

Abstract
Avidity of antigen-specific cytolytic T lymphocytes (CTL) is a critical determinant for clearing viral infection and eliminating tumor. While previous studies have demonstrated that vaccines employing enhanced costimulation will enhance the level and avidity of antigen-specific T cells from naive mice, there is conflicting data about the effect of vaccines employing enhanced costimulation (vector-based or dendritic cell–based) on the survival of memory T cells. In this study we have first extended previous observations that primary vaccination with a recombinant vaccinia virus (rV-) expressing a model antigen (LacZ) and a triad of T-cell costimulatory molecules (B7-1, ICAM-1, LFA-3, designated TRICOM) enhances the level and avidity of T cells from naive vaccinated C57BL/6 (Thy1.2) mice. Adoptive transfer of Thy1.1 memory CD8+ T cells into naive Thy1.2 C57BL/6 mice was followed with booster vaccinations with a recombinant fowlpox (rF-) expressing LacZ (rF-LacZ) or booster vaccinations with rF-LacZ/TRICOM. Analysis of levels of β-galactosidase tetramer positive T cells and functional assays (IFN-γ expression and lytic activity) determined that booster vaccinations with rF-LacZ/TRICOM were superior to booster vaccinations with rF-LacZ in terms of both maintenance and enhanced avidity of memory CD8+ T cells. Anti-tumor experiments employing a self antigen (CEA vaccines in CEA transgenic mice bearing CEA-expressing tumors) also demonstrated that the use of booster vaccinations with vaccines bearing enhanced costimulatory capacity had superior anti-tumor effects. These studies thus have implications in the design of more effective vaccine strategies.
Keywords: CTL, T-cell costimulation, vaccines, memory T cells
Introduction
Induction of efficient long-term immune memory is the aim for all vaccination protocols. Factors required to maintain memory cell populations have been controversial. In mice, memory T-cell survival does not require the persistence of cognate antigen (1–3). However, antigen-specific T cells in patients infected by HIV show a correlation between viral load and the percentage of antigen-specific cells in the blood (4). HIV-specific memory cell counts fall sharply when anti-retroviral therapy is initiated, matching the fall in viral load (4). The results suggest that the size of the memory T-cell pool in humans is highly dependent on the persistence of cognate antigen. In contrast, persistent infection of mice with lymphocytic choriomeningitis virus (LCMV)2 resulted in selective deletion or anergy of high avidity memory cytolytic T lymphocytes (CTL) (5). Administration of LCMV vaccines successfully induced lytic MHC-restricted CTL in the persistently virus infected mice; however, these CTL were of low avidity and could not clear the viral infection (5).
It has previously been shown that the proliferation and activation of naive T cells are more dependent on costimulation as compared with effector/memory T cells (6, 7). When antigen-presenting cells (APC) with decreased costimulatory capacity are used, proliferation of naive T cells requires increasingly higher peptide concentrations compared with effector/memory T cells (6). Higher peptide concentrations usually result in apoptosis of effector/memory T cells. As for the effect of costimulatory molecules on effector/memory T cells, the results are conflicting. Iezzi et al. (6) demonstrated that signaling through CD28 partially protected the effector/memory CD4 T cells (from TCR transgenic mice specific for Flu hemagglutinin peptide) from apoptosis induced by high peptide concentration/prolonged peptide stimulation. In contrast, Sabzevari et al.(8) reported that effector/memory CD4 T cells from Pigeon Cytochrome C T-cell receptor (PCC TCR) transgenic mice were more susceptible to apoptosis induced by APC expressing B7-1 in the presence of high affinity cognate peptide as compared with naive CD4 T cells. It should be pointed out, however, that both of the studies (6, 8) were performed in vitro, and both analyzed effector/memory CD4+ T cells from TCR transgenic mice.
We have previously shown that recombinant poxvirus vectors can be efficiently employed in diversified prime and boost strategies to enhance antigen-specific murine T-cell responses. Primary vaccination employed the replication-competent vaccinia (rV-) and booster vaccinations used the replication-defective avipox virus (fowlpox, rF). Subsequent studies showed that insertion of the transgenes for a triad of T-cell costimulatory molecules (B7-1, ICAM-1, and LFA-3, designated TRICOM) further enhanced the levels of CD8+ T-cell response (9, 10). These studies, however, did not address whether the multiple booster vaccinations were (a) simply generating more effector cells from naive T-cell populations, (b) expanding memory T-cell populations, or (c) both of the above. These studies also did not address the avidity of memory T cells. In the present study we have set out to answer these questions. We first vaccinated C57BL/6 Thy1.1 mice with an rV-LacZ/TRICOM vector. We then adoptively transferred antigen-specific memory T cells into Thy 1.2 C57BL/6 mice, and vaccinated those mice with either a recombinant rF-LacZ vector, or a recombinant rF-LacZ/TRICOM vector. Both the level and avidity of β-galactosidase (β-gal)–specific CD8+ memory (Thy 1.1) T cells were then analyzed. The results of these studies are of importance in the design of clinical trials in which vaccines containing costimulatory molecules (vector-based or dendritic-cell based) are used as booster vaccinations with the purpose of maintaining and/or expanding high avidity antigen-specific memory CD8+ T cells.
Materials and Methods
Mice and cell lines
Female C57BL/6J (C57BL/6) mice were obtained from the National Cancer Institute, Frederick Cancer Research Facility (Frederick, MD). Female Thy1.1 mice (formerly called C57BL/6.PL-Thy1a/Cy) were purchased from The Jackson Laboratory (Bar Harbor, ME). This C57BL/6J congenic strain carries the T lymphocyte specific Thy1a (Thy1.1) allele. The Thy1.1 and Thy1.2 mice are genetically identical mice differing only in their expression of the Thy-1 allele. Donor T cells (Thy1.1) can be easily distinguished from recipient T cells (Thy1.2) by flow cytometric analysis. Mice were housed and maintained under pathogen-free conditions in microisolator cages and used for experiments at 6–8 weeks of age.
C57BL/6 mice transgenic for human CEA (designated CEA-Tg) were originally obtained from a breeding pair provided by Dr. John Thompson (Institute of Immunobiology, University of Freiburg, Germany). The generation and characterization of the CEA-Tg mouse have been previously described (11). Polymerase chain reaction of DNA from whole blood to detect the CEA gene was used to screen for CEA-positive mice, as previously described (12). Mice were housed and maintained under pathogen-free conditions in microisolator cages. For experiments, 8-12 week old CEA-Tg mice were used. All mouse procedures were reviewed and approved by the Animal Use and Care Committee, NIH.
Murine colon adenocarcinoma cells expressing human CEA (MC-38-CEA) were generated by retroviral transduction of MC-38 cells with CEA cDNA (13). Prior to transplantation to mice, the cells were trypsinized, dispersed through a 70-μm cell strainer (Falcon, Becton Dickinson, Franklin Lakes, NJ) and washed twice in Hank's balanced salt solution (HBSS) before final suspension in HBSS.
The tumor cell line EL4 (H-2b, thymoma, ATCC TIB-39) was purchased from ATCC (Rockville, MD) and maintained in RPMI complete medium.
Recombinant viruses
The recombinant vaccinia viruses designated rV-LacZ, rV-LacZ/B7-1, and rV-LacZ/TRICOM were constructed as described (14), and contain the LacZ gene encoding β-gal. The recombinant fowlpox LacZ containing viruses rF-LacZ and rF-LacZ/TRICOM was constructed in the similar manner.
The recombinant vaccinia virus designated rV-CEA has been described (15). rV-CEA/TRICOM contains the murine B7-1, ICAM-1, and LFA-3 genes in combination with the human gene CEA as described elsewhere (16). The recombinant fowlpox virus rF-CEA/TRICOM contains the murine B7-1, ICAM-1, and LFA-3 genes in combination with the human gene CEA as described elsewhere (16). The recombinant fowlpox containing the murine GM-CSF gene (designated rF-GM-CSF) has been described (17). Therion Biologics Corporation (Cambridge, MA) kindly provided all orthopox viruses as part of a Collaborative Research and Development Agreement with NCI.
Peptide
The H-2Kb binding peptides of β-gal, β-gal96-103 (DAPIYTNV) (18), and ovalbumin (OVA) epitope (SIINFEKL) were commercially synthesized (SynPep, Dublin, CA). The purity of peptides was greater than 96%. Peptides were dissolved in DMSO and then diluted with PBS to 2 mg/ml (the final concentration of DMSO in stock solution is <5%, v/v), filtered through a 0.2 μM membrane (Millex-LG, hydrophilic PTFE membrane, Millipore, Bedford, MA) and stored at −80oC.
Antibodies, tetramer staining and flow cytometry assay
FITC, PE, PerCP or Cy-chrome-labeled anti-mouse CD2, CD3, CD4, CD8, CD11a, CD28, CD44, CD62L, IFN-γ, Thy1.1, Thy1.2, and all control antibodies were purchased from BD Pharmingen (San Diego, CA). PE-labeled H-2Kb-β-gal tetramer was provided by the Tetramer Core Facility of the National Institutes of Health (Atlanta, GA). For flow cytometric analysis of cell surface markers, 1–2 x 106 cells were incubated on ice with the appropriate Abs for 30–45 min., washed twice and then analyzed on a FACS Calibur (BD Biosciences, Mountain View, CA). Background staining was assessed by use of isotype control Abs. For tetramer staining, cells were stained with FITC- or PerCP-labeled CD8, or Thy1.1 and PE-labeled tetramer for 60 min. on ice. For intracellular IFN-γ staining, cells were stained first with surface markers and then were permeabilized with Cytofix/CytopermTM (BD Pharmingen) followed by anti-IFN-γ staining. Data were analyzed using CellQuest software.
Vaccinations and purification of memory T cells and adoptive transfer
For experiments described in Fig. 1, C57BL/6 mice (Thy 1.2) were vaccinated one time with buffer, rV-LacZ, rV-LacZ/B7-1, or rV-LacZ/TRICOM. After 30 days, splenocytes were harvested. β-gal–specific CD8+ T-cell precursor frequency and avidity were determined immediately by intracellular IFN-γ staining as described below. All viruses were administered at 1 x 108 pfu/mouse. In addition, splenic T cells from rV-LacZ and rV-LacZ/TRICOM immunized mice were stimulated with irradiated B cells pulsed with 1μg/ml β-gal peptide for 5 days and then CTL avidity was determined by lytic assay as described below.
Figure 1.
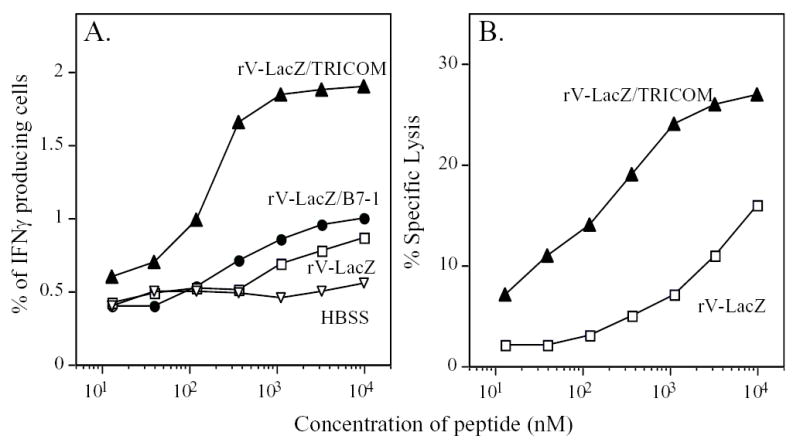
Immunization with TRICOM-based vaccines in naive mice induced high avidity antigen-specific CTL. Panel A: C57BL/6 mice were vaccinated once with buffer (HBSS, inverted open triangles), rV-LacZ (open squares), rV-LacZ/B7-1 (closed circles) or rV-LacZ/TRICOM (closed triangles). After 30 days, splenocytes were harvested. β-gal-specific CD8+ T-cell precursor frequency and avidity were determined by intracellular IFN-β staining. Panel B: β-gal-specific CD8+ T-cell avidity as determined by cytolytic assay. Splenic T cells from rV-LacZ and rV-LacZ/TRICOM immunized mice were stimulated with irradiated B cells pulsed with 1μg/ml β-gal peptide for 5 days. CTL avidity was determined using lytic assay as described in Materials and Methods. rV-LacZ (open squares), rV-LacZ/TRICOM (closed triangles).
For subsequent experiments, Thy1.1 mice were vaccinated with rV-LacZ-TRICOM (1 x 108 PFU/mouse) plus recombinant murine GM-CSF (PeproTech, Rocky Hill, NJ, 20 μg/mouse/day for 4 consecutive days) subcutaneously. After 4 weeks, Pan-T cells were isolated from spleens using a Pan-T kit and AutoMACS (Miltenyi Biotech, Bergisch Gladbach, Germany), as suggested by the manufacturer. Memory T cells were negatively isolated by depleting CD62Lhigh cells using CD62L-labeled beads (Miltenyi Biotech) as previously described (19). Purified Thy1.1 memory T cells were washed twice, re-suspended in PBS and stained with surface markers as indicated in Fig. 2 before adoptive transfer to Thy1.2 (C57BL/6) mice. All C57BL/6 (Thy 1.2) mice received the same number of Thy1.1 memory T cells (5 x 106cells/mouse) from the same source, through tail veins, and then were randomly grouped for vaccination. One week after adoptive transfer of Thy1.1 memory T cells, mice were vaccinated with PBS, rF-TRICOM, rF-LacZ or rF-LacZ-TRICOM (each at 1 x 108PFU/mouse) subcutaneously 1-3 times at 2-week intervals. β-gal tetramer cells were monitored as indicated using flow cytometry 5 days after each vaccination. For avidity studies, Thy1.1 memory T-cell recipient C57BL/6 (Thy 1.2) mice were vaccinated with either rF-LacZ or rF-LacZ/TRICOM three times. Four weeks after the last vaccination, Pan-T cells were isolated from spleens of C57BL/6 mice using Pan-T kit (Miltenyi Biotech). Thy1.1 T cells were purified from the Pan-T cells by depleting Thy1.2 T-cells, using Thy1.2 beads and AutoMACS (Miltenyi Biotech). Purified Thy1.1 T cells were used for CTL induction and avidity titration using the cytolytic method (20, 21).
Figure 2.
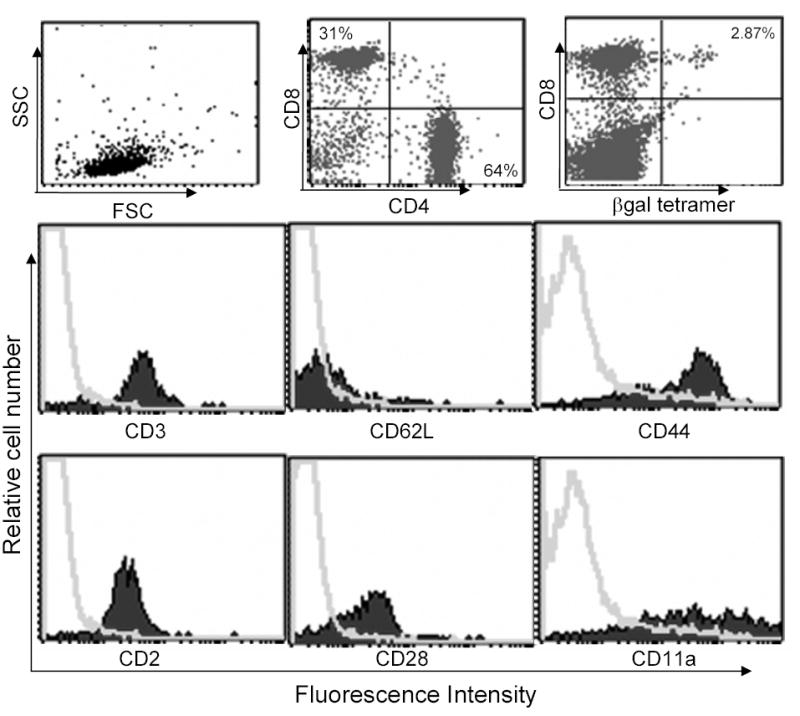
Phenotypic analysis of memory T cells for adoptive transfer. Thy1.1 mice were vaccinated with rV-LacZ/TRICOM vaccine. Four weeks later, Pan T cells were isolated using Pan T isolation kit by AutoMACS (Miltenyi Bitotech). Memory T cells were purified by depleting CD62Lhigh T cells, using CD62L-labeled beads, from the isolated Pan T cells. The purified memory T cells were stained with indicated flourochrome labeled antibodies and analyzed by flow cytometry. Top line, FSC vs. SSC, CD4 vs. CD8 and β-gal tetramer vs. CD8 were graphed as dot plot. For the histogram, isotype control (gray curves) was overlaid with specific antibody staining (solid silhouettes).
CTL generation and cytotoxicity assays
Purified Thy1.1 T cells from recipient C57BL/6 (Thy 1.2) mice were stimulated with β-gal peptide (1 μg/ml) pulsed B cells at 10:1 ratio for 5-6 days. At the end of cultures, CTL were tested for cytotoxicity in a standard 4 h 51Cr-release assay. Peptide-pulsed EL-4 targets (1 x 106/ml in the presence of graded concentrations of peptide in 1 ml of medium) were labeled with 100 μCi sodium-51Chromate for 2 h at 37oC. Target cells (5000 targets/well) were washed twice and added to wells containing effector CTL. The percent specific 51Cr release was calculated as: [(mean experimental cpm – mean spontaneous cpm) /(mean maximum cpm – mean spontaneous cpm)] x 100%, where spontaneous release represents cpm in supernatants from wells containing target cells with medium only, and maximum release represents CPM in supernatants from wells containing target cells in medium with 2% Triton X100. Spontaneous release was always <15% of maximum release.
Cytokine induction and detection
Purified Thy1.1 and Thy1.2 T cells were co-cultured with irradiated syngeneic B cells pulsed with or without β-gal peptide for 24 h, respectively. The T cells to B cells ratio was 10:1. Supernatants were collected at the end of culture and cytokine production was detected using FACS based Cytometric Bead Array (CBA) (Mouse Th1/Th2 Cytokine CBA Kit was purchased from BD Pharmingen) according to the instructions provided by the manufacturer.
Intracellular IFN-γ staining
Purified T cells were co-cultured with B cells pulsed with or without graded concentrations of cognate peptide for 6 h. GolgiStop (BD Pharmingen) was added to the cultures during the last 5 h of incubation. At the end of incubation, cells were collected, stained first with cell surface markers (Thy1.1, Thy1.2, CD8) and then permeabilized with Cytofix/CytopermTM (BD Pharmingen) followed by anti-IFN-γ staining. Data were analyzed using software CellQuest.
Avidity titration
Avidity of peptide-specific CTL was titrated by cytolytic activity against EL-4 cells pulsed with graded concentrations of β-gal peptide, or by intracellular IFN-γ staining following co-culture with B cells pulsed with graded concentrations of peptide. Avidity, expressed as MC50 in mole (M), was defined as the concentration of peptide required to achieve 50% of maximal response, and calculated using Microsoft Excel.
Tumor therapy studies
CEA-Tg mice were transplanted with 50,000 MC38-CEA cells to form experimental peripancreatic metastases, as described (22). Briefly, the spleens of anesthetized mice were exteriorized by means of a small subcostal incision. Cells were directly injected in 100 μl of HBSS using 1 ml syringes with 26 gauge 5/8 inch needles. Splenectomy was performed approximately 2 min after tumor cell injection by cauterization using a high-temperature cautery (Roboz, Rockville, MD). The abdominal cavity was closed in one layer using 9 mm wound autoclips. This dose of tumor cells is lethal to >80% of mice within 12 weeks, with the primary tumor arising in the peripancreatic environment (22).
Fourteen days following tumor transplant, mice were vaccinated subcutaneously once with 1 x 108 pfu of rV-CEA/TRICOM admixed with recombinant murine GM-CSF (20 μg; PeproTech) and human IL-2 (16,000 IU; Hoffmann-La Roche, Nutley, NJ) intraperitoneally. GM-CSF (20 μg) was administered at the injection site for the following 3 days. Concurrently, IL-2 (16,000 IU) was administered twice a day for 3 days. This vaccination schema has been previously described (23). Seven days following the primary vaccination, mice were boosted with 1 x 108 pfu of rF-CEA or rF-CEA/TRICOM, admixed with 1 x 107 pfu rF-GM-CSF. IL-2 (16,000 IU) was administered twice a day for 4 days. This booster vaccination regimen was repeated two additional times at 7-day intervals. Mice were monitored weekly for survival.
Statistical analysis
Significant differences were statistically evaluated using two-tail Student’s t test. Evaluation of survival patterns in mice bearing MC38-CEA+ tumors was performed by the Kaplan-Meier method.
Results
Vaccination with TRICOM-based vaccines induces higher levels of, and higher avidity, antigen-specific CTL in vivo
We first set out to determine whether direct vaccination with a TRICOM-based vector would induce either increased levels of antigen-specific T cells, higher avidity antigen-specific CTL, or both, in the system employed here. We vaccinated mice with rV-LacZ, rV-LacZ/B7-1, or rV-LacZ/TRICOM and monitored β-gal–specific immune responses by both an intracellular IFN-γ staining and a lytic assay. As shown in Fig. 1A, direct injection of rV-LacZ induced antigen-specific immune responses as determined by intracellular IFN-γ staining. Including the B7-1 transgene in the vaccine (rV-LacZ/B7-1) moderately enhanced immune responses to β-gal peptide. In contrast, vaccination with rV-LacZ/TRICOM increased the number of antigen-specific IFN-γ producing cells approximately 2-4 fold, depending on concentration of peptide (Fig. 1A). Initial studies have shown that mice vaccinated with rV-LacZ/TRICOM mount CD8+ T- cell responses specific for β-gal but those vaccinated with control vector rV-TRICOM do not (data not shown). Previous studies (9, 23, 24) have also shown that mice vaccinated with control rV-TRICOM vector or wild-type vector do not mount a T-cell response against specific antigens. Cytolytic activity of splenic T cells from mice vaccinated with rV-LacZ or rV-LacZ/TRICOM was also compared. Consistent with intracellular IFN-γ staining, cytolytic capacity of CTL generated from rV-LacZ/TRICOM vaccinated mice was significantly enhanced compared with CTL from rF-LacZ vaccinated animals (Fig. 1B) The avidity of T cells from vaccinated mice was also determined using previously defined methods (20, 21, 25). As measured using intracellular IFN-γ, T cells from mice vaccinated with rV-LacZ had an avidity of 9 x 10−7M compared with 2.1 x 10−7M from mice vaccinated with rV-LacZ/TRICOM (a 4-fold increase). Using the cytolytic assay, the avidities were 1.6 x 10−6M and 1 x 10−7M for rV-LacZ and rV-LacZ/TRICOM vaccinated mice, respectively (a 16-fold increase). Based on the findings of this study and others (10, 17), in which stimulation of T cells with TRICOM-based vaccines was shown to be superior to stimulation with vaccines containing B7-1, all subsequent studies utilized TRICOM-containing vectors for vaccination.
In vivo boost with TRICOM-based vaccines maintains memory T-cell populations
Although vaccination with rV-LacZ/TRICOM increased immune responses both quantitatively and qualitatively, as compared with vaccination with rV-LacZ, it could be postulated that booster vaccinations with rF-LacZ/TRICOM might cause apoptosis and delete antigen-specific memory CD8+ T cells. To investigate this, we adoptively transferred memory T cells generated from Thy1.1+ mice vaccinated with rV-LacZ/TRICOM into normal C57BL/6 mice (Thy1.2+). Memory T cells were purified from Thy1.1 mice 4 weeks after vaccination with rV-LacZ/TRICOM. As seen in Fig. 2, purified memory T cells were over 95% CD3+ T cells as judged by flow cytometry; in this population approximately 30% were CD8+ T cells and 65% were CD4+ T cells. Memory T cells purified from Thy1.1 mice demonstrated the phenotype of effector memory T cells (CD62Llow/CD44high) (Fig. 2). Antigen-specific T cells (CD8+/Tet+), as determined by β-gal-MHC tetramer staining, were approximately 2.5-4% of CD8+ T cells (Fig. 2). In addition, the memory T cells also expressed CD2, CD11a and CD28, the ligands for TRICOM.
The function of adoptively transferred memory T cells was then investigated. Four weeks after a single boost vaccination, Thy1.1 (transferred memory) and Thy1.2 (endogenous) positive T cells were isolated using AutoMACS beads. The purified T-cell populations were either not stimulated or stimulated with 1 μg/ml of β-gal peptide for 24 h and IFN-γ production was then measured. As seen from Fig. 3A, Thy1.1+ T cells (memory T cells) from either the PBS or rF-TRICOM boosted groups produced low levels of IFN-γ following peptide stimulation. Booster vaccinations with rF-LacZ increased IFN-γ production by Thy1.1+ T cells by 2.77 fold as compared with the PBS group (Fig. 3A, left panel). IFN-γ production by memory T cells (Thy1.1 population) from the rF-LacZ/TRICOM boosted group was substantially increased (5-fold) as compared with that produced by the memory T cells from the rF-LacZ group (Fig. 3A, left panel). As for the endogenous T cells (Thy1.2 population), a low but marked level of specific IFN-γ production was observed only in mice vaccinated with rF-LacZ/TRICOM (Fig. 3A, right panel; note difference in scale between panels in Fig. 3A). These data, taken together, support the hypothesis that vaccine boosting with high levels of costimulation increases the levels of both antigen-specific memory T cells as well as activated naive T cells de novo.
Figure 3.
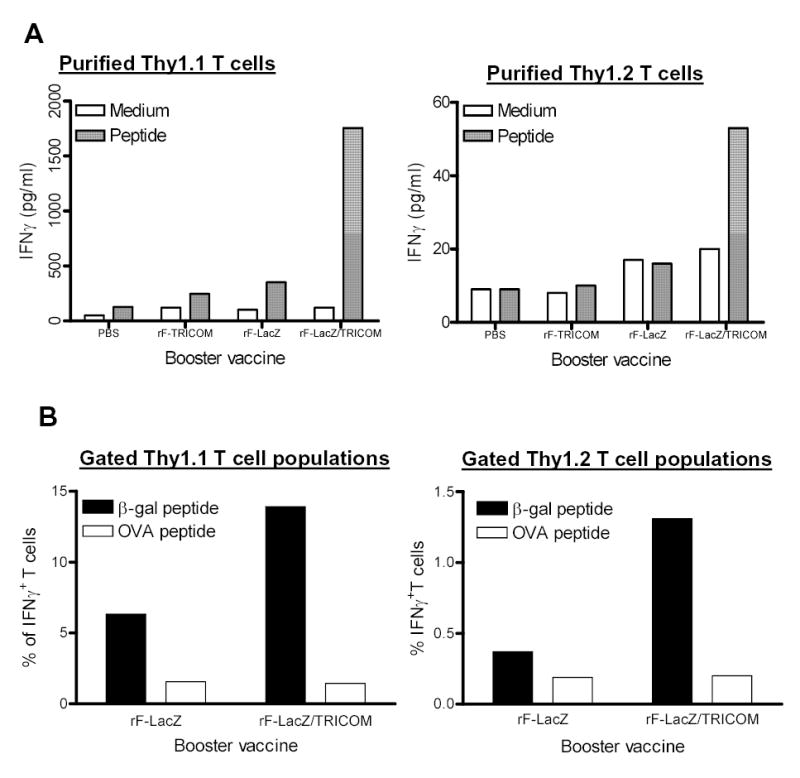
Functional analysis of antigen-specific memory T cells after booster with rF-LacZ or rF-LacZ/TRICOM vaccines. Memory T cells were prepared and adoptively transferred as described in Materials and Methods and in the legend to Fig. 2. Four weeks after one booster vaccination with the indicated vaccine, T cells were isolated for functional assay. For IFN-γ production, purified memory Thy1.1 T cells (A, left panel) and endogenous Thy1.2 T cells (A, right panel) were stimulated with irradiated B cells pulsed with β-gal peptide (1μg/ml) for 24 h. IFN-γ production was detected using a Cytometric Bead Array as described in Materials and Methods. Note the difference in scale between the panels of A. Panel B: For intracellular IFN-γ staining, purified Pan T cells were stimulated with irradiated B cells pulsed with control OVA peptide or β-gal peptide (1μg/ml) for 6 h. At the end of incubation, cells were stained with anti-CD8, Thy1.1, Thy1.2 and anti-IFN-γ antibodies. IFN-γ producing CD8+ T cells in gated Thy1.1+ and Thy1.2+ T cell populations were analyzed using CellQuest software. These data are representative of two similar experiments.
T cells from spleens of rF-LacZ and rF-LacZ/TRICOM boosted groups were also stimulated in vitro with either OVA control peptide or β-gal peptide and then analyzed by intracellular IFN-γ staining (Fig. 3B). For memory CD8+ T cells (Thy1.1 population), 4.77% of Thy1.1+/CD8+ T cells in rF-LacZ boosted mice expressed IFN-γ; 12.48% of Thy1.1+/CD8+ T cells from rF-LacZ/TRICOM boosted mice expressed IFN-γ following ex vivo cognate peptide stimulation, a 162% increase in IFN-γ–producing cells as compared with the rF-LacZ group. As for endogenous CD8+ T cells (Thy1.2 population), 0.18% of Thy1.2+/CD8+ T cells in rF-LacZ vaccinated mice expressed IFN-γ, while 1.11% of Thy1.2+/CD8+ T cells from the rF-LacZ/TRICOM group expressed IFN-γ following peptide stimulation, a 517% increase as compared with the rF-LacZ group (Fig. 3B). The results were thus consistent with IFN-γ production as described above.
Thy1.1+ memory T cells were adoptively transferred into C57BL/6 mice (Thy1.2+) and then mice were vaccinated with PBS, rF-TRICOM, rF-LacZ or rF-LacZ/TRICOM three times at 2-week intervals. Five days after the second and third booster vaccinations, expansion of adopted antigen-specific memory Thy1.1+ T cell populations (CD8+/Tet+) in spleens was monitored.
As seen from Fig. 4, antigen-specific Thy1.1+ T cells (Thy1.1+/CD8+/Tet+) in the PBS control vaccination group were ~2% of Thy1.1+ T cells after two and three boosts. Boosts with the rF-TRICOM vector devoid of the antigen transgene did not significantly change (P=0.31) the percentage of antigen-specific memory T cells compared with the control PBS booster vaccination. Compared with the PBS group, boosts with rF-LacZ or rF-LacZ/TRICOM both significantly increased β-gal–specific memory T cells (P<0.01) after two and three boosts. Moreover, boosts with rF-LacZ/TRICOM further increased the number of β-gal–specific memory CD8+ T cells compared with corresponding rF-LacZ groups after both two and three booster vaccinations (P<0.01). It is unclear at this point why the percentage of CD8+/β-gal tetramer+ T cells did not increase between the second and third booster vaccinations. This will be discussed below.
Figure 4.
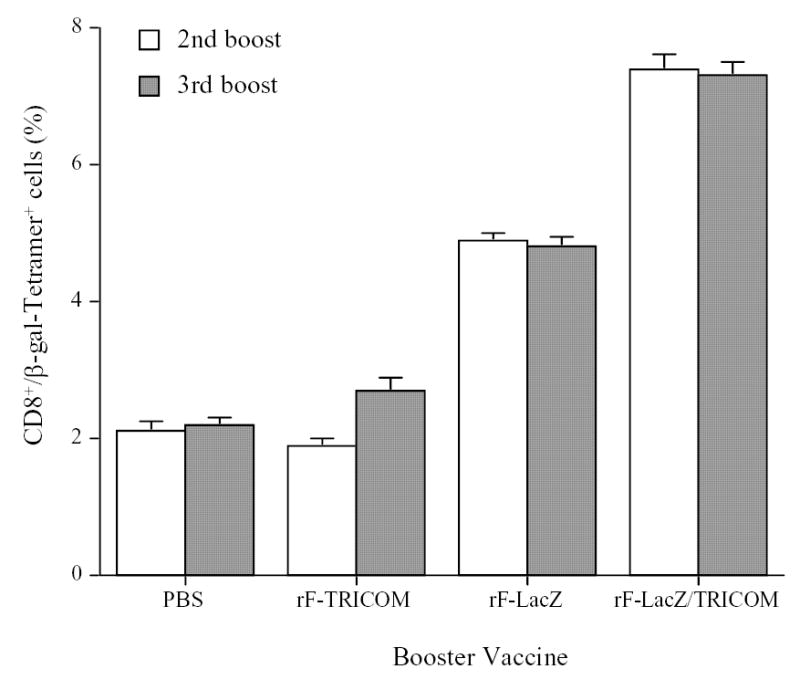
Effect of multiple booster vaccinations with rF-LacZ versus rF-LacZ/TRICOM on the expansion of antigen-specific memory T cells. Memory T cells were purified from rV-LacZ/TRICOM vaccinated Thy1.1 mice as described in Materials and Methods and in the legend to Fig. 2, and were adoptively transferred to C57BL/6 mice (Thy1.2). One week after adoptive transfer, mice were vaccinated three times with the indicated vaccines at 2-week intervals. Five days after second and third vaccinations, three mice from each group were sacrificed and splenocytes were prepared for monitoring expansion of CD8+/β-gal tetramer + T cells in the gated Thy1.1 T cell population.
Multiple boosts with TRICOM-based vaccines resulted in persistent higher avidity CD8+ T cells
After adoptive transfer of memory Thy1.1+ T cells from mice vaccinated with rV-LacZ/TRICOM into Thy1.2+ C57BL/6 mice, mice were boosted three times with either rF-LacZ or rF-LacZ/TRICOM at 2-week intervals. Four weeks after the last booster vaccination, the number and function of antigen-specific T cells were monitored. As seen in Fig. 5A, there were 1.02% and 1.65% of memory Thy1.1+ T cells in the splenocytes from rF-LacZ and rF-LacZ/TRICOM boosted mice, respectively. Among the gated Thy1.1+ T cell populations, 7.9% were β-gal tetramer positive from mice boosted with rF-LacZ, while 16.6% of Thy1.1+/CD8+ T cells from mice boosted with rF-LacZ/TRICOM were β-gal tetramer positive. The results demonstrate that TRICOM-based vaccine boosts expanded the antigen-specific memory T cells (Fig. 5A).
Figure 5.
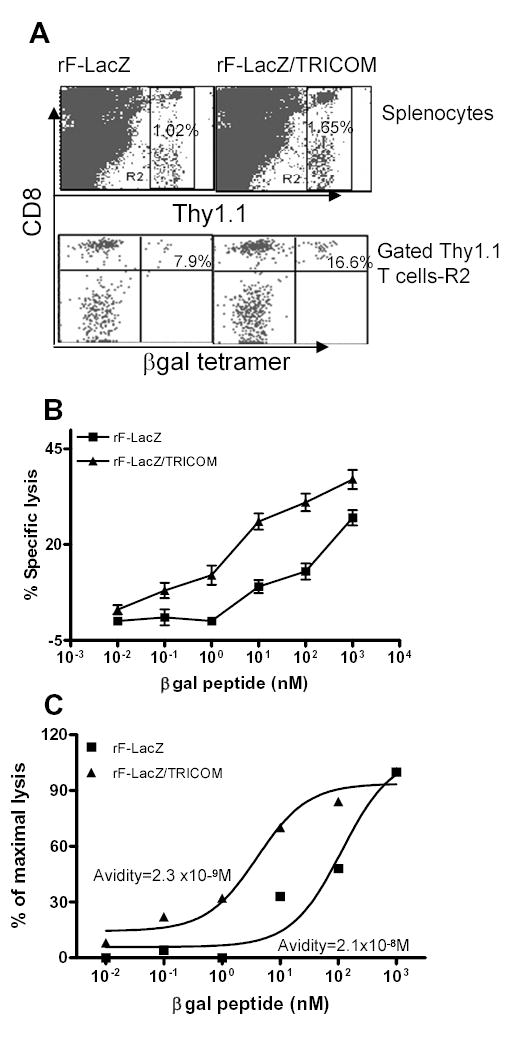
The effect of repeated booster vaccinations on the expansion of antigen-specific memory CD8+ T cells and avidity maturation. Memory T cells (Thy1.1) were prepared and adoptively transferred to Thy1.2 mice as described in Materials and Methods and in the legend to Fig. 2. Four weeks after three booster vaccinations at 2-week intervals with either rF-LacZ or rF-LacZ/TRICOM, splenocytes were purified and stained with indicated antibodies for antigen-specific memory T-cell expansion. Thy1.1+ T cells were purified, stimulated in vitro with β-gal peptide for 5 days and CTL avidity was titrated using a cytolytic assay. Panel A: Thy1.1 and β-gal tetramer staining of splenocytes from mice boosted with rF-LacZ or rF-LacZ/TRICOM. Panel B: Peptide titration of CTL avidity. Panel C: Normalization of data in B for avidity calculation. These data are representative of two similar experiments.
Thy1.1+ T cells were then purified from rF-LacZ and rF-LacZ/TRICOM boosted mice and stimulated in vitro for 5 days with β-gal peptide. CTL avidity was determined using a lytic assay. As seen from Fig. 5B, the dose-response curve of CTL from rF-LacZ/TRICOM boosted mice was more sensitive to low peptide density on target cells compared with CTL from rF-LacZ vaccinated mice. To calculate the avidity (20, 21, 25), the data shown in Fig. 5B were normalized in Fig. 5C. The avidity of CTL from rF-LacZ/TRICOM boosted mice was 2.3 x 10−9M, while that of CTL from rF-LacZ boosted mice was 2.1 x 10−8M, demonstrating a 10-fold increase in avidity following multiple boosts with TRICOM-based vaccines.
T-cell avidity was also determined by intracellular IFN-γ staining of freshly isolated Thy1.1 T cells to rule out a possible artifact of T-cell avidity measurements following a short-term in vitro peptide stimulation. Freshly isolated Thy1.1 T cells were stimulated for 6 h with autologous B cells pulsed with graded concentrations of β-gal peptide and IFN-γ–producing cells were analyzed by intracellular IFN-γ staining. As seen in Fig. 6, T cells from the rF-LacZ/TRICOM boosted group were much more sensitive to a lower concentration of peptide as compared with T cells from rF-LacZ vaccinated mice. For example, as seen in Fig. 6B, stimulation of T cells from rF-LacZ/TRICOM boosted mice with 0.1 nM of β-gal peptide elicited a substantial number of IFN-γ –producing cells, while stimulation of T cells from rF-LacZ boosted mice with even 10 nM of the peptide induced barely above the background level of IFN-γ producing cells. The avidity of T cells from rF-LacZ/TRICOM boosted mice was calculated to be 5.0 x 10−10M, which is 76-fold higher than the avidity of T cells (3.8 x 10−8M) from mice boosted with rF-LacZ vaccine. Taken together, these results demonstrate that multiple boosts with a vaccine containing high levels of T-cell costimulation have the ability to expand high avidity antigen-specific memory CD8+ T cells.
Figure 6.
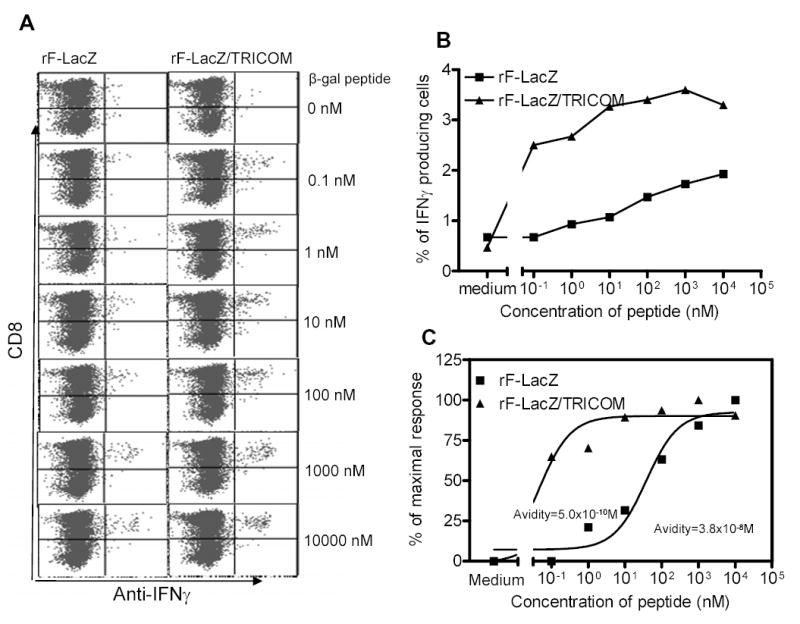
CTL avidity titration in freshly isolated memory T cells following booster vaccinations with rF-LacZ or rF-LacZ/TRICOM using intracellular IFN-γ staining. Thy1.1 mice were vaccinated with rV-LacZ/TRICOM. Thy1.1+ memory T cells were prepared and adoptively transferred to Thy1.2 mice as described in Materials and Methods and in the legend to Fig. 2. Four weeks after three booster vaccinations with either rF-LacZ or rF-LacZ/TRICOM at 2-week intervals, Thy1.1+ T cells were purified as described in Materials and Methods and stimulated with irradiated B cells pulsed with graded concentrations of β-gal peptide for 6 h. Cells were then stained with anti-Thy1.1, CD8 and anti-IFN-γ antibodies. Panel A: Phenotypic analysis of intracellular IFN-γ staining using graded concentrations of β-gal peptide. Panel B: Dose response curve of data collected from Panel A. Panel C: Avidity titration curves via normalization of data (see Refs. 20, 21, 25) from Panel B. These experiments were repeated twice with similar results.
Tumor therapy studies
The studies described above deal with a well-established experimental model antigen. β-gal, however, is a foreign antigen and does not reflect the type of situation one encounters dealing with the vast majority of “self” tumor-associated antigens. We thus designed experiments to determine if the phenomenon observed of boosting with vectors containing enhanced T-cell costimulation would result in enhanced anti-tumor effects employing vaccines to a self antigen. The CEA-Tg mouse containing a CEA-expressing carcinoma has been previously used as such a model. CEA-Tg mice express CEA in both fetal tissue and adult GI epithelium in a manner similar to that seen in humans. Fourteen days following transplant with CEA-expressing MC38 colon carcinoma cells, CEA transgenic mice received a primary vaccination with rV-CEA/TRICOM. Groups of mice (n = 10 per group) then received three booster vaccinations with either rF-CEA or rF-CEA/TRICOM. As seen in Fig. 7, there was a clear and statistical difference in the survival of mice receiving rF-CEA/TRICOM booster vaccinations (designated TTTT, Fig. 7) as compared with mice receiving booster vaccinations with rF-CEA (designated TCCC, Fig. 7) (p =0.025). Taken together these studies support the observations described above employing the β-gal antigen system that boosting with vaccines containing enhanced costimulation enhances the level of memory T cells, enhances the avidity of memory T cells, and can manifest itself in enhanced anti-tumor activity.
Figure 7.
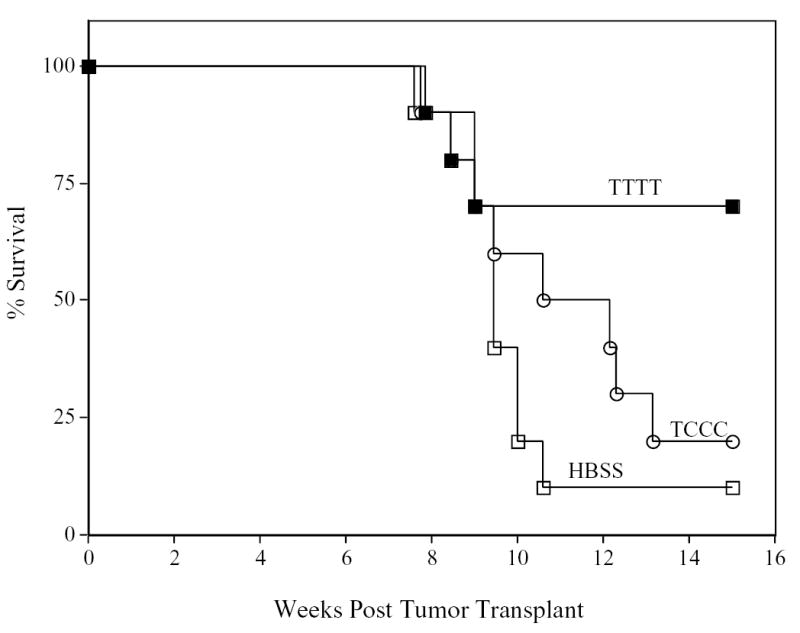
Induction of anti-tumor responses in CEA transgenic (CEA-Tg) mice by recombinant poxviral vectors. CEA-Tg mice bearing 14-day established peripancreatic metastases were divided into three treatment groups. Tumors were transplanted by intrasplenic injection of MC-38 colon carcinoma cells that were transduced with CEA (day 0). Group 1 (n = 10, closed squares) received an rV-CEA/TRICOM prime vaccination (T) followed by three weekly boosts with rF-CEA/TRICOM (T). Group 2 (n = 10, open circles) received an rV-CEA/TRICOM prime vaccination followed by three weekly boosts with rF-CEA (C). Group 3 (n = 10, open squares) received an HBSS prime vaccination followed by three weekly boosts with HBSS. Groups 1-2 prime vaccinations were administered with recombinant GM-CSF and low-dose IL-2, and all booster vaccinations were admixed with rF-GM-CSF and low-dose IL-2. Mice in each group were monitored weekly for survival.
Discussion
Previous studies have demonstrated that high avidity CTL are essential for the effective clearance of viral infections as well as for the elimination of tumor cells (21, 25–29). However, high avidity CTL are also susceptible to activation induced cell death (25, 30, 31). Therefore, the maintenance of high avidity CTL in vivo has been a challenge when designing effective vaccines for both viral infection and cancer. In the present study, we have provided evidence that multiple boosts with vaccines containing a triad of costimulatory molecules to enhance signal 2 not only expanded antigen-specific memory CD8+ T cells but also promoted the avidity maturation of the memory CTL in vivo.
CTL functional avidity, which has also been called “recognition efficiency” (28), is defined functionally, based on the peptide requirement of a CTL population. Although the term “functional avidity” may not be completely accurate to describe both the effectiveness and overall binding capacity between an antigen-specific CTL and its specific target, it is now widely used in the literature (see review article (32)). Thus, here, we have used the term “functional avidity” to describe the capacity and efficiency of a CTL to recognize and lyse target cells in an antigen-specific fashion.
Through the use of direct injection of recombinant vectors expressing TRICOM and a model antigen β-gal, the present study confirmed and extended the phenomenon observed by Oh et al. (20) and Hodge et al. (10) that primary vaccinations with vaccines containing costimulation result in increases in both the magnitude (quantity) as well as the CTL avidity (quality) of immune responses in naive hosts. It is thus clear that costimulation (especially signaling through CD28) promotes proliferation and expansion of naive T cells. However, the effects of increased costimulatory signals on the expansion and survival of memory T cells has been controversial (6–8). Sabzevari et al. (8) reported that effector/memory CD4+ T cells from PCC TCR transgenic mice were more susceptible to apoptosis induced by APC expressing CD80 in the presence of high affinity cognate peptide as compared with naive CD4+ T cells. However, Iezzi et al. (6) demonstrated that signaling through CD28 partially protected the effector/memory CD4+ T cells from TCR transgenic mice, specific for influenza hemagglutinin peptide, from apoptosis induced by high concentrations of the cognate peptide and/or prolonged peptide stimulation. A more recent report by Fontenot et al. (33) demonstrated that memory CD4+ T cells in blood from chronic beryllium disease required CD28 costimulation for proliferative and cytokine responses to beryllium. In the lung, proliferation and secretion of Th1-type cytokines by effector/memory CD4+ T cells were functionally independent of CD28 costimulation, and a proportion of the CD4+ T cells were CD28-. In addition, in some patients, CD28 signaling resulted in decreased proliferation and cytokine production by lung memory CD4+ T cells (33). Fontenot’s study (33) suggested that the effect of costimulation on memory CD4+ T cells might be dependent on the activation stages of memory T cells.
The study reported by Yu et al. (34) may provide an explanation for some of the differences observed in these different studies (6, 8) as well as those observed here. There, it was shown that the outcome of CD28 signaling on T-cell activation and expansion depended on antigen affinity. CD28 signaling enhanced T-cell activation and expansion when the TCR interacted with low and intermediate affinity alloantigen in vivo, while the same signal enhanced T-cell activation, but inhibited T-cell expansion and increased T-cell apoptosis when the TCR interacted with the high affinity alloantigen in vivo. Alhough β-gal is a foreign antigen to regular C57BL/6 mice, it is a relatively weak antigen compared with PCC. Kwok et al. (35) demonstrated that a β-gal–based vaccine was very immunogenic in Balb/c mice, while the same vaccine constructs barely induced specific immune responses in C57BL/6 mice, as used in the study reported here. One can hypothesize that signaling through costimulation protects T cells from death by upregulating survival factors (36–38), and maintains the T-cell response over a long term. On the other hand, the costimulatory signal facilitates Ag-activated T-cell apoptosis when the peptide-TCR signal exceeds a certain threshold. The results shown in Fig. 3 clearly demonstrate that rF-LacZ/TRICOM boosted memory CD8+ cells to greater levels than rF-LacZ. This observation is consistent with reports that stimulation with TRICOM-containing vectors markedly reduces the level of apoptosis in CD8+ T-cell populations (39), and is in agreement with those of previous studies, which found that costimulation through the CD28 receptor appears to play an important role in enhancing the resistance of activated T cells to undergo apoptosis in culture (38, 40).
Thus the seemingly divergent results of Sabzevari (8) and those reported here may be reconciled by the fact that the use of a strong (avid) signal 1, such as PCC, along with costimulation can result in a great degree of apoptosis of memory T cells, while the use of costimulation with a relatively weak signal 1 (β-gal), as reported here, can actually maintain or even enhance the level and avidity of memory T cells. In the present studies, we focused on the maintenance/survival of adoptively transferred memory T cells in the Thy1.2 recipient mice. As compared with the non-TRICOM vaccine, multiple boosts with TRICOM-based vaccine slightly increased the number of antigen- specific Thy1.1+ memory CTL and dramatically enhanced the functional avidity of memory CTL. Based on the modestly enhanced effect on memory T-cell maintenance/survival seen here (Figs. 3 and 4), one can argue that enhanced costimulation is not necessarily inducing apoptosis of memory CTL for all antigens, especially relatively weak ones. In addition, these two studies thus underscore the caution in generalizing the phenomenon of memory T-cell maintenance/avidity employing only one model system.
It is unclear at this point why the percentage of CD8+/β-gal tetramer+ T cells did not increase between the second and third booster vaccination with rF-LacZ/TRICOM (Fig.. 4). Perhaps the combination of apoptosis of some memory T cells along with the expansion of other memory T cells resulted in the maintenance of the number of memory T cells. However, it is clear that in mice boosted two or three times, the actual amount of memory T cells and their avidity was greater for those mice receiving rF-LacZ/TRICOM booster vaccinations than for those receiving rF-LacZ, devoid of costimulation (P<0.01). Future studies employing cytokines such as IL-15 or IL-7 (41–44) may demonstrate a greater potentiation of memory T cells employing these vaccines. Alternatively, vectors containing costimulatory molecules other than those used here may be used in booster vaccinations to further potentiate memory T cells.
In the studies reported here, avidity measured by cytolytic assay was made after 5–6 days of T-cell stimulation with peptide-pulsed APC. In vitro stimulation might potentially alter avidity. To address this issue, in our previous studies (10), avidity was measured by tetramer dissociation assay to compare avidity of T cells obtained immediately from mice vs T cells co-cultured with peptide pulsed APC for 5-6 days. Those studies showed no difference in avidity as measured by tetramer dissociation assay between the freshly isolated T cells and 5-6 day cultured T cells.
The data presented in Fig. 1 show a 4-fold increase in CTL avidity as measured by intracellular IFN-γ staining following vaccination with rV-LacZ/TRICOM vs rV-LacZ, and a 16-fold increase as determined by lytic assay using T cells cultured in vitro for 5-6 days. There are at least two possible explanations for this. First is the possibility of the alteration of CTL avidity following in vitro peptide stimulation. However, the data noted above by Hodge et al. (10) might argue against this. The second explanation is that IFN-γ production and lytic activity reflect different features of CTL. Previous studies (45) showed that different clones of Flu M1–specific T cells demonstrated different peptide sensitivity in cytolytic assay, while the different sensitivity was not seen in IFN-γ release.
The effect of TRICOM on avidity of T cells following primary vaccination has been evaluated employing both a stronger epitope (P18-I10, an HIV immunodominant epitope) and a weaker epitope (CEA peptide in CEA-Tg mice) (10, 20). The differences observed using vectors containing TRICOM vs those devoid of TRICOM on avidity are 2.5-fold for the HIV peptide and 100-fold for CEA peptide. Thus, the 16-fold difference seen in the studies reported here for β-gal peptide appears to support the concept that enhancing costimulatory molecules have a greater effect on increasing avidity of weaker antigens (such as “self” tumor-associated antigens). The actual increase in the number of antigen-specific T cells produced using the LacZ-TRICOM vector (Fig. 4) is moderate. However, the major point to consider is that the functional avidity of these cells is quite distinct from those T cells immunized with non-TRICOM–containing vector, as seen in Figs. 5 and 6.
The mechanisms underlining the enhanced avidity of CTL by increased costimulation is not clear at the present time. Membrane compartmentalization between rafts and non-rafts is required for efficient T-cell activation (46). It was reported that CD28 costimulation induced recruitment of Lck and lipid rafts as well as their accumulation at the immunological synapse (47, 48). Cawthon et al. (49) found that high avidity CTL colocalized substantially more TCR with CD8 compared with low avidity CTL. The ability of high avidity CTL to respond functionally to fewer TCR engagement events than low avidity CTL is directly related to integrating lipid rafts on their surface. Taken together, the results suggest that clustering of membrane and intracellular kinase-rich lipid rafts at the site of TCR engagements induced by costimulation may be attributed to the enhanced avidity of CTL observed in the present study.
Whereas the β-gal system employed here is a well-established model antigen, we also conducted studies to support the observations using a self antigen system. The β-gal system was employed to define numbers and avidity of antigen-specific T cells because of the availability of β-gal–specific tetramer to identify specific T cells. The CEA-Tg mice were used to define anti-tumor effects in a well-defined tumor system employing a “self” antigen. Employing the CEA transgenic mouse bearing a CEA-expressing tumor, primary vaccination with rV-CEA/TRICOM and booster vaccinations with rF-CEA/TRICOM were clearly shown to be more efficacious than the use of a primary vaccination with rV-CEA/TRICOM and booster vaccinations with rF-CEA. Taken together these studies support the observations described above employing the β-gal antigen system that boosting with vaccines containing enhanced costimulation enhances the level of memory T cells, enhances the avidity of memory T cells, and can manifest itself in enhanced anti-tumor activity.
The studies reported also provide experimental data and support the current clinical protocols using CEA/TRICOM– and PSA/TRICOM–based vaccines in patients with advanced carcinomas. In a recently completed clinical trial described by Marshall et al. (50), patients with progressive metastatic disease were first vaccinated with rV-CEA/TRICOM and then given multiple booster vaccinations with rF-CEA/TRICOM. While the primary endpoint of that trial was safety, 40% of patients were stable after 4 months, and survival—up to 2 years for some patients—appeared to correlate with CEA-specific T-cell responses. There, nine of 10 HLA-A2 positive patients demonstrated increased CEA-specific T-cell levels after the three booster vaccinations as determined by ELISPOT assay. Although it is not known if the increased levels of CD8+ T cells in these patients following vaccination expanded from a memory pool or arose from naive T cells, the data presented here (Figs. 1, 3, 5 and 6) support the hypothesis that these T cells originated from both sources. Furthermore, the stabilization of disease appeared to be correlated with continuous boosts with TRICOM-based CEA vaccines (50). Although the data in the study presented here demonstrate that in the mouse model memory T cells are maintained in both quantity and avidity by continuous boosting with TRICOM containing vectors, additional vaccine strategies may be necessary to achieve this in humans with advanced stage malignancies.
An ongoing Phase I/II trial in patients with metastatic prostate cancer involves primary vaccination with rV-PSA/TRICOM followed by multiple booster vaccinations with rF-PSA/TRICOM and has reported both clinical and biochemical (drops in serum PSA) responses (51, 52). Considering that high avidity CTL are essential for the elimination of tumor cells, these clinical trial data are consistent with the observations reported here that multiple boosts with vectors or cell-based vaccines containing relatively weak tumor antigens, but with enhanced costimulatory properties, may maintain and promote avidity maturation of antigen-specific memory T cells.
In conclusion, our results clearly demonstrate that repeated vaccinations with vectors inducing enhanced costimulation not only induced naive T cells into the effector CTL pool but also expanded antigen-specific memory CD8+ T cells. More importantly, memory CD8+T cells from mice boosted with these vaccines demonstrated higher functional avidity. The present study thus provides insight into the design of more efficient tumor vaccines.
Acknowledgments
We thank Debra Weingarten for her editorial assistance in the preparation of this manuscript.
This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Footnotes
Abbreviations used in this paper: LCMV, lymphocytic choriomeningitis virus; CTL, cytolytic T lymphocytes; APC, antigen-presenting cells; PCC, Pigeon Cytochrome C; TCR, T-cell receptor; Tg, transgenic; TRICOM, triad of costimulatory molecules; β-gal, β-galactosidase; OVA, ovalbumin
This is an author-produced version of a manuscript accepted for publication in The Journal of Immunology (The JI). This version of the manuscript has not yet been copyedited or subjected to editorial proofreading by The JI; hence, it may differ from the final version published in The JI (online and in print). The American Association of Immunologists, Inc. (AAI) (The JI) is not liable for errors or omissions in this author-produced version of the manuscript or in any version derived from it by the U.S. National Institutes of Health or any other third party. The final, citable version of record can be found at www.jimmunol.org.
References
- 1.Tanchot C, Lemonnier FA, Perarnau B, Freitas AA, Rocha B. Differential requirements for survival and proliferation of CD8 naive or memory T cells. Science. 1997;276:2057. doi: 10.1126/science.276.5321.2057. [DOI] [PubMed] [Google Scholar]
- 2.Lau LL, Jamieson BD, Somasundaram T, Ahmed R. Cytotoxic T-cell memory without antigen. Nature. 1994;369:648. doi: 10.1038/369648a0. [DOI] [PubMed] [Google Scholar]
- 3.Hou S, Hyland L, Ryan KW, Portner A, Doherty PC. Virus-specific CD8+ T-cell memory determined by clonal burst size. Nature. 1994;369:652. doi: 10.1038/369652a0. [DOI] [PubMed] [Google Scholar]
- 4.Ogg GS, Jin X, Bonhoeffer S, Dunbar PR, Nowak MA, Monard S, Segal JP, Cao Y, Rowland-Jones SL, Cerundolo V, Hurley A, Markowitz M, Ho DD, Nixon DF, McMichael AJ. Quantitation of HIV-1-specific cytotoxic T lymphocytes and plasma load of viral RNA. Science. 1998;279:2103. doi: 10.1126/science.279.5359.2103. [DOI] [PubMed] [Google Scholar]
- 5.von Herrath MG, Berger DP, Homann D, Tishon T, Sette A, Oldstone MB. Vaccination to treat persistent viral infection. Virology. 2000;268:411. doi: 10.1006/viro.1999.0130. [DOI] [PubMed] [Google Scholar]
- 6.Iezzi G, Karjalainen K, Lanzavecchia A. The duration of antigenic stimulation determines the fate of naive and effector T cells. Immunity. 1998;8:89. doi: 10.1016/s1074-7613(00)80461-6. [DOI] [PubMed] [Google Scholar]
- 7.London CA, Lodge MP, Abbas AK. Functional responses and costimulator dependence of memory CD4+ T cells. J Immunol. 2000;164:265. doi: 10.4049/jimmunol.164.1.265. [DOI] [PubMed] [Google Scholar]
- 8.Sabzevari H, Kantor J, Jaigirdar A, Tagaya Y, Naramura M, Hodge J, Bernon J, Schlom J. Acquisition of CD80 (B7-1) by T cells. J Immunol. 2001;166:2505. doi: 10.4049/jimmunol.166.4.2505. [DOI] [PubMed] [Google Scholar]
- 9.Grosenbach DW, Barrientos JC, Schlom J, Hodge JW. Synergy of vaccine strategies to amplify antigen-specific immune responses and antitumor effects. Cancer Res. 2001;61:4497. [PubMed] [Google Scholar]
- 10.Hodge JW, Chakraborty M, Kudo-Saito C, Garnett CT, Schlom J. Multiple costimulatory modalities enhance CTL avidity. J Immunol. 2005;174:5994. doi: 10.4049/jimmunol.174.10.5994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eades-Perner AM, van der Putten H, Hirth A, Thompson J, Neumaier M, von Kleist S, Zimmermann W. Mice transgenic for the human carcinoembryonic antigen gene maintain its spatiotemporal expression pattern. Cancer Res. 1994;54:4169. [PubMed] [Google Scholar]
- 12.Kass E, Schlom J, Thompson J, Guadagni F, Graziano P, Greiner JW. Induction of protective host immunity to carcinoembryonic antigen (CEA), a self-antigen in CEA transgenic mice, by immunizing with a recombinant vaccinia-CEA virus. Cancer Res. 1999;59:676. [PubMed] [Google Scholar]
- 13.Robbins PF, Kantor JA, Salgaller M, Hand PH, Fernsten PD, Schlom J. Transduction and expression of the human carcinoembryonic antigen gene in a murine colon carcinoma cell line. Cancer Res. 1991;51:3657. [PubMed] [Google Scholar]
- 14.Perkus ME, Piccini A, Lipinskas BR, Paoletti E. Recombinant vaccinia virus: immunization against multiple pathogens. Science. 1985;229:981. doi: 10.1126/science.2992092. [DOI] [PubMed] [Google Scholar]
- 15.Kantor J, Irvine K, Abrams S, Kaufman H, DiPietro J, Schlom J. Antitumor activity and immune responses induced by a recombinant carcinoembryonic antigen-vaccinia virus vaccine. J Natl Cancer Inst. 1992;84:1084. doi: 10.1093/jnci/84.14.1084. [DOI] [PubMed] [Google Scholar]
- 16.Hodge JW, Sabzevari H, Yafal AG, Gritz L, Lorenz MG, Schlom J. A triad of costimulatory molecules synergize to amplify T-cell activation. Cancer Res. 1999;59:5800. [PubMed] [Google Scholar]
- 17.Kalus RM, Kantor JA, Gritz L, Gomez Yafal A, Mazzara GP, Schlom J, Hodge JW. The use of combination vaccinia vaccines and dual-gene vaccinia vaccines to enhance antigen-specific T-cell immunity via T-cell costimulation. Vaccine. 1999;17:893. doi: 10.1016/s0264-410x(98)00275-8. [DOI] [PubMed] [Google Scholar]
- 18.Overwijk WW, Surman DR, Tsung K, Restifo NP. Identification of a Kb-restricted CTL epitope of beta-galactosidase: potential use in development of immunization protocols for "self" antigens. Methods. 1997;12:117. doi: 10.1006/meth.1997.0461. [DOI] [PubMed] [Google Scholar]
- 19.Wang LX, Chen BG, Plautz GE. Adoptive immunotherapy of advanced tumors with CD62 L-selectin(low) tumor-sensitized T lymphocytes following ex vivo hyperexpansion. J Immunol. 2002;169:3314. doi: 10.4049/jimmunol.169.6.3314. [DOI] [PubMed] [Google Scholar]
- 20.Oh S, Hodge JW, Ahlers JD, Burke DS, Schlom J, Berzofsky JA. Selective induction of high avidity CTL by altering the balance of signals from APC. J Immunol. 2003;170:2523. doi: 10.4049/jimmunol.170.5.2523. [DOI] [PubMed] [Google Scholar]
- 21.Yang S, Linette GP, Longerich S, Haluska FG. Antimelanoma activity of CTL generated from peripheral blood mononuclear cells after stimulation with autologous dendritic cells pulsed with melanoma gp100 peptide G209-2M is correlated to TCR avidity. J Immunol. 2002;169:531. doi: 10.4049/jimmunol.169.1.531. [DOI] [PubMed] [Google Scholar]
- 22.Lafreniere R, Rosenberg SA. A novel approach to the generation and identification of experimental hepatic metastases in a murine model. J Natl Cancer Inst. 1986;76:309. [PubMed] [Google Scholar]
- 23.Hodge JW, Grosenbach DW, Aarts WM, Poole DJ, Schlom J. Vaccine therapy of established tumors in the absence of autoimmunity. Clin Cancer Res. 2003;9:1837. [PubMed] [Google Scholar]
- 24.Aarts WM, Schlom J, Hodge JW. Vector-based vaccine/cytokine combination therapy to enhance induction of immune responses to a self-antigen and antitumor activity. Cancer Res. 2002;62:5770. [PubMed] [Google Scholar]
- 25.Alexander-Miller MA, Leggatt GR, Berzofsky JA. Selective expansion of high- or low-avidity cytotoxic T lymphocytes and efficacy for adoptive immunotherapy. Proc Natl Acad Sci U S A. 1996;93:4102. doi: 10.1073/pnas.93.9.4102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zeh HJ, 3rd, Perry-Lalley D, Dudley ME, Rosenberg SA, Yang JC. High avidity CTLs for two self-antigens demonstrate superior in vitro and in vivo antitumor efficacy. J Immunol. 1999;162:989. [PubMed] [Google Scholar]
- 27.Yee C, Savage PA, Lee PP, Davis MM, Greenberg PD. Isolation of high avidity melanoma-reactive CTL from heterogeneous populations using peptide-MHC tetramers. J Immunol. 1999;162:2227. [PubMed] [Google Scholar]
- 28.Rubio V, Stuge TB, Singh N, Betts MR, Weber JS, Roederer M, Lee PP. Ex vivo identification, isolation and analysis of tumor-cytolytic T cells. Nat Med. 2003;9:1377. doi: 10.1038/nm942. [DOI] [PubMed] [Google Scholar]
- 29.Derby M, Alexander-Miller M, Tse R, Berzofsky J. High-avidity CTL exploit two complementary mechanisms to provide better protection against viral infection than low-avidity CTL. J Immunol. 2001;166:1690. doi: 10.4049/jimmunol.166.3.1690. [DOI] [PubMed] [Google Scholar]
- 30.Derby MA, Snyder JT, Tse R, Alexander-Miller MA, Berzofsky JA. An abrupt and concordant initiation of apoptosis: antigen-dependent death of CD8+ CTL. Eur J Immunol. 2001;31:2951. doi: 10.1002/1521-4141(2001010)31:10<2951::aid-immu2951>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 31.Morgan DJ, Kreuwel HT, Sherman LA. Antigen concentration and precursor frequency determine the rate of CD8+ T cell tolerance to peripherally expressed antigens. J Immunol. 1999;163:723. [PubMed] [Google Scholar]
- 32.Snyder JT, Alexander-Miller MA, Berzofskyl JA, Belyakov IM. Molecular mechanisms and biological significance of CTL avidity. Curr HIV Res. 2003;1:287. doi: 10.2174/1570162033485230. [DOI] [PubMed] [Google Scholar]
- 33.Fontenot AP, Gharavi L, Bennett SR, Canavera SJ, Newman LS, Kotzin BL. CD28 costimulation independence of target organ versus circulating memory antigen-specific CD4+ T cells. J Clin Invest. 2003;112:776. doi: 10.1172/JCI18317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yu XZ, Martin PJ, Anasetti C. CD28 signal enhances apoptosis of CD8 T cells after strong TCR ligation. J Immunol. 2003;170:3002. doi: 10.4049/jimmunol.170.6.3002. [DOI] [PubMed] [Google Scholar]
- 35.Kwok LY, Lutjen S, Soltek S, Soldati D, Busch D, Deckert M, Schluter D. The induction and kinetics of antigen-specific CD8 T cells are defined by the stage specificity and compartmentalization of the antigen in murine toxoplasmosis. J Immunol. 2003;170:1949. doi: 10.4049/jimmunol.170.4.1949. [DOI] [PubMed] [Google Scholar]
- 36.Van Parijs L, Ibraghimov A, Abbas AK. The roles of costimulation and Fas in T cell apoptosis and peripheral tolerance. Immunity. 1996;4:321. doi: 10.1016/s1074-7613(00)80440-9. [DOI] [PubMed] [Google Scholar]
- 37.Grosenbach DW, Schlom J, Gritz L, Gomez Yafal A, Hodge JW. A recombinant vector expressing transgenes for four T-cell costimulatory molecules (OX40L, B7-1, ICAM-1, LFA-3) induces sustained CD4+ and CD8+ T-cell activation, protection from apoptosis, and enhanced cytokine production. Cell Immunol. 2003;222:45. doi: 10.1016/s0008-8749(03)00080-7. [DOI] [PubMed] [Google Scholar]
- 38.Boise LH, Minn AJ, Noel PJ, June CH, Accavitti MA, Lindsten T, Thompson CB. CD28 costimulation can promote T cell survival by enhancing the expression of Bcl-XL. Immunity. 1995;3:87. doi: 10.1016/1074-7613(95)90161-2. [DOI] [PubMed] [Google Scholar]
- 39.Hodge JW, Grosenbach DW, Rad AN, Giuliano M, Sabzevari H, Schlom J. Enhancing the potency of peptide-pulsed antigen presenting cells by vector-driven hyperexpression of a triad of costimulatory molecules. Vaccine. 2001;19:3552. doi: 10.1016/s0264-410x(01)00062-7. [DOI] [PubMed] [Google Scholar]
- 40.Boise LH, Noel PJ, Thompson CB. CD28 and apoptosis. Curr Opin Immunol. 1995;7:620. doi: 10.1016/0952-7915(95)80067-0. [DOI] [PubMed] [Google Scholar]
- 41.Fry TJ, Connick E, Falloon J, Lederman MM, Liewehr DJ, Spritzler J, Steinberg SM, Wood LV, Yarchoan R, Zuckerman J, Landay A, Mackall CL. A potential role for interleukin-7 in T-cell homeostasis. Blood. 2001;97:2983. doi: 10.1182/blood.v97.10.2983. [DOI] [PubMed] [Google Scholar]
- 42.Fry TJ, Mackall CL. Interleukin-7: master regulator of peripheral T-cell homeostasis? Trends Immunol. 2001;22:564. doi: 10.1016/s1471-4906(01)02028-2. [DOI] [PubMed] [Google Scholar]
- 43.Waldmann TA. The IL-2/IL-15 receptor systems: targets for immunotherapy. J Clin Immunol. 2002;22:51. doi: 10.1023/a:1014416616687. [DOI] [PubMed] [Google Scholar]
- 44.Waldmann TA. IL-15 in the life and death of lymphocytes: immunotherapeutic implications. Trends Mol Med. 2003;9:517. doi: 10.1016/j.molmed.2003.10.005. [DOI] [PubMed] [Google Scholar]
- 45.Lawson TM, Man S, Wang EC, Williams S, Amos N, Gillespie GM, Moss PA, Borysiewicz LK. Functional differences between influenza A-specific cytotoxic T lymphocyte clones expressing dominant and subdominant TCR. Int Immunol. 2001;13:1383. doi: 10.1093/intimm/13.11.1383. [DOI] [PubMed] [Google Scholar]
- 46.Xavier R, Brennan T, Li Q, McCormack C, Seed B. Membrane compartmentation is required for efficient T cell activation. Immunity. 1998;8:723. doi: 10.1016/s1074-7613(00)80577-4. [DOI] [PubMed] [Google Scholar]
- 47.Viola A, Schroeder S, Sakakibara Y, Lanzavecchia A. T lymphocyte costimulation mediated by reorganization of membrane microdomains. Science. 1999;283:680. doi: 10.1126/science.283.5402.680. [DOI] [PubMed] [Google Scholar]
- 48.Tavano R, Gri G, Molon B, Marinari B, Rudd CE, Tuosto L, Viola A. CD28 and lipid rafts coordinate recruitment of Lck to the immunological synapse of human T lymphocytes. J Immunol. 2004;173:5392. doi: 10.4049/jimmunol.173.9.5392. [DOI] [PubMed] [Google Scholar]
- 49.Cawthon AG, Alexander-Miller MA. Optimal colocalization of TCR and CD8 as a novel mechanism for the control of functional avidity. J Immunol. 2002;169:3492. doi: 10.4049/jimmunol.169.7.3492. [DOI] [PubMed] [Google Scholar]
- 50.Marshall JL, Gulley JL, Arlen PM, Beetham PK, Tsang KY, Slack R, Hodge JW, Doren S, Grosenbach DW, Hwang J, Fox E, Odogwu L, Park S, Panicali D, Schlom J. Phase I study of sequential vaccinations with fowlpox-CEA(6D)-TRICOM alone and sequentially with vaccinia-CEA(6D)-TRICOM, with and without granulocyte-macrophage colony-stimulating factor, in patients with carcinoembryonic antigen-expressing carcinomas. J Clin Oncol. 2005;23:720. doi: 10.1200/JCO.2005.10.206. [DOI] [PubMed] [Google Scholar]
- 51.Gulley J, Chen AP, Dahut W, Arlen PM, Bastian A, Steinberg SM, Tsang K, Panicali D, Poole D, Schlom J, Michael Hamilton J. Phase I study of a vaccine using recombinant vaccinia virus expressing PSA (rV-PSA) in patients with metastatic androgen-independent prostate cancer. Prostate. 2002;53:109. doi: 10.1002/pros.10130. [DOI] [PubMed] [Google Scholar]
- 52.Kaufman HL, Wang W, Manola J, DiPaola RS, Ko YJ, Sweeney C, Whiteside TL, Schlom J, Wilding G, Weiner LM. Phase II randomized study of vaccine treatment of advanced prostate cancer (E7897): a trial of the Eastern Cooperative Oncology Group. J Clin Oncol. 2004;22:2122. doi: 10.1200/JCO.2004.08.083. [DOI] [PubMed] [Google Scholar]