

Abstract
Expanded understanding of the factors that direct polypeptide ion fragmentation can lead to improved specificity in the use of tandem mass spectrometry for the identification and characterization of proteins. Like the fragmentation of peptide cations, the dissociation of whole protein cations shows several preferred cleavages, the likelihood for which is parent ion charge dependent. While such cleavages are often observed, they are far from universally observed, despite the presence of the residues known to promote them. Furthermore, cleavages at residues not noted to be common in a variety of proteins can be dominant for a particular protein or protein ion charge state. Motivated by the ability to study a small protein, turkey ovomucoid third domain, for which a variety of single amino acid variants are available, the effects of changing the identity of one amino acid in the protein sequence on its dissociation behavior were examined. In particular, changes in amino acids associated with C-terminal aspartic acid cleavage and N-terminal proline cleavage were emphasized. Consistent with previous studies, the product ion spectra were found to be dependent upon the parent ion charge state. Furthermore, the fraction of possible C-terminal aspartic acid cleavages observed to occur for this protein was significantly larger than the fraction of possible N-terminal proline cleavages. In fact, very little N-terminal proline cleavage was noted for the wild-type protein despite the presence of three proline residues in the protein. The addition/removal of proline and aspartic acids was studied along with changes in selected residues adjacent to proline residues. Evidence for inhibition of proline cleavage by the presence of nearby basic residues was noted, particularly if the basic residue was likely to be protonated.
Keywords: protein ion dissociation, ion/ion reactions, proline cleavage, top-down proteomics
Introduction
The development of electrospray ionization1 and matrix-assisted laser desorption2 has made mass spectrometry a highly suitable method for the study of large biomolecules. In particular, protein identification and characterization have greatly benefited from the rapid, sensitive, and specific nature of mass spectrometric analyses. To date, there are two general approaches to protein analysis using mass spectrometry. “Bottom-up” methods are the most widely used, and involve some form of digestion and separation followed by mass spectrometry. The peptides produced from bottom-up techniques are subjected to peptide mass fingerprinting3–5 (measurement of the peptide masses) or fragment ion analyses6,7 (tandem mass spectrometry of the peptide ions), which take advantage of the accurate mass measurements afforded by mass spectrometry. “Top-down” methodologies represent a second type of strategy for characterizing and identifying proteins, whereby tandem mass spectrometry is performed on intact protein ions. Most top-down applications have been performed using a high-resolution Fourier transform ion cyclotron resonance (FTICR) mass spectrometer.8–10 However, top-down measurements have also been performed with other types of tandem mass spectrometers11,12 including quadrupole ion traps. Ion/ion proton-transfer reactions have been used in the latter instrumentation to facilitate whole protein ion MS/MS.11–14 Gas-phase ion/ion reactions13 are useful for the manipulation of both precursor14,15 and product ion16 charge states. In particular, the manipulation of precursor ion charge states is desirable because various “preferred cleavages” can be observed from collision-induced dissociation (CID) of intact protein ions, depending on the charge state of the precursor ion.17–21
In general, very low charge states (i.e., the number of charges is less than the number of arginine residues in a protein) of protein ions predominantly lose small molecules (i.e., NH3 or H2O) when subjected to ion trap CID. Low charge states (i.e., the number of charges is equal to or slightly greater than the number of arginine residues) of protein ions fragment preferentially C-terminal to aspartic acid residues. Typically, a variety of product ions formed from nonspecific amide backbone fragmentation are observed when intermediate charge states of protein ions are dissociated. The highest charge states of protein ions formed via electrospray have been shown to produce fragment ions corresponding to cleavage N-terminal to proline residues. However, major contributions to the overall fragmentation of high charge states of protein ions by specific, uncommonly observed cleavages have been noted in prior studies.22 For example, dominant L/S cleavage (dissociation between leucine and serine residues to yield b- and y-type ions) is observed for +15 to +21 apomyoglobin,20 and dominant L/M cleavage is observed for +14 and +15 cytochrome c.21 The dissociations of highly charged proteins are therefore less subject to generalization than the dissociation of intermediate and low charge states.
It is noteworthy that ions from N-terminal proline cleavages are not as universally observed in whole protein ion MS/MS spectra as are ions from C-terminal aspartic acid cleavages. For example, in the study of ion trap CID fragmentation of 10 intact proteins, all possible C-terminal aspartic acid cleavages were observed for five proteins and only one of the possible C-terminal aspartic acid cleavages was not observed for four other proteins.18–21,24–30 In this same data set, all possible N-terminal proline cleavages were noted for only one protein and all but one proline-related cleavages were observed for 2 of the 10 proteins. Although the reason for fewer observations of N-terminal proline cleavages in dissociation of protein ions is not well understood, recent investigations of peptide fragmentation suggest that the structure and identity of the amino acid residue preceding proline play roles in determining the preference for fragmentation N-terminal to proline.31,32
When attempts are made to understand the fragmentation behavior of intact protein ions, it is desirable to assess how sensitive the dissociation process is to variations in the amino acid sequence of a protein. If the position and the identity of the variant amino acid substitutions can be chosen, then the association of “preferred cleavage sites” with specific residues can be investigated. In general, many of the preferred cleavages commonly observed in the CID of peptide ions have also been seen in CID of multiply charged, intact protein ions. Examples of these include cleavage C-terminal to aspartic acid and cleavage N-terminal to proline.33,34 Replacement of a residue with an aspartic acid residue or proline residue may result in the introduction of a new fragmentation pathway adjacent to the site of replacement when the variant is subjected to CID. Additionally, the change of one amino acid in the protein sequence could potentially alter proton mobility and protein ion structure, both of which are believed to play important roles in the dissociation of intact protein ions.20 In contrast to a previous study demonstrating the utility of ion trap CID and ion/ion chemistry for use in confirming site-directed mutagenesis products,35 the focus of this work is the fragmentation observed for whole proteins differing by a single amino acid residue at various locations in the wild-type protein sequence. The set of single amino acid substituted proteins studied here allows assessment of the effects of aspartic acid residues, proline residues, and basic residues on the fragmentation behavior of intact protein ions. This information is relevant to top-down protein analysis via the role unimolecular protein ion dissociation plays in protein identification and characterization.
The availability of 191 variants of turkey ovomucoid third domain (OMTKY3) provided the opportunity to study the effects of selected single amino acid replacements on the fragmentation of whole protein ions. The sequence of OMTKY3 is given in Chart 1. The 191 variants consist of the wild-type and single substitutions of all possible 19 amino acid residues at each of the 10 boxed positions. OMTKY3 proteins are part of the Kazal family of serine protease inhibitors. The amino acid variants were obtained by recombinant DNA technology to study the effects of single amino acid substitution on the inhibition of serine proteases.36,37 These variants were prepared using enzymology in such a way that the first five N-terminal residues were eliminated in the final protein product.37 Of particular interest for protein identification is the effect of substitutions involving residues known to be involved in preferred dissociation pathways. Therefore, emphasis has been placed on variants involving aspartic acid residues and proline residues. Fortuitously, some variants also allowed the effect of basic residues on OMTKY3 protein ion dissociation to be studied. The charge state dependent fragmentation of these OMTKY3 protein variants is also described herein.
Chart 1.
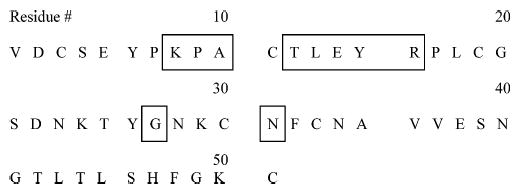
Amino Acid Sequence of the OMTKY3 Wild-Type Proteina
a The boxed areas represent positions in the sequence for which amino acid variants are available.
Materials and Methods
The single amino acid variant proteins were expressed in Escherichia coli, isolated, and purified as described previously.36 Stock solutions of the respective variants (~180 μM) were made by dissolving the protein in water. Dithiothreitol (DTT), guanidine hydrochloride, tris(hydroxymethyl)aminomethane (Tris), and iodoacetic acid were obtained from Sigma (St. Louis, MO). Ethylenediaminetetraacetic acid (EDTA) and perfluoro-1,3-dimethylcyclohexane (PDCH) were purchased from Aldrich (Milwaukee, WI). Trifluoroacetic acid (TFA) and tris(2-carboxyethyl)phosphine hydrochloride (TCEP·HCl) were obtained from Pierce (Rockford, IL). Acetonitrile, methanol, and glacial acetic acid were purchased from Mallinckrodt (Phillipsburg, NJ). A Barnstead (Dubuque, IA) Nanopure system was employed to purify water (~17.0 MΩ cm) for use in the preparation of sample and buffer solutions.
The procedure for reduction was adapted from that used previously.23 The protein of interest (0.05 mL of stock solution) was placed in a 0.5 M Tris·HCl buffer (pH 8.5) containing 6 M guanidine hydrochloride, 5 mM EDTA, and 0.5 mM DTT. The sample solution was heated in a water bath for 2 h at 50 °C to facilitate reduction of the protein disulfide bonds. Reduced protein samples were purified by reversed-phase high-performance liquid chromatography (HPLC) on a Hewlett-Packard (Palo Alto, CA) model 1090 instrument. A Perkin-Elmer Brownlee (Wellesley, MA) Aquapore RP-300 (7 μm pore size, 100 × 4.6 mm i.d.) column was used. Buffer A consisted of aqueous 0.1% TFA, while buffer B was composed of 60:40 (acetonitrile/water) containing 0.09% TFA. A linear 70 min gradient from 0 to 100% B with a flow rate of 1 mL/min was used. The column temperature was maintained at 40 °C. The UV absorbance was monitored at 215 nm. Commonly, the protein samples of interest eluted in the range of 35–37 min. Fractions containing the protein samples were collected and lyophilized to dryness using a Savant Speedvac (Holbrook, NY). Prior to mass spectrometric analysis, the protein samples were reconstituted with 50:50 (methanol/water) containing 1% acetic acid. For some of the OMTKY3 proteins studied, reduction of disulfide bonds was performed in a different manner. The reduction was accomplished by adding 50 μL of aqueous TCEP·HCl (12000 μM) to a 50 μL aqueous native protein (40 μM). The solution was allowed to react for 1 h at 45 °C. A 50 μL aliquot of the reduced protein solution was diluted with 50 μL of methanol, and 1% acetic acid was added. This protein (10 μM) solution was used for mass spectrometric analysis without further purification. Many of the same fragment ions were observed for a given protein ion regardless of the method of disulfide bond reduction.
The mass spectrometry experiments were carried out using Finnigan (San Jose, CA) ITMS38 and Hitachi (San Jose, CA) M-800039 quadrupole ion traps modified for ion/ion reactions as described previously. Proton-transfer reactions with anions derived from PDCH were used to generate protein ion charge states below +5. These reactions were also used to reduce the charge state of product ions from CID experiments. On-resonance collisional activation was used for dissociation of selected ions. Mass analysis was performed using resonance ejection. Post-ion/ion MS/MS spectra were typically the average of 500 scans. A Sciex Q-Star (Toronto, Canada) quadrupole time-of-flight instrument was used to provide beam-type fragmentation data of selected variants. The beam-type MS/MS spectra were the average of 600 scans.
The contribution of a specified fragmentation channel relative to all other fragmentation channels was determined by the following method. Using Origin (Version 6.1, OriginLab Corp., Northampton, MA), a five-point, first-degree polynomial Savitzky–Golay smoothing of the data was performed. This was followed by a baseline correction and normalization of product ion abundances. The normalization factor was based on the abundance of the most prominent product ion in each MS/MS spectrum. Next, the peak-finding function of Origin was used to identify product ions with at least 5% of the abundance of the most prominent product ion. Assignments of b and y ions corresponding to amide bond cleavage were made for this group of product ions. A fragment ion mass tolerance window of ±10 Da was used in making the b and y ion assignments. The abundances of complementary b and y ions assigned in the MS/MS spectra were summed. The summed abundances of b and y ions for a given fragmentation channel were divided by the total abundance of all assigned ions. This number was multiplied by 100 to give the percentage contribution of a specific fragmentation channel to the overall fragmentation, as assigned from the MS/MS spectra. Such calculations were performed for MS/MS data from charge states of selected variants. The resulting percentages were used when the dissociation behavior of those single amino acid variants were compared.
Results and Discussion
The amino acid sequence of the OMTKY3 wild-type is given in Chart 1. The positions in the sequence for which variants are available are indicated. Variants are referred to herein by the single-letter code for the amino acid residue in the wild-type sequence at the site of substitution, the position of substitution, and the single-letter code for the replacement amino acid residue. The variant name consistent with that in previous studies of OMTKY3 proteins is given in parentheses the first time the variant is discussed in the text and in related figures.37 For example, the variant P9H (P14H) is one in which the proline at position 9 in the wild-type protein was replaced with histidine.
The presence of disulfide bonds in these proteins allows further study of the effect of disulfide bond modifications on fragmentation patterns of intact protein ions. OMTKY3 proteins have three disulfide bonds, viz., between residues 3 and 33, 11 and 30, and 19 and 51. Thus, only two residues near the N-terminus are outside the disulfide bond linkages. Versions of the proteins with nonreduced and reduced disulfide bond linkages were studied. Protein ions with intact disulfide bonds are referred to herein as “native”, although the gas-phase ion structure may not resemble that of the native protein conformation. OMTKY3 proteins in which the disulfide bonds were cleaved are specified as “reduced”.
CID of Native OMTKY3 Protein Ions.
CID of native OMTKY3 wild-type and P9H (P14H) ions ranging in charge state from +6 to +2 was performed. Consistent with observations from previous dissociation studies of protonated protein ions with intact disulfide bonds,19,24,25 there was little or no evidence for fragmentation within the regions of the respective protein ions bound by disulfide bonds. The y49 product ion was the major product ion in the MS/MS spectra of all native OMTKY3 ions studied, regardless of the protein ion charge state. This product ion corresponds to cleavage C-terminal to the aspartic acid residue located outside the protein’s disulfide bonds (D/C). Given these observations and the fact that single amino acid variants for positions outside the part of the protein sequence protected by disulfide bonds were not available, further studies of the dissociation of multiply protonated native OMTKY3 protein ions were not pursued.
CID of Reduced OMTKY3 Wild-Type Ions.
In general, CID of protonated protein ions with reduced disulfide bonds yields more fragmentation relative to that of protein ions with intact disulfide bonds.19,24 New fragmentation channels are observed from the regions formerly protected by disulfide bonds. Since the site of amino acid substitution for the OMTKY3 variants is within disulfide bond loops, dissociation of the reduced versions of the variants should allow the effect of amino acid substitution on their fragmentation behavior to be more readily apparent. The fragmentation behavior of reduced OMTKY3 variants was investigated for charge states ranging from +3 to the highest observed in the ESI distribution for several variants. The results of this study are described below in detail. The charge state dependent fragmentation of reduced OMTKY3 wild-type ions is related first. This is followed by data from MS/MS of OMTKY3 variants that demonstrate the effect of single amino acid substitution on the dissociation of intact protein ions.
Unlike the fragmentation behavior of native OMTKY3 wild-type ions, the identities and relative abundances of the major product ions observed for reduced ions were dependent on the parent ion charge state. The charge state dependent fragmentation of reduced OMTKY3 wild-type ions is summarized in the dissociation map shown in Figure 1. The dissociation map displays the contributions of fragmentation at a particular position in the protein’s amino acid sequence relative to the overall fragmentation observed for each charge state studied. One of the noteworthy trends in Figure 1 is that the majority of fragmentation observed for OMTKY3 wild-type ions is concentrated in about six fragmentation channels for each charge state. Although they differ in abundance for the respective charge states, most of the dominant fragmentation channels are similar for all charge states. This contrasts with previous studies of charge state dependent fragmentation studies of larger intact protein ions in that major fragmentation channels observed for high charge states are not generally the same ones observed for low charge states. In most of the previous studies, there are a limited number of abundant fragmentation channels for the lowest and highest charge states, and a greater number of active fragmentation channels for intermediate charge states.20,21,26,27 Although more fragmentation channels are observed for the +5 and +6 charge states of the OMTKY3 wild type, most of the new fragmentation channels contribute <5% to the total fragmentation.
Figure 1.
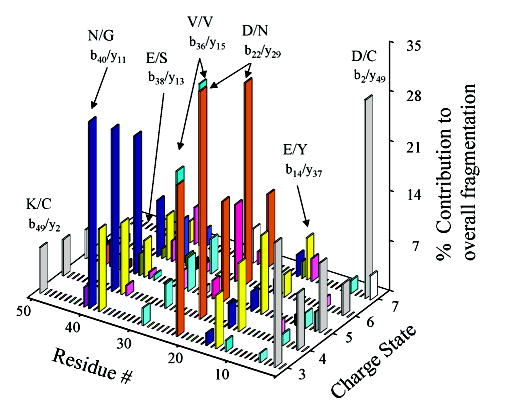
Dissociation map summarizing the charge state dependent fragmentation observed for reduced OMTKY3 wild-type ions.
When the charge state dependent fragmentation behavior of OMTKY3 wild-type ions is compared to that of intact protein ions previously examined, there are both similarities and significant differences. Fragments corresponding to cleavage C-terminal to aspartic acid residues are prominent, as is often the case for protein ions of relatively low charge,18,20,21 but they are prominent for all charge states of OMTKY3 wild-type ions investigated. The preference for the aspartic acid related cleavages may be related to proton mobility in a manner similar to that described for the fragmentation of apomyoglobin ions.20 The large contribution of the D/C cleavage for the +3 parent ion can be rationalized by the localization of protons on the basic sites of the protein such that charge-remote cleavages, such as cleavage C-terminal to acidic residues, are prevalent. The reason for the dominant D/C cleavage for +7 ions may be related to protonation of the N-terminus and the propensity for formation of a b2 ion upon fragmentation when the N-terminus is charged. It is interesting to speculate that secondary structural effects may play a role in the propensity for cleavage adjacent to D22, which accounts for more than 10% of the total fragmentation in all charge states studied. According to structures of OMTKY3 proteins obtained by NMR, D22 is situated between β sheets of residues 16–21 and 23–27, respectively.40 If the structure of the gas-phase ion in this region of the protein resembles that of the protein in solution, this position may promote facile cleavage of the amide bond to alleviate the stress present in such a position. However, little is known about the secondary structure of gas-phase protein ions, especially when the ions are at the elevated temperatures associated with collisional activation.
In terms of “preferred” cleavages for peptide and protein ions other than those C-terminal to aspartic acid residues, it is interesting that product ions from amide bond cleavage N-terminal to proline are not prominent in the MS/MS spectra of the OMTKY3 wild type for any parent ion charge state. There are three possible sites from which such product ions could arise: Y6-P7-K8-P9 and R16-P17. Fragmentation between any of the adjacent residues within the series Y6-P7-K8-P9 is observed only for +5 and +6 parent ions and represents less than 2% of the overall fragmentation. The b16+ ion, arising from cleavage of the R/P bond, is observed for all charge states, although its contribution to the overall fragmentation is less than 5% for each respective charge state. These results differ from previous dissociation studies of intact protein ions, where products from N-terminal proline cleavages are frequently observed as major ions in the MS/MS spectra.18,19,21,27 However, fragmentation N-terminal to each proline residue in a given protein sequence is not always observed. In fragmentation studies of intact protein ions, Y/P cleavage was observed to be a major fragmentation channel for high charge states of β-hemoglobin ions26 while fragmentation of adjacent lysine and proline residues (viz., K/P cleavage) was less common.19,20,26,28–30 No proteins for which the fragmentation behavior as a function of charge state has been examined, other than the OMTKY3 wild type, have had adjacent arginine and proline residues in their sequences. The reason for the minimal effect of N-terminal proline fragmentation, therefore, may be related to the presence of basic residues adjacent to proline.
In some of the previous charge state dependent fragmentation studies of whole protein ions, a preference for C-terminal lysine cleavages was noted.20,21 Active, low-abundance dissociation channels involving lysine and cysteine residues were observed for all charge states of OMTKY3 proteins other than +7. Product ions were also observed for cleavage C-terminal to lysine between residues L24 and T25. In the case of OMTKY3 wild-type ions, amide bond cleavage C-terminal to lysine residues is more prominent than cleavage N-terminal to proline residues. The trend of observing more C-terminal lysine cleavages than N-terminal proline cleavages was also noted in the fragmentation of apomyoglobin ions.20
CID of Reduced OMTKY3 Variant Ions.
Single amino acid variants of OMTKY3 were selected for dissociation studies to explore the effects of the substitutions on the fragmentation behavior of the respective variants. The charge state dependent fragmentation of OMTKY3 wild-type ions was described above to serve as a basis for comparison. Attention was focused particularly on the commonly observed cleavages C-terminal to aspartic acid and N-terminal to proline. Results are related here first for variants selected to shed light on C-terminal aspartic acid cleavages and second for variants selected to investigate N-terminal proline cleavages.
(1) Aspartic Acid Related Substitutions.
When an aspartic acid containing OMTKY3 variant was selected for the dissociation studies, the identity of the wild-type residue replaced was not a primary consideration. Observations of dominant products from cleavages C-terminal to aspartic residues, independent of the identity of neighboring residues,18–21,24–30 in protein dissociation spectra suggest that the propensity for this type of fragmentation is relatively insensitive to the local sequence of residues. Rather, it was desirable to select a variant that possessed an aspartic acid residue in an area of the protein sequence from which minimal fragmentation was observed (within the constraints of the positions for which variants were available (see Chart 1)). Having established such a criterion, the variants L13D (L18D) and N31D (N36D) were chosen and the charge state dependent fragmentation of these two aspartic acid containing variants was examined.
In the OMTKY3 variant L13D, the effect of the replacement on the fragmentation of the [M + 3H]3+ ion is demonstrated in Figure 2a. A new fragmentation channel is observed that corresponds to amide bond cleavage C-terminal to aspartic acid at the site of substitution. This fragmentation channel, representing cleavage between D13 and E14, accounts for ~30% of the total fragmentation observed for the +3 ion. The abundance of the D/C and D/N fragmentation channels is lower for the L13D ion than for the wild-type ion. Furthermore, the new D/E cleavage channel also affects the neighboring E/Y channel. A preference for D/E cleavage over E/Y cleavage is not surprising, based on the longer side chain in glutamic acid residues compared to aspartic acid residues. The increased side chain length results in less favorable proton transfer from the side chain to the amide backbone, a key step in the mechanism for cleavage C-terminal to acidic residues.41 Most likely, factors other than the mechanistic differences arising from acidic side chain length largely cancel since the aspartic and glutamic acid residues are adjacent in this portion of L13D. Despite the large contributions from cleavages associated with aspartic acid for L13D, the abundance of the N/G fragmentation channel is within 5% of that observed for the OMTKY3 wild type. The introduction of a new channel for C-terminal aspartic acid cleavage occurs at the expense of cleavages at other acidic residues but not at the expense of the preferential fragmentation of N/G observed for OMTKY3 protein ions. It is apparent that this single amino acid substitution does not significantly affect the structural and energetic features that give rise to the N/G fragmentation. Furthermore, and perhaps not surprisingly, the new fragmentation channel competes more directly with other acidic amino acid channels than it does with the N/G fragmentation channel.
Figure 2.
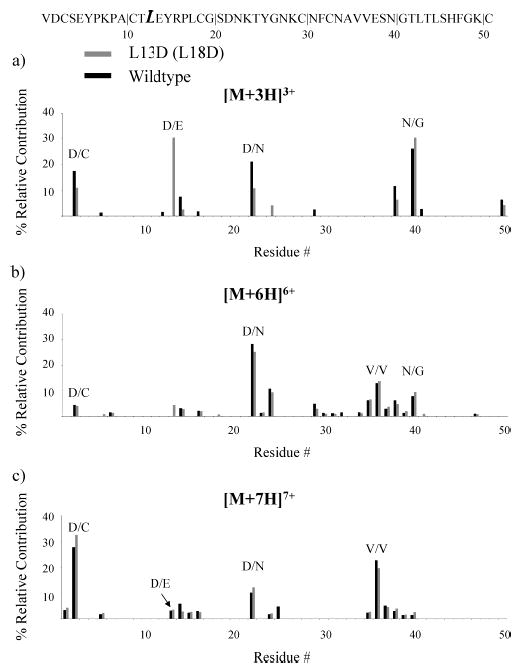
Summary of the fragmentation channels observed for reduced OMTKY3 L13D (L18D) (light) and the wild type (dark) for (a) [M + 3H]3+ ions, (b) [M + 6H]6+, and (c) [M + 7H]7+ ions.
In the dissociation behavior of [M + 4H]4+ and [M + 5H]5+ L13D ions, the C-terminal aspartic acid cleavage at the site of variation remains active (Supporting Information Figures 1 and 2). Other than this new channel, the major fragmentation channels are the same as those observed for the +4 and +5 wild-type ions. As shown in Figure 2b,c, there are no significant differences in the fragmentation patterns for the +6 and +7 ions of the OMTKY3 wild type and L13D. The D/N, V/V, and D/C (+7) cleavages are major fragmentation channels for both the L13D and wild-type ions. In light of decreasing contributions from the D/E channel that was introduced via L13D as the charge state is increased, the comparable abundances of D/N and D/C cleavages in the L13D and wild-type ions suggest that phenomena other than simply the presence of an aspartic acid residue are significant factors in the prominent D/N and D/C cleavages for high charge states.
Several key points can be discerned from the information present in Figure 2. Alteration of the protein ion sequence to include an additional location for potential aspartic acid cleavage affects the fragmentation behavior of low charge states, but does not significantly impact the fragmentation of high charge states. This is consistent with the C-terminal aspartic acid cleavage observed at the site of substitution being charge-remote, as described above. The fact that the relative contributions of the D/C cleavages are within 5% of one another for the +7 L13D and wild-type ions supports the notion that this cleavage is more likely related to protonation of the N-terminus and formation of a b2 ion than a charge-remote fragmentation process. Evidence substantiating this claim is also provided in the data for dissociation of the +3 ion, where it appears that the new D/E channel is competitive with the D/C channel. Other than the addition of the new D/E fragmentation channel, the replacement of a leucine residue with an aspartic acid residue does not dramatically alter the major dissociation pathways observed for OMTKY3 protein ions. In other words, the data in Figure 2 suggest that the impact of substitution of an aspartic acid residue is localized to the site of the replacement.
To assess the generality of the above observations, a different aspartic acid containing OMTKY3 variant (N31D) was subjected to CID studies. Consistent with the observations reported for L13D, a new fragmentation channel (D/F) representative of cleavage C-terminal to aspartic acid at the site of substitution was noted for N31D. As shown in Figure 3a, the contribution from this new channel is greater than 10% of the total fragmentation, although N/G remains the largest fragmentation channel for N31D. It is noteworthy that the abundances for the E/Y channel are similar for N31D and the wild-type ion. This contrasts with the reduced abundance of the E/Y (b14/y37) channel for +3 L13D ions (Figure 2a). In the case of N31D, however, the aspartic acid and glutamic acid residues are separated by 16 residues and are therefore likely to find themselves in dissimilar structural and charge site environments. Disregarding the new D/F fragmentation channel, the overall distribution of fragmentation is similar for +3 and +4 N31D and wild-type ions. The contribution from C-terminal aspartic acid cleavage at the site of substitution is lower for +5 N31D (Figure 3b) than for +4 and +3 N31D, which is consistent with it being a charge-remote fragmentation process.
Figure 3.
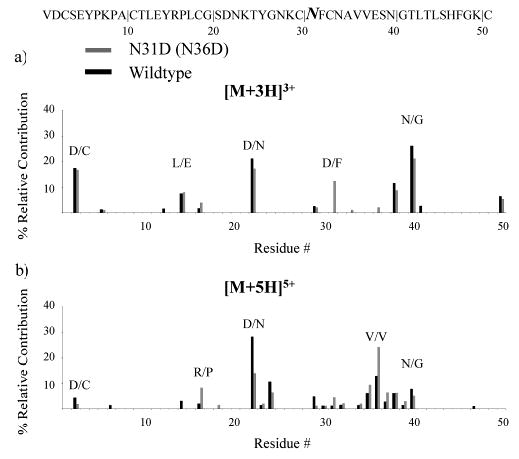
Summary of the fragmentation channels observed for reduced OMTKY3 N31D (N36D) (light) and the wild type (dark) for (a) [M + 3H]3+ ions and (b) [M + 5H]5+ ions.
For the most part, similar effects of substituting an aspartic acid residue for another residue in the sequence of OMTKY3 proteins were noted for N31D and L13D variants. In both cases, fragmentation channels corresponding to C-terminal aspartic acid cleavages at the site of substitution were “switched on” for lower charge state ions. Major fragmentation channels in the charge state dependent fragmentation of OMTKY3 ions, such as N/G or V/V, were not significantly diminished as a result of the additional aspartic acid residue in the sequence. However, the MS/MS spectra were not very similar for all charge states of both variants. For example, the fragmentation channels and respective abundances for the highest charge states of L13D correlated well with those of the wild-type ions, while the relative contributions of particular fragmentation channels for the highest charge states of N31D did not parallel closely those of the wild-type ions. This observation highlights the complex relationships between the various factors that play roles in determining protein ion fragmentation patterns. A single amino acid substitution can alter the local probability for a cleavage and can also alter secondary and tertiary structure. While the roles that the latter structural characteristics play in affecting protein ion fragmentation are not well understood, the growing body of protein ion dissociation data strongly suggests that charge state and primary structure alone cannot account for protein ion dissociation patterns.
(2) Proline Related Substitutions.
(a) Additional Proline Introduction.
A clear contrast in the charge state fragmentation behavior of wild-type OMTKY3 ions with that of many other proteins studied under similar conditions is the relative absence of N-terminal proline cleavage at any charge state. While this is not unexpected for low charge states, N-terminal proline cleavage is common at high charge states. A characteristic common to all of the proline residues in the wild-type sequence is the presence of a relatively basic residue adjacent to the proline. For example, a lysine residue is present between P7 and P9, while the only arginine residue in the sequence is N-terminal to P17 (Chart 1). Therefore, the fragmentation behavior of a variant containing an additional proline residue in a location where no basic residues were present in the primary sequence was examined. In particular, we chose L13P (L18P) to facilitate comparisons with the L13D variant described above. There were many similarities among the types of fragmentation channels observed for the L13P and wild-type ions. The major difference was that fragmentation representative of cleavage N-terminal to proline at the site of substitution was observed for +5 to +7 L13P ions. The contribution from this channel increased with parent ion charge state. A summary of the fragmentation behavior of +7 L13P, the parent ion for which the contribution from this new fragmentation channel was most abundant, is shown in Figure 4. Therefore, the addition of a proline relatively remote from a basic site in the polypeptide gave rise to the largest contribution from N-terminal proline cleavage of the OMTKY3 species described thus far.
Figure 4.
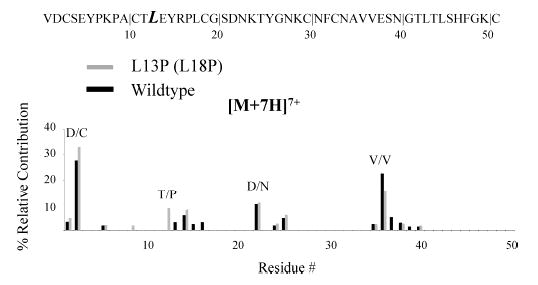
Summary of the fragmentation channels observed for reduced OMTKY3 L13P (L18P) (light) and the wild type (dark) for [M + 7H]7+ ions.
(b) Removal of Arginine Adjacent to Proline.
The results for L13P lend credence to the possibility that the presence of nearby basic residues inhibits N-terminal cleavages for the proline residues present in the wild-type protein. To further explore this possibility, the fragmentation of R16A (R21A) was studied. In this variant, the most basic amino acid residue, arginine, was replaced with an aliphatic amino acid residue. Unlike the other OMTKY3 variants, the fragmentation patterns of R16A ions are quite different from those of the corresponding wild-type ions. A summary of the fragmentation observed for +5 and +6 wild-type and R16A ions is shown in Figure 5. The fragmentation channel corresponding to cleavage N-terminal to proline adjacent to the site of substitution is dominant for +5 R16A (Figure 5a). The trend continues in a comparison of the +6 R16A and wild-type ions (Figure 5b) as the fragmentation of R16A is concentrated in fewer and different channels relative to that of the wild type. The D/N, K/T, and V/V channels are the largest for the +6 wild type, while L/E/Y, A/P, and V/V are the largest for +6 R16A (Figure 5b). This variant is noteworthy in that, unlike most of the others, the change of arginine to alanine at residue 16 effectively shut down more than one of the major fragmentation channels for +6. Since the alanine residue replaced one of the basic sites, no +7 parent ions were observed for R16A. One of the major differences in the fragmentation of R16A relative to the other variants is the reduced contribution from D/N to the fragmentation of R16A. It is the major cleavage site for the +6 wild-type ion, but this fragmentation channel is not active for +6 R16A. Clearly, the substitution of alanine for arginine demonstrates that replacement of the most basic residue adjacent to proline can have a dramatic impact on the observed fragmentation for OMTKY3 protein ions. This result lends further support to the possibility that the presence of a basic residue near a proline can inhibit N-terminal proline cleavage.
Figure 5.
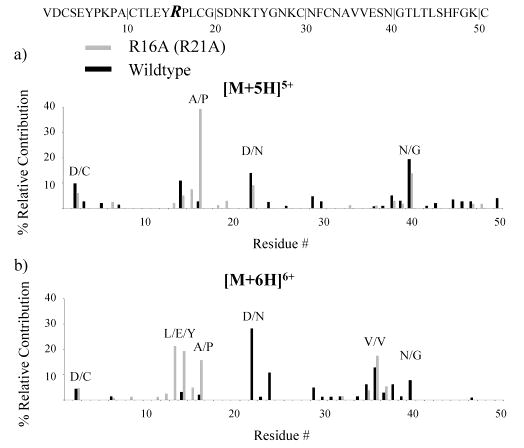
Summary of the fragmentation channels observed for reduced OMTKY3 R16A (R21A) (light) and the wild type (dark) for (a) [M + 5H]5+ ions and (b) [M + 6H]6+ ions.
Cleavage of the A/P bond in tryptic peptides is not particularly favored.31,42 However, it is more commonly observed than fragmentation of the R/P bond. This may not be entirely due to the basicity of arginine since preferential cleavage of the H (second most basic amino acid residue)/P bond has been noted.31,32,42,43 Adjacent arginine and proline residues have not been present in the sequences of other intact proteins for which charge state dependent fragmentation has been characterized. The A/P linkage was part of the sequence for four intact proteins studied previously.21,28,29 Fragmentation N-terminal to proline was only observed for two of the four proteins, and the corresponding product ions were not dominant ones in the MS/MS spectra.21,29 Therefore, the presence or absence of a proximate basic residue in the primary sequence does not appear to be the only factor in determining the extent to which N-terminal proline cleavage is observed in highly charged protein ions. However, in the context of observations from the dissociation of OMTKY3 ions, replacement of arginine with alanine resulted in “turning on” a dominant fragmentation channel associated with N-terminal proline cleavage.
(c) Replacement of Lysine or Arginine Adjacent to Proline with a Histidine Residue.
Given the reportedly high tendency for H/P cleavages in peptide ions31,32,42,43 and the observation that cleavage of the R/P bond in OMTKY3 is not dominant for any charge state and that replacement of arginine with an alanine residue yields an abundant A/P cleavage for high charge states of OMTKY3, we examined variants of OMTKY3 in which H/P bonds were present. Specifically, we examined the K8H (K13H) and R16H (R21H) variants. Furthermore, both ion trap and beam-type MS/MS spectra were obtained with these variants to determine if there is a significant dissociation time frame dependence to the preferred cleavages. Product ions resulting from the cleavage of the H/P bond are not observed in the ion trap CID spectra of K8H for charge states +5 to +7 (Supporting Information Figures 3–5). Similar results were obtained from the beam-type dissociation experiments. Fragment ions from the H/P linkage in R16H are observed from the +5 ions, but not from the highest charge states of this variant (Supporting Information Figures 6–8). As an example, the beam-type fragmentation spectra of the highest charge state, +7, of the OMTKY3 wild type and R16H are shown in Figure 6. The major product ions for the wild-type (Figure 6a) and R16H (Figure 6b) ions are the same, and neither show N-terminal proline cleavage products. The observation of N-terminal proline cleavage of the H/P bond in the +5 ion of the R16H variant and the lack of such cleavage of the R/P bond in the +5 ion of the wild-type protein suggest that there may be a greater tendency for H/P cleavage than for R/P cleavage. However, the fact that the higher charge states of both H protein variants and the wild type lack N-terminal proline cleavages may be revealing as to the nature of the influence of a basic residue in inhibiting N-terminal proline cleavage. The H/P fragmentation channel in the R16H variant occurs at an intermediate charge state, in which it is less likely that the histidine is protonated. Therefore, inhibition of N-terminal proline cleavage by a basic residue may be dependent upon it being protonated.
Figure 6.
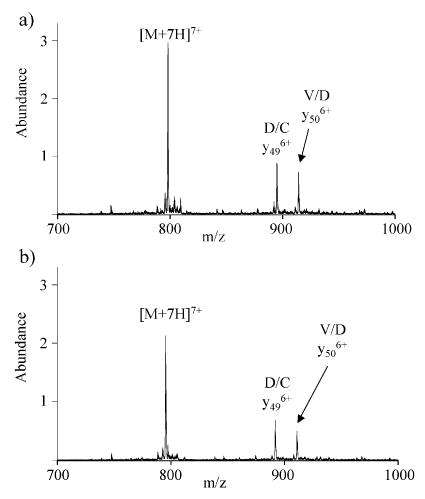
Beam-type fragmentation of (a) wild-type [M + 7H]7+, 33 eV, and (b) R16H (R21H) [M + 7H]7+, 33 eV.
Figure 8.

Summary of the fragmentation channels observed for reduced OMTKY3 P9H (P14H) (light) and thewild type (dark) for [M + 7H]7+ ions.
(d) Replacement of Lysine or Arginine Adjacent to Proline with Aspartic Acid.
The most strongly favored cleavage of collisionally activated protonated peptide and protein ions is observed for aspartic acid residues positioned N-terminal to proline residues. The factors that favor C-terminal aspartic acid cleavage and N-terminal proline cleavage are both present. The dissociation behavior of K8D (K13D) and R16D (R21D) variants was therefore studied to compare with results for the K8H, R16H, and R16A variants.
Product ions corresponding to cleavage of the newly introduced D/P bonds were present in both ion trap MS/MS and beam-type MS/MS product ion spectra for some, but not all, of the charge states of K8D and R16D, respectively (Supporting Information Figures 9–16). The data from these experiments provided further evidence that introduction of an aspartic acid residue in the protein sequence can promote preferential cleavage C-terminal to the new aspartic acid residue regardless of the location of introduction in the amino acid sequence. The fragmentation pattern observed for +6 K8D was particularly noteworthy, as shown in the beam-type fragmentation spectrum of this ion in Figure 7. Interestingly, the major fragment ion was not from cleavage of the D/P bond (although abundant products from this cleavage were observed) as was the case for +6 R16D). Rather, cleavage of the bond between the Y6 and P7 residues yields the most abundant product ion. Therefore, the replacement of the basic lysine residue in the Y6-P7-K8-P9 sequence with the acidic aspartic acid residue eliminated the condition that inhibits proline cleavage. This result is therefore consistent with that observed with the R16A variant in which the replacement of a basic residue (arginine) adjacent to a proline residue gave rise to a prominent N-terminal proline cleavage. This suggests that the presence of lysine in the wild-type sequence (Y6-P7-K8-P9) influences fragmentation N-terminal to these proline residues. Overall, the results related above for the fragmentation of the R16A, R16D, R16H, K8D, and K8H ions provide a consistent picture. Replacement of a basic residue with a nonbasic residue adjacent to proline “turns on” and/or enhances fragmentation channels associated with cleavage N-terminal to proline near the site of replacement. The data from the histidine variants imply that the reason for no or low-abundance fragments associated with proline is not related to the presence of a lysine at position 8 or an arginine at position 16 specifically but more generally to the presence of an amino acid residue having a basic site. Furthermore, the charge state dependent fragmentation of the R16H variant suggests that inhibition of N-terminal proline cleavage by the presence of a nearby basic residue may depend on its being protonated.
Figure 7.

Beam-type fragmentation of K8D (K13D) [M + 6H]6+, 35 eV.
(e) Removal of Proline and Lysine/Histidine Introduction
As part of the investigation of proline related cleavages, one of the proline residues was removed. Specifically, P9 was replaced with H (OMTKY3 P9H (P14H)). This substitution creates a K/H pair. Cleavage of amide bonds C-terminal to lysine in whole protein ions has been observed in previous dissociation studies of whole protein ions. In particular, K/H cleavages yielded prominent product ions in the MS/MS of apomyoglobin20 and cytochrome c21 ions. The fragmentation patterns of +3 to +6 P9H ions have many similarities to the fragmentation patterns of +3 to +6 wild-type ions. Low-abundance fragmentation corresponding to cleavage of the newly introduced L8/H9 bond was observed for +5 and +6 P9H ions. When the fragmentation behaviors of +7 P9H and wild-type ions are compared, as shown in Figure 8, there are dramatic differences in the dominant fragmentation channels. The b2/y49 aspartic acid cleavage (D/C) contributes ~20% more for the wild-type ion than for the P9H ion, while the b22 aspartic acid cleavage (D/N) is ~20% more abundant for the P9H ion than it is for the wild-type ion. This comparison highlights the fact that a single amino acid substitution may have little effect on the fragmentation of some charge states but may dramatically impact the fragmentation of other charge states. In this particular case, the fact that a basic residue was substituted for a proline may have affected the degree to which the N-terminus of the protein was protonated in the +7 parent ion charge state. If the appearance of abundant product ions associated with the b2 fragmentation channel is dependent upon protonation of the N-terminus, the provision of an additional basic site can affect this channel via its role in determining sites of protonation.
Conclusions
The systematic study of the fragmentation of reduced OMTKY3 ions and their variants provides further insight into the fragmentation behavior of intact protein ions. In some ways, each protein that has been subjected to charge state dependent fragmentation has exhibited unique behavior. A major difference in the fragmentation observed for OMTKY3 ions relative to other protein ions studied to date was the minor contribution from fragmentation N-terminal to proline residues. Furthermore, for all OMTKY3 proteins examined, abundant product ions corresponding to C-terminal acidic residues were observed for intermediate and higher charge states. Such cleavages are not unusual for relatively low charge states, but they are rarely dominant at high charge states. The large contributions from fragmentation of N/G and V/V for all OMTKY3 proteins is also unusual, as neither of these cleavages has been found to be dominant in other studies of protein ion dissociation. Although cleavages corresponding to N/G and V/V have been observed for other proteins when subjected to CID, the abundances of their respective product ions in the MS/MS spectra were relatively low. However, recent examination of a database of 5500 unique peptide MS/MS spectra revealed enhanced N/G cleavages for some doubly charged tryptic peptides.42 A preference for cleavage of the V/V amide bond was not reported in previous assessments of peptide fragmentation.42,43 The unique dissociation behavior of OMTKY3 ions, relative to many other proteins, presumably arises from structural characteristics of the protein ions that tend to override many of the other factors known to influence dissociation behavior. Nevertheless, C-terminal aspartic acid cleavages are prominent, as has been noted with many other proteins. Furthermore, the relative abundances of the product ions are sensitive to the parent ion charge state, as has been noted for essentially all other protein ions studied to date.
Investigations of the fragmentation of reduced OMTKY3 variants demonstrated the impact that changing one amino acid residue can have on MS/MS spectra. In general, differences in the fragmentation behavior were localized to the site/region of substitution. However, in some cases, fragmentation channels more than four residues away from the site of substitution were impacted by changing one amino acid in the protein sequence. On the basis of the relatively small subset of data presented here, the extent of the effect of the exchange depends more on the identities of the amino acid residues involved in the substitution than on the position of the substitution in the amino acid sequence. For example, replacement of a nonacidic residue with an aspartic acid residue at positions 8, 13, 16, and 31, independently, resulted in turning on a fragmentation channel at the position of substitution for those variants. In most cases, the charge state dependent behavior of these new fragmentation channels was consistent with that expected for C-terminal aspartic cleavages. Substitution of a proline residue at position 13 introduced fragmentation N-terminal to this proline residue at higher charge states, as anticipated. However, this channel was not as dominant as the one observed for the aspartic acid substitution at position 13. The fragmentation behavior of the other proline related variants suggested that the close proximity of prolines to basic residues in the sequences of OMTKY3 proteins may be responsible for the minimal contribution of N-terminal proline cleavages to OMTKY3 protein dissociation.
Acknowledgments
Dr. Gavin Reid is acknowledged for helpful discussions. This research was sponsored by the National Institutes of Health under Grant GM 45372 (S.A.M.). The ovomucoid third domain variant library was acquired and is maintained under Grants GM 10831 and GM 63539 (M.L.).
Footnotes
Supporting Information Available: Fragmentation summaries of +4 and +5 L13D (L18D) as well as post-ion/ion MS/MS spectra of various charge states of the variants K8H (K13H), R16H (R21H), K8D (K13D), and R16D (R21D) (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Fenn JB, Mann M, Meng CK, Wong SF, Whitehouse CM. Science. 1990;246:64–71. doi: 10.1126/science.2675315. [DOI] [PubMed] [Google Scholar]
- 2.Karas M, Hillenkamp F. Anal Chem. 1988;60:2299–2301. doi: 10.1021/ac00171a028. [DOI] [PubMed] [Google Scholar]
- 3.James P, Quadroni M, Carafoli E, Gonnet G. Biochem Biophys Res Commun. 1993;214:58–64. doi: 10.1006/bbrc.1993.2009. [DOI] [PubMed] [Google Scholar]
- 4.Yates JR, III, Speicher S, Griffin PR, Hunkapiller T. Anal Biochem. 1993;214:397–408. doi: 10.1006/abio.1993.1514. [DOI] [PubMed] [Google Scholar]
- 5.Henzel WJ, Billeci TM, Stults JT, Wong SC, Grimley C, Watanabe C. Proc Natl Acad Sci USA. 1993;90 doi: 10.1073/pnas.90.11.5011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goodlett DR, Bruce JE, Anderson GA, Rist B, Pasa-Tolic L, Fiehn O, Smith RD, Aebersold R. Anal Chem. 2000;72 doi: 10.1021/ac9913210. [DOI] [PubMed] [Google Scholar]
- 7.Hunt DF, Yates JR, III, Shabanowitz J, Winston S, Hauer CH. Proc Natl Acad Sci USA. 1986;83:6233–6237. doi: 10.1073/pnas.83.17.6233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Horn DM, Zubarev RA, McLafferty FW. Proc Natl Acad Sci USA. 2000;97:10313–10317. doi: 10.1073/pnas.97.19.10313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kelleher NL, Lin HY, Valaskovic GA, Aaserud DJ, Fridricksson EK. J Am Chem Soc. 1999;121:806–812. [Google Scholar]
- 10.Mortz E, O’Connor PB, Ropestorff P, Kelleher NL, Wood TD, McLafferty FW, Mann M. Proc Natl Acad Sci USA. 1996;93:8264–8267. doi: 10.1073/pnas.93.16.8264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nemeth-Cawley JF, Tangarone BS, Rouse JC. J Proteome Res. 2003;2:495–505. doi: 10.1021/pr034008u. [DOI] [PubMed] [Google Scholar]
- 12.Nemeth-Cawley JF, Rouse JC. J Mass Spectrom. 2002;3:270–282. doi: 10.1002/jms.281. [DOI] [PubMed] [Google Scholar]
- 13.McLuckey SA, Stephenson JL., Jr Mass Spectrom Rev. 1998;17:369–407. doi: 10.1002/(SICI)1098-2787(1998)17:6<369::AID-MAS1>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 14.Reid GE, Shang H, Hogan JM, Lee GU, McLuckey SA. J Am Chem Soc. 2002;124:7353–7362. doi: 10.1021/ja025966k. [DOI] [PubMed] [Google Scholar]
- 15.McLuckey SA, Reid GE, Wells JM. Anal Chem. 2002;74:336–346. doi: 10.1021/ac0109671. [DOI] [PubMed] [Google Scholar]
- 16.Stephenson JL, Jr, McLuckey SA. Anal Chem. 1998;70:3533–3544. doi: 10.1021/ac9802832. [DOI] [PubMed] [Google Scholar]
- 17.Pitteri SJ, Reid GE, McLuckey SA. J Proteome Res. 2004;3:46–54. doi: 10.1021/pr034054u. [DOI] [PubMed] [Google Scholar]
- 18.Reid GE, Wu J, Chrisman PA, Wells JM, McLuckey SA. Anal Chem. 2001;73:3274–3281. doi: 10.1021/ac0101095. [DOI] [PubMed] [Google Scholar]
- 19.Hogan JM, McLuckey SA. J Mass Spectrom. 2003;38:245–256. doi: 10.1002/jms.458. [DOI] [PubMed] [Google Scholar]
- 20.Newton KA, Chrisman PA, Reid GE, Wells JM, McLuckey SA. Int J Mass Spectrom. 2001;212:359–376. doi: 10.1002/rcm.512. [DOI] [PubMed] [Google Scholar]
- 21.Engel BJ, Pan P, Reid GE, Wells JM, McLuckey SA. Int J Mass Spectrom. 2002;219:171–187. [Google Scholar]
- 22.Iavarone AT, Williams ER. Anal Chem. 2003;75:4525–4533. doi: 10.1021/ac034144i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wieczorek M, Laskowski M., Jr Biochemistry. 1983;22:2630–2636. doi: 10.1021/bi00280a006. [DOI] [PubMed] [Google Scholar]
- 24.Stephenson JL, Jr, Cargile BJ, McLuckey SA. Rapid Commun Mass Spectrom. 1999;13:2040–2048. doi: 10.1002/(SICI)1097-0231(19991030)13:20<2040::AID-RCM754>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 25.Wells JM, Stephenson JL, Jr, McLuckey SA. Int J Mass Spectrom. 2000;203:A1–A9. [Google Scholar]
- 26.Schaaff TG, Cargile BJ, Stephenson JL, Jr, McLuckey SA. Anal Chem. 2000;72:899–907. doi: 10.1021/ac991344e. [DOI] [PubMed] [Google Scholar]
- 27.He M, Reid GE, Shang H, Lee GU, McLuckey SA. Anal Chem. 2002;74:4653–4661. doi: 10.1021/ac025587+. [DOI] [PubMed] [Google Scholar]
- 28.Hogan JM, Pitteri SJ. Anal Chem. 2003;75:6509–6516. doi: 10.1021/ac034410s. [DOI] [PubMed] [Google Scholar]
- 29.Watson, D. J.; McLuckey, S. A. Unpublished data.
- 30.Reid GE, Stephenson JL, Jr, McLuckey SA. Anal Chem. 2002;74:577–583. doi: 10.1021/ac015618l. [DOI] [PubMed] [Google Scholar]
- 31.Breci LA, Tabb DL, Yates JR, III, Wysocki VH. Anal Chem. 2003;75:1963–1971. doi: 10.1021/ac026359i. [DOI] [PubMed] [Google Scholar]
- 32.Huang Y, Triscari JM, Pasa-Tolic L, Anderson GA, Lipton MS, Smith RD, Wysocki VH. J Am Chem Soc. 2004;126:3034–3035. doi: 10.1021/ja038041t. [DOI] [PubMed] [Google Scholar]
- 33.Reid GE, McLuckey SA. J Mass Spectrom. 2002;37:663–675. doi: 10.1002/jms.346. [DOI] [PubMed] [Google Scholar]
- 34.Stephenson JLJ, McLuckey SA, Reid GE, Wells JM, Bundy JL. Curr Opin Biotechnol. 2002;13:57–64. doi: 10.1016/s0958-1669(02)00285-9. [DOI] [PubMed] [Google Scholar]
- 35.VerBerkmoes NC, Strader MB, Smiley RD, Howell EE, Hurst GB, Hettich RL, Stephenson JL., Jr Anal Biochem. 2002;395:68–61. doi: 10.1006/abio.2002.5636. [DOI] [PubMed] [Google Scholar]
- 36.Lu W, Apostol I, Qasim MA, Warne N, Wynn R, Zhang WL, Anderson S, Chiang YW, Ogin E, Rothberg I, Ryan K, Laskowski M., Jr J Mol Biol. 1997;266:441–461. doi: 10.1006/jmbi.1996.0781. [DOI] [PubMed] [Google Scholar]
- 37.Lu SM, Lu W, Qasim MA, Anderson S, Apostol I, Ardelt W, Bigler T, Chiang YW, Cook J, James MNG, Kato I, Kelly C, Kohr W, Komiyama T, Lin TY, Ogawa M, Otlewski J, Park SJ, Qasim S, Ranjbar M, Tashiro M, Warne N, Whatley H, Wieczorek A, Wieczorek M, Wilusz T, Wynn R, Zhang W, Laskowski MJ. Proc Natl Acad Sci USA. 2001;98:1410–1415. doi: 10.1073/pnas.031581398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stephenson JL, Jr, McLuckey SA. Int J Mass Spectrom Ion Processes. 1997;162:89–106. [Google Scholar]
- 39.Reid GE, Wells JM, Badman ER, McLuckey SA. Int J Mass Spectrom. 2003;222:243–258. [Google Scholar]
- 40.Krezel AM, Darba P, Robertson AD, Fejzo J, Macura S, Markley JL. J Mol Biol. 1994;242:203–214. doi: 10.1006/jmbi.1994.1573. [DOI] [PubMed] [Google Scholar]
- 41.Dongré AR, Jones JL, Somogyi A, Wysocki VH. J Am Chem Soc. 1996;118 [Google Scholar]
- 42.Kapp EA, Schutz F, Reid GE, Eddes JS, Moritz RL, O’Hair RAJ, Speed TP, Simpson RJ. Anal Chem. 2003;75:6251–6264. doi: 10.1021/ac034616t. [DOI] [PubMed] [Google Scholar]
- 43.Tabb DL, Smith LL, Breci LA, Wysocki VH, Lin D, Yates JR., III Anal Chem. 2003;75:1155–1163. doi: 10.1021/ac026122m. [DOI] [PMC free article] [PubMed] [Google Scholar]