing expression of human complement regulatory proteins will undoubtedly improve the outcome of xenografts (the best survival of porcine organ xenografts before genetic engineering was 23 days, and since is longer than 100 days), daunting problems, such as production of antibodies against other antigens [31] and incompatibilities between coagulant and inflammatory cascades [32], may preclude enduring graft function, as discussed elsewhere [33]. These concerns appear to be confirmed by anecdotal reports that organs transplanted from α1-3GT-deficient pigs into non-human primates still undergo acute vascular rejection.
Applications of xenotransplantation for organ replacement
Given the hurdles discussed above and the emerging of new technologies reviewed below, we think that xenotransplantation may be less suitable than other modalities to replace heart and kidney function. Of particular concern to us is the possibility that immune-induced thrombosis in coronary blood vessels could cause life threatening arrhythmia. On the other hand we think xenotransplantation might eventually become the preferred approach to treating severe chronic pulmonary failure. Although, the hurdles to successful pulmonary xenotransplantation are formidable, including profound resistance to blood flow caused by immune reactants [34], significant progress has been made in reversing this problem and other means of replacing the lungs appear remote.
Xenotransplantation may also be used to treat liver disease. We had once thought that incompatibilities of coagulation and complement cascades between species would preclude hepatic xenotransplantation [13]. However, recent experiments in which porcine liver were transplanted with apparent success into baboons argues against our view [35]. Furthermore, Ito and co-worker recently found that porcine hepatocyte xenografts restore physiology and prolong the life of animals with chemically induced cirrhosis [36]. Since porcine hepatocytes resist infection by hepatitis c virus, the most common cause of hepatic failure in many parts of the world, xenografts might soon be used to treat this and other types of viral hepatitis.
Another application of xenotransplantation may be as a means of expanding human cells or growing human organs for transplantation in human subjects. We discuss this subject in the section on organogenesis below.
Stem cells for augmentation and replacement of organ function
Stem cells are cells capable of self-renewal and of generating at least one, and often more than one, differentiated line of cells. Stem cells obtained from the inner cell mass of the blastocysts are called embryonic stem (ES) cells. Stem cells may also be isolated from mature individuals or generated by transfer of nuclei from mature cells to immature cell bodies, i.e. cloning. Stem cells are thought to be capable of regenerating diseased or damaged tissues [37, 38] and of being coaxed to generate tissues and organs de novo [5, 39, 40]. However, the application of stem cells for tissue repair and regeneration is limited by: (i) the proliferative potential of the cells; (ii) the ability of the cells to differentiate into mature, functional tissues; (iii) the possibility of transformation in culture; and (iv) immune response against foreign antigens expressed by the cells. How these limitations apply in the replacement of organs will be discussed below in relation to organ replacement.
The source of stem cells determines the potentiality and the limitations to use. Embryonic stem cells have the capacity to proliferate indefinitely and generate all tissues and organs [41] and thus the use of these cells has been greeted with much enthusiasm. However, embryonic stem cells express allogeneic histocompatibility antigens and their use would therefore require immunosuppression (Figure 1A). The ideal way to avoid immune mediated rejection is to generate tissues and organs from cells of the individual needing treatment. This might be accomplished by using “adult” stem cells or by cloning.
Stem cells from adult subjects can migrate through the blood, take up residence, differentiate and function in tissues in which injury has been induced [37, 42]. Thus, stem cells would appear to have a broadly construed mission for regenerating diseased tissues. However, regeneration of tissue by stem cells does not evidently prevent any of the diseases most responsible for human mortality and morbidity. Failure of stem cells to naturally avert such diseases may reflect: (i) the low frequency of such cells in adults; (ii) the kinetics of tissue injury exceeding the regenerative potential of stem cells; and (iii) pathogenic mechanisms that compromise the regenerative capacity of stem cells. Effective application of stem cells for treatment of disease may require identifying which of these factors limits the natural regenerative functions of stem cells and the development of means to provide the cells in large numbers, and with the capacity to survive and differentiate under conditions of disease.
Cloning as means of generating autologous stem cells.
The main challenges to using adult stem cells for organ replacement are obtaining sufficient numbers of stem cells and overcoming the limited ability of these cells to proliferate {in contrast to embryonic stem cells (ES cells), which have unlimited proliferative potential}. An ideal way of obtaining an unlimited supply of stem cells of any individual might however be provided by cloning of somatic nuclei to obtain ES cells.
The feasibility of obtaining ES cells by nuclear cloning was demonstrated by recent successes in cloning large animals [43] (reproductive cloning). Cloning was carried out by transfer of the nucleus from a somatic cell to a recipient enucleated cell capable of reverting the epigenetic modifications of chromatin characteristic of the differentiated state. Reproductive cloning is intrinsically inefficient. In mice, only 0.5% to 2% of implanted blastocysts lead to newborn animals [44]. Part of the reason for loss of efficiency is that creation of a new individual requires recapitulation of early embryogenesis and, thus, reprogramming of the chromatin to revert the epigenetic alterations of differentiation: (i) silencing of differentiated transcripts; (ii) activation of early genes; (iii) reactivation of both X chromosomes in female nuclei; (iv) maintenance of imprinting of the maternal and paternal chromosomes; and (v) the reversal of shortened telomeres. While these events may occur, allowing development to adulthood [44], reprogramming, as such, is often faulty. In addition, the nuclei used to generate embryos may have accumulated mutations that preclude normal development. The molecular basis for reprogramming of chromatin to allow de-differentiation and subsequent re-differentiation has only now began to be understood [45]. A provocative report suggests that faulty imprinting of such genes as Igf2, Igf2r, and H19 in the extra-embryonic tissue rather than in the embryo may underlie low efficiency in cloning [46].
Despite the aforementioned limitations, cloning might be used as a source of cells for bioengineering of tissues and organs, i.e. therapeutic cloning. Recent studies have suggested that efficiency in generating blastocysts by nuclear transfer may be inversely related to the state of differentiation of the donor nucleus source. In one report, nuclei from ES cells generated blastocysts in 10–20% of cases while nuclei from cumulus cells and fibroblasts generated blastocysts in 70 % and 58 % respectively [47]. Successful therapeutic cloning for functional tissue engineering does not require the orchestration of gene expression needed for development of the body plan, and faulty reprogramming and accumulation of mutations may not preclude use of nuclear transfer to generate new tissues. One critical advantage of therapeutic cloning is that tissues or cells so derived are genetically similar to the source of the nuclei. However, mitochondria and hence mitochondria-encoded proteins are provided by the oocyte/zygote that receives the nuclei and, thus, may be foreign. This problem can be solved if nuclear reprogramming can be accomplished without nuclear transfer. Some progress toward this has been made [45].
So far, therapeutic cloning has only been accomplished by transferring the nucleus from a somatic cell to an oocyte, egg or zygote. To generate customized ES cells from a patient, nuclei obtained from somatic cells, such as hematopoietic cells, would be transferred to an enucleated zygote capable of supporting growth to the blastocyst stage. Custom made ES cells can then be isolated from the inner cell mass of the blastocyst, propagated in culture and made to differentiate into the specialized tissues and organs, as needed. As more is learned about nuclear reprogramming, we anticipate that mature cells might be fashioned into pluripotent or conditionally totipotent stem cells, thus avoiding ethical problems associated with ES cells and the immune responses to mitochondrial antigens, as mentioned above.
Tissue engineering
While embryonic stem cells have the capacity to differentiate and contribute to formation of mature tissues and organs, realizing that capacity depends on cell-cell and cell-matrix interactions to deliver requisite cues. One way to deliver such cues is through tissue engineering, the use of scaffolds consisting of synthetic or biological polymers, to coax growth and development [48]. Tissue engineering has been used to generate blood vessels [49–52], heart valves [53–55], cardiac muscle [56–58], bone [59], liver [60–62], nerve [63] and islets [64]. The most successful applications have been engineered cartilage [65, 66] and skin [67–70]. Tissue engineering is not generally thought to be applicable for organ replacement because the matrices in current use do not permit the growth of cells into a sufficient mass or anatomic complexity to yield a whole organ.
Organogenesis
What is needed for organ replacement is a method to grow organs de novo, i.e. organogenesis. Fetal mesenchyme and human embryonic stem cells cultured under suitable conditions can develop into organ-like structures in vitro [40, 71]. Since human fetal cells will not be available, any effort to apply organogenesis must be based on the driving of stem cells to form the organ of interest. Ideally, organogenesis using autologous stem cells stimulated in such a way to give rise to the organ needed, would be undertaken in the individual needing treatment to avoid vascularization by foreign blood vessels. How stem cells can be driven in this way is not yet clear and organs grown in vitro are too small to achieve physiological impact and lack blood vessels [72].
Some of the limitations of in vitro organogenesis might be circumvented if organogenesis could be carried out in vivo. Indeed, fetal tissues of various types have been found to mature after implantation into adult animals [73-78]. Organs grown in this way might achieve physiologic size because the organs are vascularized by in-growth of blood vessels of the “recipient.” The ideal source of cells for organogenesis would be stem cells originating from the affected individual grown in the natural environment of the organ, for example, the thorax in the case of the lungs or the abdomen in the case of the kidney. Growing an organ de novo in an individual with severe disease might be difficult to envision; however, as an alternative, organogenesis might be carried out using an animal as a temporary recipient for the human cells [14]. Thus, human stem cells could be introduced into fetal animals in which the local microenvironment supports and directs the development of the organ of interest. One limitation to applying this approach is that the temporary graft of human cells might be subject to immune-mediated injury [79]. This problem could be overcome by using immunodeficient animals as temporary hosts. The use of a temporary host for organogenesis does, however, engender another problem, the blood vessels in the organ derive from the animal host [73] and upon transfer to a human, these blood vessels would subject to vascular rejection [10, 80]. Unless vascular rejection is avoided, e.g. by genetic engineering [81] or unless human blood vessels can be induced to grow [82], this problem may limit application of organogenesis as it has organ xenotransplantation.
Application of cell transplantation, tissue engineering and organogenesis for augmentation and replacement of organ function
The prospects for effective application of cell transplantation, tissue engineering and organogenesis for replacement of organ function vary widely. Recent experiments in animals and humans suggest that muscle cells or stem cells capable of developing into muscle cells injected into the heart can improve cardiac function. For example, skeletal myoblasts, precursors of myocytes, were recently shown to engraft in myocardium [83] and take on the function of cardiac myocytes [84]. Skeletal myoblasts have been implanted in the heart of an individual with ischemic heart disease, and improvement in cardiac function has been ascribed to the cellular graft [85]. One limitation of cellular transplantation, rejection of heterologous myoblasts, might be averted by using autologous skeletal myoblasts [85], or stem cells as a source of cells for the procedure. Another limitation is that the transplanted cells may not engraft in the optimal anatomic orientation or in the most severely affected regions. Anatomic orientation might be improved by tissue engineering, i.e. growing myocytes as sheets or patches for repairing focal defects. However, sheets of cells cannot replace en entire organ and cells or engineered tissues may engraft poorly or be subject to ischemia in damaged myocardium. Since vascular disease is the most common cause of cardiac failure, engraft might require revascularization, which might in turn be achieved by co-implanting precursors of vascular cells derived from hematopoietic stem cells [82]. Neither transplanted cells nor engineered tissues will be suitable for replacing the function of diffusely injured heart. For this purpose, an artificial device, allograft or xenograft will be needed.
Augmenting or replacing function of the lung or kidneys is a far greater challenge. The complex anatomy of these organs (a branching system of ducts associated with air sacs or glomeruli each with paired blood vessels) makes it difficult to imagine how injection of cells of any type could give rise to functional tissue. Still, recent studies connecting small defects in kidney function with heightened susceptibility cardiovascular disease [86] raise in our minds the possibility that metabolic or endocrine functions of the kidney (or lung) may be critical to health and these functions in turn might one day be augmented by cellular transplants. These aspects notwithstanding, full replacement of kidney or lung function will require organogenesis or xenotransplantation.
Considering how organogenesis might be undertaken for replacement of the lungs or kidneys is a useful exercise. We have recently discussed this subject in detail [87, 88]. In individuals with failing lungs, organogenesis would likely add prohibitively to respiratory requirements. Hence, we would envision organogenesis being undertaken in an animal host using human stem cells – such an organ would have xenogeneic blood vessels but autologous airways and might thus be less susceptible to acute and chronic rejection. On the other hand, the availability of dialysis should make it reasonable to consider developing the means to undertake organogenesis in humans with renal failure.
Cellular transplantation has shown promise for the treatment of liver disease. Hepatocyte transplantation has been successfully performed in experimental animals [89–92] and in a few human subjects [93] with metabolic diseases of the liver, such as Crigler-Najar syndrome. In this setting, the transplanted hepatocytes are infused into the portal vein, which drains into the liver or into the splenic artery. Once engrafted in the liver or spleen, the hepatocytes need not replace all of the functions of the liver but only must augment the one deficient function.
Much more difficult would be the treatment of diffuse, destructive liver diseases, such as cirrhosis, by hepatocyte transplantation. Yet, recent work in experimental animals suggests that even cirrhosis can potentially be treated by hepatocyte transplantation [36]. The preeminent challenge to transplantation of hepatocytes is finding a plentiful source of hepatocytes. Human livers are in short supply, and so hepatocytes are likewise. One approach to producing large numbers of hepatocytes is to reversibly transform the cells by introducing “immortalizing” genes, such as the Simian virus large T antigen, flanked by loxP sites, that can be excised [94]. Another approach is to use hepatic or hematopoietic stem cells [95, 96], which can be driven to proliferate, although the proliferative potential of stem cells from mature individuals may not suffice to generate a sufficient number of hepatocytes.
Transplanted human hepatocytes (or stem cells) would of course be subject to infection by hepatitis viruses. Thus, individuals with viral-induced cirrhosis might be treated by xenogeneic hepatocyte transplantation, or by engineering human hepatocytes to prevent viral entry and/or replication. How human cells could be so engineered is uncertain, but likely to be known within a few years. Another potential limitation of transplanted hepatocytes is the lack of a biliary tree and thus the likelihood of bile accumulation leading to injury in the site of implantation. The problem of bile excretion might, in principal, be addressed by tissue engineering. Efforts are underway to engineer an artificial liver tissue [61, 62] by growing hepatocytes on a scaffold that would promote formation of tracts through which bile could be drained.
Concluding remarks
The past few years have brought to the fore new technologies such as xenotransplantation, stem cell biology, tissue engineering, cloning and organogenesis, which are widely seen as solutions to scourges of human-kind. These technologies are generally studied by distinct groups of investigators and therefore little consideration has been given to the limitations of the technologies or how the technologies might be combined. Given the importance of organ failure a cause of death and disability, we think that this consideration is due and should be based on the distinct needs and challenges of each organ system.
Acknowledgments
Work in the laboratories of the authors is supported by grants from the National Institutes of Health, AI48602 to MC, HL46810, HL52297 to JLP. We thank Tom Hostetter for very helpful information on the consequences of renal disease.
References
- 1.Oberman A. Principles of Preventive Health Care. In: Goldman L, Bennett JC, Drazen JM et al. Eds, Cecil Textbook of Medicine. 21st ed. Philadelphia, W.B. Saunders Company 2000; 26–28.
- 2.Evans RW. Coming to terms with reality: why xenotransplantation is a necessity. In: Platt JL Ed, Xenotransplantation. Washington, D. C., ASM Press 2001; 29–51.
- 3.Krause DS, Theise ND, Collector MI, et al. Multi-organ, multi-lineage engraftment by a single bone marrow-derived stem cell. Cell. 2001;105:369–77. doi: 10.1016/s0092-8674(01)00328-2. [DOI] [PubMed] [Google Scholar]
- 4.Evans MJ, Kaufman MH. Establishment in culture of pluripotential cells from mouse embryos. Nature. 1981;292:154–6. doi: 10.1038/292154a0. [DOI] [PubMed] [Google Scholar]
- 5.Liechty KW, MacKenzie TC, Shaaban AF, et al. Human mesenchymal stem cells engraft and demonstrate site-specific differentiation after in utero transplantation in sheep. Nat Med. 2000;6:1282–6. doi: 10.1038/81395. [DOI] [PubMed] [Google Scholar]
- 6.Li M, Carpio DF, Zheng Y, et al. An essential role of the NF-kappa B/Toll-like receptor pathway in induction of inflammatory and tissue-repair gene expression by necrotic cells. J Immunol. 2001;166:7128–35. doi: 10.4049/jimmunol.166.12.7128. [DOI] [PubMed] [Google Scholar]
- 7.Dai Y, Vaught TD, Boone J, et al. Targeted disruption of the alpha1,3-galactosyltransferase gene in cloned pigs. Nat Biotechnol. 2002;20:251–5. doi: 10.1038/nbt0302-251. [DOI] [PubMed] [Google Scholar]
- 8.Guthrie CC. Blood-vessel surgery and its applications. New York, Longmans, Green & Co., 1912.
- 9.Evans RW, Orians CE, Ascher NL. The potential supply of organ donors: an assessment of the efficiency of organ procurement efforts in the United States. JAMA. 1992;267:239–46. [PubMed] [Google Scholar]
- 10.Cascalho M, Platt JL. The immunological barrier to xenotransplantation. Immunity. 2001;14:437–46. doi: 10.1016/s1074-7613(01)00124-8. [DOI] [PubMed] [Google Scholar]
- 11.White D, Wallwork J. Xenografting: probability, possibility, or pipe dream? Lancet. 1993;342:879–80. doi: 10.1016/0140-6736(93)91939-j. [DOI] [PubMed] [Google Scholar]
- 12.Buhler L, Friedman T, Iacomini J, Cooper DKC. Xenotransplantation--state of the art--update 1999. Front Biosci. 1999;4:D416–32. doi: 10.2741/A438. [DOI] [PubMed] [Google Scholar]
- 13.Platt JL. New directions for organ transplantation. Nature. 1998;392:11–17. doi: 10.1038/32023. [DOI] [PubMed] [Google Scholar]
- 14.Cascalho M, Platt JL. Xenotransplantation and other means of organ replacement. Nat Rev Immunol. 2001;1:154–60. doi: 10.1038/35100578. [DOI] [PubMed] [Google Scholar]
- 15.Ricordi C, Lacy PE, Sterbenz K, Davie JM. Low-temperature culture of human islets or in vivo treatment with L3T4 antibody produces a marked prolongation of islet human-to-mouse xenograft survival. Proc Natl Acad Sci USA. 1987;84:8080–84. doi: 10.1073/pnas.84.22.8080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marchetti P, Scharp DW, Finke EH, et al. Prolonged survival of discordant porcine islet xenografts. Transplantation. 1996;61:1100–02. doi: 10.1097/00007890-199604150-00019. [DOI] [PubMed] [Google Scholar]
- 17.Groth CG, Korsgren O, Tibell A, et al. Transplantation of porcine fetal pancreas to diabetic patients. Lancet. 1994;344:1402–04. doi: 10.1016/s0140-6736(94)90570-3. [DOI] [PubMed] [Google Scholar]
- 18.Deacon T, Schumacher J, Dinsmore J, et al. Histological evidence of fetal pig neural cell survival after transplantation into a patient with Parkinson's disease. Nat Med. 1997;3:350–53. doi: 10.1038/nm0397-350. [DOI] [PubMed] [Google Scholar]
- 19.Platt JL, Vercellotti GM, Dalmasso AP, et al. Transplantation of discordant xenografts: a review of progress. Immunol Today. 1990;11:450–56. doi: 10.1016/0167-5699(90)90174-8. [DOI] [PubMed] [Google Scholar]
- 20.Cozzi E, White DJG. The generation of transgenic pigs as potential organ donors for humans. Nat Med. 1995;1:964–66. doi: 10.1038/nm0995-964. [DOI] [PubMed] [Google Scholar]
- 21.McCurry KR, Kooyman DL, Diamond LE, Byrne GW, Logan JS, Platt JL. Human complement regulatory proteins in transgenic animals regulate complement activation in xenoperfused organs. Transplant Proc. 1995;27:317–18. [PubMed] [Google Scholar]
- 22.McCurry KR, Kooyman DL, Alvarado CG, et al. Human complement regulatory proteins protect swine-to-primate cardiac xenografts from humoral injury. Nat Med. 1995;1:423–27. doi: 10.1038/nm0595-423. [DOI] [PubMed] [Google Scholar]
- 23.Diamond LE, Quinn CM, Martin MJ, Lawson J, Platt JL, Logan JS. A human CD46 transgenic pig model system for the study of discordant xenotransplantation. Transplantation. 2001;71:132–42. doi: 10.1097/00007890-200101150-00021. [DOI] [PubMed] [Google Scholar]
- 24.Cozzi E, Yannoutsos N, Langford GA, Pino-Chavez G, Wallwork J, White DJG. Effect of transgenic expression of human decay-accelerating factor on the inhibition of hyperacute rejection of pig organs. In: Cooper DKC, Kemp E, Platt JL and White DJG Eds, Xenotransplantation: the transplantation of organs and tissues between species. 2nd ed. Berlin, Springer 1997; 665-82.
- 25.Byrne GW, McCurry KR, Martin MJ, McClellan SM, Platt JL, Logan JS. Transgenic pigs expressing human CD59 and decay-accelerating factor produce an intrinsic barrier to complement-mediated damage. Transplantation. 1997;63:149–55. doi: 10.1097/00007890-199701150-00027. [DOI] [PubMed] [Google Scholar]
- 26.Lin SS, Hanaway MJ, Gonzalez-Stawinski GV, et al. The role of anti-Galα1-3Gal antibodies in acute vascular rejection and accommodation of xenografts. Transplantation (Rapid Communication) 2000;70:1667–74. doi: 10.1097/00007890-200012270-00002. [DOI] [PubMed] [Google Scholar]
- 27.Lai L, Kolber-Simonds D, Park KW, et al. Production of α-1,3-galactosyltransferase knockout pigs by nuclear transfer cloning. Science. 2002;295:1089–92. doi: 10.1126/science.1068228. [DOI] [PubMed] [Google Scholar]
- 28.Phelps CJ, Koike C, Vaught TD, et al. Production of alpha 1,3-galactosyltransferase-deficient pigs. Science. 2003;299:411–4. doi: 10.1126/science.1078942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Collins BH, Cotterell AH, McCurry KR, et al. Cardiac xenografts between primate species provide evidence for the importance of the α-galactosyl determinant in hyperacute rejection. J Immunol. 1995;154:5500–10. [PubMed] [Google Scholar]
- 30.Li SF, Neethling FA, Taniguchi S, et al. Glycans derived from porcine stomach mucin are effective inhibitors of natural anti-α-galactosyl antibodies in vitro and after intravenous infusion in baboons. Transplantation. 1996;62:1324–31. doi: 10.1097/00007890-199611150-00026. [DOI] [PubMed] [Google Scholar]
- 31.McCurry KR, Parker W, Cotterell AH, et al. Humoral responses in pig-to-baboon cardiac transplantation: implications for the pathogenesis and treatment of acute vascular rejection and for accommodation. Hum Immunol. 1997;58:91–105. doi: 10.1016/s0198-8859(97)00229-2. [DOI] [PubMed] [Google Scholar]
- 32.Alwayn IPJ, Buhler L, Appel IIIJZ, et al. Mechanisms of thrombotic microangiopathy following xenogeneic hematopoietic progenitor cell transplantation. Transplantation. 2001;71:1601–09. doi: 10.1097/00007890-200106150-00020. [DOI] [PubMed] [Google Scholar]
- 33.Platt JL. Knocking out xenograft rejection. Nat Biotechnol. 2002;20:231–32. doi: 10.1038/nbt0302-231. [DOI] [PubMed] [Google Scholar]
- 34.Lau CL, Daggett WC, Yeatman MF, et al. The role of antibodies in dysfunction of pig-to-baboon pulmonary transplants. J Thorac Cardiovasc Surg. 2000;120:29–38. doi: 10.1067/mtc.2000.106841. [DOI] [PubMed] [Google Scholar]
- 35.Ramirez P, Chavez R, Majado M, et al. Life-supporting human complement regulator decay accelerating factor transgenic pig liver xenograft maintains the metabolic function and coagulation in the nonhuman primate for up to 8 days. Transplantation. 2000;70:989–98. doi: 10.1097/00007890-200010150-00001. [DOI] [PubMed] [Google Scholar]
- 36.Nagata H, Ito M, Cai J, Edge A, Platt JL, Fox IJ. Treatment of cirrhosis and liver failure in rats by hepatocyte xenotransplantation. Gastroenterology. 2003;124:422–31. doi: 10.1053/gast.2003.50065. [DOI] [PubMed] [Google Scholar]
- 37.Blau HM, Brazelton TR, Weimann JM. The evolving concept of a stem cell: entity or function? Cell. 2001;105:829–41. doi: 10.1016/s0092-8674(01)00409-3. [DOI] [PubMed] [Google Scholar]
- 38.Anversa P, Nadal-Ginard B. Myocyte renewal and ventricular remodeling. Nature. 2002;415:240–3. doi: 10.1038/415240a. [DOI] [PubMed] [Google Scholar]
- 39.Lumelsky N, Blondel O, Laeng P, Valasco I, Ravin R, McKay R. Differentiation of embryonic stem cells to insulin-secreting structures similar to pancreatic islets. Science. 2001;292:1389–94. doi: 10.1126/science.1058866. [DOI] [PubMed] [Google Scholar]
- 40.Bianco P, Robey PG. Stem cells in tissue engineering. Nature. 2001;414:118–21. doi: 10.1038/35102181. [DOI] [PubMed] [Google Scholar]
- 41.Thomson JA, Itskovitz-Eldor J, Shapiro SS, et al. Embryonic stem cell lines derived from human blastocysts. Science. 1998;282:1145–47. doi: 10.1126/science.282.5391.1145. [DOI] [PubMed] [Google Scholar]
- 42.Quaini F, Urbanek K, Beltrami AP, et al. Chimerism of the transplanted heart. N Engl J Med. 2002;346:5–15. doi: 10.1056/NEJMoa012081. [DOI] [PubMed] [Google Scholar]
- 43.Wilmut I, Schnieke AE, McWhir J, Kind AJ, Campbell KH. Viable offspring derived from fetal and adult mammalian cells. Nature. 1997;385:810–13. doi: 10.1038/385810a0. [DOI] [PubMed] [Google Scholar]
- 44.Rideout IIIWM, Eggan K, Jaenisch R. Nuclear cloning and epigenetic reprogramming of the genome. Science. 2001;293:1093–98. doi: 10.1126/science.1063206. [DOI] [PubMed] [Google Scholar]
- 45.Hansis C, Barreto G, Maltry N, Niehrs C. Nuclear reprogramming of human somatic cells by xenopus egg extract requires BRG1. Curr Biol. 2004;14:1475–80. doi: 10.1016/j.cub.2004.08.031. [DOI] [PubMed] [Google Scholar]
- 46.Inoue K, Kohda T, Lee J, et al. Faithful expression of imprinted genes in cloned mice. Science. 2002;295:297. doi: 10.1126/science.295.5553.297. [DOI] [PubMed] [Google Scholar]
- 47.Humphreys T, Reinherz EL. Invertebrate immune recognition, natural immunity and the evolution of positive selection [see comments] Immunol Today. 1994;15:316–20. doi: 10.1016/0167-5699(94)90079-5. [DOI] [PubMed] [Google Scholar]
- 48.Griffith LG, Naughton G. Tissue engineering--current challenges and expanding opportunities. Science. 2002;295:1009–16. doi: 10.1126/science.1069210. [DOI] [PubMed] [Google Scholar]
- 49.Ogle BM, Mooradian DL. The role of vascular smooth muscle cell integrins in the compaction and mechanical strengthening of a tissue-engineered blood vessel. Tissue Eng. 1999;5:387–402. doi: 10.1089/ten.1999.5.387. [DOI] [PubMed] [Google Scholar]
- 50.Niklason LE, Gao J, Abbott WM, et al. Functional arteries grown in vitro. Science. 1999;284:489–93. doi: 10.1126/science.284.5413.489. [DOI] [PubMed] [Google Scholar]
- 51.L'Heureux N, Paquet S, Labbe R, Germain L, Auger FA. A completely biological tissue-engineered human blood vessel. FASEB J. 1998;12:47. doi: 10.1096/fasebj.12.1.47. [DOI] [PubMed] [Google Scholar]
- 52.Shum-Tim D, Stock U, Hrkach J, et al. Tissue engineering of autologous aorta using a new biodegradable polymer. Ann Thorac Surg. 1999;68:2298–304. doi: 10.1016/s0003-4975(99)01055-3. [DOI] [PubMed] [Google Scholar]
- 53.Shin-oka T, Breuer CK, Tanel RE, et al. Tissue engineering heart valves: valve leaflet replacement study in a lamb model. Ann Thorac Surg. 1995;60:S513–S16. doi: 10.1016/0003-4975(95)00733-4. [DOI] [PubMed] [Google Scholar]
- 54.Stock UA, Nagashima M, Khalil PN, et al. Tissue engineered valved conduits in the pulmonary circulation. J Thorac Cardiovasc Surg. 2000;119:732–40. doi: 10.1016/s0022-5223(00)70008-0. [DOI] [PubMed] [Google Scholar]
- 55.Jockenhoevel S, Zund G, Hoerstrup SP, et al. Fibrin gel - advantages of a new scaffold in cardiovascular tissue engineering. Eur J Cardiothorac Surg. 2001;19:424–30. doi: 10.1016/s1010-7940(01)00624-8. [DOI] [PubMed] [Google Scholar]
- 56.Li RK, Yau TM, Weisel RD, et al. Construction of a bioengineered cardiac graft. J Thorac Cardiovasc Surg. 2000;119:368–75. doi: 10.1016/S0022-5223(00)70193-0. [DOI] [PubMed] [Google Scholar]
- 57.Li RK, Weisel RD, Mickle DA, et al. Autologous porcine heart cell transplantation improved heart function after a myocardial infarction. J Thorac Cardiovasc Surg. 2000;119:62–68. doi: 10.1016/s0022-5223(00)70218-2. [DOI] [PubMed] [Google Scholar]
- 58.Leor J, Aboulafia-Etzion S, Dar A, et al. Bioengineered cardiac grafts: a new approach to repair the infarcted myocardium. Circulation. 2000;102:III-56–III-61. doi: 10.1161/01.cir.102.suppl_3.iii-56. [DOI] [PubMed] [Google Scholar]
- 59.Yoshikawa T, Ohgushi H, Nakajima H, et al. In vivo osteogenic durability of cultured bone in porous ceramics: a novel method for autologous bone graft substitution. Transplantation. 2000;69:128–34. doi: 10.1097/00007890-200001150-00022. [DOI] [PubMed] [Google Scholar]
- 60.Kaufmann PM, Sano K, Uyama S, et al. Evaluation of methods of hepatotrophic stimulation in rat heterotopic hepatocyte transplantation using polymers. J Pediatr Surg. 1999;34:1118–23. doi: 10.1016/s0022-3468(99)90580-8. [DOI] [PubMed] [Google Scholar]
- 61.Kim SS, Kaihara S, Benvenuto MS, Kim BS, Mooney DJ, Vacanti JP. Small intestinal submucosa as a small caliber venous graft: a novel model for hepatocyte transplantation on synthetic biodegradable polymer scaffolds with direct access to the portal venous system. J Pediatr Surg. 1999;34:124–28. doi: 10.1016/s0022-3468(99)90241-5. [DOI] [PubMed] [Google Scholar]
- 62.Mayer J, Karamuk E, Akaike T, Wintermantel E. Matrices for tissue engineering-scaffold structure for a bioartificial liver support system. J Control Release. 2000;64:81–90. doi: 10.1016/s0168-3659(99)00136-4. [DOI] [PubMed] [Google Scholar]
- 63.Chamberlain LF, Yannas IV, Hsu HP, Spector M. Connective tissue response to tubular implants for peripheral nerve regeneration: the role of myofibroblasts. J Comp Neurol. 2000;417:415–30. doi: 10.1002/(sici)1096-9861(20000221)417:4<415::aid-cne3>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 64.Pollok JM, Begemann JF, Jaufmann PM. Long-term insulin-secretory function of islets of Langerhans encapsulated with a layer of confluent chondrocytes for immunoisolation. Pediatr Surg Int. 1999;15:164–67. doi: 10.1007/s003830050546. [DOI] [PubMed] [Google Scholar]
- 65.Vacanti CA, Langer R, Schloo B, Vacanti JP. Synthetic polymers seeded with chondrocytes provide a template for new cartilage formation. Plast Reconstr Surg. 1991;88:753–59. doi: 10.1097/00006534-199111000-00001. [DOI] [PubMed] [Google Scholar]
- 66.Vanjak-Novakovic G, Freed L, Biron RJ, Langer R. Effects of mixing on the composition and morphology of tissue engineered cartilage. AIChE J. 1996;42:850–60. [Google Scholar]
- 67.Burke JF, Yannas IV, Quimby WCJ, Bondo CC, Jung WK. Successful use of a physiologically acceptable artificial skin in the treatment of extensive burn injury. Ann Surg. 1981;194:413–48. doi: 10.1097/00000658-198110000-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Winfrey ME, Cochran M, Hegarty MT. A new technology in burn therapy: INTEGRA artificial skin. Dimens Crit Care Nurs. 1999;18:14–20. doi: 10.1097/00003465-199901000-00003. [DOI] [PubMed] [Google Scholar]
- 69.Boyce ST, Supp AP, Wickett RR, Hoath SB, Warden GD. Assessment with the dermal torque meter of skin pliability after treatment of burns with cultured skin substitutes. J Burn Care Rehabil. 2000;21:55–63. doi: 10.1097/00004630-200021010-00011. [DOI] [PubMed] [Google Scholar]
- 70.Waymack P, Duff RG, Sabolinski M, Group TABS. The effect of a tissue engineered bilayered living skin analog, over meshed split-thickness autografts on the healing of excised burn wounds. Burns. 2000;26:609–19. doi: 10.1016/s0305-4179(00)00017-6. [DOI] [PubMed] [Google Scholar]
- 71.Grobstein C. Inductive epithelio-mesenchymal interaction in cultured organ rudiments of the mouse. Science. 1953;118:52–55. doi: 10.1126/science.118.3054.52. [DOI] [PubMed] [Google Scholar]
- 72.Ekblom P. Formation of basement membranes in the embryonic kidney: an immunohistological study. J Cell Biol. 1981;91:1–10. doi: 10.1083/jcb.91.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sariola H. Interspecies Chimeras: An Experimental Approach for Studies on Embryonic Angiogenesis. Med Biol. 1985;63:43–65. [PubMed] [Google Scholar]
- 74.Dekel B, Burakova T, Ben-Hur H, et al. Engraftment of human kidney tissue in rat radiation chimera: II. Human fetal kidneys display reduced immunogenicity to adoptively transferred human peripheral blood mononuclear cells and exhibit rapid growth and development. Transplantation. 1997;64:1550–8. doi: 10.1097/00007890-199712150-00008. [DOI] [PubMed] [Google Scholar]
- 75.Rogers SA, Lowell JA, Hammerman NA, Hammerman MR. Transplantation of developing metanephroi into adult rats. Kidney Int. 1998;54:27–37. doi: 10.1046/j.1523-1755.1998.00971.x. [DOI] [PubMed] [Google Scholar]
- 76.Rogers SA, Hammerman MR. Transplantation of rat metanephroi into mice. Am J Physiol. 2001;280:R1865–R69. doi: 10.1152/ajpregu.2001.280.6.R1865. [DOI] [PubMed] [Google Scholar]
- 77.Dekel B, Burakova T, Arditti FD, et al. Human and porcine early kidney precursors as a new source for transplantation. Nat Med. 2003;9:53–60. doi: 10.1038/nm812. [DOI] [PubMed] [Google Scholar]
- 78.Hammerman MR. Transplantation of embryonic organs. Am J Transplant. 2004;4 (Suppl 6):14–24. doi: 10.1111/j.1600-6135.2004.0341.x. [DOI] [PubMed] [Google Scholar]
- 79.Dekel B, Marcus H, Herzel BH, Bocher WO, Passwell JH, Reisner Y. In vivo modulation of the allogeneic immune response by human fetal kidneys: the role of cytokines, chemokines, and cytolytic effector molecules. Transplantation. 2000;69:1470–8. doi: 10.1097/00007890-200004150-00044. [DOI] [PubMed] [Google Scholar]
- 80.Bach FH, Winkler H, Ferran C, Hancock WW, Robson SC. Delayed xenograft rejection. Immunol Today. 1996;17:379–84. doi: 10.1016/0167-5699(96)10024-4. [DOI] [PubMed] [Google Scholar]
- 81.Ogle BM, Platt JL. Genetic therapies and xenotransplantation. Expert Opin Biol Ther. 2002;2:299–310. doi: 10.1517/14712598.2.3.299. [DOI] [PubMed] [Google Scholar]
- 82.Kocher AA, Schuster MD, Szabolcs MJ, et al. Neovascularization of ischemic myocardium by human bone-marrow-derived angioblasts prevents cardiomyocyte apoptosis reduces remodeling and improves cardiac function. Nat Med. 2001;7:430–36. doi: 10.1038/86498. [DOI] [PubMed] [Google Scholar]
- 83.Murry CE, Wiseman RW, Schwartz SM, Hauschka SD. Skeletal myoblast transplantation for repair of myocardial necrosis. J Clin Invest. 1996;98:2512–23. doi: 10.1172/JCI119070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Taylor DA, Atkins BZ, Hungspreugs P, et al. Regenerating functional myocardium: improved performance after skeletal myoblast transplantation. Nat Med. 1998;4:929–33. doi: 10.1038/nm0898-929. [DOI] [PubMed] [Google Scholar]
- 85.Menasche P, Hagege AA, Scorsin M, et al. Myoblast transplantation for heart failure. Lancet. 2001;357:279–80. doi: 10.1016/S0140-6736(00)03617-5. [DOI] [PubMed] [Google Scholar]
- 86.Kasiske BL. The kidney in cardiovascular disease. Ann Intern Med. 2001;134:707–09. doi: 10.7326/0003-4819-134-8-200104170-00014. [DOI] [PubMed] [Google Scholar]
- 87.Ogle BM, Cascalho M, Platt JL. Fusion of approaches to the treatment of organ failure. Am J Transplant. 2004;4 (Suppl 6):74–77. doi: 10.1111/j.1600-6135.2004.0347.x. [DOI] [PubMed] [Google Scholar]
- 88.Cascalho M, Ogle BM, Platt JL. Xenotransplantation and the future of renal replacement. J Am Soc Nephrol. 2004;15:1106–12. doi: 10.1097/01.asn.0000113298.28480.7e. [DOI] [PubMed] [Google Scholar]
- 89.Rugstad HE, Robinson SH, Yannoni C, Tashjian AH. Transfer of bilirubin uridine diphosphate-glucuronyltransferase to enzyme-deficient rats. Science. 1970;170:553–55. doi: 10.1126/science.170.3957.553. [DOI] [PubMed] [Google Scholar]
- 90.Matas AJ, Sutherland DER, Steffes MW, et al. Hepatocellular transplantation for metabolic deficiencies: decrease of plasma bilirubin in Gunn rats. Science. 1976;192:892–94. doi: 10.1126/science.818706. [DOI] [PubMed] [Google Scholar]
- 91.Sutherland DER, Numata M, Matas AJ, Simmons RL, Najarian JS. Hepatocellular transplantation in acute liver failure. Surgery. 1977;82:124–32. [PubMed] [Google Scholar]
- 92.Vons C, Loux N, Simon L, et al. Transplantation of hepatocytes in nonhuman primates: a preclinical model for the treatment of hepatic metabolic diseases. Transplantation. 2001;72:811–18. doi: 10.1097/00007890-200109150-00012. [DOI] [PubMed] [Google Scholar]
- 93.Fox IJ, Chowdhury JR, Kaufman SS, et al. Treatment of the Crigler-Najjar syndrome type I with hepatocyte transplantation. New Eng J Med. 1998;338:1422–26. doi: 10.1056/NEJM199805143382004. [DOI] [PubMed] [Google Scholar]
- 94.Kobayashi N, Fujiwara T, Westerman KA, et al. Prevention of acute liver failure in rats with reversibly immortalized human hepatocytes. Science. 2000;287:1258–62. doi: 10.1126/science.287.5456.1258. [DOI] [PubMed] [Google Scholar]
- 95.Reid LM. Stem cell/lineage biology and lineage-dependent extracellular matrix chemistry: keys to tissue engineering of quiescent tissues such as liver. Lanza R, Langer R, Chick W Eds. In: Textbook of tissue engineering. New York, Landes 1996; 481-518.
- 96.Lagasse E, Connors H, Al-Dhalimy M, et al. Purified hematopoietic stem cells can differentiate into hepatocytes in vivo. Nat Med. 2000;6:1229–34. doi: 10.1038/81326. [DOI] [PubMed] [Google Scholar]