

Abstract
In yeast, the Origin Recognition Complex (ORC) is bound to replication origins throughout the cell-cycle, but in animal cells, there are conflicting data as to whether and when ORC is removed from chromatin. We find ORC1, 2 and ORC4 to be metabolically stable proteins that co-fractionate with chromatin throughout the cell-cycle in Chinese hamster fibroblasts. Since cellular extraction methods cannot directly examine the chromatin binding properties of proteins in vivo, we examined ORC:chromatin interactions in living cells. Fluorescence loss in photobleaching (FLIP) studies revealed ORC1 and ORC4 to be highly dynamic proteins during the cell-cycle with exchange kinetics similar to other regulatory chromatin proteins. In vivo interaction with chromatin was not significantly altered throughout the cell-cycle, including S-phase. These data support a model in which ORC subunits dynamically interact with chromatin throughout the cell-cycle.
Keywords: Cell-cycle, DNA replication, GFP, Origin Recognition Complex, Photobleaching
Introduction
Eukaryotic DNA replication requires the ordered assembly of protein complexes on or near origins of replication prior to the start of S-phase. In mammalian cells, these pre-replicative complexes (pre-RCs) are assembled in telophase, coincident with the degradation of B-type cyclins, and are completed by early G1-phase [1–4]. The first pre-RC component associated with the origin is the Origin Recognition Complex (ORC), a heterohexameric complex that is conserved from S. cerevisiae through human. ORC recruits the Cdc6 protein that, in conjunction with Cdt1, loads the minichromosome maintenance proteins (MCM) to form a functional pre-RC [5]. Upon entering S-phase, Cdt1 is degraded by the proteosome via the SCF (Skp/Cullin/F-box) protein–ubiquitin ligase [6], and MCM proteins are gradually dissociated from chromatin, while Cdc6 remains chromatin associated [2,3,7]. The exact fate of the ORC subunits during the mammalian cell-cycle, particularly that of the largest subunit, ORC1, has not yet been fully resolved. ORC1 is of particular interest as a potential regulatory subunit as it: contains the only ATP binding motif that is essential for the DNA binding activity of ORC [8–10], makes direct contacts with DNA [10,11] and is required for the loading of several regulatory proteins, including Cdc6 [12–15].
In both budding and fission yeast, all six ORC subunits remain in a complex, bound to replication origins throughout the cell-cycle and have functions in both pre-RC formation and intra-S-phase checkpoint control [16–22]. In metazoa, there is evidence that ORC1 may play a unique regulatory role as transcription of ORC1 is developmentally regulated and downregulated during quiescence via an E2F-responsive promoter [23–25]. In mammalian cells, ORC1 appears loosely associated with the core complex of ORC2–5 subunits [26,27], and several recent reports have concluded that ORC1 is degraded upon entry into S-phase and re-accumulates after mitosis [28–30]. In addition, many studies have observed the amount of ORC1 present in various types of chromatin preparations to decrease during S-phase, including chromatin immunoprecipitation experiments [31–34]. These results have led to the “ORC-cycle” model [35] in which ORC1 availability is regulated to prevent re-initiation of replication during S-phase, while the core complex remains chromatin-associated.
However, this model does not account for all reported results. Several laboratories do not observe fluctuation of total ORC1 levels during the mammalian cell-cycle [3,26,36,37] and in one study, protein synthesis was inhibited throughout the prophase to telophase period, with no effect on the assembly of functional pre-RCs [3]. Importantly, it has been shown that ORC-depleted Xenopus egg extracts can initiate replication within G2-phase Hela cell nuclei [38], which presumably must require the presence of mammalian ORC1 during G2-phase. Moreover, some studies have found the majority of ORC1 bound to chromatin throughout S-phase [3,15]. In fact, every study has reported that at least 30–50% of the detectable ORC1 is bound to chromatin during S-phase, and none of these studies have detected the predicted remainder of unbound ORC1 [29,30,37]. These results leave open the possibility that at least half of the ORC1 present within a cell is, in fact, interacting with the origin-bound complex throughout the cell-cycle. Indeed, it is unclear how the core ORC subunits could remain chromatin-associated without ORC1, as human ORC1 is required for recombinant ORC DNA binding [10] and recombinant yeast and Drosophila ORC exhibit reduced ATP-dependent DNA binding when ORC1 is catalytically inactive [8,9]. Since all the disparate results with mammalian cells have been carried out with variations of cellular extraction methods, there is clearly a need for novel methods in order to clarify ORC subunit interactions during the cell-cycle.
Here, we show that ORC1 is a metabolically stable, chromatin-associated protein throughout the cell-cycle in Chinese hamster ovary (CHO) cells. Using in vivo photobleaching techniques with stable GFP-CgORC (Cricetus griseus, Chinese hamster) expressing cell lines, we find that ORC1 and a core ORC subunit, ORC4, dynamically associate with chromatin throughout all phases of the cell-cycle.
Results
ORC1, 2 and 4 are stable proteins that co-fractionate with chromatin throughout the cell-cycle
The ‘ORC-cycle’ model predicts that ORC1 is degraded or released from chromatin during S-phase, a prediction that is not consistent with our previously reported results [3]. To further evaluate this prediction, we first repeated our experiments using a newly generated anti-CgORC1 antibody (Antibody characterization shown in Supplemental Fig. 1) and a widely used chromatin fractionation method [2,29,37]. In order to determine if there were changes occurring at or near the time of the origin decision point (ODP), a G1-phase hallmark at which origins of replication are specified [3,39], we first examined ORC–chromatin association in mitosis and G1-phase using CHO cells synchronized with a brief nocodazole block and release. Consistent with our studies and those of others utilizing the same cell line [3,36,40], we did not detect any significant change in the amounts of ORC1, 2 or 4 in the chromatin-containing fraction during mitosis and throughout G1-phase, although a small amount of ORC1 was consistently found in the soluble fraction during mitosis (Fig. 1A). The degree of soluble ORC1 present in mitosis was variable between experiments and was independent of the nocodazole block (data not shown). In addition, we did not detect any increase in either the steady state levels or chromatin-association of the ORC1 subunit throughout G1-phase. Consistent with our previous findings [3], these results indicate that there are no apparent changes in ORC–chromatin association occurring at the ODP.
Fig. 1.
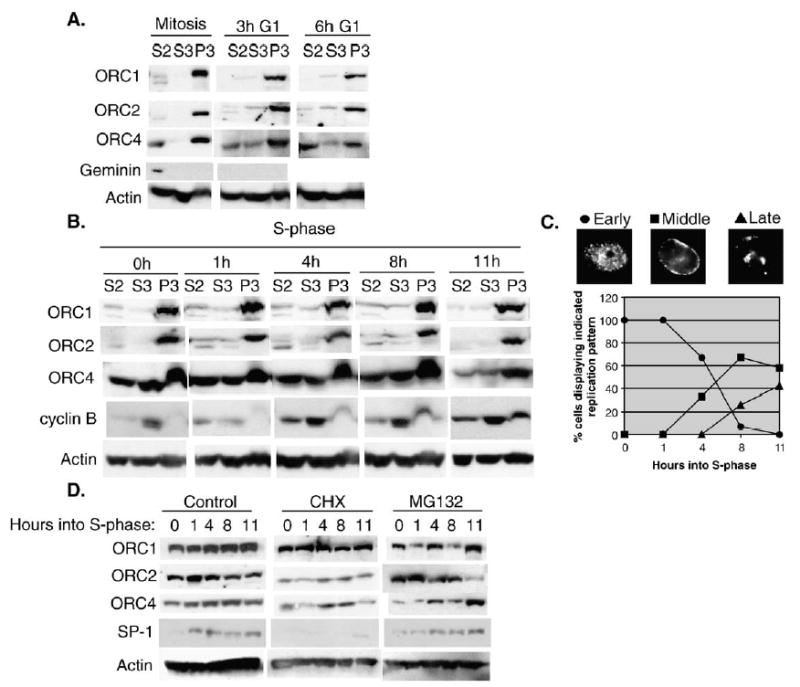
ORC is stable and chromatin-associated throughout the cell-cycle in CHO cells. (A) CHO cells were synchronized in mitosis with a brief (4 h) nocodazole block and collected and fractionated at the indicated timepoints as described in Materials and methods. Cell extracts were subjected to electrophoresis on 8% SDS-PAGE gels and immunoblotted with anti-ORC1, 2 and 4. Anti-geminin and anti-actin were used as controls for mitotic release and protein levels. The S2 and S3 fractions represent soluble protein fractions and the P3 fraction represents the chromatin-enriched fraction. (B) CHO cells were synchronized in S-phase and cells fractionated at 0, 1, 4, 8 or 11 h as described in Materials and methods. As a control for S-phase progression, extracts were immunoblotted with anti-cyclin B1. Anti-actin was used as a loading control. Similar results were obtained in 4 independent experiments (3 times with aphidicolin and once with mimosine arrested cells). The results of two of these experiments were additionally confirmed using an anti-HsORC1 antibody (gift of J. Mendez and B. Stillman, not shown). (C) To monitor S-phase progression, aliquots of cells from (B) were pulse labeled with BrdU for 30 min, fixed and stained with anti-BrdU. The percentage of cells displaying the indicated replication pattern (^ Early, ▪ Middle, ▴ Late) was scored. (D) CHO cells synchronized in S-phase as in panel (B) were released into media containing: DMSO (control), 50 μg/ml cycloheximide or 25 μM MG132 for 0, 1, 4, 8 and 11 h. Whole cell extracts were prepared and immunoblotted for ORC1, 2, 4, SP1 and actin. Similar results were obtained in two independent experiments.
To examine changes in ORC–chromatin association during S-phase, CHO cells were synchronized at the G1/S boundary with aphidicolin, and chromatin was prepared at 0, 1, 4, 8 and 11 h after release (Fig. 1B). S-phase progression (Fig. 1C) was monitored by quantifying the well-characterized spatial changes in DNA synthesis as well as by the increase in chromatin-associated cyclin B (Fig. 1B) [1]. ORC1 and 2 largely co-fractionated with chromatin throughout this period, although a minor fraction of both subunits was present in the soluble fraction or eluted during the washing step (Fig. 1B). In most experiments, the soluble ORC1 and ORC2 appeared as a doublet, potentially indicating the post-translational modification of these subunits. The S-phase ORC1 results were verified using an anti-human ORC1 antibody [30] that cross-reacts with hamster ORC1 (data not shown). In contrast, similar amounts of ORC4 were detected in both the soluble and chromatin-containing fractions (Fig. 1B), consistent with our previous results using a different fractionation method [3]. Hence, we find no evidence for the degradation of ORC1 during S-phase, and we detect only a minor population being selectively released from chromatin during S-phase in CHO cells.
Our previous results demonstrated that ORC1 in CHO cells is a stable protein throughout mitosis and into early G1-phase and that the assembly of functional pre-RCs during telophase is unaffected by protein synthesis inhibition throughout the M to G1-phase transition [3]. However, we did not examine the stability of ORC1 during S-phase progression. To determine the metabolic stability of ORC1 during S-phase, we released cells synchronized at the G1/S boundary into media containing either the protein synthesis inhibitor cycloheximide or the proteosome inhibitor MG132. These drugs are general inhibitors and provide an indirect measure of protein stability. This method is commonly used for studies of pre-RC component stability, including ORC subunits [3,28,30,41]. There was no significant change in the steady state levels of ORC1, 2, 4 or actin detected throughout the course of S-phase (Fig. 1D). As a control, we examined the levels of the SP1 transcription factor, which was prevented from accumulating in the presence of cycloheximide and accumulated normally in the presence of MG132. Together with our previous results, we conclude that ORC1, 2 and 4 are metabolically stable proteins throughout the cell-cycle. Interestingly, in the presence of MG132, no additional modified forms of ORC1 were detected that would be consistent with ubiquitination of ORC1 during S-phase.
In vivo cell-cycle kinetics of ORC-chromatin association
Cellular extraction experiments are limited by potential artifacts occurring during cell lysis and only measure the average behavior of a population of molecules. Photo-bleaching of GFP-tagged proteins has recently emerged as an alternative means to evaluate the interaction of proteins with chromatin in intact living cells [42]. Apart from the analysis of living cells, these single-cell methods have the advantage of resolving differential binding properties of protein sub-populations. These studies have revealed the dynamic nature and prevalence of transient chromatin binding by nuclear proteins [43]. For example, transcriptional regulatory proteins interact dynamically with chromatin and recover quickly (3–30 s) [43], while proteins that are involved in active RNA or DNA synthesis such as RNA pol I [44], pol II [45] and PCNA [46] recover more slowly (2–15 min).
To systematically analyze the in vivo binding dynamics of ORC1, we constructed a stable, tetracycline inducible GFP-ORC1 CHO cell line in which 100% of the cells express GFP-ORC1 at a homogenous level (Fig. 2A and [3]). This cell line (3KA4) was carefully selected to express GFP-ORC1 at trace levels compared to that of endogenous ORC1 in order to avoid the effects of over-expression. ORC1 over-expression is toxic to cell growth, and high levels of ORC1 in transient transfections result in insoluble aggregates (L. Archibold and DMG unpublished observation). We previously demonstrated that GFP-ORC1 associates with chromatin throughout mitosis and co-localizes with ORC4 on metaphase chromosomes [3]. During interphase, GFP-ORC1 is eluted from chromatin at the same range of salt concentrations as endogenous ORC1 (Fig. 2B and [3]). While we were unable to reproducibly immunoprecipitate either ORC1 or GFP-ORC1 with other endogenous ORC subunits, GFP-ORC1 was found to co-fractionate with endogenous ORC1 and ORC2 in a chromatin-associated complex of greater than 300 kDa (Fig. 2C). Chromatin proteins were solubilized with micro-coccal nuclease [2] and fractionated using an ultrafiltration device with a 300 kDa molecular weight cut-off. This result demonstrates that GFP-ORC1 interacts with a chromatin-associated protein complex, consistent with the reported size of the 6 subunit ORC [27,47]. In addition, GFP-ORC1 co-fractionated with endogenous ORC1 and chromatin throughout the cell-cycle (Fig. 2D). The levels of GFP-ORC1 do not appear to fluctuate during the cell-cycle, consistent with our previous findings using fluorescence intensity measurements of individual cells [3]. GFP-ORC1 was predominantly nuclear (Fig. 4A), with accumulation in the nucleolus and at sites of heterochromatin (e.g. nuclear periphery).
Fig. 2.
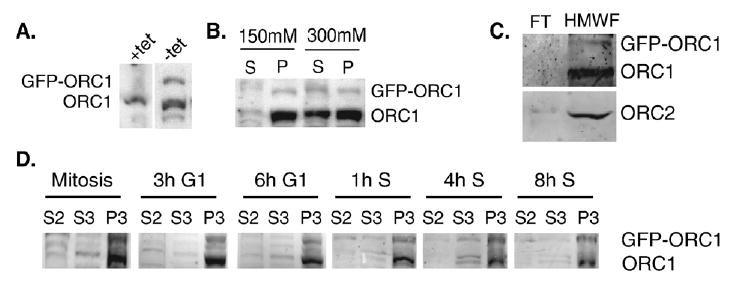
GFP-ORC1 co-fractionates with endogenous ORC1 and chromatin. (A) Immunoblots of whole cell extracts prepared from a GFP-ORC1 expressing cell line (3KA4) grown either in the presence or absence of tetracycline were probed with anti-CgORC1 antibodies. (B) Nuclei isolated from 3KA4 cells grown in the absence of tetracycline were incubated with different concentrations of salt (150 mM v 300 mM), and soluble and insoluble fractions were probed with anti-CgORC1. Additional characterization of this cell line can be found in Okuno et al. [3]. (C) Isolated chromatin from 3KA4 was digested with micrococcal nuclease and the soluble fraction concentrated with a 300 kDa molecular weight cut-off ultrafiltration device. GFP-ORC1, endogenous ORC1 and ORC2 were all present in the greater than 300 kDa fraction. Shown are the flow-through (FT) and high molecular weight fraction (HMWF). (D) GFP-ORC1 co-fractionates with endogenous ORC1 and chromatin throughout the cell-cycle. Induced 3KA4 cells were synchronized and fractionated as described in Fig. 1 and immunoblots probed with anti-CgORC1.
Fig. 4.
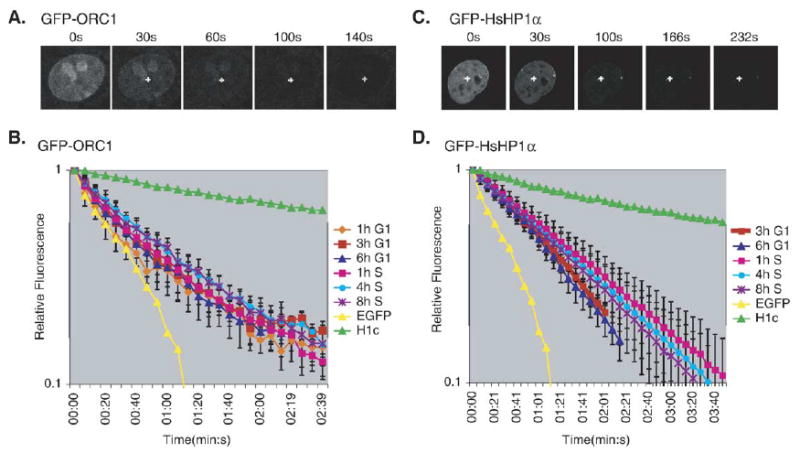
FLIP of GFP-CgORC1 and GFP-HP1α, corrected for nuclear volume. (A) FLIP analysis of GFP-CgORC1 during the cell-cycle. GFP-CgORC1 expressing cells were subjected to a repeating cycle of an image scan, a 0.5 s bleach pulse at 100% laser power and a 5 s recovery period. The images are from a typical FLIP experiment showing the loss of GFP fluorescence over time as a result of the repeated bleach pulse (indicated by the cross). (B) FLIP kinetic plot of GFP-CgORC1 fluorescence loss as a function of time. Each line represents the average fluorescence loss from at least 5 cells, with the loss quantified at 2–3 distinct sites within the nucleus of each cell, at the indicated times during the cell-cycle. The error bars indicate the standard deviation among the individual measurements for each timepoint. Similar results were obtained in three independent experiments. Results with EGFP and histone H1c are provided as reference marks for in vivo protein mobility. (C) FLIP analysis of a stable CHO cell line expressing GFP-HP1α, performed as in panel (A). (D) FLIP kinetic plot for GFP-HP1α during the cell-cycle. Data are displayed as in panel (B). Similar results were obtained in three independent experiments.
Initial FRAP (Fluorescence recovery after photobleaching) studies indicated that GFP-ORC1 was surprisingly mobile, with bleach spots recovering in less than 5 s (not shown), which is similar to the mobility of numerous chromatin binding proteins [43] but inconsistent with models of a static ORC. Given the rapid recovery time, we applied the more sensitive FLIP ( Fluorescence loss in photobleaching) method in order to determine if the kinetics of GFP-ORC1 change during the cell-cycle. In FLIP, a single spot in the nucleus is repeatedly bleached and the loss of fluorescence is quantified at locations away from the bleach spot [48]. To follow ORC dynamics during G1-phase, we synchronized cells in mitosis and released them into G1-phase for 1, 3 and 6 h. S-phase cells were obtained by release from a G1/S-phase aphidicolin block for 1, 4 and 8 h.
The initial FLIP results revealed that ORC1 is highly mobile throughout the cell-cycle and exhibited several kinetic shifts over the course of the cell-cycle (Fig. 3A). However, when free EGFP was analyzed in a control cell line, it also exhibited similar kinetic shifts over the course of the cell-cycle (Fig. 3B). Since the volume of the eukaryotic nucleus expands during the course of the cell-cycle, we examined whether or not nuclear volume could affect the kinetic mobility. When the nuclear area of multiple cells from each timepoint was measured, a correlation between the timing of the observed kinetic shifts and increasing nuclear size became apparent (Fig. 3C). We conclude from this experiment that, since the diameter of the laser is fixed and the concentration of GFP molecules within the cell is relatively constant, a lower percentage is bleached in larger nuclei, giving the appearance of reduced mobility (Fig. 3D).
Fig. 3.
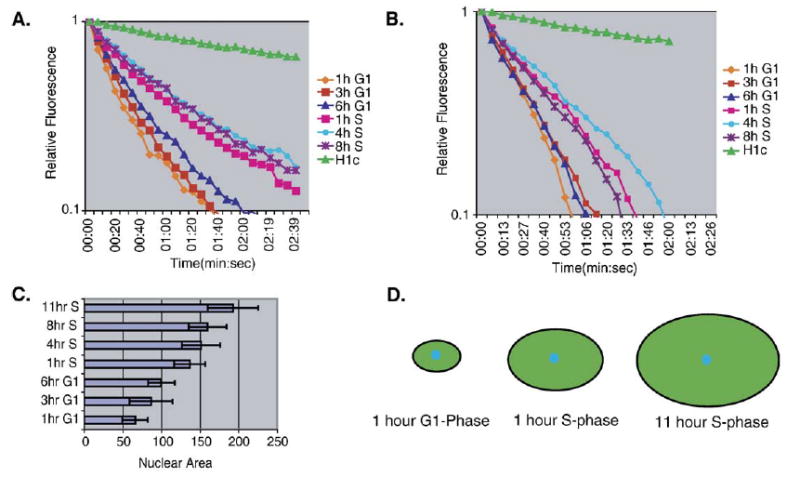
The effect of nuclear volume on Fluorescence loss in photobleaching kinetics. (A) Initial FLIP kinetic plot of the GFP-ORC1 expressing cell line, 3KA4 demonstrating fluorescence loss as a function of time. 3KA4 cells were synchronized and released at the indicated timepoints as described in Materials and methods. Cells were subjected to a repeating cycle of an image scan, a 0.5 s bleach pulse at 100% laser power and a 5 s recovery period. Each line represents the average fluorescence loss from at least 5 cells, with the loss quantified at 2–3 distinct sites within the nucleus of each cell, at the indicated time during the cell-cycle. Similar results were obtained in three independent experiments. Results with GFP-tagged histone H1c are provided as a reference mark for in vivo protein mobility. (B) FLIP analysis of a stable cell line expressing free-EGFP was performed as described in panel (B). (C) The relative size of the nucleus expands as cells progress through the cell-cycle. The area of the nucleus at its largest diameter was measured at each timepoint from multiple cell lines and averaged. (D) Schematic representation of the effect of increasing nuclei size on photobleaching kinetics. Details are contained within the text.
Fig. 4 shows the results of a complete FLIP analysis of GFP-ORC1, corrected for nuclear volume, revealing that the mobility of GFP-ORC1 does not exhibit any significant changes during the cell-cycle (Fig. 4B). GFP-ORC1 exhibited significantly slower kinetics than EGFP alone, reflecting its transient binding interaction with immobile structures in the nucleus (e.g. chromatin), but exchanged much faster than linker histone H1 which, interestingly, elutes from chromatin under the same cellular extraction conditions as ORC1 [40]. As a control, a separate stable CHO cell line expressing the heterochromatin protein GFP-HP1α [49] was similarly examined during the cell-cycle, revealing exchange kinetics comparable to GFP-ORC1 (Fig. 4D).
To determine if the rapid mobility observed with ORC1 was unique to the ORC1 subunit or represented the mobility of the entire ORC, we examined a core ORC subunit. Based upon studies of the interaction of recombinant ORC subunits, ORC2–4 form a tightly associated core complex, with which ORC1, 5 and 6 interact [27]. Hence, we cloned the CgORC4 and CgORC5 cDNAs (sequence analysis in Supplemental Fig. 2) (Genbank accession #AB183827, AB183828) and constructed GFP-tagged cell lines. The expression of GFP-ORC5, even after tet-regulatable induction in stable cell lines, was toxic to cells (not shown), and thus we focused on GFP-ORC4. The initial GFP-ORC4 cell line exhibited FLIP kinetics similar to EGFP alone (Fig. 5E and Supplemental Fig. 3). This result suggested that the GFP-tag was preventing the fusion protein from properly interacting with the other ORC subunits, thus we introduced a 16 amino acid linker between ORC4 and EGFP to make GFP-P-ORC4 and we reconstructed the stable cell line [46,50]. GFP-P-ORC4 exhibited identical salt extractability as endogenous ORC4 (Figs. 5A and B). GFP-P-ORC4 was predominantly nuclear, with a visible cytoplasmic pool consistent with the presence of soluble ORC4 in cell fractionation experiments (Fig. 5C). Cell-cycle FLIP analysis of GFP-ORC4 revealed ORC4 to be a highly dynamic protein with kinetics similar to ORC1 (Fig. 5D). Fig. 5E shows a comparative summary of the FLIP kinetics for all proteins in this study. Based upon these findings, we conclude that ORC is a highly dynamic protein complex with subunits that interact with chromatin throughout the cell-cycle.
Fig. 5.
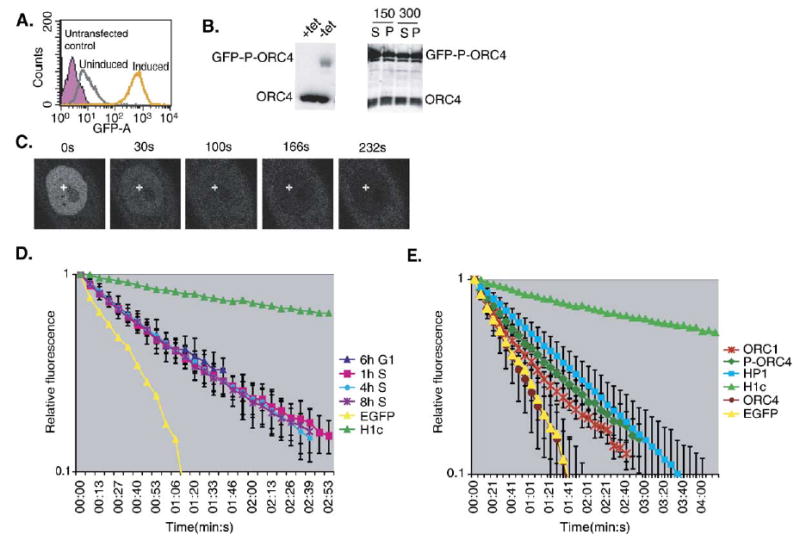
Cell line characterization and FLIP of ORC4. (A) Homogeneity and inducibility of GFP-P-ORC4. PC1–11 cells (CHO cells stably expressing GFP-P-ORC4) were grown either in the presence (uninduced—gray) or absence (induced—orange) of tetracycline and analyzed for EGFP expression by FACS. Untransfected CHO cells (purple) were used as a negative control. (B) The expression of GFP-P-ORC4 is inducible (left panel) and GFP-P-ORC4 exhibits similar salt extractability as endogenous ORC4 (right panel). Whole cell extracts from induced and uninduced PC1-11 cells were run on 10% SDS-PAGE gels and immunoblotted with anti-ORC4 demonstrating induction of full-length GFP-P-ORC4 (left panel). PC1-11 cells were induced and isolated nuclei fractionated using increasing levels of NaCl (150 mM and 300 mM) and the resulting fractions were probed with anti-ORC4 antibodies (right panel). (C) Typical images from the cell-cycle FLIP of GFP-P-ORC4 in PC1-11 as in Figs. 4A and C. (D) FLIP kinetic plot from the cell-cycle FLIP analysis of GFP-P-ORC4, as described in Fig. 4. The error bars represent the standard deviation from one experiment. For each experiment, at least 5 cells were photobleached with 2–3 measurements taken per cell. EGFP and histone H1c are provided as reference marks for mobility. Similar results were obtained in three independent experiments. (E) Comparative summary of the relative FLIP kinetics for GFP-ORC1 (ORC1), GFP-P-ORC4 (P-ORC4), ORC4 (without the peptide linker) and HsHP1α.
Discussion
Our results provide evidence that mammalian ORC consists of metabolically stable subunits that co-fractionate with chromatin throughout the cell-cycle. We did not detect any changes in chromatin affinity or degradation of the ORC1 subunit during S-phase. We demonstrate that ORC1 and ORC4 are highly dynamic in their interaction with chromatin throughout the cell-cycle, with a residence time of not more than a few seconds by photobleaching methods with GFP-tagged subunits stably expressed in living cells. These results necessitate a reconsideration of our original views about the interactions of ORC subunits with replication origins and origin binding proteins.
Dynamic ORC-chromatin interaction
It has become apparent in recent years that many chromatin-binding proteins exchange on and off chromatin much more rapidly than would be predicted by their in vitro binding constants. The majority of chromatin-associated proteins examined to date exhibit transient binding with short residence times (less than 30 s), including proteins involved in modifying chromatin structure and transcription [43], whereas as linker and core histones exhibit much longer residence times [50–52]. We find that the exchange rate of ORC is very similar to that of other regulatory proteins such as HMG and HP1 proteins [43] and was not significantly altered during DNA synthesis (i.e. during S-phase). This implies that ORC can sample many sites throughout the nucleus within a very short period of time [43]. ORC is best known for its role in pre-RC formation during telophase, and many reports have cited the number of ORC molecules per cell to be on the order of the number of active replication origins [28,36,53]. However, there are many more pre-RCs assembled in a eukaryotic cell than there are active replication origins [3,54,55]. Moreover, ORC is known to have additional roles in the formation of heterochromatin [56], chromosome condensation [57,58] and checkpoint control [21,59]. Therefore, a dynamic ORC could explain why estimates of the number of ORC molecules in a cell could not account for all the activities of ORC or the number of pre-RCs in a eukaryotic cell.
A question that arises from recent in vivo protein dynamic studies is how a protein can reside on chromatin for only a few seconds, yet remain stably bound to chromatin during biochemical isolation and generate efficient in vivo footprints. The rapid in vivo exchange rates appear to be a result of several factors, including: the active displacement of proteins by another copy of the same protein, the presence of chromatin remodeling complexes that actively displace proteins, ATP levels and post-translation modifications [60–69]. In addition, decreasing the temperature at which the cells are photobleached results in reduced protein mobility [60,66,70]. In biochemical applications, cells are permeabilized at low temperatures, which would result in both the dilution of soluble factors and a decrease in mobility, giving the impression of a statically bound protein.
We cannot rule out the possibility that the observed FLIP kinetics are unique to GFP-tagged proteins, a caveat that applies to all photobleaching methods that rely on this tag. However, several considerations make this possibility unlikely. First, we carefully selected cell lines that express trace levels of exogenous protein in order to avoid potential artifacts due to over-expression. We determined that the fusion proteins behave identically to their endogenous counterparts biochemically and by their sub-cellular localization. Unfortunately, we were unable to reproducibly immunoprecipitate the fusion proteins with endogenous ORC subunits. The conditions necessary to remove ORC from chromatin result in the disruption of the complex, as only individual subunits, either endogenous or GFP-tagged, were able to be immunoprecipitated. However, we were able to show that GFP-ORC1 co-fractionated with ORC1 and 2 in a >300 kDa complex that could be selectively solubilized by micrococcal nuclease digestion of a chromatin-enriched preparation, strongly suggesting that GFP-ORC1 does interact with the other ORC subunits. Furthermore, we show that, without a flexible linker separating the GFP moiety from the ORC4 protein, GFP-ORC4 exhibits a kinetic mobility similar to free GFP (Fig. 5E and Supplemental Fig. 3). This control strongly suggests that the observed mobility of GFP-P-ORC4 is due to an interaction that is disrupted in the absence of the linker.
Dynamic complex vs. dynamic subunits
Due to the similarity in the kinetics of ORC1 and ORC4, we cannot distinguish whether ORC is a dynamic, mobile complex or a stable complex consisting of dynamic subunits (Fig. 6). However, there are several reasons to consider a model in which a stable complex consists of dynamic individual subunits. First, ORC1 and ORC4 partly differ in their sub-cellular localizations (Figs. 4A and 5C), and several ORC subunits interact independently with complexes that are not directly involved in the regulation of DNA replication [71–73]. Secondly, the rate at which the entire Xenopus ORC can be competed off of one chromatin template and on to another in vitro is considerably longer (~15 min) and is even longer (up to 128 min) when ORC is prevented from loading MCM proteins [74]. These results imply a much longer exchange rate for the entire complex and predict a slower exchange rate for ORC during S-phase when MCM loading is inhibited. Both data sets could be accounted for if individual subunits exchange faster with the complex than the complex exchanges with the chromatin (Fig. 6B). This is analogous to transcription complexes and heterochromatin domains, both of which are stable chromatin-associated complexes consisting of rapidly exchanging components [44,49]. Hence, it is possible that the concept of stability generated from dynamic components can be extended from transcriptional holoenzymes and chromatin domains to small multi-protein complexes such as ORC.
Fig. 6.
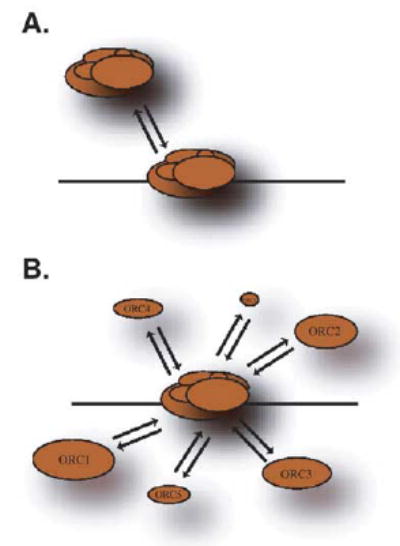
Model of ORC dynamics. (A) ORC may exist as a stable complex that dynamically associates with chromatin. (B) Alternatively, ORC may exist as a more stable chromatin-associated complex, consisting of dynamic subunits. Details are contained within the text.
What is the “ORC-cycle”?
We find ORC1 to be metabolically stable throughout the cell-cycle in both whole cell and fractionated extracts in CHO cells. These results are consistent with all other reports in CHO cells [3,36,40] and some reports in human cells [26,37]. These differences may be due to technical differences, the particular cell lines utilized in different reports or to variable degradation of ORC1 during cellular lysis. Recently, it has become apparent that both protein levels and gene regulation of replication proteins are affected by cellular transformation. The levels of geminin and Cdt1 were recently shown to be dramatically different in primary versus cancer cell lines [75,76]. In addition, when a primary human cell line, an isogenic transformed line and a Hela cancer cell line are directly compared, the protein levels of ORC1 as well as several core ORC subunits are up-regulated following transformation (A. McNairn and D.M. Gilbert, manuscript in preparation). The over-expression of ORC in Hela cells may result in the observed degradation of excess ORC1 as the cell contains more ORC than is necessary to support DNA replication. In our hands, we find that transient transfection of ORC1 in CHO cells results in the accumulation of an insoluble pool of ectopic ORC1 protein that is a likely target for protein degradation (L. Archibold and DMG, unpublished observation). Many conclusions with mammalian ORC1 derive from studies using transiently transfected cells [30,77]. In any case, it is clear that endogenous ORC1 is not degraded in CHO cells. Indeed, one might question the cellular rationale for degrading ORC1 during S-phase since in Xenopus it is required for the re-binding of CDC6 to chromatin following the initiation of DNA replication to facilitate an intra-S-phase checkpoint [14].
Our results demonstrate that the majority of ORC1 co-fractionates with chromatin throughout the cell-cycle. While these results are consistent with other reports using this cell line [3], our results differ from many reports in both human [28–30] and CHO [40] cells claiming that ORC1 is released from chromatin during S-phase. These reports suggest that up to 70% of S-phase ORC1 is soluble in human cells [28–30] and up to 50% in CHO cells [40]. We detect a minor soluble fraction of ORC1 in both S-phase and mitosis, however, this amount was variable from experiment to experiment. We have detected the hyper-phosphorylation of ORC1 in mitosis in both CHO cells and Hela cells (not shown), consistent with a previous report [78], however, we only detected a minor soluble fraction of ORC1 during S-phase rather than the significant pool described by Li and DePamphilis [40]. Furthermore, histone H1, a nuclear protein with a relatively long residence time on chromatin [51,52], is released from chromatin using these same extraction methods [36,40]. Hence, it is clear that detergent-extraction does not necessarily reflect the in vivo binding characteristics of a protein. Live cell photobleaching enables the analysis of the in vivo kinetic mobility of a protein and provides insight into the chromatin-binding characteristics. Through the use of photobleaching, we were able to detect only one population of GFP-ORC1 during the cell-cycle. A significant population of soluble molecules would be revealed as a rapid initial loss in fluorescence followed by a slower rate or a bi-phasic FLIP kinetics plot [48]. We conclude that, in CHO cells, there is little or no change in the chromatin interaction kinetics of ORC1 during the cell-cycle.
In summary, we provide evidence that both ORC1 and ORC4 are metabolically stable proteins that rapidly cycle on and off chromatin throughout interphase. Models of a static ORC:DNA interaction should be modified to account for this rapid cycling. Our results also raise questions regarding the well-accepted model by which ORC1 is selectively removed from chromatin and degraded during S-phase. Previous reports regarding the degradation or release from chromatin of ORC1 during S-phase may have resulted from ORC1 over-expression, degradation during sample preparation or to characteristics particular to the cell lines used. Hence, while it is clear that each of the ORC subunits has unique properties and may participate in roles separate from the regulation of DNA replication and although some properties of ORC1 may change during the cell-cycle, we propose that a significant population of ORC1 continues to interact with the entire complex throughout the cell-cycle.
Materials and methods
Cloning of CgORC4 and CgORC5
To clone CgORC4 and CgORC5, a combination of PCR and colony hybridization was used to obtain full-length cDNAs. To obtain the N-terminus of CgORC4, two sets of PCR primers were generated based upon regions of high homology from the published cDNA sequences of human and mouse ORC4 (ORC4C1: 5′-TGCGCGGCCGCTAA-CATCTGTAGGGCAGTTGG-3′, ORC4N1: 5′-TACGGAT-CCTGTCAGTCTTGGAAATCTGTC-3′ and ORC4C2: 5′-TGCGCGGCCGCTAACATCTGTAGGGCAGTTGG-3′, ORC4N2: 5′-ATCGGATCCGACACCAGTAGCAGTTATTGG-3′). Primers ORC4C1 and ORC4N1 amplified a 335 bp fragment from a CHOC400 cDNA pool. Primers ORC4C2 and ORC4N2 amplified a 745 bp fragment. Both PCR products were cloned in pBluescript II KS+. A total of five independent clones were sequenced: ORC4S-2, S-7, ORC4L-1, L-2 and L-3. A similar strategy was used to clone CgORC5 using primers ORC5N1: 5′-TTGGATCCCCTGTTTGGAGAGAGAC-3′ and ORC5C1: 5′-CAACTCGAGATACAAGTATTTTATTATG-3′. A 1.3 kb PCR product was amplified and cloned into pBluescript II KS+, and an independent clone was sequenced. To obtain the 5′-end of CgORC5, asymmetric PCR amplification was performed using a primer set consisting of a T7 primer and ORC5C1 [79]. PCR products were cloned into pBluescript II KS+, and two independent clones (ORC5-2, 3) were sequenced and verified.
To obtain C-terminus end of the cDNAs, the positive clones for CgORC4 and CgORC5 were used as probes to screen a CHO cDNA plasmid library (Invitrogen) by colony hybridization following an enrichment step using the CloneCapture Kit (Clontech) according to the manufacturer’s instructions. After the 2nd round of screening, 3 independent positive clones for ORC4 (ORC4-1, 2,3) and 1 clone for ORC5 (ORC5-1) were isolated. All clones were sequenced and confirmed to contain both an ATG start and poly-A tail. ClustalW analysis was performed using MacVector (Oxford Molecular Ltd.).
Construction of expression plasmids
An in frame fusion to the C-terminus of EGFP was generated by PCR amplifying the CgORC4 sequence with primers 5′-ATTGGTACCATGAGCAATCGTAAAAC-CAAGGGT-3′ and 5′-GCGGGATCCTTAGTTCTTTACAGAAATGTACTT-3′. The resulting PCR product was digested with KpnI/BamHI and ligated to ptetGFP ins1/1 [80]. To generate EGFP-CgORC5, the CgORC5 cDNA was PCR amplified using primers: 5′-AAACTCGAGATGTCTCACTTGGAGGGCGCGGTGCTC-3′ and 5′-GCACTGCCAGATGTCTGAGCTCTTTCAGATCTCGA-3′, digested with XhoI/SacI and ligated to ptetEGFP ins1/1. To introduce the peptide linker (GEGQGQGQGPGRGYAYRS) into pEGFP-CgORC4 ins1/1, two oligos containing the sequence: 5′-CCGGAGGAGAAGGACAAGGACAAGGACAAGGACCAGGAAGAGGATATGCATATAGATCAAC-3′ and 5′-TCGAGTTGATCTATATGCATATCCTCTTCCTGGTCCTTGTCCTTGTCCTTGTCCTTCTCCT-3′ were annealed and ligated into the BstEII/XhoI site of ptetEGFP-C1 ins1/1 to form ptetEGFP-PC1 ins1/1. The ORC4 cDNA was then ligated in frame using the SalI/BamHI sites of ptetEGFP-PC1 ins1/1 to form ptetEGFP-PC1-CgORC4.
Cell line construction and synchrony
ptetEGFP ins1/1, ptetEGFP-CgORC4 and ptetEGFP-PC1-CgORC4 vectors were linearized and transfected into Hyg16, a CHOC400 cell line stably expressing tTA [80], using Lipofectamine 2000 (Invitrogen). Stable cell lines were selected and screened as described [3]. The construction and characterization of the GFP-HP1α and GFP-CgORC1 cell line (3KA4) have been described [3,49]. In the course of this study, we identified a point mutation in the published CgORC1 cDNA that results in an amino acid change in the N-terminus region, R483M. This mutation has been confirmed by the sequencing of the genomic CgORC1 locus (T. Sasaki and D.M. Gilbert, unpublished) and is currently being characterized.
CHOC400 cells were grown in DMEM supplemented with 5% FBS, penicillin–streptomycin and non-essential amino acids. Tet-regulatable GFP-fusion protein cell lines were maintained in DMEM supplemented with 5% FBS, penicillin–streptomycin, non-essential amino acids, 0.5 mg/ml G418, 0.5 mg/ml hygromycin B and 2 μg/ml tetracy-cline. To synchronize cells in mitosis, 0.05 μg/ml nocodazole was added for 4 h followed by mechanical shake-off. Metaphase spread analysis indicated that greater than 95% mitotic cells were routinely obtained. For isoleucine starvation, cells were grown to near confluency, washed with PBS, and isoleucine minus media with 5% Dialyzed FBS was added. After 36 h, a coverslip was removed and pulsed for 30 min with BrdU to confirm that the cells were not actively entering S-phase (less than 5% BrdU positive). For S-phase synchrony, cells were first synchronized by isoleucine starvation (CHOC400) and released into either: 5 μg/ml aphidicolin, 2.5 mM hydroxyurea or 0.5 mM mimosine for 12–14 h.
Preparation of cellular extracts
Chromatin enrichment was carried out as described by Mendez and Stillman [2], with the following modifications, 25 μM MG132 (Calbiochem) and 1 mM ATP were added to all buffers. Whole cell extracts were prepared by lysing cells with boiling SDS-buffer containing 5% SDS and 8 M urea. For the salt extraction experiments, nuclei from asynchronously growing GFP-CgORC1 or GFP-CgORC4 expressing cells were prepared as described in [27]. The isolated nuclei were divided into two fractions and placed in OPB2 buffer (50 mM HEPES at pH 7.5, 5 mM MgCl2, 0.1 mM EDTA, 0.1 mM EGTA, 1 mM DTT, 10% glycerol, 0.01% Tween 20, PMSF, pepstatin, leupeptin, aprotinin and MG132) containing 150 mM or 300 mM NaCl for 30 min on ice to solubilize proteins. Soluble and insoluble fractions were separated by centrifugation.
For immunoprecipitations, 5 μl of either anti-CgORC1 or purified rabbit IgG (Santa Cruz) was added to a CHO cell nuclear extract and incubated overnight at 4°C. Protein G magnetic beads (Dynal) were then added and immunocomplexes collected. The beads were washed three times with OPB2 buffer containing 250 mM NaCl prior to re-suspending the beads in SDS-loading buffer.
Chromatin was fractionated as described above and treated with 2 U micrococcal nuclease (Sigma) at 28°C for 30 min. The reaction was stopped by the addition of EDTA to 5 mM. The soluble fraction was removed and placed in a Vivaspin 2 300 kDa molecular weight cut-off ultrafiltration device (Vivascience) and centrifuged at 16,000 × g for 30 min. The flow-through and high molecular weight fraction was then utilized for Western blots.
Antibody purification and immunoblotting
To generate anti-CgORC1 antibodies, an N-terminal fragment of CgORC1 was expressed in E. coli and purified using an Ni-NTA column (Amersham). The purified protein was then used for rabbit immunization and antibody development (Covance). Anti-CgORC1 antibody was purified from crude serum using Hi-Trap protein A columns according to the manufacturer’s instructions (Amersham).
Cell extracts were normalized to cell number and run on either 8% or 10% SDS-PAGE gels and transferred to PVDF membrane (Immobilion, Millipore) by wet transfer using Bolt and Mahoney buffer (40 mM Tris, 10 mM sodium acetate, 2 mM EDTA, 0.05% SDS, 20% MeOH) at 1A for 45 min [81]. Blots were blocked with 1% non-fat milk in TBS (150 mM NaCl, 10 mM Tris, pH 8.0, 0.05% Tween 20) for 1 h at room temperature. All primary antibody incubations were carried out overnight at 4°C in blocking buffer. The following antibodies were used: anti-CgORC1 NY674 (1:10,000), anti-ORC2 (1:500) (Santa Cruz), anti-ORC4 (1:500) (BD Transduction Labs) and anti-cyclin B1 (1:2000) (Cell Signaling Technology). Detection was with Affinipure goat anti-rabbit HRP or goat anti-mouse HRP (Jackson Immunolabs) and Supersignal West Pico ECL substrate for all antibodies.
FLIP
EGFP-ORC fusion cells were induced for 96 h, synchronized in mitosis and plated into LabTec II chambers (Nalgene). After mitotic shake-off, cells were released into G1-phase and either used as G1-phase cells or synchronized at the G1/S boundary with 5 μg/ml aphidicolin. S-phase progression was measured by labeling cells with BrdU at each timepoint. FLIP was performed using a Leica TCS 2 confocal microscope with a 488 nm laser line as described [82]. Cell nuclei were repeatedly imaged and bleached at 5 s intervals. The bleach pulse consisted of a 0.5 s pulse at 100% power. All experiments were performed at 37°C using an air stream incubator (Nevtek). The quantification of fluorescence loss was performed using Leica TCS software.
Acknowledgments
We thank J. Mendez and B. Stillman for their generous gift of the anti-human ORC1 antibody used in their studies and T. Karpova for technical support. Imaging was performed at the NCI Core Fluorescence Imaging Facility. This work was supported by NIH grant GM-57233-01 and American Cancer Society grant RPG-97-098-04-CCG to D.M.G.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.yexcr. 2005.05.009.
References
- 1.Dimitrova DS, Gilbert DM. The spatial position and replication timing of chromosomal domains are both established in early G1 phase. Mol Cell. 1999;4:983–993. doi: 10.1016/s1097-2765(00)80227-0. [DOI] [PubMed] [Google Scholar]
- 2.Mendez J, Stillman B. Chromatin association of human origin recognition complex, cdc6, and minichromosome maintenance proteins during the cell cycle: assembly of prereplication complexes in late mitosis. Mol Cell Biol. 2000;20:8602–8612. doi: 10.1128/mcb.20.22.8602-8612.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Okuno Y, McNairn AJ, den Elzen N, Pines J, Gilbert DM. Stability, chromatin association and functional activity of mammalian pre-replication complex proteins during the cell cycle. EMBO J. 2001;20:4263–4277. doi: 10.1093/emboj/20.15.4263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dimitrova DS, Prokhorova TA, Blow JJ, Todorov IT, Gilbert DM. Mammalian nuclei become licensed for DNA replication during late telophase. J Cell Sci. 2002;115:51–59. doi: 10.1242/jcs.115.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kearsey SE, Cotterill S. Enigmatic variations: divergent modes of regulating eukaryotic DNA replication. Mol Cell. 2003;12:1067–1075. doi: 10.1016/s1097-2765(03)00441-6. [DOI] [PubMed] [Google Scholar]
- 6.Li X, Zhao Q, Liao R, Sun P, Wu X. The SCF(Skp2) ubiquitin ligase complex interacts with the human replication licensing factor Cdt1 and regulates Cdt1 degradation. J Biol Chem. 2003;278:30854–30858. doi: 10.1074/jbc.C300251200. [DOI] [PubMed] [Google Scholar]
- 7.Alexandrow MG, Hamlin JL. Cdc6 chromatin affinity is unaffected by serine-54 phosphorylation, S-phase progression, and overexpression of cyclin. A Mol Cell Biol. 2004;24:1614–1627. doi: 10.1128/MCB.24.4.1614-1627.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Klemm RD, Austin RJ, Bell SP. Coordinate binding of ATP and origin DNA regulates the ATPase activity of the origin recognition complex. Cell. 1997;88:493–502. doi: 10.1016/s0092-8674(00)81889-9. [DOI] [PubMed] [Google Scholar]
- 9.Chesnokov I, Remus D, Botchan M. Functional analysis of mutant and wild-type Drosophila origin recognition complex. Proc Natl Acad Sci U S A. 2001;98:11997–12002. doi: 10.1073/pnas.211342798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vashee S, Cvetic C, Lu W, Simancek P, Kelly TJ, Walter JC. Sequence-independent DNA binding and replication initiation by the human origin recognition complex. Genes Dev. 2003;17:1894–1908. doi: 10.1101/gad.1084203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chastain PD, II, Bowers JL, Lee DG, Bell SP, Griffith JD. Mapping subunit location on the S. cerevisiae ORC free and bound to DNA using a novel nanoscale biopointer. J Biol Chem. 2004:36354–36362. doi: 10.1074/jbc.M403501200. [DOI] [PubMed] [Google Scholar]
- 12.Liang C, Weinreich M, Stillman B. ORC and Cdc6p interact and determine the frequency of initiation of DNA replication in the genome. Cell. 1995;81:667–676. doi: 10.1016/0092-8674(95)90528-6. [DOI] [PubMed] [Google Scholar]
- 13.Madine MA, Swietlik M, Pelizon C, Romanowski P, Mills AD, Laskey RA. The roles of the MCM, ORC, and Cdc6 proteins in determining the replication competence of chromatin in quiescent cells. J Struct Biol. 2000;129:198–210. doi: 10.1006/jsbi.2000.4218. [DOI] [PubMed] [Google Scholar]
- 14.Oehlmann M, Score AJ, Blow JJ. The role of Cdc6 in ensuring complete genome licensing and S phase checkpoint activation. J Cell Biol. 2004;165:181–190. doi: 10.1083/jcb.200311044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rowles A, Blow JJ. Chromatin proteins involved in the initiation of DNA replication. Curr Opin Genet Dev. 1997;7:152–157. doi: 10.1016/s0959-437x(97)80123-2. [DOI] [PubMed] [Google Scholar]
- 16.Diffley JF, Cocker JH, Dowell SJ, Rowley A. Two steps in the assembly of complexes at yeast replication origins in vivo. Cell. 1994;78:303–316. doi: 10.1016/0092-8674(94)90299-2. [DOI] [PubMed] [Google Scholar]
- 17.Fujita M, Hori Y, Shirahige K, Tsurimoto T, Yoshikawa H, Obuse C. Cell cycle dependent topological changes of chromosomal replication origins in Saccharomyces cerevisiae. Genes Cells. 1998;3:737–749. doi: 10.1046/j.1365-2443.1998.00226.x. [DOI] [PubMed] [Google Scholar]
- 18.Liang C, Stillman B. Persistent initiation of DNA replication and chromatin-bound MCM proteins during the cell cycle in cdc6 mutants. Genes Dev. 1997;11:3375–3386. doi: 10.1101/gad.11.24.3375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lygerou Z, Nurse P. The fission yeast origin recognition complex is constitutively associated with chromatin and is differentially modified through the cell cycle. J Cell Sci. 1999;112(Pt 21):3703–3712. doi: 10.1242/jcs.112.21.3703. [DOI] [PubMed] [Google Scholar]
- 20.Kong D, DePamphilis ML. Site-specific DNA binding of the Schizosaccharomyces pombe origin recognition complex is determined by the Orc4 subunit. Mol Cell Biol. 2001;21:8095–8103. doi: 10.1128/MCB.21.23.8095-8103.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shimada K, Pasero P, Gasser SM. ORC and the intra-S-phase checkpoint: a threshold regulates Rad53p activation in S phase. Genes Dev. 2002;16:3236–3252. doi: 10.1101/gad.239802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Murakami H, Yanow SK, Griffiths D, Nakanishi M, Nurse P. Maintenance of replication forks and the S-phase checkpoint by Cdc18p and Orp1p. Nat Cell Biol. 2002;4:384–388. doi: 10.1038/ncb789. [DOI] [PubMed] [Google Scholar]
- 23.Thome KC, Dhar SK, Quintana DG, Delmolino L, Shahsafaei A, Dutta A. Subsets of human origin recognition complex (ORC) subunits are expressed in non-proliferating cells and associate with non-ORC proteins. J Biol Chem. 2000;275:35233–35241. doi: 10.1074/jbc.M005765200. [DOI] [PubMed] [Google Scholar]
- 24.Asano M, Wharton RP. E2F mediates developmental and cell cycle regulation of ORC1 in Drosophila. EMBO J. 1999;18:2435–2448. doi: 10.1093/emboj/18.9.2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ohtani K, DeGregori J, Leone G, Herendeen DR, Kelly TJ, Nevins JR. Expression of the HsOrc1 gene, a human ORC1 homolog, is regulated by cell proliferation via the E2F transcription factor. Mol Cell Biol. 1996;16:6977–6984. doi: 10.1128/mcb.16.12.6977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Saha P, Chen J, Thome KC, Lawlis SJ, Hou ZH, Hendricks M, Parvin JD, Dutta A. Human CDC6/Cdc18 associates with Orc1 and cyclin-cdk and is selectively eliminated from the nucleus at the onset of S phase. Mol Cell Biol. 1998;18:2758–2767. doi: 10.1128/mcb.18.5.2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vashee S, Simancek P, Challberg MD, Kelly TJ. Assembly of the human origin recognition complex. J Biol Chem. 2001;276:26666–26673. doi: 10.1074/jbc.M102493200. [DOI] [PubMed] [Google Scholar]
- 28.Tatsumi Y, Ohta S, Kimura H, Tsurimoto T, Obuse C. The ORC1 cycle in human cells: I. Cell cycle-regulated oscillation of human ORC1. J Biol Chem. 2003;278:41528–41534. doi: 10.1074/jbc.M307534200. [DOI] [PubMed] [Google Scholar]
- 29.Ohta S, Tatsumi Y, Fujita M, Tsurimoto T, Obuse C. The ORC1 cycle in human cells: II. Dynamic changes in the human ORC complex during the cell cycle. J Biol Chem. 2003;278:41535–41540. doi: 10.1074/jbc.M307535200. [DOI] [PubMed] [Google Scholar]
- 30.Mendez J, Zou-Yang XH, Kim SY, Hidaka M, Tansey WP, Stillman B. Human origin recognition complex large subunit is degraded by ubiquitin-mediated proteolysis after initiation of DNA replication. Mol Cell. 2002;9:481–491. doi: 10.1016/s1097-2765(02)00467-7. [DOI] [PubMed] [Google Scholar]
- 31.Ritzi M, Baack M, Musahl C, Romanowski P, Laskey RA, Knippers R. Human minichromosome maintenance proteins and human origin recognition complex 2 protein on chromatin. J Biol Chem. 1998;273:24543–24549. doi: 10.1074/jbc.273.38.24543. [DOI] [PubMed] [Google Scholar]
- 32.Schaarschmidt D, Baltin J, Stehle IM, Lipps HJ, Knippers R. An episomal mammalian replicon: sequence-independent binding of the origin recognition complex. EMBO J. 2004;23:191–201. doi: 10.1038/sj.emboj.7600029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Keller C, Ladenburger EM, Kremer M, Knippers R. The origin recognition complex marks a replication origin in the human TOP1 gene promoter. J Biol Chem. 2002;277:31430–31440. doi: 10.1074/jbc.M202165200. [DOI] [PubMed] [Google Scholar]
- 34.Ladenburger EM, Keller C, Knippers R. Identification of a binding region for human origin recognition complex proteins 1 and 2 that coincides with an origin of DNA replication. Mol Cell Biol. 2002;22:1036–1048. doi: 10.1128/MCB.22.4.1036-1048.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.DePamphilis ML. The ‘ORC cycle’: a novel pathway for regulating eukaryotic DNA replication. Gene. 2003;310:1–15. doi: 10.1016/s0378-1119(03)00546-8. [DOI] [PubMed] [Google Scholar]
- 36.Natale DA, Li CJ, Sun WH, DePamphilis ML. Selective instability of Orc1 protein accounts for the absence of functional origin recognition complexes during the M–G(1) transition in mammals. EMBO J. 2000;19:2728–2738. doi: 10.1093/emboj/19.11.2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ritzi M, Tillack K, Gerhardt J, Ott E, Humme S, Kremmer E, Hammerschmidt W, Schepers A. Complex protein–DNA dynamics at the latent origin of DNA replication of Epstein–Barr virus. J Cell Sci. 2003;116:3971–3984. doi: 10.1242/jcs.00708. [DOI] [PubMed] [Google Scholar]
- 38.Romanowski P, Madine MA, Rowles A, Blow JJ, Laskey RA. The Xenopus origin recognition complex is essential for DNA replication and MCM binding to chromatin. Curr Biol. 1996;6:1416–1425. doi: 10.1016/s0960-9822(96)00746-4. [DOI] [PubMed] [Google Scholar]
- 39.Wu JR, Gilbert DM. A distinct G1 step required to specify the Chinese hamster DHFR replication origin. Science. 1996;271:1270–1272. doi: 10.1126/science.271.5253.1270. [DOI] [PubMed] [Google Scholar]
- 40.Li CJ, DePamphilis ML. Mammalian Orc1 protein is selectively released from chromatin and ubiquitinated during the S-to-M transition in the cell division cycle. Mol Cell Biol. 2002;22:105–116. doi: 10.1128/MCB.22.1.105-116.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Petersen BO, Wagener C, Marinoni F, Kramer ER, Melixetian M, Denchi EL, Gieffers C, Matteucci C, Peters JM, Helin K. Cell cycle- and cell growth-regulated proteolysis of mammalian CDC6 is dependent on APC-CDH1. Genes Dev. 2000;14:2330–2343. doi: 10.1101/gad.832500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Phair RD, Gorski SA, Misteli T. Measurement of dynamic protein binding to chromatin in vivo, using photobleaching microscopy. Methods Enzymol. 2004;375:393–414. doi: 10.1016/s0076-6879(03)75025-3. [DOI] [PubMed] [Google Scholar]
- 43.Phair RD, Scaffidi P, Elbi C, Vecerova J, Dey A, Ozato K, Brown DT, Hager G, Bustin M, Misteli T. Global nature of dynamic protein–chromatin interactions in vivo: three-dimensional genome scanning and dynamic interaction networks of chromatin proteins. Mol Cell Biol. 2004;24:6393–6402. doi: 10.1128/MCB.24.14.6393-6402.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dundr M, Hoffmann-Rohrer U, Hu Q, Grummt I, Rothblum LI, Phair RD, Misteli T. A kinetic framework for a mammalian RNA polymerase in vivo. Science. 2002;298:1623–1626. doi: 10.1126/science.1076164. [DOI] [PubMed] [Google Scholar]
- 45.Kimura H, Sugaya K, Cook PR. The transcription cycle of RNA polymerase II in living cells. J Cell Biol. 2002;159:777–782. doi: 10.1083/jcb.200206019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sporbert A, Gahl A, Ankerhold R, Leonhardt H, Cardoso MC. DNA polymerase clamp shows little turnover at established replication sites but sequential de novo assembly at adjacent origin clusters. Mol Cell. 2002;10:1355–1365. doi: 10.1016/s1097-2765(02)00729-3. [DOI] [PubMed] [Google Scholar]
- 47.Chesnokov I, Gossen M, Remus D, Botchan M. Assembly of functionally active Drosophila origin recognition complex from recombinant proteins. Genes Dev. 1999;13:1289–1296. doi: 10.1101/gad.13.10.1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Phair RD, Misteli T. Kinetic modelling approaches to in vivo imaging. Nat Rev Mol Cell Biol. 2001;2:898–907. doi: 10.1038/35103000. [DOI] [PubMed] [Google Scholar]
- 49.Cheutin T, McNairn AJ, Jenuwein T, Gilbert DM, Singh PB, Misteli T. Maintenance of stable heterochromatin domains by dynamic HP1 binding. Science. 2003;299:721–725. doi: 10.1126/science.1078572. [DOI] [PubMed] [Google Scholar]
- 50.Kimura H, Cook PR. Kinetics of core histones in living human cells: little exchange of H3 and H4 and some rapid exchange of H2B. J Cell Biol. 2001;153:1341–1353. doi: 10.1083/jcb.153.7.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lever MA, Th’ng JP, Sun X, Hendzel MJ. Rapid exchange of histone H1.1 on chromatin in living human cells. Nature. 2000;408:873–876. doi: 10.1038/35048603. [DOI] [PubMed] [Google Scholar]
- 52.Misteli T, Gunjan A, Hock R, Bustin M, Brown DT. Dynamic binding of histone H1 to chromatin in living cells. Nature. 2000;408:877–881. doi: 10.1038/35048610. [DOI] [PubMed] [Google Scholar]
- 53.Blow JJ, Gillespie PJ, Francis D, Jackson DA. Replication origins in Xenopus egg extract Are 5–15 kilobases apart and are activated in clusters that fire at different times. J Cell Biol. 2001;152:15–25. doi: 10.1083/jcb.152.1.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wyrick JJ, Aparicio JG, Chen T, Barnett JD, Jennings EG, Young RA, Bell SP, Aparicio OM. Genome-wide distribution of ORC and MCM proteins in S. cerevisiae: high-resolution mapping of replication origins. Science. 2001;294:2357–2360. doi: 10.1126/science.1066101. [DOI] [PubMed] [Google Scholar]
- 55.Walter J, Newport JW. Regulation of replicon size in Xenopus egg extracts. Science. 1997;275:993–995. doi: 10.1126/science.275.5302.993. [DOI] [PubMed] [Google Scholar]
- 56.Shareef MM, Badugu R, Kellum R. HP1/ORC complex and heterochromatin assembly. Genetica. 2003;117:127–134. doi: 10.1023/a:1022963223220. [DOI] [PubMed] [Google Scholar]
- 57.Loupart ML, Krause SA, Heck MS. Aberrant replication timing induces defective chromosome condensation in Drosophila ORC2 mutants. Curr Biol. 2000;10:1547–1556. doi: 10.1016/s0960-9822(00)00844-7. [DOI] [PubMed] [Google Scholar]
- 58.Pflumm MF, Botchan MR. Orc mutants arrest in metaphase with abnormally condensed chromosomes. Development. 2001;128:1697–1707. doi: 10.1242/dev.128.9.1697. [DOI] [PubMed] [Google Scholar]
- 59.Watanabe K, Morishita J, Umezu K, Shirahige K, Maki H. Involvement of RAD9-dependent damage checkpoint control in arrest of cell cycle, induction of cell death, and chromosome instability caused by defects in origin recognition complex in Saccharomyces cerevisiae. Eukaryotic Cell. 2002;1:200–212. doi: 10.1128/EC.1.2.200-212.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wagner S, Chiosea S, Ivshina M, Nickerson JA. In vitro FRAP reveals the ATP-dependent nuclear mobilization of the exon junction complex protein SRm160. J Cell Biol. 2004;164:843–850. doi: 10.1083/jcb.200307002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Stavreva DA, Muller WG, Hager GL, Smith CL, McNally JG. Rapid glucocorticoid receptor exchange at a promoter is coupled to transcription and regulated by chaperones and proteasomes. Mol Cell Biol. 2004;24:2682–2697. doi: 10.1128/MCB.24.7.2682-2697.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nagaich AK, Walker DA, Wolford R, Hager GL. Rapid periodic binding and displacement of the glucocorticoid receptor during chromatin remodeling. Mol Cell. 2004;14:163–174. doi: 10.1016/s1097-2765(04)00178-9. [DOI] [PubMed] [Google Scholar]
- 63.Hager GL. The dynamics of intranuclear movement and chromatin remodeling by the glucocorticoid receptor. Ernst Schering Res Found Workshop. 2002:111–129. doi: 10.1007/978-3-662-04660-9_8. [DOI] [PubMed] [Google Scholar]
- 64.Fletcher TM, Xiao N, Mautino G, Baumann CT, Wolford R, Warren BS, Hager GL. ATP-dependent mobilization of the glucocorticoid receptor during chromatin remodeling. Mol Cell Biol. 2002;22:3255–3263. doi: 10.1128/MCB.22.10.3255-3263.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Harrer M, Lührs H, Bustin M, Scheer U, Hock R. Dynamic interaction of HMGA1a proteins with chromatin. J Cell Sci. 2004:3459–3471. doi: 10.1242/jcs.01160. [DOI] [PubMed] [Google Scholar]
- 66.Elbi C, Walker DA, Romero G, Sullivan WP, Toft DO, Hager GL, DeFranco DB. Molecular chaperones function as steroid receptor nuclear mobility factors. Proc Natl Acad Sci U S A. 2004;101:2876–2881. doi: 10.1073/pnas.0400116101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Contreras A, Hale TK, Stenoien DL, Rosen JM, Mancini MA, Herrera RE. The dynamic mobility of histone H1 is regulated by cyclin/CDK phosphorylation. Mol Cell Biol. 2003;23:8626–8636. doi: 10.1128/MCB.23.23.8626-8636.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Catez F, Yang H, Tracey KJ, Reeves R, Misteli T, Bustin M. Network of dynamic interactions between histone H1 and high-mobility-group proteins in chromatin. Mol Cell Biol. 2004;24:4321–4328. doi: 10.1128/MCB.24.10.4321-4328.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Becker M, Baumann C, John S, Walker DA, Vigneron M, McNally JG, Hager GL. Dynamic behavior of transcription factors on a natural promoter in living cells. EMBO Rep. 2002;3:1188–1194. doi: 10.1093/embo-reports/kvf244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Reits EA, Neefjes JJ. From fixed to FRAP: measuring protein mobility and activity in living cells. Nat Cell Biol. 2001;3:E145–E147. doi: 10.1038/35078615. [DOI] [PubMed] [Google Scholar]
- 71.Prasanth SG, Prasanth KV, Stillman B. Orc6 involved in DNA replication, chromosome segregation, and cytokinesis. Science. 2002;297:1026–1031. doi: 10.1126/science.1072802. [DOI] [PubMed] [Google Scholar]
- 72.Chesnokov IN, Chesnokova ON, Botchan M. A cytokinetic function of Drosophila ORC6 protein resides in a domain distinct from its replication activity. Proc Natl Acad Sci U S A. 2003;100:9150–9155. doi: 10.1073/pnas.1633580100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Prasanth SG, Prasanth KV, Siddiqui K, Spector DL, Stillman B. Human Orc2 localizes to centrosomes, centromeres and heterochromatin during chromosome inheritance. EMBO J. 2004:2651–2653. doi: 10.1038/sj.emboj.7600255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Harvey KJ, Newport J. Metazoan origin selection: origin recognition complex chromatin binding is regulated by CDC6 recruitment and ATP hydrolysis. J Biol Chem. 2003;278:48524–48528. doi: 10.1074/jbc.M307661200. [DOI] [PubMed] [Google Scholar]
- 75.Karakaidos P, Taraviras S, Vassiliou LV, Zacharatos P, Kastrinakis NG, Kougiou D, Kouloukoussa M, Nishitani H, Papavassiliou AG, Lygerou Z, Gorgoulis VG. Overexpression of the replication licensing regulators hCdt1 and hCdc6 characterizes a subset of non-small-cell lung carcinomas: synergistic effect with mutant p53 on tumor growth and chromosomal instability—evidence of E2F-1 transcriptional control over hCdt1. Am J Pathol. 2004;165:1351–1365. doi: 10.1016/S0002-9440(10)63393-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Xouri G, Lygerou Z, Nishitani H, Pachnis V, Nurse P, Taraviras S. Cdt1 and geminin are down-regulated upon cell cycle exit and are over-expressed in cancer-derived cell lines. Eur J Biochem. 2004;271:3368–3378. doi: 10.1111/j.1432-1033.2004.04271.x. [DOI] [PubMed] [Google Scholar]
- 77.Lidonnici MR, Rossi R, Paixao S, Mendoza-Maldonado R, Paolinelli R, Arcangeli C, Giacca M, Biamonti G, Montecucco A. Subnuclear distribution of the largest subunit of the human origin recognition complex during the cell cycle. J Cell Sci. 2004;117:5221–5231. doi: 10.1242/jcs.01405. [DOI] [PubMed] [Google Scholar]
- 78.Li CJ, Vassilev A, DePamphilis ML. Role for Cdk1 (Cdc2)/cyclin A in preventing the mammalian origin recognition complex’s largest subunit (Orc1) from binding to chromatin during mitosis. Mol Cell Biol. 2004;24:5875–5886. doi: 10.1128/MCB.24.13.5875-5886.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.S. Huang, A.Y. Jong, W. Yang, J. Holcenberg, Amplification of gene ends from gene libraries by PCR with single sided specificity, in: B.A. White (Ed.), PCR Protocols: Current Methods and Applications, Human Press, 1993. [DOI] [PubMed]
- 80.Izumi M, Gilbert DM. Homogeneous tetracycline-regulatable gene expression in mammalian fibroblasts. J Cell Biochem. 1999;76:280–289. doi: 10.1002/(sici)1097-4644(20000201)76:2<280::aid-jcb11>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 81.Bolt MW, Mahoney PA. High-efficiency blotting of proteins of diverse sizes following sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Anal Biochem. 1997;247:185–192. doi: 10.1006/abio.1997.2061. [DOI] [PubMed] [Google Scholar]
- 82.Phair RD, Misteli T. High mobility of proteins in the mammalian cell nucleus. Nature. 2000;404:604–609. doi: 10.1038/35007077. [DOI] [PubMed] [Google Scholar]