

Abstract
The arginine dehydrogenase (or oxidase) pathway catabolically converts arginine to succinate via 2-ketoglutarate and 4-guanidinobutyrate (4-GB) with the concomitant formation of CO2 and urea. Guanidinobutyrase (GBase; EC 3.5.3.7) catalyzes the conversion of 4-guanidinobutyrate to 4-aminobutyrate and urea in this pathway. We investigated the structure and regulation of the gene for GBase (designated gbuA) of Pseudomonas aeruginosa PAO1 and characterized the gbuA product. The gbuA and the adjacent gbuR genes were cloned by functional complementation of a gbuA9005 mutant of strain PAO1 defective in 4-GB utilization. The deduced amino acid sequence of GbuA (319 amino acids; Mr 34,695) assigned GBase to the arginase/agmatinase family of C-N hydrolases. Purified GbuA was a homotetramer of 140 kDa that catalyzed the specific hydrolysis of 4-GB with Km and Kcat values of 49 mM and 1,012 s−1, respectively. The divergent gbuR gene, which shared the intergenic promoter region of 206 bp with gbuA, encoded a putative regulatory protein (297 amino acids; Mr 33,385) homologous to the LysR family of proteins. Insertional inactivation of gbuR by a gentamicin resistance cassette caused a defect in 4-GB utilization. GBase and gbuA′::′lacZ fusion assays demonstrated that this gbuR mutation abolishes the inducible expression of gbuA by exogenous 4-GB, indicating that GbuR participates in the regulation of this gene. Northern blotting located an inducible promoter for gbuA in the intergenic region, and primer extension localized the transcription start site of this promoter at 40 bp upstream from the initiation codon of gbuA. The gbuRA genes at the genomic map position of 1547000 are unlinked to the 2-ketoarginine utilization gene kauB at 5983000, indicative of at least two separate genetic units involved in the arginine dehydrogenase pathway.
Pseudomonads utilize arginine and related guanidino compounds such as agmatine and 4-guanidinobutyrate (4-GB) as carbon and nitrogen sources (33, 35). Four arginine catabolic pathways have been identified in P. aeruginosa PAO1 (6, 9) (Fig. 1). The arginine deiminase (ADI) pathway encoded by the arcDABC operon is induced under anaerobic conditions with an auxiliary induction effect by exogenous arginine (8, 9, 21). This pathway supports slow anaerobic cell growth of PAO1 by providing ATP from arginine (9) (Fig. 1). The arginine succinyltransferase (AST) pathway (encoded by the aru operon) is a major route of arginine utilization as carbon and nitrogen sources under aerobic conditions (6, 9, 15, 17). Exogenous arginine induces the expression of the aru and aot-argR operons (encoding an arginine/ornithine transport system and the ArgR regulatory protein) through the ArgR function (26, 28). Glutamate, the product of this pathway, is finally channeled into the tricarboxylic acid cycle by catabolic glutamate dehydrogenase, which is coordinately induced by ArgR in the presence of exogenous arginine (20). The arginine decarboxylase (ADC) pathway consists of arginine decarboxylase, agmatine deiminase (aguA gene product), and N-carbamoylputrescine amidohydrolase (aguB gene product). These enzymes successively convert arginine to putrescine, which is further catabolized into 4-aminobutyrate (6, 9, 11, 25) (Fig. 1).
FIG. 1.

Arginine catabolic pathways in P. aeruginosa PAO1. Only relevant intermediates and genes are shown. ADI, arginine deiminase pathway; AST, arginine succinyltransferase pathway; ADC, arginine decarboxylase pathway; ADH, arginine dehydrogenase (or oxidase) pathway; TCA, tricarboxylic acid. l-Arginine oxidase is present in P. putida P2 (36) but not in P. aeruginosa PAO1 (16).
The arginine dehydrogenase (ADH; also called arginine oxidase) pathway was first discovered in Streptomyces griseus (36a), then in Pseudomonas putida and Pseudomonas aeruginosa (4, 9, 16, 36). In P. putida, l-arginine is initially oxidized to 2-ketoarginine by l-arginine oxidase. Subsequent intermediates are 4-GB and 4-aminobutyrate, which, at the confluence of the ADC pathway, is finally channeled into the tricarboxylic acid cycle (4, 9, 36, 38) (Fig. 1). The presence of d-arginine dehydrogenase instead of l-arginine oxidase in P. aeruginosa PAO1 implies that this pathway may be primarily involved in d-arginine catabolism (16). The enzymes of this pathway have been established in strain PAO1 (9, 16), as well as in P. putida (4, 36) (Fig. 1), and two genes of this pathway have been identified in PAO1 but not yet cloned.
The kauB gene (at 10 min) encodes a bifunctional enzyme with both 4-guanidinobutyraldehyde dehydrogenase and 4-aminobutyraldehyde dehydrogenase activities (14, 16), and the unmapped gbu-9005 gene locus is required for guanidinobutyrase (GBase) synthesis (16). Expression of kauB is inducible by 2-ketoarginine, putrescine or agmatine, but only weakly by d-arginine, while GBase synthesis is induced by 4-GB, 2-ketoarginine, and d-arginine (in that order) (16). Thus, unlike the arc and aru operons, the ADH genes are regulated differently by distinct intermediates of the pathway, suggesting that the ADH genes could be located at separate loci on the genome.
To understand the gene organization and regulatory mechanisms of the ADH pathway, we cloned the gbu-9005 locus of P. aeruginosa PAO1. Sequencing and characterization of this locus identified the structural gene for GBase (designated gbuA) and identified the neighboring gbuR gene, encoding a LysR-type transcriptional regulator. Experiments with a gbuR knockout mutant established that GbuR mediates the inducible expression of gbuA by exogenous 4-GB. The deduced GbuA sequence assigns GBase to the arginase/agmatinase family.
MATERIALS AND METHODS
Strains, plasmids, and media.
Strains and plasmids are listed in Table 1. Escherichia coli and P. aeruginosa PAO strains were cultured in Luria-Bertani (LB) and nutrient yeast broth (NYB), respectively, supplemented with antibiotics when appropriate (10, 29). P. aeruginosa PAO strains were cultured in minimal medium P (MMP) (10) containing specific carbon and nitrogen sources at 20 mM for enzyme and lacZ fusion assays.
TABLE 1.
Strains and plasmids used in this study
| Strain or plasmid | Relevant characteristics | Reference or source |
|---|---|---|
| P. aeruginosa | ||
| PAO1 | Wild type | 10 |
| PAO4156 | gbuA9005 (=gbu-9005) | 16 |
| PAO4173 | gbuA9005 (=gbu-9005) gpu-9018 | N. Gotoh (Matsumoto collection) |
| PAO4508 | gbuR::Gm | This study |
| PAO4509 | PA1418::Gm | This study |
| PAO4511 | PA1419::Gm | This study |
| PAO4513 | PA1420::Gm | This study |
| PAO4518 | gbuA::Gm | This study |
| E. coli | ||
| DH5α | F− endA1 hsdR17(rk− mk+) supE44 thi-1 recA1 gyrA relA1 Δ(laclZYA-argF)U169 deoR [φ80dlacΔ(lacZ)M15] | Bethesda Research Laboratories |
| HB101 | supE44 hsdS20(rB−mB−) recA13 ara-14 proA2 lacY galK2 rpsL20 xyl-5 mtl-1 | 29 |
| S17-1 | pro thi hsdR Tpr Smr; chromosome::RP4-2 Rc::Mu-Km::Tn7 | 32 |
| XL1-Blue | endA1 gyrA96 thi-1 hsdR17 supE44 recA1 lac [F′ proAB lacIqZΔM15 Tn10 (Tc)] | Stratagene |
| Plasmids | ||
| pEX18Ap | Apr, ColE1 replicon, oriT sacB | 13 |
| pME6013 | Tcr, pACYC177 and pVS1 replicons | 27 |
| pMMB67EH | Apr Cbr, RSF1010 derivative | 7 |
| pNIC6011 | Apr Cbr, pACYC177 and pVS1 replicons | 27 |
| pPS858 | Apr Gmr, ColE1 replicon carrying a Gmr cassette | 13 |
| pRK2013 | Kmr, ColE1 replicon, tra (RK2) | 5 |
| pUC118 | Apr, ColE1 replicon | 37 |
| pUC119 | Apr, ColE1 replicon | 37 |
| pYJ103 | Apr Cbr, pNIC6011 derivative carrying the 3.4-kb gbuRA region of the PAO1 chromosome | This study |
DNA techniques.
DNA purification, restriction enzyme analyses, and other DNA manipulations proceeded as described (15, 29). PCR was performed with KOD Dash DNA polymerases (Toyobo Biochemicals) under the reaction conditions recommended by the supplier. The nucleotide sequences were determined on both strands with an ABI371 DNA sequencer (Perkin-Elmer) and ABI Big-Dye terminator cycle sequencing kits (Perkin-Elmer).
Cloning of the gbuA locus.
To clone the gbu-9005 (= gbuA9005) locus, the chromosomal DNA of strain PAO1 was partially digested with the restriction endonuclease Sau3AI and ligated to the BamHI site of plasmid pNIC6011, a mobilizable Escherichia coli-Pseudomonas shuttle vector (12, 27). The ligated DNA was subsequently introduced into E. coli XL1-Blue (Stratagene) by electroporation, and cells were mated with strain PAO4173 (gbu-9005) with helper E. coli HB101/pRK2013 as described previously (25, 27). PAO4173 transconjugants that acquired a gbuA+ recombinant pNIC6011 were selected on MMP agar containing 125 μg of carbenicillin/ml and 20 mM 4-GB as the sole source of carbon and nitrogen. Plasmid DNA was then isolated from several transconjugants to analyze the cloned sequences by restriction enzyme digestion and nucleotide sequencing. Plasmid pYJ103 having a gbuA region of 3.4 kb was selected for further studies.
Plasmid and strain construction.
We localized gbuA on the chromosomal DNA region cloned in plasmid pYJ103 by complementation tests (Fig. 2B). The 2.0-kb EcoRI-HindIII fragment of the insert was subcloned into pNIC6011 at the same sites to produce plasmid pYI1009 (Fig. 2B). The 1.1-kb KpnI fragment on the insert was removed by cutting pYJ103 with KpnI and subsequent circularization with a DNA ligation kit (Takara Shuzo) to yield plasmid pYI1010 (Fig. 2B). To maximize GbuA production, the gbuA region (2.0 kb) was recloned from pYJ103 as a PstI-HindIII fragment between the corresponding sites downstream of the tac promoter (Ptac) on plasmid pMMB67EH (7) to generate plasmid pYI1011 (Fig. 2B).
FIG. 2.

Structure of gbuRA region of the PAO1 genome and insertion site of Gmr cassette in mutant genes (A) and plasmids used in complementation tests (B). (A) The arrow below the map indicates the transcription of gbuA. Gmr cassettes and the Ptac promoter are not drawn to scale. (B) Complementation tests were performed with MMP agar plus 4-GB. +, growth in 24 h; −, no growth in 24 h. Abbreviations for restriction sites: A, ApaI; AL, ApaLI; Ba, BamHI; Bb, BbsI; Be, BstEII; Bg, BglII; Bs, BstBI; E, EcoRI; H, HindIII; K, KpnI; P, PstI; S, SmaI; Sp, SphI; Su, Sau3AI; X, XhoI.
To construct lacZ fusions, the intergenic promoter region between gbuR and gbuA was amplified by PCR with plasmid pYJ103 as the template and the following primers designed to create HindIII sites (underlined) at both ends of the amplified fragment: 5′-AAGCTTCCACGGCGACTCCGGTTTGCG-3′ (+4 to −17 of gbuR) and 5′-AAGCTTCCACGGGGTGGCCTCACGGTC-3′ (complementary to +5 to −17 of gbuA). The nucleotides of the amplified DNA fragment were confirmed by sequencing after cloning into the SmaI site of plasmid pUC119 (37). This promoter sequence was then excised from the plasmid as a HindIII fragment and inserted into the HindIII site on plasmid pME6013, a lacZ translational fusion vector (12, 27), to generate the fusion plasmids pYI1016 (gbuA′::′lacZ) and pYI1017 (gbuR′::′lacZ).
To construct a knockout mutant of gbuR, the 2.8-kb SphI-HindIII fragment covering gbuRA was excised from plasmid pYJ103 and recloned into the mobilizable suicide plasmid pEX18Ap (ColE1 replicon, mob+ sacB) (13) at the corresponding sites, resulting in plasmid pYI1012. A gentamicin resistance (Gmr) cassette isolated from plasmid pPS858 (13) as a SmaI fragment was then inserted into the blunt-ended BstEII site on pYI1012 to create plasmid pYI1013, which was introduced into strain PAO1 by conjugation via E. coli S17-1 (32). The gbuR::Gm mutant PAO4508 was selected on MMP agar containing 100 μg of gentamicin/ml and 20 mM 4-GB and purified on LB agar containing 5% sucrose, which prevents the growth of cells containing pYI1013 (sacB) (13).
To construct knockout mutants of the downstream PA1418, PA1419, and PA1420 genes (Fig. 2A), we first amplified the 4.8-kb region containing these genes by PCR with KOD Dash DNA polymerase, PAO1 chromosomal DNA as the template, and primers 5′-TGGAGATCACCAGCATCGGAATCGAGT-3′ (nucleotides 1542455 to 1542481 of the PAO1 genome) (34) and 5′-ATTTGTAGCCAGCGATTTGACTCGCTAC-3′ (complementary to nucleotides 1547291 to 1547318). The amplified DNA fragment was purified through agarose gel electrophoresis, digested doubly with KpnI and EcoRI, and cloned into the corresponding sites of plasmid pUC118 (37) to construct plasmid pYI1018. After confirming the sequence, the 3.5-kb ApaLI fragment having the PA1418 and PA1419 regions (Fig. 2A) was excised from pYI1018 and cloned into plasmid pEX18Ap to produce pYI1019. Likewise, cloning of the 2.4-kb KpnI-PstI fragment (which covers from the 5′ half of PA1419 to the 3′ half of PA1421; Fig. 2A) into pEX18Ap created plasmid pYI1021.
Insertion of the Gmr cassette into the BglII site on pYI1019 as a BamHI fragment yielded pYI1020. Insertion of this cassette as a SmaI fragment into blunt-ended ApaI, BstBI, and BbsI sites on pYI1021 produced plasmids pYI1022, pYI1024, and pYI1025, respectively. These plasmids were introduced into strain PAO1, and then Gmr knockout mutants of the corresponding genes (Fig. 2A) were selected as described above. Correct insertion of the Gmr cassettes was verified by Southern blotting (15) with the 4.7-kb KpnI fragment of pYI1018 as the probe.
Northern blotting and primer extension.
RNA was isolated from cells growing exponentially in MMP containing the carbon and nitrogen sources described before (27). RNA samples (50 μg) in 10% (wt/vol) glyoxal were resolved along with RNA markers (Toyobo Biochemicals) on 1.0% agarose HS (Nippon Gene) and blotted onto Hybond N+ nylon membranes (Amersham Pharmacia Biotech) with a GenVac blotter (Pharmacia LKB). The gbuA transcripts on the membranes were detected with the 0.8-kb KpnI-SmaI fragment of gbuA (Fig. 2B) labeled with fluorescein-dUTP and the ECL detection system, version II (Amersham Pharmacia Biotech).
In primer extension experiments, RNA samples (20 μg) were annealed with the oligonucleotide 5′-CGATGCCGCCGAAGCGGGGCATTTCATTG-3′, complementary to nucleotides 33 to 61 of gbuA labeled with 32P at the 5′ end with [γ-32P]ATP (220 Bq/nmol; Amersham Pharmacia Biotech) and polynucleotide kinase (Takara Shuzo). A complementary strand was synthesized with avian reverse transcriptase (RAV-2; Takara Shuzo) in the presence of deoxyribonucleotides as described previously (27) and resolved on 6% denaturing polyacrylamide gels along with a sequence ladder generated with the BcaBEST sequencing kit (Takara Shuzo), the template plasmid pYJ103, and the 32P-end-labeled oligonucleotide primer. Radioactive DNA fragments on the gels were visualized on X-ray film.
Purification of GbuA.
Strain PAO4156 harboring plasmid pYI1011 (Ptac::gbuA) was cultured in 600 ml of MMP containing 20 mM 4-GB. When the optical density of the culture at 600 nm (OD600) reached 0.5, isopropyl-β-thiogalactopyranoside (IPTG; 0.5 mM) was added, and the culture was incubated for a further 2 h. Cells were harvested by centrifugation at 5,000 × g at 4°C for 10 min, suspended in 15 ml of TMM buffer (20 mM Tris-HCl [pH 8.0], 1 mM 2-mercaptoethanol, 0.1 mM MnSO4) and disrupted by passage through a French pressure cell (LSM Instruments). After cells and debris had been removed by centrifugation as described above, membrane proteins were further removed by centrifugation at 100,000 × g at 4°C for 1 h. The supernatant was then eluted through a column containing High Prep 16/10 DEAE-agarose (Amersham Pharmacia Biotech) with a 0 to 0.5 M KCl gradient in Tris-HCl buffer at a flow rate of 2.0 ml/min.
Active fractions were pooled, dialyzed against TMM buffer containing 1 M KCl, and then eluted from a Butyl-Toyopearl 650S column (8 mm by 75 mm; Tosoh) with a descending gradient of KCl (1 to 0.5 M) in TMM buffer (0.5 ml/min). After dialysis against TMM buffer, GbuA fractions from the preceding hydrophobic column chromatography were eluted from a MonoQ 5/5 column (Amersham Pharmacia Biotech) with a KCl gradient (0.2 to 0.5 M) in TMM buffer (0.5 ml/min). To determine the molecular mass of purified GbuA, a portion of the purified GbuA sample was gel filtered through a Superose 12 column (Amersham Pharmacia Biotech) with TMM buffer containing 0.1 M KCl (0.3 ml/min).
The protein concentration was determined with a protein assay kit (Bio-Rad Laboratories) and bovine serum albumin as the standard. GbuA purity was determined by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) (19) with 10% Ready gel J (Bio-Rad Laboratories). The amino-terminal amino acid sequence of purified GbuA was determined with an HP G1000A protein sequencer (Hewlett-Packard).
Enzyme assays.
The reaction mixture (0.5 ml) for GBase contained 0.3 M glycine (pH 9.5), 0.1 M 4-GB, 134 μM MnSO4, and enzyme (5 × 10−3 to 50 × 10−3 U). After an incubation at 37°C for 20 min, the reaction was terminated by adding 0.3 ml of 10% HClO4, and urea generated by the reaction was measured according to the method of Chou and Rodwell (4). One enzyme unit was defined as the amount of enzyme that forms 1.0 μmol of urea per min. β-Galactosidase activity in toluenized whole cells with o-nitro-β-galactopyranoside as the substrate is expressed as Miller units (23).
RESULTS
Cloning and structure of gbuA and gbuR.
To clone the gbu-9005 locus, a Sau3AI library of PAO1 chromosomal DNA constructed in E. coli XL1-Blue was transferred into strain PAO4173 (gbu-9005) by triparental mating employing E. coli HB101/pRK2013 as the helper (5). Plasmids were isolated from several Gbu+ transconjugants of strain PAO4173 selected as described in Materials and Methods, and DNA inserts were analyzed by restriction digestion followed by nucleotide sequencing. Plasmid pYJ103 contained an insert of 3.4 kb, which corresponded perfectly to the genomic region of PAO1 from nucleotides 1545999 to 1549483 (34) (Fig. 2A and B).
According to the gene annotation by the Pseudomonas Genome Project (www.pseudomonas.com), this insert contains two complete and two truncated genes of unknown or putative function (Fig. 2A and B). The first truncated gene (PA1423) encodes a probable chemotaxis transducer similar to the methyl-accepting chemotaxis McpA protein. The second gene (PA1422; designated here gbuR) specifies a putative protein (297 amino acids) with a calculated Mr of 33,385. This protein has similarity to LysR-type transcriptional regulators (30). The third gene (PA1421), located opposite gbuR, was designated speB2, according to the homology (58% similarity) of its product to E. coli agmatinase (the speB product). As we will show below, this gene corresponds to the gbu-9005 locus and encodes GBase. The last truncated gene (PA1420) on pYJ103 encoded a hypothetical protein of 139 amino acids.
To demonstrate that gbu-9005 is an allele of PA1421, we constructed deletions from plasmid pYJ103 and examined their ability to complement the gbu-9005 mutation. A deletion including the 5′ half of PA1422, as in pYI1009, did not affect the Gbu+ complementation of strain PAO4156 (gbu-9005) (Fig. 2B), whereas a deletion of part of PA1421, as in plasmid pYI1010, abolished the complementation (Fig. 2B). Moreover, a knockout mutant (PAO4518) having an insertion of a Gmr cassette on PA1421 (at the BbsI site on nucleotide 532) (Fig. 2A) did not grow on 4-GB and failed to form GBase (Table 2). These results and the determined amino-terminal sequence of purified GBase (see below) established that gbu-9005 is an allele of the PA1421 gene encoding GBase. We accordingly designated this gene gbuA and renamed gbu-9005 gbuA9005.
TABLE 2.
Regulation of GBase synthesis
| Enzyme source | GBase activitya (U/mg of protein)
|
|||
|---|---|---|---|---|
| Glu | 4-GB | Glu + 4-GB | 3-GP | |
| PAO1 (wild type) | <0.1 | 4.7 ± 0.4 | 3.1 ± 0.2 | <0.1 |
| PAO4156 (gbuA9005) | <0.1 | ng | <0.1 | nd |
| PAO4508 (gbuR::Gm) | <0.1 | ng | <0.1 | nd |
| PAO4518 (gbuA::Gm) | <0.1 | ng | <0.1 | nd |
| PAO4156/pYl1011 (Ptac::gbuA) | <0.1 | 5.7 ± 0.2 | nd | nd |
| PAO4156/pYl1011 (Ptac::gbuA) | 1.4 ± 0.1b | 10.2 ± 0.5b | nd | nd |
Cells were cultured to mid-log phase (OD600 = 0.5) in MMP containing the indicated supplements as sole C and N sources at 20 mM. Cell extracts were prepared and enzymes were assayed as described in Materials and Methods. Glu, glutamate; 4-GB, 4-guanidinobutyrate; 3-GP, 3-guanidinopropionate.
When cultures reached an OD600 of 0.5, 0.5 mM IPTG was added, and incubation was continued for an additional 2 h. Values are averages of three independent measurements with standard errors. ng, no growth; nd, not determined.
Purification and properties of GBase.
We purified GbuA to characterize it and to determine the translation initiation site of gbuA. The 2.0-kb DNA region containing gbuA as a PstI-HindIII fragment was fused to the tac promoter on plasmid pMMB67EH (7), and the resultant plasmid, pYI1011 (Ptac::gbuA) (Fig. 2B), was introduced into strain PAO4156 (gbuA9005) to increase GbuA synthesis as described in Materials and Methods. Strain PAO4156 (gubA9005) harboring pYI1011 had the Gbu+ phenotype and produced GbuA to levels comparable to those of PAO1 when cultured on 4-GB: no copy number effect of pMMB67EH (20 to 40 copies per chromosome) (18) was observed. Neither PAO1 nor PAO4156/pYI1011 grown in MMP containing 20 mM glutamate (noninducible conditions) produced detectable amounts of GBase (Table 2). IPTG induced GBase synthesis by PAO4156/pYI1011 cultured in MMP plus glutamate, but only to a level of 1.4 U/mg of protein (Table 2). However, when this recombinant strain was cultured in the presence of both 4-GB and IPTG, these inducers exerted a synergistic effect on GBase synthesis to produce 10.2 U of GBase/mg of protein (Table 2).
We therefore purified GBase from this recombinant strain grown in the presence of both 4-GB and IPTG. Three steps of column chromatography (see Materials and Methods) purified GbuA 30-fold, with a yield of 40% (0.6 mg from a 600-ml culture). As judged by SDS-PAGE (Fig. 3), the GBase purified in this manner was apparently homogeneous and had a measured mass of 34 kDa, which is in good agreement with the calculated Mr (34,695) of GbuA. The determined amino-terminal sequence (M-D-K-N-L-H-Q-P-L) of the purified GBase was identical to that deduced for GbuA (Fig. 4), confirming that the initiation codon of GbuA is GTG (34).
FIG. 3.
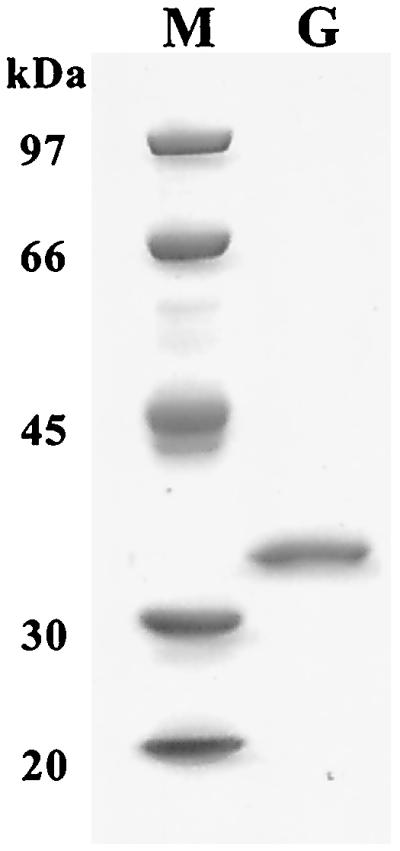
SDS-PAGE of purified GbuA. GbuA (1 μg) from MonoQ column chromatography (lane G) was analyzed along with molecular mass markers (lane M) by SDS-10% PAGE. Molecular markers were phosphorylase b (97 kDa), bovine serum albumin (66 kDa), ovalbumin (45 kDa), carbonic anhydrase (30 kDa), and catalase (20 kDa).
FIG. 4.

(A) Primer extension analysis of gbuA transcript and (B) promoter structure of intergenic region between gbuA and gbuR. (A) Complementary strands were synthesized with primer 32P-end-labeled oligonucleotide (corresponding to nucleotides 33 to 61 of gbuA) and RNA samples from PAO1 cells cultured in MMP plus 20 mM glutamate (lane 1), MMP plus 20 mM glutamate and 4-GB (lane 2), MMP plus 20 mM 4-GB (lane 3), or MMP plus 20 mM 3-guanidinopropionate (lane 4). Synthesized DNA strands were resolved on 6% denaturing polyacrylamide gels along with sequence ladders (G, A, T, and C).
Superose 12 gel filtration column chromatography indicated an apparent mass of 140 kDa for the native form, suggesting that this enzyme is a tetramer of identical 34-kDa subunits. EDTA inhibited the activity of GbuA, and Mn2+ ions but not Co2+ ions restored the full activity at 0.2 mM (data not shown). The apparent Km and Kcat values for 4-GB were 49 mM and 1,012 s−1, respectively. This enzyme showed the highest activity at pH 9.5. No enzyme activity was detected with other guanidino compounds, including arginine, agmatine, 3-guanidinopropionate, and guanidinoacetate.
Downstream genes.
According to the gene annotation by the Pseudomonas Genome Project (www.pseudomonas.com), PA1418 and PA1419 are located downstream of PA1420 and encode putative membrane transport proteins (Fig. 2A). They are oriented in the same direction as gbuA and are separated by short intergenic spacers (14 bp between gbuA and PA1420, 18 bp between PA1419 and PA1420, and 93 bp between PA1418 and PA1419) with no detectable ρ factor-independent transcriptional termination signal. Strain PAO1 growing on 4-GB excretes urea generated from the substrate by GBase (35). We therefore speculated that they might participate in 4-GB uptake and/or urea excretion. To test this hypothesis, we amplified the 4.8-kb region covering PA1418 to PA1420 by PCR and used it to construct knockout strains of each gene (Fig. 2A). However, none of the constructed mutants, PAO4509 (PA1418::Gm), PAO4511 (PA1419::Gm), or PAO4513 (PA1420::Gm), had obvious defects in growth on 4-GB or excretion of urea (data not shown).
Regulation of gbuA by GbuR.
To establish the role of GbuR in 4-GB utilization, we inactivated the gbuR gene by inserting a Gmr cassette. The resultant knockout mutant, PAO4508 (gbuR::Gm) (Fig. 2A), could not grow on 4-GB, indicating that this gene plays an essential role in 4-GB utilization. In agreement with previous findings (16), strain PAO1 produced GBase to a concentration of 4.7 U/mg of protein in MMP plus 4-GB and 3.1 U/mg in MMP plus glutamate plus 4-GB (Table 2). As reported previously (41, 42), 3-guanidinopropionate did not induce GBase synthesis (Table 2). Because strain PAO4508 did not grow in MMP plus 4-GB, we cultured it in MMP plus glutamate plus 4-GB (inducible conditions) and prepared cell extracts. GBase activity was undetectable in cell extracts of PAO4508 (Table 2), indicating the importance of GbuR in GBase synthesis induced by exogenous 4-GB.
We further examined the regulation of gbuA and gbuR expression with gbuA′-′lacZ and gbuR′-′lacZ fusions, respectively. The intergenic region (206 bp) between gbuR and gbuA was fused in frame to the lacZ gene at the HindIII site on plasmid pME6013 (12, 27) in both orientations to produce plasmids pYI1016 (gbuA′-′lacZ; fused to ′lacZ at the second codon of gbuA) and pYI1017 (gbuR′-′lacZ; fused at the second codon of gbuR). In agreement with the results of the GBase assays, β-galactosidase activity was negligible (<10 Miller units) in the gbuA′-′lacZ fusion harbored by strain PAO1 cells cultured on glutamate or 3-guanidinopropionate, but reached 1,500 and 1,300 Miller units when harbored by wild-type PAO1 cultured in MMP plus 4-GB and in MMP plus glutamate plus 4-GB, respectively. The amount of β-galactosidase activity produced by PAO4508 (gbuR::Gm) cells carrying pYI1010 was below detection (<10 U) after growth in MMP plus glutamate plus 4-GB. Similar assays of the gbuR′-′lacZ fusion failed to detect activity in PAO1 cells cultured in MMP plus glutamate or MMP plus 4-GB, indicating that the level of gbuR expression was very low.
Northern blot and primer extension analyses of the gbuA transcript.
We used Northern blots to estimate the size of the gbuA transcript. Figure 5 shows that the gbuA transcript was undetectable in strain PAO1 cultured in MMP plus glutamate. In contrast, a transcript of about 2,100 nucleotides was detected in PAO1 cultured in MMP plus 4-GB or MMP plus glutamate plus 4-GB (Fig. 5). The relative amounts of the transcripts in the PAO1 cells were proportional to the GBase (Table 2) and GbuA′-′LacZ levels under the corresponding growth conditions.
FIG. 5.

Northern blot of gbuA transcript. RNA was extracted from PAO1 cells cultured in MMP plus 20 mM glutamate (lane 1), MMP plus 20 mM glutamate and 4-GB (lane 2), and MMP plus 20 mM 4-GB (lane 3), blotted onto Hybond N+ nylon membranes, and then probed with the gbuA sequence between KpnI and SmaI (nucleotides [nt] 166 to 939). Numbers to the left of the gel indicate sizes of RNA markers.
We next determined the 5′ end of the gbuA transcript by primer extension. In agreement with the results of Northern blotting, the cDNA fragments were detected in RNA samples extracted from PAO1 cells cultured in MMP plus 4-GB or MMP plus glutamate plus 4-GB, but not in MMP plus glutamate (Fig. 4A). The size of the synthesized cDNA, determined by comparison with the sequence ladders, localized the 5′ end of the gbuA transcript at 40 bp upstream of the initiation codon of gbuA (Fig. 4A and B). The inferred gbuA promoter contained elements similar to the -10 and -35 consensus sequences of the σ70 RNA polymerase holoenzyme (Fig. 4B).
DISCUSSION
This study has identified and characterized the gbuRA genes of the ADH pathway. GBases have been purified and characterized from P. putida P2, Pseudomonas sp. strain ATCC 14676, and Brevibacterium helvolum (4, 39, 40). Yorifuji et al. (41) also purified GBase from P. aeruginosa PAO1, without, however, characterizing this enzyme in detail. The GbuA enzyme of strain PAO1 purified in this study has essentially the same properties as the GBases of P. putida P2 and Pseudomonas sp. strain ATCC 14676 in terms of substrate specificity, Mn2+ requirement, and high optimum pH (4, 39, 40). However, the size of the tetrameric PAO1 enzyme (140 kDa) appears to be smaller than that of its hexameric counterparts from P. putida P2 (178 to 190 kDa) (4) and Pseudomonas sp. strain ATCC 14676 (180 to 186 kDa) (40).
The deduced GbuA sequence assigns GBase to the arginase/agmatinase family (31). In accordance with their amino acid sequence homology (31), all enzymes of this family have common properties with respect to catalytic reactions (ureohydrolysis of the corresponding guanidino substrates), Mn2+ requirement, high pH optimum, and heat stability (1-4, 24, 31). Among three phylogenetic groups of ureohydrolases (31), a group consisting of some plant and bacterial enzymes, which has been suggested to be involved in secondary metabolism (31), includes the GBase of P. aeruginosa, suggesting that this enzyme group may consist of guanidino acid ureohydrolases (amidinohydrolases). The other two phylogenetic groups correspond to enzymes hydrolyzing a guanidino amino acid (arginine) or a guanidino amine (agmatine). Thus, the previous assignment of the PA1421 gene to speB2 (agmatinase) has to be revised, and there may be further enzymes, particularly in plants, that have been designated agmatinases (31) but may have a guanidino acid ureohydrolase activity in this group.
The inducible expression of gbuA depends essentially on the function of GbuR (Table 2), which is one of the ubiquitous transcriptional regulators of the LysR family (30). The LysR family regulators have a helix-turn-helix DNA-binding motif at the amino-terminal domain and an inducer-binding domain in a central region (30). LysR-type regulators can bind to regions near the −35 RNA polymerase binding site, resulting in transcription activation (30). GbuR also has a helix-turn-helix DNA-binding motif at amino acid positions 21 to 42, probably mediating the binding of GbuR to a regulatory region of the gbuA promoter. The primer extension experiments demonstrated that gbuA is transcribed from a σ70-dependent promoter in the presence of 4-GB (Fig. 4A and B). The gbuA transcript appears to cover the 2.1-kb region and hence to include PA1420, having no obvious function (34) (Fig. 2A and 5).
The products of PA1418 and PA1419 downstream of PA1420 show 46% similarity with the sodium/pantothenate symporter of E. coli and 52% similarity with a putative transmembrane transport protein of Streptomyces coelicolor, respectively (34). However, Northern blotting indicated that the transcription of gbuA terminates within the PA1419 coding region (Fig. 2A and 5), suggesting that this gene is not transcribed from the gbuA promoter. In fact, knockout of the PA1418 and PA1419 genes (as in strains PAO4509 and PAO4511; Fig. 2A) caused no detectable defect in 4-GB utilization or in urea excretion. These results imply that the major transport system of 4-GB is not closely linked to gbuRA. The genetically uncharacterized gbu-9001 locus at 19 min (14) is a possible candidate for a 4-GB transport gene.
The ADH enzymes are induced by different substrates: d-arginine dehydrogenase is induced by d-arginine, the KauB aldehyde dehydrogenase by 2-ketoarginine, and the GbuA enzyme by 4-GB (Table 2) (16, 22, 42, 43). The gbuRA pair is located on the PAO1 genome at genome position 1547000 (34), which is separate from kauB (position 5983000) (34). These findings support the notion that the ADH pathway genes are not clustered and that they are regulated independently and successively by distinct pathway intermediates. The Pseudomonas GBases show high Km values for 4-GB, ranging from 32 to 49 mM (4, 40). When arginine or 2-ketoarginine is catabolized via this pathway, the low affinity of GBase for 4-GB might lead to accumulation of 4-GB to levels that are sufficient to induce the synthesis of this enzyme.
Acknowledgments
We are grateful to D. Haas for valuable suggestions on this study, careful review of the manuscript, and plasmids. We also thank N. Gotoh for providing strains and S. Heeb and H. Schweizer for plasmids.
Y.N. is a domestic research fellow supported by the Japan Science and Technology Corporation.
REFERENCES
- 1.Ash, D. E., J. D. Cox, and D. W. Christianson. 2000. Arginase: a binuclear manganese metalloenzyme. Metal Ions Biol. Syst. 37:407-428. [PubMed] [Google Scholar]
- 2.Campo, M. L., J. M. Fuentes, and G. Soler. 1992. An arginine regulated γ-guanidinobutyrate ureahydrolase from tench liver (Tinca tinca L.). Arch. Int. Physiol. Biochim. Biophys. 100:55-60. [DOI] [PubMed] [Google Scholar]
- 3.Carvajal, N., V. López, M. Salas, E. Uribe, P. Herrera, and J. Cerpa. 1999. Manganese is essential for catalytic activity of Escherichia coli agmatinase. Biochem. Biophys. Res. Commun. 258:808-811. [DOI] [PubMed] [Google Scholar]
- 4.Chou, C. S., and V. W. Rodwell. 1972. Metabolism of basic amino acids in Pseudomonas putida. γ-guanidinobutyrate amidinohydrolase. J. Biol. Chem. 247:4486-4490. [PubMed] [Google Scholar]
- 5.Comai, L., C. Schilling-Cordaro, A. Mergia, and C. M. Houck. 1983. A new technique for genetic engineering of Agrobacterium Ti plasmid. Plasmid 10:21-30. [DOI] [PubMed] [Google Scholar]
- 6.Cunin, R., N. Glansdorff, A. Piérard, and V. Stalon. 1986. Biosynthesis and metabolism of arginine in bacteria. Microbiol. Rev. 50:314-352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fürste, J. P., W. Pansegrau, R. Frank, H. Blöcker, P. Scholz, M. Bagdasarian, and M. Lanka. 1986. Molecular cloning of the plasmid RP4 primase region in a multi-host-range tacP expression vector. Gene 48:119-131. [DOI] [PubMed] [Google Scholar]
- 8.Gamper, M., A. Zimmermann, and D. Haas. 1991. Anaerobic regulation of transcription initiation in the arcDABC operon of Pseudomonas aeruginosa. J. Bacteriol. 173:4742-4750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haas, D., M. Galimand, M. Gamper, and A. Zimmermann. 1990. Arginine network of Pseudomonas aeruginosa: specific and global controls, p. 303-316. In S. Silver, A. M. Chakrabarty, B. Iglewski, and S. Kaplan (ed.), Pseudomonas. American Society for Microbiology, Washington, D.C.
- 10.Haas, D., B. W. Holloway, A. Schamböck, and T. Leisinger. 1977. The genetic organization of arginine biosynthesis in Pseudomonas aeruginosa. Mol. Gen. Genet. 154:7-22. [DOI] [PubMed] [Google Scholar]
- 11.Haas, D., H. Matsumoto, P. Moretti, V. Stalon, and A. Mercenier. 1984. Arginine degradation in Pseudomonas aeruginosa mutants blocked in two arginine catabolic pathways. Mol. Gen. Genet. 193:437-444. [DOI] [PubMed] [Google Scholar]
- 12.Heeb, S., Y. Itoh, T. Nishijyo, U. Schnider, C. Keel, J. Wade, U. Walsh, F. O'Gara, and D. Haas. 2000. Small, stable shuttle vectors based on the minimal pVS1 replicon for use in gram-negative, plant-associated bacteria. Mol. Plant-Microbe Interact. 13:232-237. [DOI] [PubMed] [Google Scholar]
- 13.Hoang, T. T., R. R. Karkhoff-Schweizer, A. J. Kutchma, and H. P. Schweizer. 1998. A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally located DNA sequence: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene 212:77-86. [DOI] [PubMed] [Google Scholar]
- 14.Holloway, B. W., U. Römling, and B. Tümmler. 1994. Genomic mapping of Pseudomonas aeruginosa PAO. Microbiology 140:2907-2929. [DOI] [PubMed] [Google Scholar]
- 15.Itoh, Y. 1997. Cloning and characterization of the aru genes encoding enzymes of the catabolic arginine succinyltransferase pathway in Pseudomonas aeruginosa. J. Bacteriol. 179:7280-7290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jann, A., H. Matsumoto, and D. Haas. 1988. The fourth arginine catabolic pathway of Pseudomonas aeruginosa. J. Gen. Microbiol. 134:1043-1053. [DOI] [PubMed] [Google Scholar]
- 17.Jann, A., V. Stalon, C. Vander Wauven, T. Leisinger, and D. Haas. 1986. N2-succinylated intermediates in an arginine catabolic pathway of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 83:4937-4941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jeenes, D. J., L. Soldati, H. Baur, J. M. Watson, A. Mercenier, C. Reimmann, T. Leisinger, and D. Haas. 1986. Expression of biosynthetic genes from Pseudomonas aeruginosa and Escherichia coli in the heterologous host. Mol. Gen. Genet. 203:421-429. [DOI] [PubMed] [Google Scholar]
- 19.Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680-685. [DOI] [PubMed] [Google Scholar]
- 20.Lu, C. D., and A. T. Abdelal. 2001. The gdhB of Pseudomonas aeruginosa encodes an arginine-inducible NAD-dependent glutamate dehydrogenase which is subject to allosteric regulation. J. Bacteriol. 183:490-499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lu, C. D., H. Winteler, A. Abdelal, and D. Haas. 1999. The ArgR regulatory protein, a helper to the anaerobic regulator ANR during transcriptional activation of the arcD promoter in Pseudomonas aeruginosa. J. Bacteriol. 181:2459-2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marshall, V. P., and J. R. Sokatch. 1968. Oxidation of d-amino acids by a particulate enzyme from Pseudomonas aeruginosa. J. Bacteriol. 95:1419-1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miller, J. H. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
- 24.Mora, J., R. Tarrab, J. Martuscelli, and G. Soberon. 1965. Characteristics of arginases from ureotelic and nonureotelic animals. Biochem. J. 96:588-594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nakada, Y., Y. Jiang, T. Nishijyo, Y. Itoh, and C. D. Lu. 2001. Molecular characterization and regulation of the aguBA operon, responsible for agmatine utilization in Pseudomonas aeruginosa PAO1. J. Bacteriol. 183:6517-6524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nishijyo, T., S. M. Park, C. D. Lu, Y. Itoh, and A. T. Abdelal. 1998. Molecular characterization and regulation of an operon encoding a system for transport of arginine and ornithine and the ArgR regulatory protein in Pseudomonas aeruginosa. J. Bacteriol. 180:5559-5566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nishijyo, T., D. Haas, and Y. Itoh. 2001. The CbrA-CbrB two-component regulatory system controls the utilization of multiple carbon and nitrogen sources in Pseudomonas aeruginosa. Mol. Microbiol. 40:917-931. [DOI] [PubMed] [Google Scholar]
- 28.Park, S.-M., C.-D. Lu, and A. T. Abdelal. 1997. Purification and characterization of an arginine regulatory protein, ArgR, from Pseudomonas aeruginosa and its interactions with the control regions for the car, argF, and aru operons. J. Bacteriol. 179:5309-5317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
- 30.Schell, M. A. 1993. Molecular biology of the LysR family of transcriptional regulators. Annu. Rev. Microbiol. 47:597-626. [DOI] [PubMed] [Google Scholar]
- 31.Sekowska, A., A. Danchin, and J. L. Risler. 2000. Phylogeny of related functions: the case of polyamine biosynthetic enzymes. Microbiology 146:1815-1828. [DOI] [PubMed] [Google Scholar]
- 32.Simon, R., U. Priefer, and A. Pühler. 1983. A broad-host range mobilization system for in vivo genetic engineering: transposon mutagenesis in Gram-negative bacteria. Bio/Technology 1:784-790. [Google Scholar]
- 33.Stalon, V., and A. Mercenier. 1984. L-arginine utilization by Pseudomonas species. J. Gen. Microbiol. 130:69-76. [DOI] [PubMed] [Google Scholar]
- 34.Stover, C. V., et al. 2000. Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature 406:959-964. [DOI] [PubMed] [Google Scholar]
- 35.Tricot, C., A. Piérard, and V. Stalon. 1990. Comparative studies on the degradation of guanidino and ureido compounds by Pseudomonas. J. Gen. Microbiol. 136:2307-2317. [DOI] [PubMed] [Google Scholar]
- 36.Vanderbilt, A. S., N. S. Gaby, and V. W. Rodwell. 1975. Intermediates and enzymes between α-ketoarginine and γ-guanidinobutyrate in the L-arginine catabolic pathway of Pseudomonas putida. J. Biol. Chem. 250:5322-5329. [PubMed] [Google Scholar]
- 36a.van Thoai, N., F. Thome-Beau, and A. Olomucki. 1966. Induction et spécificité des enzymes de la nouvelle voie catabolique de l'arginine. Biochim. Biophys. Acta 115:73-80. [PubMed] [Google Scholar]
- 37.Vieira, J., and J. Messing. 1987. Production of single-stranded plasmid DNA. Methods Enzymol. 153:3-11. [DOI] [PubMed] [Google Scholar]
- 38.Voellmy, R., and T. Leisinger. 1976. Role of 4-aminobutyrate aminotransferase in the arginine metabolism of Pseudomonas aeruginosa. J. Bacteriol. 128:722-729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yorifuji, T., M. Kaneoke, T. Okazaki, and E. Shimizu. 1995. Degradation of 2-ketoarginine by guanidinobutyrase in arginine aminotransferase pathway of Brevibacterium helvolum. Biosci. Biotechnol. Biochem. 59:512-513. [DOI] [PubMed] [Google Scholar]
- 40.Yorifuji, T., M. Kato, T. Kobayashi, S. Ozaki, and S. Ueno. 1980. 4-Guanidinobutyrase amidinohydrolase from Pseudomonas sp. ATCC14676: purification to homogeneity and property. Agric. Biol. Chem. 44:1127-1134. [Google Scholar]
- 41.Yorifuji, T., T. Kobayshi, A. Tabuchi, Y. Shiritani, and K. Yonaha. 1983. Distribution of amidinohydrolases among Pseudomonas and comparative studies of some purified enzymes by one-dimensional peptide mapping. Agric. Biol. Chem. 47:2825-2830. [Google Scholar]
- 42.Yorifuji, T., and I. Sugai. 1978. 3-Guanidinopropionate amidinohydrolase and 4-guanidinobutyrate amidinohydrolase of Pseudomonas aeruginosa strain PAO1. Agric. Biol. Chem. 42:1789-1790. [Google Scholar]
- 43.Yorifuji, T., I. Sugai, H. Matsumoto, and A. Tabuchi. 1982. Characterization of 3-guanidinopropionate amidinohydrolase from Pseudomonas aeruginosa and a comparative study with 4-guanidinobutyrate amidinohydrolase from another Pseudomonas. Agric. Biol. Chem. 46:1361-1367. [Google Scholar]