

Abstract
In dividing cells, the RNA-binding protein HuR associates with and stabilizes labile mRNAs encoding proliferative proteins, events that are linked to the increased cytoplasmic presence of HuR. Here, assessment of HuR levels in various vascular pathologies (intimal hyperplasia, atherosclerosis and neointimal proliferation, sclerosis of arterialized saphenous venous graft, and fibromuscular dysplasia) revealed a distinct increase in HuR expression and cytoplasmic abundance within the intima and neointima layers. On the basis of these observations, we postulated a role for HuR in promoting the proliferation of vascular smooth muscle cells. To test this hypothesis directly, we investigated the expression, subcellular localization, and proliferative influence of HuR in human vascular smooth muscle cells (hVSMCs). Treatment of hVSMCs with platelet-derived growth factor increased HuR levels in the cytoplasm, thereby influencing the expression of metabolic, proliferative, and structural genes. Importantly, knockdown of HuR expression by using RNA interference caused a reduction of hVSMC proliferation, both basally and following platelet-derived growth factor treatment. We propose that HuR contributes to regulating hVSMC growth and homeostasis in pathologies associated with vascular smooth muscle proliferation.
In adult individuals, vascular smooth muscle cells (VSMCs)1 exert remarkable plasticity and can undergo profound phenotypic changes in response to environmental cues (1). In pathological conditions, vascular injury can trigger phenotypic alterations such as proliferation, migration, and synthesis of extracellular matrix that contribute to the onset and progression of vascular diseases. The accumulation of VSMCs is central to many vascular pathologies including vein bypass failure, in-stent restenosis, transplant vasculopathy, and atherosclerosis (2). Underlying the proliferative phenotype of VSMCs in both physiological and pathological settings are critical changes in the gene expression patterns of the proliferating cells (3). Stimulation of VSMCs with growth-promoting factors such as angiotensin II or oxidized low density lipoprotein caused the increased expression of genes coding for ion transporters, extracellular matrix components, cell-cell adhesion molecules, cytoskeletal proteins, transcription factors, and cell cycle regulatory proteins (4,5). Similar changes in gene expression patterns were observed when comparing healthy VSMCs and VSMCs isolated from various disease conditions, such as primary atherosclerosis and in-stent stenosis (6).
Alterations in gene expression programs are strongly influenced by transcriptional events, but the essential contribution of posttranscriptional events (such as mRNA processing, transport, turnover, and translation) is becoming increasingly recognized. In particular, regulated mRNA stability critically contributes to the implementation of gene expression patterns during the cellular response to mitogens, immunological triggers, stressful stimuli, and differentiation agents (7,8). Indeed, a growing number of proteins central to the execution of such responses (p21, Hsp70, MnSOD, catalase, Cdc25, cyclin A, cyclin B1, c-Fos, c-Jun, c-Myc, Egr-1, etc.) are encoded by labile mRNAs, which have tightly regulated half-lives. The best understood determinants of transcript stability are U-rich or A + U-rich elements (collectively termed AREs) generally present in the 3′-untranslated regions of labile mRNAs (9). Several RNA-binding proteins have been reported to bind to AREs and cause transcript decay, including BRF1, AUF1, and KSRP (10-12). Among the RNA-binding proteins that bind to AREs and instead promote transcript stabilization is HuR, a member of the Hu/ELAV protein family. HuR stabilizes many mRNAs, including those that encode growth factors, cell division proteins, and cytokines. The role of HuR during the response to immune factors and proliferative signals has been described in several cell systems (13-17).
In two recent studies, HuR was found to regulate cell proliferation in a mouse model of skeletal muscle development and regeneration, purportedly through its influence on the expression of proteins governing cell growth and differentiation (18,19). These observations along with the documented influence of RNA turnover on the expression of genes involved in VSMC pathology (as triggered by angiotensin II, platelet-derived growth factor (PDGF-BB, hereafter PDGF) and C-reactive protein (20-24)) led us to postulate a role for HuR in modulating the response of VSMCs to growth-promoting stimuli. Here, we employed primary vascular smooth muscle cells from human aorta (hVSMCs) to investigate the involvement of HuR in the response of VSMCs to proliferative agents in culture. Our results highlight a critical role for HuR in cell proliferation following PDGF treatment and point to HuR as a likely contributing factor in vascular pathologies.
EXPERIMENTAL PROCEDURES
Immunohistochemistry—Specimens of fibromuscular dysplasia, arterialized saphenous vein graft, atherosclerosis, neointimal hyperplasia, and control coronary and renal arteries were obtained from the pathology archives of The Johns Hopkins Hospital. Anonymous hematoxylin and eosin slides were evaluated for vascular pathologies. Appropriate formalin-fixed, paraffin-embedded blocks were cut to make slides for anti-HuR immunohistochemistry. Slides were subjected to heat-induced epitope retrieval, incubation with primary antibody, and detection with the LSAB2 system (Dako, Carpinteria, CA). A monoclonal anti-HuR antibody (Molecular Probes Inc., Eugene, OR) was used at 0.25 μg/ml. An institutional review board exemption was granted for this anonymous collection of human tissues.
Immunofluorescence—hVSMCs seeded on coverslips were either stimulated with PDGF (10 ng/ml) for the times indicated or left untreated, then fixed for 15 min in 4% paraformaldehyde, and permeabilized for 15 min in phosphate-buffered saline containing 0.4% Triton X-100. Following incubation in blocking buffer (phosphate-buffered saline containing 2% bovine serum albumin and 0.1% Tween 20) for 16 h, coverslips were incubated for 1 h in either a 1:1000 dilution of rabbit polyclonal heterogeneous nuclear ribonucleoprotein C1/C2 (Santa Cruz Biotechnology, Santa Cruz, CA) or a 1:500 dilution of mouse anti-HuR (Santa Cruz Biotechnology) prepared in blocking buffer, washed with blocking buffer, incubated for 1 h with either a rhodamine Red-Xconjugated donkey anti-rabbit IgG (1:250, Jackson ImmunoResearch Laboratories, Inc., West Grove, PA) or a horse anti-mouse Texas Red IgG (1:200; Jackson Laboratories, Bar Harbor, ME), and washed again with blocking buffer. Cell images were acquired with a Zeiss LSM-410 inverted confocal microscope (Carl Zeiss, Inc.) using a 63 ×/1.4 N.A. oil immersion lens. Images were processed by MetaMorph image analysis software version 4.6r6 (Universal Imaging Corp., Inc.).
Cell Culture, Treatment, and Transfection—Human aortic smooth muscle cells were purchased from Cambrex Bio Science (Walkersville, MD) and Cell Applications (San Diego, CA) and were cultured at 37 °C in smooth muscle cell basal medium (SmBM, Cambrex Bio Science) containing 5% fetal bovine serum supplemented with insulin (100 ng/ml), human fibroblast growth factor (200 ng/ml), human epidermal growth factor (100 ng/ml), gentamicin (50 μg/ml), and amphotericin B (50 ng/ml) (complete SmBM). For growth inhibition, DMEM was supplemented with 2 mM L-glutamine, 1% antibiotics, and 0.2% fetal bovine serum. For all experiments, hVSMCs were used from passages between 4 and 8. Human embryonic lung fibroblasts (WI-38, Coriell Cell Repositories) were grown in DMEM supplemented with 1% minimum Eagle's medium nonessential amino acids, 5% fetal calf serum, 2 mM L-gluta-mine, and 1% antibiotics. Both cell types were maintained in a humidified atmosphere at 37 °C and 5% CO2. PDGF-BB was from Calbiochem. For transfection, 106 hVSMCs were trypsinized and resuspended in 100 μl of Nucleofector solution (Amaxa Biosystems, Koeln, Germany), and the siRNA duplexes (10 μM, Qiagen) electroporated as advised by the manufacturer were as follows: HuR siRNA, AAGAGGCAATTACCAGTTTCA; control siRNA (targeting genes expressed in fungus (Ustilago maydis) and bacteria (Thermotoga maritima)), TTCTCCGAACGTGTCACGT. Immediately after transfection, VSMCs were seeded in 6-well plates for further experiments.
Cell Proliferation—To monitor cell growth, 104 hVSMCs were plated into 24-well cluster plates and cultured in complete SmBM overnight. hVSMCs were then rendered quiescent by overnight culture in DMEM containing 0.2% fetal bovine serum and then cultured for an additional 3 days in the absence or presence of 10 ng/ml PDGF (PDGF was replenished 48 h after the beginning of treatment), whereupon cells in each treatment group were counted using a hemacytometer. To assess [3H]thymidine incorporation, 25,000 hVSMCs were plated/well and incubated for 24 h in complete SmBM. Cells were then incubated in DMEM containing 0.2% fetal bovine serum for 24 h and were either left without further treatment or treated with 10 ng/ml PDGF for an additional 24 h. Following the addition of 2 μCi of [3H]thymidine for 16 h at 37 °C, cells were washed with phosphate-buffered saline, lysed, and maintained at 4 °C for 30 min in 5% ice-cold trichloroacetic acid. Following washes, the cell material was resuspended in 0.5 N NaOH and 0.5% SDS, and radioactivity was measured by liquid scintillation (25).
Microarray—RNA was isolated using the STAT-60 reagent and was reverse-transcribed in the presence of [α-33 P]dCTP. The radiolabeled product was used to hybridize cDNA arrays (MGC arrays, 9,600 genes, www.grc.nia.nih.gov), employing previously reported methodologies (26). All of the data were analyzed by using the Array Pro software (Media Cybernetics, Carlsbad, CA), normalized by Z score transformation, and used to calculate differences in signal intensities. The complete cDNA array dataset is available from the authors.
Real-time PCR—RNA was reverse-transcribed using oligo-dT and reverse transcriptase (Invitrogen), and the resulting material was used for PCR amplification using gene-specific primer pairs (supplemental material) and SYBR Green PCR master mix (Applied Biosystems, Foster City, CA). For real-time PCR, amplification conditions were 50 °C (2 min), 95 °C (10 min), and then 40 cycles at 95 °C (15 s) and 60 °C (1 min).
Western Blotting—For Western blot analysis, whole-cell lysates were size-fractionated by electrophoresis in SDS-containing polyacrylamide gels (SDS-PAGE) and transferred onto polyvinylidene difluoride membranes. Monoclonal antibodies recognizing either HuR or β-actin were from Santa Cruz Biotechnology. Following secondary antibody incubations, signals were detected by enhanced chemiluminescence (Amer-sham Biosciences AB, Uppsala, Sweden).
Synthesis of Biotinylated Transcripts and Analysis of HuR Bound to Biotinylated RNA—For in vitro synthesis of biotinylated transcripts, genomic DNA was used as a template for PCR reactions. Whole-cell and cytoplasmic lysates were prepared according to previously described protocols (27). All 5′ oligonucleotides contained the T7 RNA polymerase promoter sequence; primers were used for the amplification of sequences 2063-2447 of SLC7A7 (NM_003982),2 2070-2669 of OSBPL2 (NM_144498), 682-1033 of SAT (NM_002970), 1706-2335 of CDK2 (NM_001798), 7072-7537 of CCNA2 (X68303), 518-1128 of CALM2 (NM_001743), 1095-1720 of RPA2 (NM_002946), 850-1017 of PSMA6 (NM_002791), 982-1181 of TAF9 (NM_003187) and 182-515 of PTMA coding region (negative control, NM_002823) (see supplemental material). PCR-amplified products were used as templates for the synthesis of corresponding biotinylated RNAs using T7 RNA polymerase and biotin-CTP. Biotin pull-down assays were carried out by incubating either whole-cell hVSMC lysates (25 μg) or WI-38 cytoplasmic fractions (12.5 μ g) with purified biotinylated transcripts (1 μg/incubation) for 1 h at 25 °C. Complexes were isolated with paramagnetic streptavidinconjugated Dynabeads (Dynal, Oslo, Norway), and bound proteins in the pull-down material were analyzed by Western blotting using monoclonal antibodies recognizing either HuR or β-actin (Santa Cruz Bio-technology). After secondary antibody incubations, signals were visualized by enhanced chemiluminescence (Super Signal West Femto Substrate, Pierce Biotechnology).
RESULTS
HuR Expression Is Increased in Proliferative Vascular Pathologies—Given our previous findings that HuR promoted the proliferation of normal fibroblasts and cancer cells (13-15), we sought to investigate whether HuR contributed to the proliferative phenotype underlying certain VSMC pathologies. Immunohistochemical staining of normal coronary samples showed low HuR expression in medial and intimal VSMCs, localizing primarily in the nuclei, with negligible cytoplasmic staining (Fig. 1A). By contrast, samples from patients with intimal hyperplasia, neointimal proliferation in atherosclerotic plaque, and sclerosis of arterialized saphenous vein graft all showed an overall increase in HuR signal with both nuclear and cytoplasmic staining (Fig. 1A). HuR signal was moderately increased in both subcellular compartments of intimal hyperplasia specimens, was stronger at the sites of neointimal proliferation in the atherosclerotic plaque, and was most prominent in the arterialized saphenous vein graft, wherein cytoplasmic HuR staining was particularly abundant (Fig. 1A). In normal renal VSMCs, HuR staining was low and predominantly nuclear. However, the hyperplastic intimal VSMCs present in a fibromuscular dysplasia specimen showed elevated HuR staining in both subcellular compartments (Fig. 1A). As anticipated, more intense staining in both the nucleus and the cytoplasm was seen in two positive control samples: Grawitz (renal cell) carcinoma (a neoplastic focus) and the epithelium of the ureter (in which epithelial cells undergo relatively high turnover rates) (Fig. 1B).
FIG. 1.
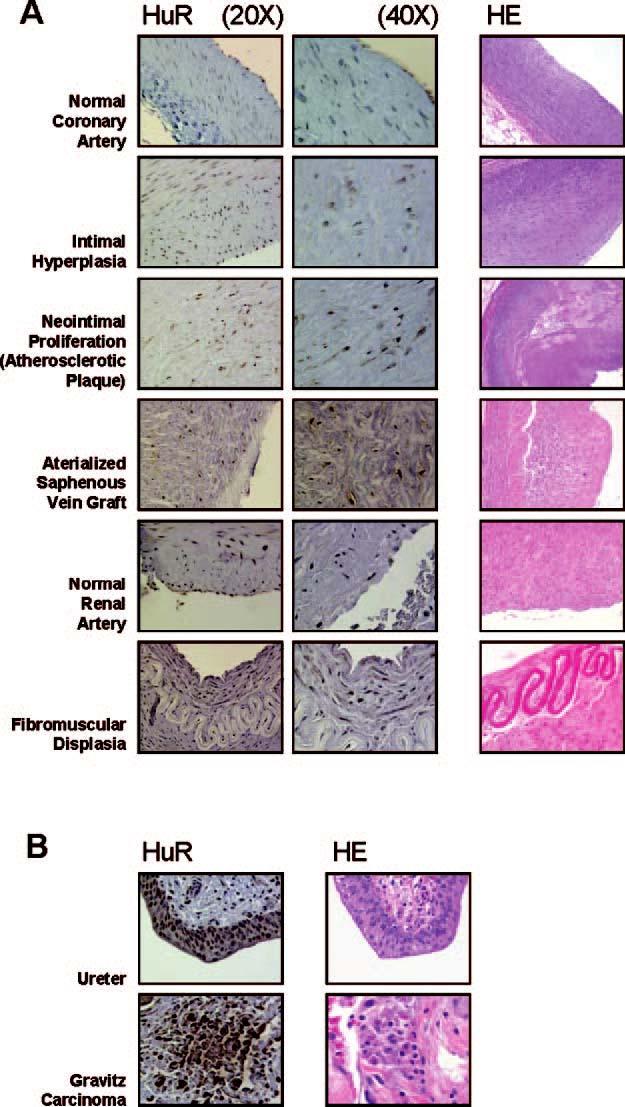
Immunohistochemical detection of HuR in healthy and diseased vascular tissues. A, HuR expression (brown signal) was examined in the indicated vascular specimens (left) at either ×20 or ×40. Corresponding hematoxylin-eosin (HE) staining is shown (right). B, positive controls for HuR signals are shown.
PDGF Treatment of Cultured hVSMCs Triggered a Marked Increase in Cytoplasmic HuR—The subcellular distribution of HuR in cultured hVSMCs was studied by confocal microscopy. In quiescent hVSMCs, HuR was predominantly nuclear at all times examined; the addition of 10 ng/ml PDGF-BB (hereafter PDGF), an inducer of VSMC proliferation and migration, caused a slight increase in nuclear HuR abundance by 1 h of treatment. By contrast, cytoplasmic HuR levels increased markedly following PDGF treatment, reaching peak levels by 6 h of treatment (Fig. 2). It is generally believed that increases in cytoplasmic HuR are largely due to the export of nuclear HuR (16,28), but there was no concomitant nuclear signal reduction when the cytoplasmic HuR increased. This discrepancy is likely explained by the fact that HuR comprised 95% of the total cellular HuR, so the export of a small proportion of nuclear HuR was not readily detectable in the nucleus but caused a pronounced increase in cytoplasmic HuR as reported previously (13,29). In control incubations, the heterogeneous nuclear ribonucleoprotein C1/C2 (Fig. 2, hnRNP C1/C2) was found unchanged following PDGF treatment. The intensity of HuR fluorescence in the nucleus and cytoplasm was quantified and represented numerically (Fig. 2, graphs).
FIG. 2.
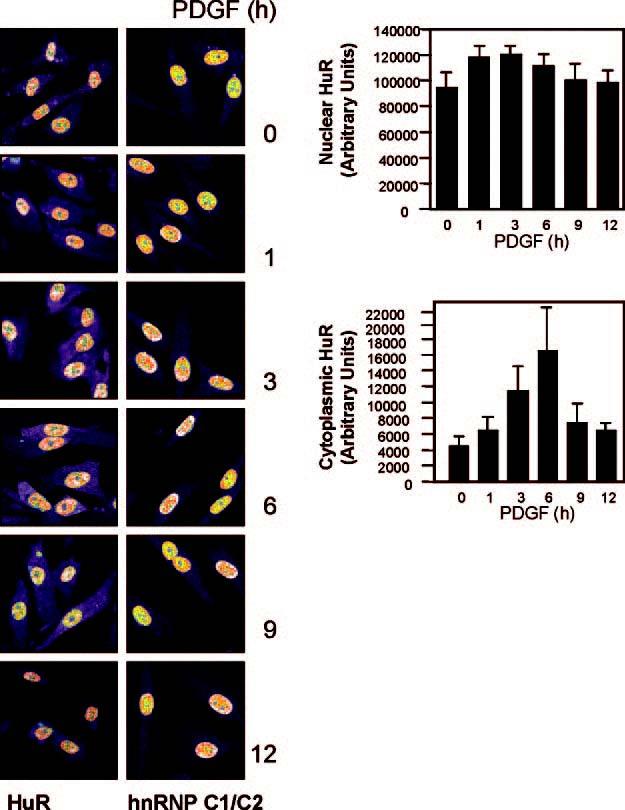
Left, subcellular distribution of HuR in hVSMCs as detected by immunofluorescence at the times shown after stimulation with 10 ng/ml PDGF. Control heterogeneous nuclear ribonucleoprotein C1/C2 (hnRNP C1/C2), also detected by immunofluorescence, remained in the nucleus throughout the treatment. Right, quantitation of HuR immunofluorescent signals; data represent the means ± S.E. from three independent experiments. Significant differences (p < 0.05) were found at 3 and 6 h.
HuR Levels Influence Gene Expression Profiles in PDGF-stimulated hVSMCs—To assess the influence of HuR on gene expression patterns in hVSMCs, HuR levels were reduced by RNA interference. Western blot analysis revealed the effective reduction of HuR (only 10% remaining) in the HuR siRNA transfection group compared with the control transfection group (Fig. 3, Ctrl. siRNA). In each population, cells either were left untreated or were treated with PDGF for 8 h, whereupon RNA was extracted and used to hybridize cDNA microar-rays (7,26). Subsequent analysis of the genes differentially regulated by PDGF in the two transfection groups revealed the influence of HuR on the expression of several groups of genes, including those regulating cell proliferation, metabolism, organization of the cytoskeleton and extracellular matrix, mitochondrial function, transcription, translation, and signal transduction (Table I). As anticipated, for some genes (e.g. FLNA, VAPB, TIMM17A), PDGF induced mRNA abundance (Z ratios ≥ 1.00, the significance value chosen for this analysis) in control populations, but this induction was significantly reduced in HuR knockdown populations (Z ratios ∼ 1.00 or lower than 1.00). The entire array dataset is available from the authors. These observations suggested that, following PDGF stimulation, HuR prevented the degradation of mRNA subsets in the cells, thereby enhancing their accumulation.
FIG. 3.
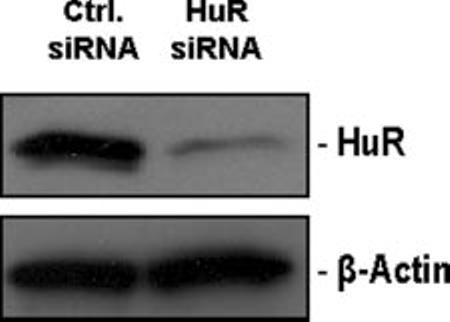
Silencing of HuR expression in hVSMCs by RNA interference. Following transfection of hVSMCs with 10 μM HuR siRNA, HuR levels were monitored by Western blot analysis. β-Actin signals served to monitor the evenness in sample loading and transfer.
TABLE I.
Influence of HuR levels on gene expression profiles of h VSMC upon PDGF stimulation for 8 h. This is a partial list of genes differentially expressed between Ctrl, siRNA- and HuR siRNA-transfected h VSMC populations, as evaluated by cDNA microarray analysis; the complete list of genes is available from the authors. The data were normalized using Z score transformation (see “Experimental Procedures”); the Z ratios indicate the difference in abundance between PDGF-treated cells and untreated cells; Z ratios ≥ 1.00 were considered as significantly elevated in the PDGF treatment groups. In parentheses, the number of genes is identified in each functional category.
| Genes and classification | Symbol | Z ratio |
GenBank™ accession no. | |
|---|---|---|---|---|
| Control siRNA | HuR siRNA | |||
| Proliferation (15) | ||||
| v-myb-like 2 | MYBL2 | 1.25 | 0.92 | NM_002466 |
| Cyclin-dependent kinase 2 | CDK2 | 1.00 | 0.77 | NM_001798 |
| v-raf viral oncogene homolog 1 | ARAF1 | 1.44 | 1.27 | NM_001654 |
| Replication factor C (activator 1) 5 | RFC 5 | 1.10 | 0.57 | NM_007370 |
| Metabolism (22) | ||||
| Arginase, type II | ARG2 | 1.27 | 0.54 | NM_001172 |
| Cationic amino acid transporter (y+) member 7 | SLC7A7 | 1.62 | 1.28 | NM_003982 |
| Spermidine/spermine acetyltransferase | SAT | 1.45 | 0.32 | NM_002970 |
| Oxysterol binding protein-like 2 | OSBPL2 | 1.05 | 0.48 | NM_144498 |
| Glutathione synthetase | GSS | 1.35 | 1.10 | NM_000178 |
| Procollagen C-endopeptidase enhancer | PCOLCE | 1.08 | 0.23 | NM_002593 |
| Diacylglycerol O-acyltransferase homolog 1 | DGAT 1 | 1.07 | 0.69 | NM_012079 |
| Ribophorin II | RPN2 | 1.26 | 1.00 | NM_002951 |
| Cytoskeleton and ECMa organization (18) | ||||
| Decorin | DCN | 1.03 | 0.17 | NM_001920 |
| Filamin A, α (actin binding protein 280) | FLNA | 1.41 | 1.05 | NM_001456 |
| Tara-like protein | HRIHFB2122 | 1.45 | 1.10 | NM_007032 |
| VAMP-associated protein B and C | VAPB | 1.34 | 0.88 | NM_004738 |
| Mitochondria (8) | ||||
| Translocase of inner mitochondrial membrane 17A | TIMM17A | 1.64 | 0.89 | NM_006335 |
| Solute carrier family 25, member 11 | SLC25A11 | 1.23 | 0.93 | NM_003562 |
| NADH dehydrogenase 1 β subcomplex | NDUFB8 | 1.91 | 1.69 | NM_005004 |
| Ribosome and translation (22) | ||||
| Ribosomal protein S6 | RPS6 | 1.43 | 0.63 | NM_001010 |
| Ribosomal protein L13 | RPL13 | 1.27 | 0.53 | NM_033251 |
| Eukaryotic translation initiation factor 4 γ, 1 | EIF4G1 | 1.02 | 0.71 | NM_004953 |
| Glutaminyl-tRNA synthetase | QARS | 1.20 | 0.68 | NM_005051 |
| Ribosomal protein S14 | RPS14 | 1.37 | 1.11 | NM_005617 |
| Eukaryotic translation initiation factor 4E-like 3 | EIF4EL3 | 1.46 | 1.27 | NM_004846 |
| Signal transduction (20) | ||||
| Calmodulin 2 (phosphorylase kinase, δ) | CALM2 | 1.04 | 0.58 | NM_001743 |
| Homer homolog 2 (Drosophila) | Homer-2 | 1.15 | 0.89 | NM_004839 |
| ADP-ribosylation factor 5 | ARF5 | 1.83 | 1.46 | NM_001662 |
| Guanine nucleotide binding protein β 2-like 1 | GNB2L1 | 1.21 | 1.00 | NM_006098 |
| Phospholipase C γ 2 | PLCG2 | 1.03 | 0.86 | NM_002661 |
| Transcription machinery and splicing (22) | ||||
| TAF9 TATA box binding protein-associated factor | TAF9 | 1.07 | 0.56 | NM_003187 |
| Splicing factor, arginine/serine-rich 7 | SFRS7 | 1.00 | 0.58 | NM_006276 |
| Zinc finger protein 144 (Mel-18) | ZNF144 | 1.30 | 0.99 | NM_007144 |
| Signal transducer and activator of transcription 3 | STAT3 | 1.02 | 0.80 | NM_139276 |
| Achaete-scute complex-like 1 (Drosophila) | ASCL 1 | 1.29 | 0.90 | NM_004316 |
| Miscellaneous (47) | ||||
| Nuclear RNA export factor 1 | NXF1 | 1.46 | 1.01 | NM_006362 |
| Small nuclear ribonucleoprotein polypeptide G | SNRPG | 1.14 | 0.87 | NM_003096 |
| Annexin A11 | ANXA 11 | 1.33 | 1.09 | NM_145869 |
| Proteasome subunit β type 4 | PSMB 4 | 1.32 | 0.77 | NM_002796 |
ECM, extracellular matrix.
Initial verification of the array data was pursued by direct measurement of mRNA abundance using reverse transcription plus real-time PCR with gene-specific primers. Accordingly, the levels of mRNAs encoding SAT, CDK2, SLC7A7, and OSBLP2 were elevated in PDGF-treated hVSMCs but only under normal HuR levels (Fig. 3, Ctrl. siRNA); cells expressing reduced HuR (HuR siRNA) did not exhibit increased levels of these mRNAs and instead displayed a small reduction in the abundance of SAT and CDK2 mRNAs after PDGF treatment. The levels of positive control p21 mRNA (a PDGF-inducible gene that is also an HuR target (29,30)) increased only after PDGF treatment of the Ctrl. siRNA population but failed to do so in the HuR siRNA group (Fig. 4 A); expression levels of the housekeeping gene GAPDH were not altered by the treatments (not shown). Subsequent experiments were aimed at directly assessing if the differentially regulated mRNAs were direct targets of HuR. To monitor the existence of such putative ribonucleoprotein interactions, the mRNAs of interest were synthesized as biotinylated transcripts in vitro (described under “Experimental Procedures”) and incubated with lysates prepared from hVSMCs to allow the formation of (HuR-biotinylated RNA) associations. The resulting complexes were pulled down using streptavidin-conjugated paramagnetic beads (“Experimental Procedures”), and the presence of HuR was tested by Western blotting. As shown (Fig. 4 B), the CDK2 3′-untranslated region RNA appeared to bind HuR very strongly, but CALM2, RPA2, SLC7A7, OSBLP2, PSMA6, and TAF9 were also found to associate with HuR; a transcript corresponding to the cyclin A 3′-untranslated region, previously reported to bind HuR (13), was used as a positive control, whereas the coding region of prothymosin α, which does not bind HuR, was included as a negative control. We then sought to examine whether binding of HuR to these target transcripts was dependent on PDGF stimulation but were unable to use hVSMC because this cell system provides too few cells for analysis. Therefore, we employed an alternative system of primary mesenchymal cells, WI-38 human diploid fibroblasts. As seen with hVSMCs, PDGF treatment caused WI-38 human diploid fibroblasts to increase proliferation (31) and elevated HuR presence in the cytoplasm (Fig. 4 C), whereas nuclear and overall HuR levels remained unchanged (not shown). Biotin pull-down using cytoplasmic lysates from WI-38 cells that were either left untreated (Fig. 4 C, Unt.) or were treated with PDGF for 8 h showed that, upon PDGF stimulation, HuR association with all of the target transcripts was increased (Fig. 4 D, PDGF lanes).
FIG. 4.
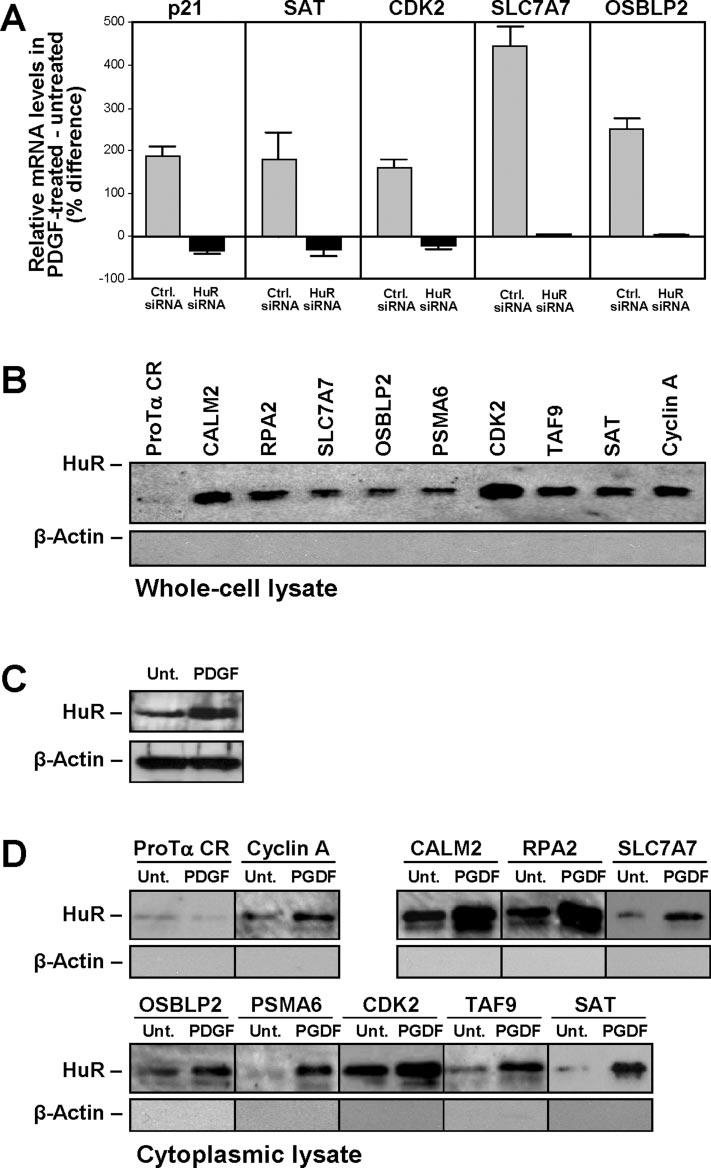
Validation of HuR target mRNAs showing differential regulation by PDGF. A, reverse transcription + real-time PCR analysis of the levels of mRNAs encoding SAT, CDK2, SLC7A7, OSBLP2, and p21 (positive control). Initial normalization was performed by comparing the levels of these mRNAs with those of housekeeping gene SDHA for each treatment group (Ctrl. siRNA, HuR siRNA, each following either no treatment or PDGF stimulation). The percentage difference in mRNA abundance in PDGF-treated relative to untreated cultures was then calculated for each transfection group. The data reflect the means ± S.E. from three independent experiments. Ctrl., control. B, biotin pull-down assays using hVSMC whole-cell ly-sates (details under “Experimental Procedures”). C, Western blot of HuR in the cytoplasm of WI-38 cells that were either untreated (Unt.) or treated with PDGF for 8 h. D, biotin pull-down assay using cytoplasmic lysates from WI-38 cells that were either left untreated or were stimulated with PDGF for 8 h.
HuR Silencing Led to Impaired Proliferation of hVSMCs in Response to PDGF—To examine the effect of HuR on hVSMC proliferation, cells expressing either normal or knockdown HuR levels were treated with PDGF, and the changes in cell number and DNA synthesis were studied. As shown, PDGF treatment triggered a marked increase in cell numbers in both transfection groups, but the increase was lower in the HuR knockdown population (Fig. 5 A). Likewise, [3H]thymidine incorporation was higher after PDGF treatment, but in the HuR knockdown populations, the increase was attenuated (Fig. 5 B). Together, these observations support the notion that HuR contributes to the proliferation of hVSMCs stimulated by PDGF.
FIG. 5.
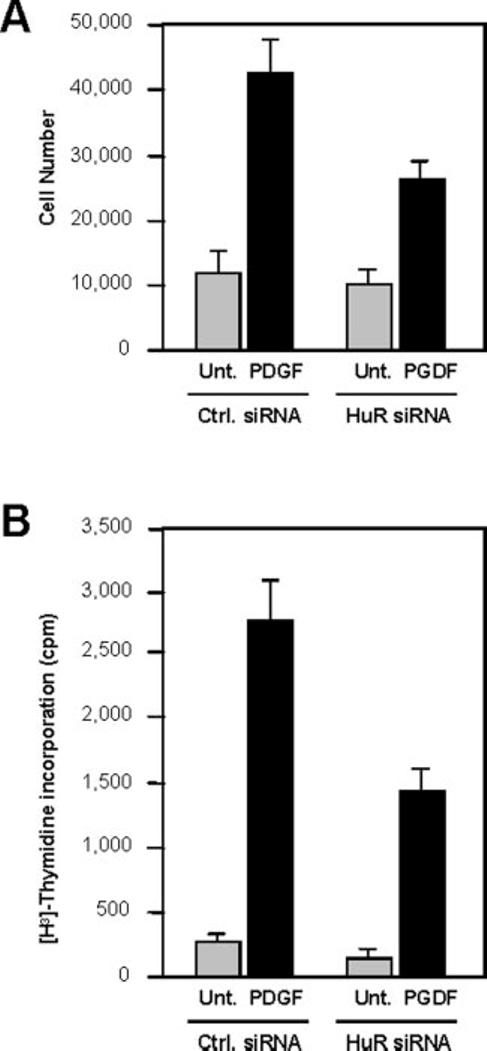
Impact of HuR knockdown on PDGF-mediated proliferation of hVSMCs. A, changes in cell numbers after incubation of cell populations in each transfection group with PDGF for 3 days. B, incorporation of [3H]thymidine for 16 h in populations expressing either wild-type (Ctrl. siRNA) or reduced (HuR siRNA) HuR levels. The data represent the means ± S.E. from three independent experiments. Unt., untreated.
DISCUSSION
In this study, we present evidence that HuR contributes to the proliferation of hVSMCs in response to PDGF treatment. The starting point for this investigation was the discovery that HuR expression was elevated in histological specimens of hyperplastic smooth muscle pathologies, including intimal hyper-plasia and neointimal proliferation, vein bypass graft failure, and fibromuscular dysplasia (Fig. 1). The relative contribution of VSMC proliferation and migration to these vascular pathologies remains to be definitively elucidated. However, based on the ability of HuR to promote the proliferation of various primary and transformed cells (13, 15-19), we hypothesized that HuR might also promote the proliferation of the smooth muscle layer of the vasculature. To investigate this question, we employed a model of PDGF stimulation of cultured hVSMCs, a cell system that has been shown to recapitulate key phenotypic traits of intact VSMCs (2). HuR was essentially nuclear in unstimulated cells, but it increased rapidly and robustly in the cytoplasm of hVSMCs following treatment with PDGF. Similar elevations in cytoplasmic HuR have been described in other cell types in response to growth factors, DNA-damaging agents, differentiation inducers, and immune stimuli (13, 17-19, 29, 32). Because the mRNA-stabilizing function of HuR has been linked to its translocation to the cytoplasm (16, 28), the PDGF-induced accumulation of cytoplasmic HuR indeed suggested that PDGF increased HuR function. After knocking down HuR expression in hVSMCs by RNA interference, cDNA array analysis revealed the influence of HuR on the expression of many PDGF-regulated genes in hVSMCs. This analysis further uncovered novel target mRNAs that were subsequently confirmed to associate with HuR (Fig. 4). Cultured hVSMCs are a powerful system to investigate many biological questions but unfortunately provide limited cell numbers for analysis. Consequently, the influence of PDGF stimulation on the binding of HuR to the proposed target mRNAs was tested in human WI-38 fibroblasts; WI-38 is an untransformed cell type of mesenchymal origin that responds to PDGF and provides greater quantities of cell material for analysis. Using this system, we found that HuR binding to the putative target mRNAs (Fig. 4 D) increased following PDGF stimulation.
Among the plausible signaling events whereby PDGF treatment induces cytoplasmic HuR levels in hVSMCs are those leading to the activation of Akt (33), which in turn inhibits AMP-activated protein kinase (AMPK), an enzyme involved in sensing metabolic stresses that perturb ATP/AMP ratios (34). Increased AMPK activity has been reported to cause a reduction of cytoplasmic HuR, probably because it promotes the nuclear import of HuR (34, 35). Conversely, inhibition of AMPK activity, as triggered by activated Akt, elicited an increase in cytoplasmic HuR followed by the enhanced stabilization and translation of HuR target transcripts (35-37). Given that no inhibitors of HuR have been identified to date and that AMPK may prove to be an effective target to modulate VSMC growth, it will be interesting to study whether AMPK activity changes following PDGF treatment. In this regard, it is important to note that the angiotensin II-triggered proliferation of VSMCs was recently shown to be suppressed by AMPK (38).
The finding that lowering HuR expression reduced the proliferation of cultured hVSMCs (Fig. 5) is in keeping with an emerging role for HuR in promoting cell growth. HuR increased the rate of proliferation of human diploid fibroblasts (15), shortened the cell division time of colon cancer cells (13), and accelerated the development of tumors in nude mice (14). HuR is thought to stimulate cell proliferation by increasing the stability of several mRNAs, including those that encode cyclin A, cyclin B1, c-Fos, c-Myc, and cyclin D1, thereby enhancing the expression of the corresponding proteins, which promote progression through the cell division cycle (27). Here, we have identified the CDK2 mRNA as a novel target of HuR; this observation may be particularly significant in light of the regulatory influence of Cdk2 and cyclin A on rat carotid artery VSMC proliferation (39). Although the effects of HuR on individual mRNAs encoding Cdk2 and other proliferative factors remain to be formally studied in hVSMCs, we propose that HuR may be a contributing factor to smooth muscle cell and neointima proliferation and consequently to atherosclerosis. In addition to influencing cell proliferation, HuR likely contributes to other aspects of vascular pathology. Many genes that regulate processes such as cell migration (e.g. MMP-9), inflammation (TNF-α, G-CSF or GM-CSF, eotaxin), muscle contraction (myosin), signaling (nitric-oxide synthase, the α 1 subunit of soluble guanylate cyclase), and redox balance (MnSOD) are all regulated by HuR (17-19, 40-46). Efforts are under way to establish whether HuR regulates these other functions of hVSMCs.
In summary, our study underscores the importance of HuR in hVSMC proliferation and points to possible downstream mediators of this effect. Given the influence of HuR on the expression of additional proteins that influence VSMC-related pathology (inflammation, oxidative damage, chemotaxis, extra-cellular matrix remodeling, etc.), further study of the influence of HuR in these events is warranted. Ultimately, interventions to suppress HuR function or block expression of its downstream targets may provide valuable avenues of intervention in atherosclerosis and its complications.
Acknowledgments
—We are grateful to K. G. Becker and the NIA Array Facility for supplying the cDNA arrays for analysis. K. Mazan-Mamczarz, A. Lal, K. Abdelmohsen, I. López de Silanes, T. Kawai, and J. Fan provided invaluable assistance with experiments. We thank S. M. Schwartz and S. J. Sollott for enlightening discussions.
Footnotes
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbs.org) contains supplemental material.
Accession numbers in parentheses are from GenBank™.
The abbreviations used are: VSMC, vascular smooth muscle cell; hVSMC, human vascular smooth muscle cell; PDGF, platelet-derived growth factor; SmBM, smooth muscle cell basal medium; DMEM, Dulbecco's modified Eagle's medium; siRNA, small interfering RNA; AMPK, AMP-activated protein kinase.
REFERENCES
- 1.Berk BC. Physiol. Rev. 2001;81:999–1030. doi: 10.1152/physrev.2001.81.3.999. [DOI] [PubMed] [Google Scholar]
- 2.Dzau VJ, Braun-Dullaeus RC, Sedding DG. Nat. Med. 2002;8:1249–1256. doi: 10.1038/nm1102-1249. [DOI] [PubMed] [Google Scholar]
- 3.Boehm M, Nabel EG. Prog. Cell Cycle Res. 2003;23:555–565. [PubMed] [Google Scholar]
- 4.Sukhanov S, Song YH, Delafontaine P. Biochem. Biophys. Res. Comm. 2003;306:443–449. doi: 10.1016/s0006-291x(03)00990-2. [DOI] [PubMed] [Google Scholar]
- 5.Campos AH, Zhao Y, Pollman MJ, Gibbons GH. Circ. Res. 2003;92:111–118. doi: 10.1161/01.res.0000049100.22673.f6. [DOI] [PubMed] [Google Scholar]
- 6.Zhang QJ, Goddard M, Shanaha C, Shapiro L, Bennett M. Arterioscler. Thromb. Vasc. Biol. 2002;22:2030–2036. doi: 10.1161/01.atv.0000042206.98651.15. [DOI] [PubMed] [Google Scholar]
- 7.Fan J, Yang X, Wang W, Wood WH, III, Becker KG, Gorospe M. Proc. Natl. Acad. Sci. U. S. A. 2002;99:10611–10616. doi: 10.1073/pnas.162212399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wilusz CJ, Wormington M, Peltz SW. Nat. Rev. Mol. Cell. Biol. 2001;2:237–246. doi: 10.1038/35067025. [DOI] [PubMed] [Google Scholar]
- 9.Xu N, Chen CY, Shyu A-B. Mol. Cell. Biol. 1997;17:4611–4621. doi: 10.1128/mcb.17.8.4611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gherzi R, Lee KY, Briata P, Wegmuller D, Moroni C, Karin M, Chen C-Y. Mol. Cell. 2004;14:571–583. doi: 10.1016/j.molcel.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 11.Zhang W, Wagner BJ, Ehrenman K, Schaefer AW, DeMaria CT, Crater D, DeHaven K, Long L, Brewer G. Mol. Cell. Biol. 1993;13:7652–7665. doi: 10.1128/mcb.13.12.7652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stoecklin G, Colombi M, Raineri I, Leuenberger S, Mallaun M, Schmidlin M, Gross B, Lu M, Kitamura T, Moroni C. EMBO J. 2002;21:4709–4718. doi: 10.1093/emboj/cdf444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang W, Caldwell MC, Lin S, Furneaux H, Gorospe M. EMBO J. 2000;19:2340–2350. doi: 10.1093/emboj/19.10.2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.López de Silanes I, Fan J, Yang X, Potapova O, Zonderman AB, Pizer ES, Gorospe M. Oncogene. 2003;22:7146–7154. doi: 10.1038/sj.onc.1206862. [DOI] [PubMed] [Google Scholar]
- 15.Wang W, Yang X, Cristofalo VJ, Holbrook NJ, Gorospe M. Mol. Cell. Biol. 2001;21:5889–5898. doi: 10.1128/MCB.21.17.5889-5898.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brennan CM, Steitz JA. Cell. Mol. Life Sci. 2001;58:266–277. doi: 10.1007/PL00000854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Atasoy U, Curry SL, López de Silanes I, Shyu A-B, Casolaro V, Gorospe M, Stellato C. J. Immunol. 2003;171:4369–4378. doi: 10.4049/jimmunol.171.8.4369. [DOI] [PubMed] [Google Scholar]
- 18.Figueroa A, Cuadrado A, Fan J, Atasoy U, Muscat GE, Muñoz-Canoves P, Gorospe M, Muñoz A. Mol. Cell. Biol. 2003;23:4991–5004. doi: 10.1128/MCB.23.14.4991-5004.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Van der Giessen K, Di-Marco S, Clair E, Gallouzi IE. J. Biol. Chem. 2003;278:47119–47128. doi: 10.1074/jbc.M308889200. [DOI] [PubMed] [Google Scholar]
- 20.Ohnaka K, Numaguchi K, Yamakawa T, Inagami T. Hypertension. 2000;35:68–75. doi: 10.1161/01.hyp.35.1.68. [DOI] [PubMed] [Google Scholar]
- 21.Blaschke F, Bruemmer D, Yin F, Takata Y, Wang W, Fishbein MC, Okura T, Higaki J, Graf K, Fleck E, Hsueh WA, Law RE. Circulation. 2004;110:579–587. doi: 10.1161/01.CIR.0000136999.77584.A2. [DOI] [PubMed] [Google Scholar]
- 22.Corjay MH, Blank RS, Owens GK. J. Cell. Physiol. 1990;145:391–397. doi: 10.1002/jcp.1041450302. [DOI] [PubMed] [Google Scholar]
- 23.Corjay MH, Thompson MM, Lynch KR, Owens GK. J. Biol. Chem. 1989;264:10501–10506. [PubMed] [Google Scholar]
- 24.Holycross BJ, Blank RS, Thompson MM, Peach MJ, Owens GK. Circ. Res. 1992;71:1525–1532. doi: 10.1161/01.res.71.6.1525. [DOI] [PubMed] [Google Scholar]
- 25.Coward P, Wada HG, Falk MS, Chan SD, Meng F, Akil H, Conklin BR. Proc. Natl. Acad. Sci. U. S. A. 1998;95:352–357. doi: 10.1073/pnas.95.1.352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kawai T, Fan J, Mazan-Mamczarz K, Gorospe M. Mol. Cell. Biol. 2004;24:6773–6787. doi: 10.1128/MCB.24.15.6773-6787.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.López de Silanes I, Zhan M, Lal A, Yang X, Gorospe M. Proc. Natl. Acad. Sci. U. S. A. 2004;101:2987–2992. doi: 10.1073/pnas.0306453101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Keene JD. Proc. Natl. Acad. Sci. U. S. A. 1999;96:5–7. doi: 10.1073/pnas.96.1.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang W, Furneaux H, Cheng H, Caldwell MC, Hutter D, Liu Y, Holbrook NJ, Gorospe M. Mol. Cell. Biol. 2000;20:760–769. doi: 10.1128/mcb.20.3.760-769.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moon SK, Jung SY, Choi YH, Lee YC, Kim CH. FEBS Lett. 2003;552:130–134. doi: 10.1016/s0014-5793(03)00935-9. [DOI] [PubMed] [Google Scholar]
- 31.Phillips PD, Kaji K, Cristofalo VJ. J. Gerontol. 1984;39:11–17. doi: 10.1093/geronj/39.1.11. [DOI] [PubMed] [Google Scholar]
- 32.Esnault S, Malter JS. J. Immunol. 2003;171:6780–6787. doi: 10.4049/jimmunol.171.12.6780. [DOI] [PubMed] [Google Scholar]
- 33.Rubin LJ, Magliola L, Feng X, Jones AW, Hale CC. J. Appl. Physiol. 2005;98:296–306. doi: 10.1152/japplphysiol.00075.2004. [DOI] [PubMed] [Google Scholar]
- 34.Hardie DG, Carling D, Carlson M. Annu. Rev. Biochem. 1998;67:821–855. doi: 10.1146/annurev.biochem.67.1.821. [DOI] [PubMed] [Google Scholar]
- 35.Wang W, Fan J, Yang X, Furer-Galban S, López de Silanes I. von, Kobbe C, Guo J, Georas SN, Foufelle F, Hardie DG, Carling D, Gorospe M. Mol. Cell. Biol. 2002;22:3425–3436. doi: 10.1128/MCB.22.10.3425-3436.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang W, Yang X, Kawai T, López de Silanes I, Mazan-Mamczarz K, Chen P, Chook YM, Quensel C, Kohler M, Gorospe M. J. Biol. Chem. 2004;279:48376–48388. doi: 10.1074/jbc.M409014200. [DOI] [PubMed] [Google Scholar]
- 37.Wang W, Yang X, López de Silanes I, Carling D, Gorospe M. J. Biol. Chem. 2003;278:27016–27023. doi: 10.1074/jbc.M300318200. [DOI] [PubMed] [Google Scholar]
- 38.Nagata D, Takeda R, Sata M, Satonaka H, Suzuki E, Nagano T, Hirata Y. Circulation. 2004;110:444–451. doi: 10.1161/01.CIR.0000136025.96811.76. [DOI] [PubMed] [Google Scholar]
- 39.Chen D, Krasinski K, Sylvester A, Chen J, Nisen PD, Andres V. J. Clin. Investig. 1997;99:2334–2341. doi: 10.1172/JCI119414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kirigiti P, Bai Y, Yang YF, Li X, Li B, Brewer G, Machida CA. Mol. Pharmacol. 2001;60:1308–1324. doi: 10.1124/mol.60.6.1308. [DOI] [PubMed] [Google Scholar]
- 41.Levy NS, Chung S, Furneaux H, Levy AP. J. Biol. Chem. 1998;273:6417–6423. doi: 10.1074/jbc.273.11.6417. [DOI] [PubMed] [Google Scholar]
- 42.Dixon DA, Tolley ND, King PH, Nabors LB, McIntyre TM, Zimmerman GA, Prescott SM. J. Clin. Investig. 2001;108:1657–1665. doi: 10.1172/JCI12973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Skalweit A, Doller A, Huth A, Kahne T, Persson PB, Thiele BJ. Circ. Res. 2003;92:419–427. doi: 10.1161/01.RES.0000059300.67152.4E. [DOI] [PubMed] [Google Scholar]
- 44.Akool el-S., Kleinert H, Hamada FM, Abdelwahab MH, Forstermann U, Pfeilschifter J, Eberhardt W. Mol. Cell. Biol. 2003;23:4901–4916. doi: 10.1128/MCB.23.14.4901-4916.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pilz RB, Casteel DE. Circ. Res. 2003;93:1034–1046. doi: 10.1161/01.RES.0000103311.52853.48. [DOI] [PubMed] [Google Scholar]
- 46.Kloss S, Furneaux H, Mulsch A. J. Biol. Chem. 2003;278:2377–2383. doi: 10.1074/jbc.M206453200. [DOI] [PubMed] [Google Scholar]