

Abstract
Mitochondrial aldehyde dehydrogenase (ALDH2) is responsible for the metabolism of acetaldehyde and other toxic lipid aldehydes. Despite many reports about the inhibition of ALDH2 by toxic chemicals, it is unknown whether nitric oxide (NO) can alter the ALDH2 activity in intact cells or in vivo animals. The aim of this study was to investigate the effects of NO on ALDH2 activity in H4IIE-C3 rat hepatoma cells. NO donors such as S-nitrosoglutathione (GSNO), S-nitroso-N-acetylpenicillamine, and 3-morpholinosydnonimine significantly increased the nitrite concentration while they inhibited the ALDH2 activity. Addition of GSH-ethylester (GSH-EE) completely blocked the GSNO-mediated ALDH2 inhibition and increased nitrite concentration. To directly demonstrate the NO-mediated S-nitrosylation and inactivation, ALDH2 was immunopurified from control or GSNO-treated cells and subjected to immunoblot analysis. The anti-nitrosocysteine antibody recognized the immunopurified ALDH2 only from the GSNO-treated samples. All these results indicate that S-nitrosylation of ALDH2 in intact cells leads to reversible inhibition of ALDH2 activity.
Keywords: Aldehyde dehydrogenase, Glutathione, Nitric oxide, NO donors, S-Nitrosylation
Abbreviations: ALDH2, mitochondrial aldehyde dehydrogenase; BSO, l-buthionine-sulphoximine; DTT, dithiothreitol; GSH-EE, glutathioneethylester; GSNO, S-nitrosoglutathione; HNE, 4-hydroxy-2-nonenal; MDA, malondialdehyde; NO, nitric oxide; S-NO-Cys, S-nitroso-cysteine; ROS/RNS, reactive oxygen/nitrogen species; SIN-1, 3-morpholinosydnonimine · HCl; SNAP, S-nitroso-N-acetyl-d, l-penicillamine
1. Introduction
Mitochondrial class 2 aldehyde dehydrogenase (EC 1.2.1.3; ALDH2) is one of the key enzymes in the NAD+-dependent oxidation of various aldehydes produced during intermediary metabolisms. This enzyme exhibits a very low Km for acetaldehyde, and thus efficiently metabolizes acetaldehyde produced during ethanol metabolism [1–3]. ALDH2 is also known to metabolize toxic lipid aldehydes such as 4-hydroxy-2-nonenal (HNE) and malondialdehyde (MDA), which are produced after exposure to toxic chemicals [4,5]. ALDH2 is expressed in many mammalian tissues [1–3]. Catalytically active ALDH2 is also present in certain cell lines such as H4IIE-C3 rat hepatoma cells [6,7] and WIF-B cells [8]. Because of the role of ALDH2 in the metabolism of potentially toxic aldehydes, inactivation of ALDH2 by various chemicals [9–11] or under various disease states [12–15] is likely to cause marked accumulation of toxic aldehydes, leading to increased susceptibility towards irreversible damage.
Nitric oxide (NO) plays an important role in normal physiological processes and pathological conditions, largely depending on the location, quantity, timing and duration of its presence in the target cells [16,17]. In small quantities, NO can be involved in normal signal transduction pathways, while NO produced in large quantities can cause cellular damage [18] by producing a potent oxidant peroxynitrite (ONOO−) in the presence of superoxide anion. Both peroxynitrite and NO donors including S-nitrosoglutathione (GSNO) can modify cysteine (Cys) residues of target proteins, and thus negatively affect their functions in many cases [16,17]. Since Cys302 is critical for the catalytic activity of ALDH2 [19], ALDH2 may be inhibited by increased reactive oxygen and nitrogen species (ROS/RNS), possibly through modification of the active site Cys. In fact, earlier reports showed that the active site Cys in the purified yeast ALDH2 enzyme can be modified in NO-independent [20] and NO-dependent mechanisms [21]. In the latter study, Cys residues in the purified yeast ALDH2 protein were oxidized to intra-subunit disulfides rather than S-nitrosylation in the presence of NO donors, and this modification was not reversed by the addition of glutathione (GSH) [21]. However, it is still unknown whether ALDH2 in intact cells or in vivo animals can be inhibited by NO and whether ALDH2 inhibition can be reversed by reducing agents such as GSH. The aim of this study was to investigate potential S-nitrosylation and the inactivation of mammalian ALDH2 after cells were exposed to NO donors. We also studied whether the inactivation of ALDH2 could be reversed by a cell-permeable glutathione provider such as GSH-ethylester (GSH-EE).
2. Materials and methods
2.1. Chemicals and other materials
Anti-S-nitrosocysteine (S-NO-Cys) antibody, l-buthionine-sulphoximine (BSO), cyanamide, dithiothreitol (DTT), GSH-EE, GSNO, NAD+, propionyl aldehyde, pyrazole, and sodium ascorbate were purchased from Sigma (St. Louis, MO, USA). S-Nitroso-N-acetyl-d, l-penicillamine (SNAP), and 3-morpholinosydnonimine · HCl (SIN-1) were from Biomol (Plymouth Meeting, PA, USA). AminoLink® Plus immobilization kit from Pierce (Rockford, IL, USA) was used in preparing the immunoaffinity columns. Polyclonal antibody to mitochondrial ALDH2 was prepared in rabbits as previously described [22]. This antibody was used to detect ALDH2 in washed mitochondria or immunopurified ALDH2 since cross-detection of recombinant ALDH1 and ALDH3 or other cytosolic ALDH isozymes by the anti-ALDH2 antibody has not been tested.
2.2. Cell culture
H4IIE-C3 rat hepatoma cells, purchased from the American Type Culture Collection (Manassa, VA, USA), were grown in Dulbecco’s modified Eagle’s medium supplemented with 5% fetal bovine serum, 10% horse serum, and antibiotics as described [7]. After treatment with various agents for indicated times, the attached cells were washed twice with cold PBS and harvested by centrifugation at 2500 × g for 5 min. Mitochondrial pellets from the harvested cells were freshly prepared by diffierential centrifugation [23], washed twice with cold PBS to remove contaminating cytosolic proteins before being rapidly frozen, and stored at −80 °C until use. To prepare solubilized mitochondrial proteins, frozen mitochondrial pellets were quickly thawed and incubated in the mitochondria extraction buffer [23] with 1% CHAPS for 5 min. Protein concentration was determined using the BioRad protein assay kit, as described [23,24].
2.3. Measurement of ALDH activities
ALDH2 activity was measured by increased production of NADH at 340 nm by the method of Tank et al. [25]. The reaction mixture contained: 60 mM Na-phosphate buffer (pH 8.5), 1 mM NAD+, 1mM EDTA and mitochondrial or cytosolic proteins (0.5 mg/assay). After the reaction mixture was kept for 2 min at room temperature, the enzyme reaction initiated by adding the substrate (10 μM propionyl aldehyde). The absorbance change was monitored for 1 or 2 min to calculate the rate of NADH production. Activities of cyosolic ALDH1 were determined by the same method, except that 10 mM pyrazole was added to inhibit alcohol dehydrogenase activity [22,25] with two different concentrations of propionyl aldehyde (0.05 and 1.0 mM). Specific activity of ALDH2 was calculated by using the molar extinction coefficient of reduced NAD(P) of 6.22 × 106 cm2 at 340 nm (Merck Index) and 1 unit represents a reduction of 1 nmol NAD+/min/mg protein at room temperature.
2.4. Determination of NO concentration
Total NO concentration was determined as nitrite by the method of Green et al. [26] with slightly modifications. Briefly, samples were diluted 4-fold with deionized water and deproteinized by adding 1/20th volume of 30% (w/v) ZnSO4. After centrifugation at 1500 × g for 5 min at room temperature, the supernatant was transferred into the microcentrifuge tubes containing the same volume of Griess reagent (1 g/L sulfanilamide, 25 g/L phosphoric acid, and 0.1 g/L N-1-naphthylethylenediamine). After 10 min of color development at room temperature, the absorbance at 540 nm was measured to determine the nitrite content calculated from the standard curve using sodium nitrite.
2.5. Immunoaffinity purification and immunoblot analysis
The IgG fraction of anti-ALDH2 antibody was covalently immobilized onto AminoLink® agarose beads by following the manufacturer’s instruction. Immunoaffinity purification of ALDH2 proteins from differently treated samples was performed by the method [27]. The unpurified mitochondrial proteins and immunopurified ALDH2 proteins were resolved on SDS–polyacrylamide gels and subjected to immunoblot analysis with the specific anti-ALDH2 antibody [22] or anti-S-nitroso-Cys antibody and enhanced chemiluminescence detection. The statistical analysis and other methods not described here were the same as described [22–24].
3. Results
3.1. Selective inhibition of ALDH2 in intact cells by BSO
To study the potential inhibition of ALDH2 in intact cells by NO, we used H4IIEC3 cells as a model, since these cells contain considerable amounts of catalytically active ALDH2 [6,7]. To generate increased production of NO, we followed the method of Corrales et al. [28] by using BSO through selective inhibition of GSH synthesis. Under our conditions, the specific activity of ALDH2, which is the major mitochondrial ALDH enzyme with a very low Km for acetaldehyde, in H4IIE-C3 cells was 1.8 units, which are comparable to those values reported earlier [6]. Our results show that ALDH2 activity rapidly declined after the BSO treatment. For instance, 30 min after BSO exposure, approximately 50% of the ALDH2 activity was inhibited while more than 70% activity was inhibited at 1 h. The inhibition persisted up to 4 h after BSO treatment (Fig. 1). In contrast, the specific activity (1.7 units) of cytosolic ALDH1 was not inhibited by the BSO treatment, indicating a selective inhibition of mitochondrial ALDH2 by BSO, possibly through reduction of intracellular GSH and NO production [28]. These results further suggest that the mitochondrial ALDH2 may contain a local microenvironment with a higher affinity towards NO [17] than that of the cytosolic ALDH1 enzyme.
Fig. 1.
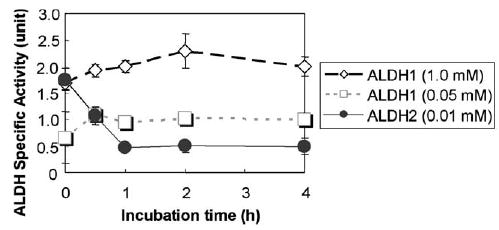
Selective inhibition of ALDH2 activity after BSO exposure. H4IIE-C3 cells, grown in large culture dishes (150 mm diameter), were treated with 10 mM BSO up to 4 h, before cells were harvested. Cytosolic and mitochondrial proteins (0.5 mg/assay) were used to determine the activities of ALDH1 and ALDH2, respectively. Each point represents the average ± S.D. of three determinations.
3.2. Reversible inhibition of ALDH2 by NO donors
To further evaluate the NO-mediated ALDH2 inhibition, we studied whether ALDH2 activity can be inhibited by various NO donors such as GSNO, SIN-1, and SNAP. Although the relative levels of ALDH2 inhibition differed slightly, all NO donors tested in this study significantly inhibited the ALDH2 activity, in a manner similar to that by cyanamide [1,7], a classical inhibitor of ALDH2 (Fig. 2A). In contrast, immunoblot analysis showed similar amounts of ALDH2 proteins recognized by the anti-ALDH2 antibody (Fig. 2B). These results suggest that the ALDH2 inhibition by NO donors is likely to be a result of NO-mediated modification of ALDH2.
Fig. 2.
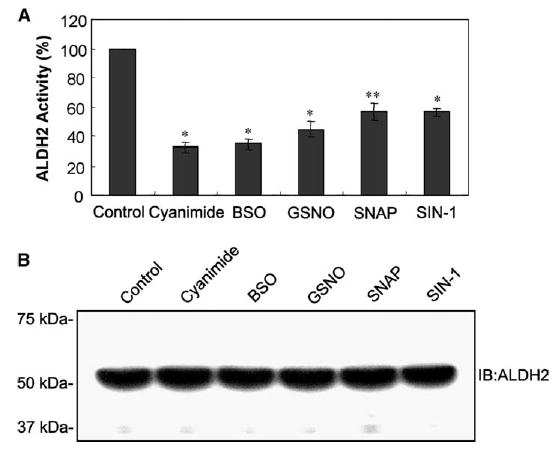
Inactivation of ALDH2 by various compounds including NO donors. (A) H4IIE-C3 cells were exposed to various agents: 1 mM cyanamide, 10 mM BSO, 0.5 mM GSNO, 1.0 mM SNAP, or 1.0 mM SIN-1 for 4 h, before cells were harvested. Mitochondrial proteins (0.5 mg/assay) were used to measure the ALDH2 activity. ALDH2 activity in each sample is compared to that of the untreated control. Each point represents the average ± S.D. of three determinations. * and **Significant difference (*P < 0.005 and **P < 0.01) from the control sample. (B) Equal amounts of mitochondrial proteins (20 μg/well) from the same set of samples were separated on 12% SDS–PAGE and subjected to immunoblot analysis using the anti-ALDH2 antibody. Migration of molecular weight marker proteins is shown in the left.
It is known that S-nitrosylated Cys residues are reversibly reduced to Cys especially in the presence of GSH, sodium ascorbate, or DTT [16,17,23,28]. To determine whether NOmediated modification and the subsequent inhibition of ALDH2 were reversible, we studied the effects of GSH-EE, a cell-permeable GSH provider, on NO-mediated ALDH2 inhibition (Fig. 3A). Simultaneous addition of GSH-EE completely blocked the BSO- or GSNO-mediated ALDH2 inhibition. Furthermore, addition of 10 mM DTT and 5 mM sodium ascorbate significantly restored the ALDH2 activity suppressed in GSNO-treated cells (data not shown). All these results indicate that reversible regulation of ALDH2 in intact H4IIE-C3 cells could occur by the cellular levels of NO and GSH.
Fig. 3.
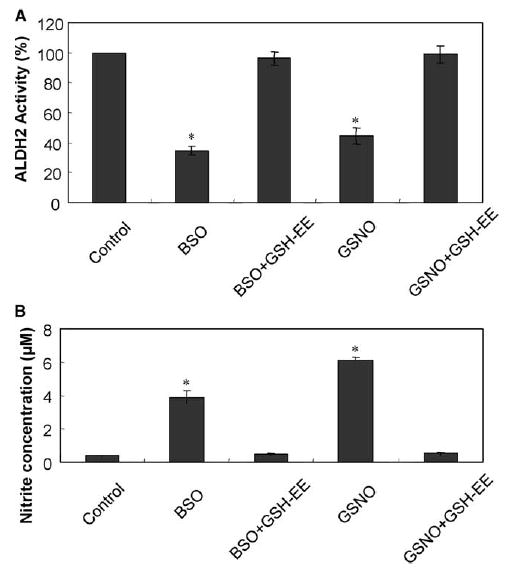
Complete blockade of BSO- or GSNO-mediated inhibition of ALDH2 activity and increased nitrite concentrations by GSH-EE. H4IIE-C3 cells were exposed to the following agents: either 10 mM BSO or 0.5 mM GSNO in the absence and presence of 5 mM GSH-EE for 4 h, before cells were harvested. (A) Mitochondrial proteins (0.5 mg/assay) were used to measure the activities of ALDH2. ALDH2 activity in each sample is compared to that of untreated control. Each point represents the average ± S.D. of three determinations. (B) Mitochondrial proteins (0.4 mg/assay) were used to determine the nitrite concentrations. Each point represents the average ± S.D. of three determinations. *Significant difference (P < 0.005) from the control sample.
3.3. Increased nitrite concentrations by NO donor compounds
To verify whether nitrite concentrations were increased under our conditions, we further measured the levels of nitrite concentration. Both BSO and GSNO significantly increased the nitrite concentrations (Fig. 3B), although a higher nitrite concentration was observed with GSNO than with BSO, which reduces the intracellular GSH content and thus indirectly affects the nitrite level [28]. GSH-EE also completely blocked the increased nitrite concentrations caused by BSO or GSNO. These results suggest that the increased nitrite concentrations are likely to inhibit the ALDH2 activity through the modification of its Cys residue(s).
3.4. Evidence for NO-mediated S-nitrosylation of Cys residue(s) of ALDH2
To directly demonstrate the NO-mediated S-nitrosylation of ALDH2, the mitochondrial ALDH2 proteins in untreated control and GSNO-treated cells in the absence and presence of GSH-EE, respectively, were purified by immunoaffinity columns using the IgG fraction of anti-ALDH2 antibody. Similar amounts of ALDH2 protein (54 kDa) were detected by immunoblot analysis for the immunopurified ALDH2 proteins from different treatments (Fig. 4, top panel). Immunoblot analysis with the anti-S-NO-Cys antibody showed that one immunoreactive band (54 kDa) was recognized for the immunopurified ALDH2 only from the GSNO-treated cells (Fig. 4, bottom panel, lane 3). However, the immunoreactive S-NO-Cys band disappeared (lane 2) when GSH-EE was added, consistent with the restoration of ALDH2 activity (Fig. 3A). These results provide direct evidence for NO-mediated S-nitrosylation of Cys residue(s) of ALDH2, causing the reversible inhibition of ALDH2 activity.
Fig. 4.
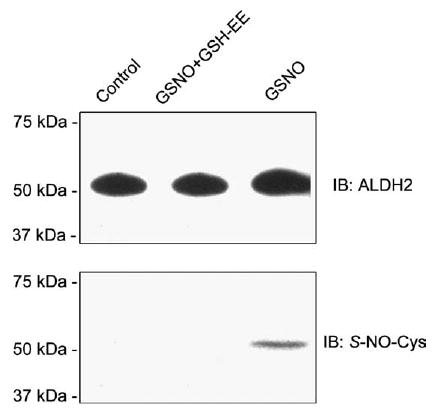
Immunoblot analysis for immunopurified ALDH2 proteins. H4IIE-C3 cells were treated with different agents as indicated. Mitochondrial ALDH2 proteins from the differently treated cells were purified by immunoaffinity chromatography and subjected to immunoblot analysis using the anti-ALDH2 antibody (top) or the anti-S-NO- Cys antibody (bottom).
4. Discussion
Despite many reports on the inhibition of ALDH2 following exposure to toxic chemicals [9–11] and pathological conditions [12–15], the mechanism of ALDH2 inhibition is poorly understood. We hypothesized that the active site Cys302 [19] must be modified and thus inhibited by increased ROS/RNS and/or peroxynitrite produced under stressful conditions [9–15] due to decreased ALDH2 activities. It is known that Cys residues can undergo the following modifications: oxidation to disulfides [21,29], sulfenic acid [30], sulfinic acid and sulfonic acid [31], S-glutathionylation [32], S-nitrosylation [16,17], ADP-ribosylation [20], etc. In addition, Cys residues in ALDH2 were shown to be covalently modified with the reactive metabolites of acetaminophen [10] or disulfiram [33], thus becoming inactivated. Therefore, it is possible that the active site Cys in the ALDH2 protein underwent one of these modifications and became inactivated after cells were exposed to BSO or GSNO. Our current results show that ALDH2 in intact cells is modified by NO-dependent S-nitrosylation based on the following facts: the inhibition of ALDH2 activity by BSO and GSNO; the increased nitrite concentrations after BSO and GSNO treatments; the recognition of the immunopurified ALDH2 with the anti-S-NO-Cys antibody; and the complete blockade of BSO- or GSNO-mediated ALDH2 inhibition, increased nitrite concentrations, and S-nitrosylation in the presence of GSH-EE. To our knowledge, our results represent the first report about the inhibition of ALDH2 in intact cells via NO-mediated S-nitrosylation.
DeMaster et al. [21] reported that Cys residues of the purified ALDH2 can be oxidized to intra-subunit disulfides rather than S-nitrosylation in the presence of NO donors while the oxidized enzyme was not reduced by the addition of GSH. Cys residues of the purified ALDH2 can also be modified by ADP-ribosylation in an NO-independent manner [20]. However, these kinds of modifications of ALDH2 in intact cells and potential S-nitrosylation of ALDH2 have not been studied, despite the well-characterized S-nitrosylation of Cys residues in many other proteins [16,17,28]. Our current results showed that ALDH2 in intact cells is S-nitrosylated by NO with the concurrent inhibition of its activity. We do not know the reason why we observed the GSH-mediated blockade of S-nitrosylation and inhibition of ALDH2 caused by NO donors, whereas DeMaster et al. [21] did not. The difference could simply result from the different status of Cys residue(s) of ALDH2: S-nitrosylation versus disulfide bond formation. These results warrant precaution in interpreting the data with the purified enzyme.
Although it is theoretically possible to identify the S-nitrosylated cysteine residues, we were unsuccessful in this effort because of small amounts of the cultured H4IIE-C3 cells. Despite the failure to identify the modified Cys residue(s) by protein sequencing analysis, we believe that Cys162 may not be the S-nitrosylated Cys since mutation of this residue decreases the ALDH2 stability [19] and we did not observe reduced amounts of ALDH2 after GSNO treatment (Fig. 2B). It was also shown that Cys49 is not involved in the catalytic activity [19]. Subsequently, S-nitrosylation of Cys49 is not likely to change the ALDH2 activity after GSNO treatment. Because of the reduced ALDH2 activity after treatment with NO donors (Fig. 2), we conclude that Cys302 of ALDH2 must be the S-nitrosylated cysteine residue. Our preliminary results also showed that ALDH2 activity was inhibited and S-nitrosylated (detected with the anti-S-NO-Cys antibody after immunopurification) in alcohol-exposed rats. To directly demonstrate S-nitrosylation of the active site Cys302, the immunopurified ALDH2 from alcohol-treated rat livers is being analyzed by proteomics approaches.
Under pathological conditions or after exposure to toxic chemicals, increased ROS/RNS production leads to elevated lipid peroxidation with increased levels of lipid aldehydes [4]. Reactive acetaldehyde and lipid aldehydes such as HNE and MDA are usually metabolized by ALDH2, alcohol dehydrogenase, and glutathione S-transferase [4,5]. However, ALDH2 can be inhibited by various potentially toxic chemicals such as CCl4 [9] and acetaminophen [10]. Although the molecular mechanism of the ALDH2 inhibition by these agents has not been elucidated further, it is likely that these chemicals can inhibit the ALDH2 activity by S-nitrosylation of Cys residue( s), as shown in the current study, because these chemicals are also known to activate the inducible NO synthase and/or increase peroxynitrite production [34]. Inhibition of ALDH2 subsequently leads to marked elevations of reactive aldehydes including HNE and MDA. These conditions are likely to increase susceptibility toward irreversible cell damage, unless these conditions are properly treated by suitable measures. In fact, markedly elevated levels of acetaldehyde (as protein adducts) [22,35] and lipid aldehydes such as HNE are frequently observed in alcoholic hepatitis and other chronic disease states [13,35,36]. These data may suggest an important role of ALDH2 in cellular protection against potential damage by reactive aldehydes as demonstrated [37]. In addition, potential inhibition of ALDH2 by various chemicals [9–11] or pathological conditions [12–15] may reduce the efficacy of nitroglycerins used in treating acute angina and congestive heart failure, since ALDH2 plays a critical role in bioactivation of nitroglycerins [38,39]. Alternatively inhibition of ALDH2 may cause tolerance toward nitroglycerin-mediated relaxation of blood vessels [40]. Based on these views, it would be of interest to find out whether S-nitrosylation and subsequent inhibition of ALDH2 activity can be observed in animal models of stressful or pathological conditions.
Acknowledgments
We are grateful to Dr. Norman Salem, Jr. for his support throughout the experiments.
Footnotes
Edited by Vladimir Skulachev
References
- 1.Svanas GW, Weiner H. Aldehyde dehydrogenase activity as the rate limiting factor for aldehyde metabolism in rat liver. Arch Biochem Biophys. 1985;236:36–46. doi: 10.1016/0003-9861(85)90603-4. [DOI] [PubMed] [Google Scholar]
- 2.Hafer G, Agarwal DP, Goedde HW. Human brain aldehyde dehydrogenase: activity with DOPAL and isozyme distribution. Alcohol. 1987;4:413–418. doi: 10.1016/0741-8329(87)90077-2. [DOI] [PubMed] [Google Scholar]
- 3.Day CP, Bashir R, James OF, Bassendine MF, Crabb DW, Thomasson HR, Li TK, Edenberg HJ. Investigation of the role of polymorphisms at the alcohol and aldehyde dehydrogenase loci in genetic predisposition to alcohol-related end organ damage. Hepatology. 1991;14:798–801. doi: 10.1002/hep.1840140509. [DOI] [PubMed] [Google Scholar]
- 4.Esterbauer H, Schaur RJ, Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malondialdehyde and related aldehydes. Free Radic Biol Med. 1991;11:81–128. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- 5.Hartley DP, Ruth JA, Petersen DR. The hepatocellular metabolism of 4-hydroxynonenal by alcohol dehydrogenase, aldehyde dehydrogenase, and glutathione S-transferase. Arch Biochem Biophys. 1995;316:197–205. doi: 10.1006/abbi.1995.1028. [DOI] [PubMed] [Google Scholar]
- 6.Crabb DW, Stewart MJ, Xiao Q. Hormonal and chemical influences on the expression of class 2 aldehyde dehydrogenases in rat H4IIEC3 and human HuH7 hepatoma cells. Alcohol Clin Exp Res. 1995;19:1414–1419. doi: 10.1111/j.1530-0277.1995.tb01000.x. [DOI] [PubMed] [Google Scholar]
- 7.Moncada C, Fuentes N, Lladser A, Encina G, Sapag A, Karahanian E, Israel Y. Use of an ‘‘acetaldehyde clamp’’ in the determination of low-Km aldehyde dehydrogenase activity in H4-II-E-C3 rat hepatoma cells. Alcohol. 2003;31:19– 24. doi: 10.1016/j.alcohol.2003.06.004. [DOI] [PubMed] [Google Scholar]
- 8.Schaffert CS, Todero SL, McVicker BL, Tuma PL, Sorrell MF, Tuma DJ. WIF-B cells as a model for alcohol-induced hepatocyte injury. Biochem Pharmacol. 2004;67:2167–2174. doi: 10.1016/j.bcp.2004.01.022. [DOI] [PubMed] [Google Scholar]
- 9.Hjelle JJ, Grubbs JH, Beer DG, Petersen DR. Time course of the carbon tetrachloride-induced decrease in mitochondrial aldehyde dehydrogenase activity. Toxicol Appl Pharmacol. 1983;67:159–165. doi: 10.1016/0041-008x(83)90220-x. [DOI] [PubMed] [Google Scholar]
- 10.Landin JS, Cohen SD, Khairallah EA. Identification of a 54-kDa mitochondrial acetaminophen-binding protein as aldehyde dehydrogenase. Toxicol Appl Pharmacol. 1996;141:299– 307. doi: 10.1006/taap.1996.0287. [DOI] [PubMed] [Google Scholar]
- 11.Mitchell DY, Petersen DR. Inhibition of rat hepatic mitochondrial aldehyde dehydrogenase-mediated acetaldehyde oxidation by trans-4-hydroxy-2-nonenal. Hepatology. 1991;13:728–734. doi: 10.1016/0270-9139(91)92572-p. [DOI] [PubMed] [Google Scholar]
- 12.Palmer KR, Jenkins WJ. Aldehyde dehydrogenase in alcoholic subjects. Hepatology. 1985;5:260–263. doi: 10.1002/hep.1840050218. [DOI] [PubMed] [Google Scholar]
- 13.Traverso N, Menini S, Odetti P, Pronzato MA, Cottalasso D, Marinari UM. Diabetes impairs the enzymatic disposal of 4-hydroxynonenal in rat liver. Free Radic Biol Med. 2002;32:350–359. doi: 10.1016/s0891-5849(01)00811-5. [DOI] [PubMed] [Google Scholar]
- 14.Yokoyama A, Muramatsu T, Ohmori T, Yokoyama T, Okuyama K, Takahashi H, Hasegawa Y, Higuchi S, Maruyama K, Shirakura K, Ishii H. Alcohol-related cancers and aldehyde dehydrogeanse-2 in Japanese alcoholics. Carcinogenesis. 1998;19:1383–1387. doi: 10.1093/carcin/19.8.1383. [DOI] [PubMed] [Google Scholar]
- 15.Park KS, Cho SY, Kim H, Paik YK. Proteomic alterations of the variants of human aldehyde dehydrogenase isozymes correlate with hepatocellular carcinoma. Int J Cancer. 2002;97:261–265. doi: 10.1002/ijc.1585. [DOI] [PubMed] [Google Scholar]
- 16.Hess DT, Matsumoto A, Kim SO, Marshall HE, Stamler JS. Protein S-nitrosylation: purview and parameters. Nat Rev Mol Cell Biol. 2005;6:150–166. doi: 10.1038/nrm1569. [DOI] [PubMed] [Google Scholar]
- 17.Lane P, Hao G, Gross SS. S-nitrosylation is emerging as a specific and fundamental posttranslational protein modification: head-to-head comparison with O-phosphorylation. Sci STKE. 2001;2001;86:RE1–RE9. doi: 10.1126/stke.2001.86.re1. [DOI] [PubMed] [Google Scholar]
- 18.Lin KT, Xue JY, Nomen M, Spur B, Wong PYK. Peroxynitrite induced apoptosis in HL-60 cells. J Biol Chem. 1995;270:16487–16490. doi: 10.1074/jbc.270.28.16487. [DOI] [PubMed] [Google Scholar]
- 19.Farres J, Wang TT, Cunningham SJ, Weiner H. Investigation of the active site cysteine residue of rat liver mitochondrial aldehyde dehydrogenase by site-directed mutagenesis. Biochemistry. 1995;34:2592–2598. doi: 10.1021/bi00008a025. [DOI] [PubMed] [Google Scholar]
- 20.McDonald LJ, Moss J. Nitric oxide-independent, thiol-associated ADPribosylation inactivates aldehyde dehydrogenase. J Biol Chem. 1993;268:17878–17882. [PubMed] [Google Scholar]
- 21.DeMaster EG, Redfern B, Quast BJ, Dahlseid T, Nagasawa HT. Mechanism for the inhibition of aldehyde dehydrogenase by nitric oxide. Alcohol. 1997;14:181–189. doi: 10.1016/s0741-8329(96)00142-5. [DOI] [PubMed] [Google Scholar]
- 22.Jeong KS, Soh Y, Jeng J, Felder MR, Hardwick JP, Song BJ. Cytochrome P450 2E1 (CYP2E1)-dependent production of a 37-kDa acetaldehyde protein adduct in the rat liver. Arch Biochem Biophys. 2000;384:81–87. doi: 10.1006/abbi.2000.2119. [DOI] [PubMed] [Google Scholar]
- 23.Suh SK, Hood BL, Kim BJ, Conrads TP, Veenstra TD, Song BJ. Identification of oxidized mitochondrial proteins in alcohol-exposed human hepatoma cells and mouse liver. Proteomics. 2004;4:3401–3412. doi: 10.1002/pmic.200400971. [DOI] [PubMed] [Google Scholar]
- 24.Bae MA, Song BJ. Critical role of c-Jun N-terminal protein kinase activation in troglitazone-induced apoptosis of human HepG2 hepatoma cells. Mol Pharmacol. 2003;63:401–408. doi: 10.1124/mol.63.2.401. [DOI] [PubMed] [Google Scholar]
- 25.Tank AW, Weiner H, Thurman JA. Enzymology and subcellular localization of aldehyde oxidation in rat liver. Oxidation of 3,4-dihydroxyphenylacetaldehyde derived from dopamine to 3,4-dihydroxyphenylacetic acid. Biochem Pharmacol. 1981;30:3265–3275. doi: 10.1016/0006-2952(81)90598-0. [DOI] [PubMed] [Google Scholar]
- 26.Green LC, Wagner DA, Glogowski J, Skipper PL, Wishnok JS, Tannenbaum SR. Analysis of nitrate, nitrite, and [15N] nitrate in biological fluids. Anal Biochem. 1982;126:131–138. doi: 10.1016/0003-2697(82)90118-x. [DOI] [PubMed] [Google Scholar]
- 27.Song BJ, Veech RL, Park SS, Gelboin HV, Gonzalez FJ. Induction of rat hepatic N-nitrosodimethylamine demethylase by acetone is due to protein stabilization. J Biol Chem. 1989;264:3568–3572. [PubMed] [Google Scholar]
- 28.Corrales FJ, Ruiz F, Mato JM. In vivo regulation by glutathione of methionine adenosyltransferase S-nitrosylation in rat liver. J Hepatol. 1999;31:887–894. doi: 10.1016/s0168-8278(99)80291-8. [DOI] [PubMed] [Google Scholar]
- 29.Cumming RC, Andon NL, Haynes PA, Park M, Fischer WH, Schubert D. Protein disulfide bond formation in the cytoplasm during oxidative stress. J Biol Chem. 2004;279:21749– 21758. doi: 10.1074/jbc.M312267200. [DOI] [PubMed] [Google Scholar]
- 30.DeMaster EG, Quast BJ, Redfern B, Nagasawa HT. Reaction of nitric oxide with the free sulfhydryl group of human serum albumin yields a sulfenic acid and nitrous oxide. Biochemistry. 1995;34:11494–11499. doi: 10.1021/bi00036a023. [DOI] [PubMed] [Google Scholar]
- 31.Woo HA, Jeong W, Chang TS, Park KJ, Park SJ, Yang JS, Rhee SG. Reduction of cysteine sulfinic acid by sulfiredoxin is specific to 2-cys peroxiredoxins. J Biol Chem. 2005;280:3125–3128. doi: 10.1074/jbc.C400496200. [DOI] [PubMed] [Google Scholar]
- 32.Mohr S, Hallak H, de Boitte A, Lapetina EG, Brune B. Nitric oxide-induced S-glutathionylation and inactivation of glyceraldehyde-3-phosphate dehydrogenase. J Biol Chem. 1999;274:9427–9430. doi: 10.1074/jbc.274.14.9427. [DOI] [PubMed] [Google Scholar]
- 33.Shen ML, Johnson KL, Mays DC, Lipsky JJ, Naylor S. Determination of in vivo adducts of disulfiram with mitochondrial aldehyde dehydrogenase. Biochem Pharmacol. 2001;61:537–545. doi: 10.1016/s0006-2952(00)00586-4. [DOI] [PubMed] [Google Scholar]
- 34.Jaeschke H, Knight TR, Bajt ML. The role of oxidant stress and reactive nitrogen species in acetaminophen hepatotoxicity. Toxicol Lett. 2003;144:279–288. doi: 10.1016/s0378-4274(03)00239-x. [DOI] [PubMed] [Google Scholar]
- 35.Viitala K, Israel Y, Blake JE, Niemela O. Serum IgA, IgG, and IgM antibodies directed against acetaldehyde-derived epitopes: relationship to liver disease severity and alcohol consumption. Hepatology. 1997;25:1418–1424. doi: 10.1002/hep.510250619. [DOI] [PubMed] [Google Scholar]
- 36.Pawlosky RJ, Flynn BM, Salem N., Jr The effects of low dietary levels of polyunsaturates on alcohol-induced liver disease in rhesus monkeys. Hepatology. 1997;26:1386– 1392. doi: 10.1002/hep.510260602. [DOI] [PubMed] [Google Scholar]
- 37.Soh Y, Jeong KS, Lee IJ, Bae MA, Kim YC, Song BJ. Selective activation of the c-Jun N-terminal protein kinase pathway during 4-hydroxynonenal induced apoptosis of PC12 cells. Mol Pharmacol. 2000;58:535–541. doi: 10.1124/mol.58.3.535. [DOI] [PubMed] [Google Scholar]
- 38.Sydow K, Daiber A, Oelze M, Chen Z, August M, Wendt M, Ullrich V, Mulsch A, Schulz E, Keaney JF, Jr, Stamler JS, Munzel T. Central role of mitochondrial aldehyde dehydrogenase and reactive oxygen species in nitroglycerin tolerance and cross-tolerance. J Clin Invest. 2004;113:482–489. doi: 10.1172/JCI19267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen Z, Foster MW, Zhang J, Mao L, Rockman HA, Kawamoto T, Kitagawa K, Nakayama KI, Hess DT, Stamler JS. An essential role for mitochondrial aldehyde dehydrogenase in nitroglycerin bioactivation. Proc Natl Acad Sci USA. 2005;102:12159–12164. doi: 10.1073/pnas.0503723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Parker JD. Nitrate tolerance, oxidative stress, and mitochondrial function: another worrisome chapter on the effects of organic nitrates. J Clin Invest. 2004;113:352–354. doi: 10.1172/JCI21003. [DOI] [PMC free article] [PubMed] [Google Scholar]