

Abstract
The homo- and heterodimerization of Bcl-2 family proteins is important for transduction and integration of apoptotic signals and control of the permeability of mitochondria and endoplasmic reticulum membranes. Here we mapped the interface of the Bcl-2 homodimer in a cell-free system using site-specific photocross-linking. Bcl-2 homodimer-specific photoadducts were detected from 11 of 17 sites studied. When modeled into the structure of Bcl-2 core, the interface is composed of two distinct surfaces: an acceptor surface that includes the hydrophobic groove made by helices 2 and 8 and the loop connecting helices 4 and 5 and a donor surface that is made by helices 1–4 and the loop connecting helices 2 and 3. The two binding surfaces are on separate faces of the three-dimensional structure, explaining the formation of Bcl-2 homodimers, homo-oligomers, and Bcl-2/Bax hetero-oligomers. We show that in vitro the Bcl-2 dimer can still interact with activated Bax as a larger oligomer. However, formation of a Bax/Bcl-2 heterodimer is favored, since this interaction inhibits Bcl-2 homodimerization. Our data support a simple model mechanism by which Bcl-2 interacts with activated Bax during apoptosis in an effective manner to neutralize the proapoptotic activity of Bax.
Bcl-2 family proteins are key regulators of apoptosis. These proteins share sequence homology in Bcl-2 homology (BH)1 domains and function to promote or prevent apoptosis. Antiapoptotic proteins such as Bcl-2 and Bcl-xL show homology in four BH domains (BH1 to −4). Proapoptotic proteins can be grouped into “multidomain” and “BH3-only” subfamilies. Multidomain proapoptotic proteins such as Bax and Bak display homology in BH domains 1–3, whereas BH3-only proteins such as Bid and Bim are similar structurally to multidomain family members, but sequence similarity is limited to only the BH3 domain. The current model for how Bcl-2 family proteins regulate apoptosis involves three sequential processes: (i) BH3-only proteins are activated by various death signals; (ii) the active BH3-only proteins then either activate multidomain proapoptotic proteins or inhibit antiapoptotic proteins or both; and (iii) unless inhibited by antiapoptotic Bcl-2 proteins, activated multidomain proapoptotic proteins form oligomers in the mitochondrial outer membrane that release proapoptotic proteins such as cytochrome c and Smac/DIABLO from the mitochondrial intermembrane space. The released proteins trigger activation of the caspases and nucleases that eventually dismantle the cell (1–4).
The interaction between Bcl-2 family proteins is obviously important for coupling apoptotic signals to the alteration of mitochondrial membrane permeability. Three-dimensional structures of monomeric antiapoptotic proteins such as Bcl-xL, Bcl-2, Bcl-w, and CED-9 as well as proapoptotic proteins such as Bax and Bid have been determined (5–12). A hydrophobic surface groove common to the structures of all of the antiapoptotic proteins is encircled by five helices (helices 2–5 and 8) and connecting loops. The equivalent hydrophobic groove in the multidomain proapoptotic protein Bax is occluded by its C-terminal hydrophobic helix (helix 9). Although the overall structure is similar, the BH3-only protein Bid does not contain this hydrophobic groove. Further, a complementary hydrophobic surface that would interact with the hydrophobic groove is not obvious in the structures of monomeric Bcl-2 family proteins, making modeling of a dimeric complex difficult (13). However, one possible interface for the heterodimer between anti- and proapoptotic proteins has been deduced from structures of Bcl-xL in complex with a BH3 domain peptide from a proapoptotic protein such as Bak, Bad, or Bim (14–16). In all of these complex structures, the hydrophobic groove of Bcl-xL binds the peptide as an α-helix.
Modeling a heterodimer between an anti- and a proapoptotic protein based on these complex structures provided explanations for some early mutagenesis data. For example, replacement of glycine 145 with alanine (G145A) in the BH1 domain of Bcl-2 and equivalent G138 mutations in Bcl-xL both diminish heterodimerization with Bax and antiapoptotic function (17–21). In both cases, the glycine replaced is located in the bottom of the hydrophobic groove that has been proposed to constitute one binding interface. Conversely, some mutations in the BH3 domain of Bax, Bad, Bid, and Bim abolish the binding of these proteins to Bcl-2 or Bcl-xL as well as their proapoptotic activity (22–29). These mutations usually alter the hydrophobic face of the BH3 helix predicted to make critical contacts with the hydrophobic groove of Bcl-xL based on the peptide-bound Bcl-xL structures. Thus, a model for heterodimerization between pro-and antiapoptotic proteins exists that satisfactorily accounts for the existing structural and functional data.
Much less is known about homodimerization of Bcl-2 family proteins. Most single point mutations created in several Bcl-2 family proteins have failed to disrupt homodimer formation (17, 18). Only Jeong et al. (21) reported recently that G138A mutation in Bcl-xL reduced the homodimer formation. More dramatic deletion mutagenesis has been used to disrupt Bcl-2 homodimerization (30, 31), but the principle caveat of this approach is that the overall structure of the protein is probably being altered, making molecular interpretation of the mutagenesis results difficult. It is surprising that the structure of the homodimer remains unknown, since homodimers have been reported as the major form of intracellular Bcl-2 family proteins for more than 10 years (e.g. see Refs. 17 and 21). To determine the mechanisms by which Bcl-2 family proteins interact and function under normal as well as apoptotic conditions, it is essential to determine how both homo- and heterodimerization are regulated. As an essential step in understanding these interactions, we have mapped the binding surfaces involved for two functionally well characterized molecules, Bcl-2 and Bax. To identify binding surfaces, we performed cross-linking experiments using both purified and in vitro synthesized proteins, each having a cross-linker attached to a single site. Our physical map for the Bcl-2 homodimer interface confirms a known binding surface, reveals an additional binding surface, and suggests a mechanism by which homodimers can form higher oligomers as well as how homodimers can dissociate to form heterodimers with activated Bax.
EXPERIMENTAL PROCEDURES
Plasmids and Proteins
To construct Bcl-2 mutants, we used a plasmid that contains the coding region of full-length Bcl-2 protein in the vector pSPUTK (32). The lysine-null Bcl-2 mutant (K0) was generated by mutating all four lysine codons in the Bcl-2 coding region to codons for arginine. Other Bcl-2 mutants with a single lysine codon at a particular position were generated by mutating the corresponding codon of the K0 mutant to the lysine codon and are designated here as K followed by a number that indicates the codon position in the sequence. For example, the mutant Bcl-2 K108 has a single lysine codon at position 108 of the Bcl-2 coding sequence. The last 22 codons of the bcl-2 gene were then replaced with a single asparagine codon to create Bcl-2ΔTM mutants with a single lysine at the desired location. These mutants are named similarly; thus, the C-terminal deletion of Bcl-2 Lys108 is designated Bcl-2ΔTM K108.
All mutagenesis was done using appropriate primers and an overlapping PCR-based method (33). The sequence of all plasmids was verified by DNA sequencing. For transfection of MCF-7 cells, the coding region of Bcl-2 K0 or other single lysine mutants was shuttled from the pSPUTK-based plasmid to the vector pRc/CMV (Invitrogen). Details of all plasmid preparations will be provided upon request.
Expression and purification of human Bcl-2 protein with a sequence of Leu-Gln-His6, replacing the C-terminal 22 residues (designated 6H-Bcl-2ΔTM) were carried out as described (34). A similar protocol was used to express and purify the 6H-Bcl-2ΔTM protein with mutation G145A, C158A, or C158A/P168C (designated 6H-Bcl-2ΔTM G145A, C0 or C168, respectively). The purified proteins were stored at −80 °C in small 240 μm aliquots in a buffer containing 50 mm Tris-HCl (pH 8.5), 50 mm NaCl, 2 mm EDTA, 5 mm dithiothreitol, and 10% (v/v) glycerol. Expression and purification of recombinant Bax protein were described previously (35).
Prevention of Apoptosis by Bcl-2 K0 in MCF-7 Cells
MCF-7 cell culture, transfection, adriamycin treatment, and focal adhesion kinase (FAK) and poly(ADP-ribose) polymerase (PARP) cleavage assays were conducted as described (36, 37). Briefly, MCF-7 cells were grown in α-minimal essential medium supplemented with 10% fetal bovine serum. The pRc/CMV vector, or a pRc/CMV plasmid encoding either wild type Bcl-2 (WT Bcl-2) or Bcl-2 K0 was used to transfect MCF-7 cells, and clones that stably express Bcl-2 proteins were selected. The expression of endogenous Bcl-2 in the MCF-7 cells was reduced by growing the cells without estrogen (α-minimal essential medium without phenol red, supplemented with charcoal-filtered fetal bovine serum) for 6 days prior to adriamycin treatment. After the addition of 10 μm adriamycin, the cells were incubated for 48 h and then harvested for FAK and PARP cleavage assays. 10 million cells were lysed in 1.5 ml of ice-cold lysis buffer (37) by passing through a 26-gauge needle four times. Cellular debris and nuclei were removed by centrifugation at 13,000 rpm for 10 min in a microcentrifuge. Aliquots of the supernatant corresponding to equal cell numbers were analyzed by SDS-PAGE followed by immunoblotting using antibodies specific for Bcl-2 (36), FAK (Pharmingen), or PARP (Biomol).
Preparation of ANB-labeled Bcl-2 Proteins
The Nε-(5-azido-2-nitro-benzoyl)-Lys-tRNALys (εANB-Lys-tRNALys) was prepared as described (38, 39). RNAs encoding the Bcl-2 mutants were prepared from the corresponding pSPUTK-based plasmids using SP6 RNA polymerase as described (40). Translations of these RNAs were carried out in a rabbit reticulocyte lysate-based in vitro translation system with the εANB-Lys-tRNALys as described (41), except that the reactions contained 110 mm KOAc and were conducted for 50 min at 26 °C.
Dimerization, Photocross-linking, and Characterization of Photoadducts
For photocross-linking of 5-azido-2-nitrobenzol (ANB)-labeled Bcl-2ΔTM and 6H-Bcl-2ΔTM, purified 6H-Bcl-2ΔTM was added to 10 μl of in vitro translated ANB-Bcl-2ΔTM to a final concentration of 2.5 μm. The sample was shielded from light and incubated at 4 °C for 1 h with occasional mixing to allow the proteins to interact. The sample was then photolyzed for 10 min at 0 °C as before (42). A 2-μl aliquot was removed and analyzed directly by SDS-PAGE (as the total). To the remaining sample, 200 μl of Ni2+-nitrilotriacetic acid-agarose (Qiagen; 3% (v/v) suspension in buffer A: 20 mm HEPES (pH 7.5), 100 mm KOAc, 5 mm Mg(OAc)2, 10% (v/v) glycerol, 0.5% (v/v) Triton X-100, and 5 mm imidazole) were added and mixed at 4 °C for 1.5 h to allow binding of 6H-Bcl-2ΔTM and its adducts to Ni2+-beads. The beads were then washed three times with 400 μl of buffer A and one time with 400 μl of phosphate buffer (140 mm NaCl, 2.7 mm KCl, 10 mm Na2HPO4, and 1.8 mm KH2PO4 (pH 7.4)). The proteins bound to Ni2+-beads (as the Ni2+-bound) were eluted at 65 °C for 30 min with a reducing SDS-sample buffer as before (40). The samples were loaded onto a 15% gel for SDS-PAGE. The radioactivity in dried gels was detected using an Amersham Biosciences PSI PhosphorImager.
For photocross-linking of ANB-labeled Bcl-2ΔTM mutants to Bax, purified Bax and Triton X-100 were added to 10 μl of in vitro translated ANB-Bcl-2ΔTM to final concentrations of 0.5 μm and 0.25% (v/v), respectively. After binding and photolysis as described above, Bax was immunoprecipitated from the samples using Bax-specific antiserum as before (36). The immunoprecipitates were analyzed by SDS-PAGE and phosphorimaging as described above.
For photocross-linking of ANB-labeled Bax to Bcl-2ΔTM dimers, 32 μm 6H-Bcl-2ΔTM C168 proteins were first cross-linked with 167 μm homobifunctional sulfhydryl-reactive cross-linker bis-maleimidohexane (BMH) (Pierce) in 120 μl of 51 mm Na2HPO4, 4 mm citric acid, and 10 mm EDTA (pH 7.4) at 25 °C for 1 h in the dark. The reaction was stopped by a 10-min incubation with 50 mm μm β-mercaptoethanol. Control reactions were done in parallel using Me2SO instead of BMH or 6H-Bcl-2ΔTM C0 instead of C168 protein. After a brief centrifugation in a microcentrifuge to remove particulate material, the sample was dialyzed three times in 330 ml of 25 mm HEPES (pH 7.5), 110 mm KOAc, 1 mm Mg(OAc)2, 10 mm EDTA, and 25% (v/v) glycerol at 4 °C for 2 h. Then 3 μl of the dialyzed sample were incubated with 10 μl of in vitro translated ANB-Bax in the presence of 0.25% (v/v) Triton X-100, photolyzed, and analyzed as described above.
For Bax inhibition of photocross-linking of ANB-labeled Bcl-2ΔTM to 6H-Bcl-2ΔTM, 1 μl of 210, 42, 4.2, or 0 μm 6H-Bcl-2ΔTM and 10 μl of 1 μm Bax were added to 10 μl of in vitro translated ANB-Bcl-2ΔTM to bring the molar ratio of 6H-Bcl-2ΔTM and Bax to 10, 2, 0.2, or 0, respectively. After the addition of 0.25% (v/v) Triton X-100, all samples were processed as described above.
Gel Filtration Chromatography of Bcl-2 Protein
100 μl of 40 μm 6H-Bcl-2ΔTM protein in buffer B (50 mm Tris-HCl (pH 8.5), 200 mm NaCl, 2 mm EDTA, and 0.5% (v/v) glycerol) were incubated at 25 °C for 1 h before injection into a Superdex 200 HR10/30 column in an AKTA-FPLC (Amersham Biosciences) in buffer B with a flow rate of 0.25 ml/min at 25 °C. Protein peaks (determined by A280) were collected, and 100 μl of oligomer fractions and 7 μl of the monomer fraction were precipitated with Cl3CCOOH and analyzed by 15% SDS-PAGE and silver staining. Protein standards and 2000-kDa dextran molecules were used to calibrate the column.
RESULTS
Experimental Design
The Bcl-2 homodimer interface was mapped using site-specific photocross-linking. RNAs encoding a mutant Bcl-2 containing a lysine codon at a single specific location (Fig. 1A) were translated in vitro in the presence of εANB-Lys-tRNALys and [35S]Met (to facilitate product detection). The εANB-Lys-tRNALys is a functional aminoacyl-tRNA analog that recognizes the lysine codon in RNA and incorporates a lysine analog with a photocross-linker covalently attached to the lysine side chain into the polypeptide being synthesized in vitro. In this way, the 35S-labeled Bcl-2 proteins, each with a photocross-linker attached to a specific location, were produced.
Fig. 1. Sequence and function of Bcl-2 mutants.
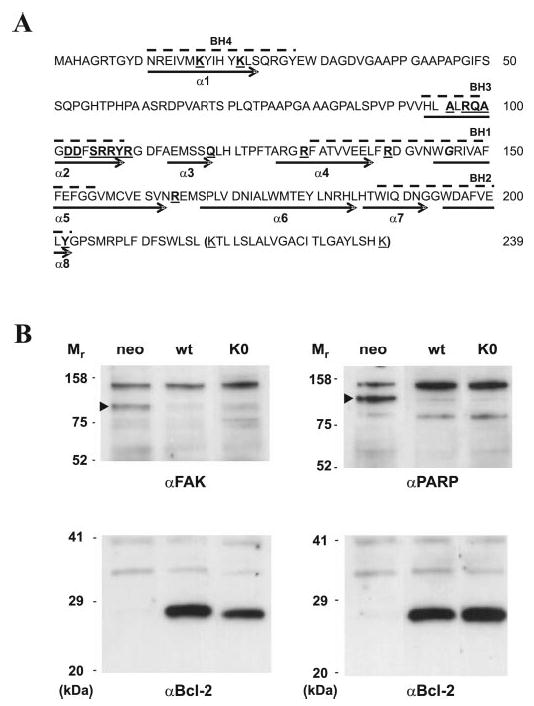
A, the Bcl-2 sequence is shown with BH domain sequences highlighted by dashed lines above and helices by arrows below. All four lysines (underlined) were changed to arginines to create Bcl-2 K0. The last 22 residues in parentheses were deleted in all Bcl-2ΔTM mutants. The residues replaced individually by ANB-Lys are in boldface type and underlined. The Gly145 is in boldface italic type. B, adriamycin-treated MCF-7 cells that stably express exogenous Bcl-2 WT and Bcl-2 K0 as well as trans-fect with the empty vector (neo) were lysed. Aliquots of lysate from equal numbers of cells were analyzed by SDS-PAGE followed by immunoblotting using antibodies specific for FAK, PARP, or Bcl-2, as indicated below each blot. The caspase cleavage product of FAK or PARP is indicated by an arrowhead beside the relevant bands. The relative molecular masses (Mr) of standards are indicated to the left of each panel.
The ANB- and 35S-labeled Bcl-2 was then incubated with nonradioactive His-tagged recombinant Bcl-2 to allow dimerization, and upon exposing the sample to UV light, a nitrene was generated from the ANB moiety (43). The nitrene is a powerful electrophile that rapidly reacts with any nearby heteroatoms possessing nonbonding electron pairs (S, O, N, etc.), double bonds, or even C–H bonds, including such heteroatoms or bonds from the bound His-tagged Bcl-2. The presence of each Bcl-2 in a covalently linked dimer photoadduct was shown by its affinity to Ni2+-chelating beads and by the detection of 35S in the photoadduct. Since the ANB probe is located at a single specific site, the detection of a His-tagged Bcl-2-specific photoadduct demonstrates that the ANB-labeled site in the Bcl-2 synthesized in vitro is located at or adjacent to the Bcl-2 dimerization interface. Therefore, sites of contact or close approach can be mapped for the two proteins by altering the location of the photoreactive lysine. The precision of this mapping approach will be limited by the long flexible lysine side chain that links the ANB probe to the protein surface site. The distance between the nitrene and the Cα of the lysine is roughly 12 Å if the entire side chain assumes a fully extended, rigid conformation. However, most photocross-linking reactions will occur at a shorter distance because of the randomness of probe motion and photochemical reaction. Therefore, the maximum uncertainty for the location of a site in the interface map will be much less than 12 Å.
Unlike soluble Bcl-2 family proteins such as Bcl-w, the hydrophobic tail of Bcl-2 expressed in cells is not found in the hydrophobic binding groove on the protein surface. Instead, the protein is constitutively anchored to mitochondrial, ER, and nuclear membranes with the C-terminal hydrophobic helix inserted into the lipid bilayer and the rest of the protein exposed to the cytoplasm (29). Thus, the cytosolic domain of Bcl-2 is most likely involved in homodimerization. Therefore, as a first step toward determining the native Bcl-2 homodimer interface, we examined the interaction of the cytosolic domains of Bcl-2 using the Bcl-2ΔTM proteins described above.
Since a single Lys residue must be positioned at a specific site in Bcl-2 to achieve specific ANB-labeling, we substituted all four Lys residues in wild type Bcl-2 with Arg, generating a Lys-null Bcl-2 protein (Fig. 1A, Bcl-2 K0). To ensure that the substitution did not inactivate the protein, the antiapoptotic activity of Bcl-2 K0 was compared with that of Bcl-2 WT after constructing stable MCF-7 cell lines expressing similar amounts of each of the proteins. MCF-7 cells were used for these studies, because the endogenous Bcl-2 expression can be reduced by growing the cells in estrogen-free medium, thereby sensitizing the cells to apoptosis and allowing measurement of the activity of the exogenous Bcl-2 mutants specifically (compare expression of the endogenous protein (neo) with the exogenous proteins (WT and K0) in Fig. 1B). Both Bcl-2 and Bcl-2 K0 inhibited the cleavage of the well characterized caspase 3/7 substrates FAK and PARP equally, confirming that Bcl-2 K0 retains close to wild type activity in MCF-7 cells. Consistent with these observations, both proteins also inhibited cytochrome c release from mitochondria (data not shown). Therefore, the Lys-null Bcl-2 is fully functional in MCF-7 cells and hence can be used as the template for constructing the single Lys Bcl-2 mutants.
The selection of the other positions in Bcl-2 that were suitable for incorporating a photocross-linker to map the interface was based on the result of a modeling experiment using the Bcl-2 core structure (6). The incorporation of an ANB-Lys at these positions would have minimal or no effect on the rest of Bcl-2 structure. The only exception is that an ANB-Lys at position 96 might clash with residues from the C terminus. But since these C-terminal residues in the Bcl-2 structure are in a flexible region, the ANB-Lys at position 96 may be accommodated without having a dramatic effect on the overall structure of Bcl-2. Unlike lysine, the side chain of ANB-lysine is neutral and therefore does not leave a charge in the hydrophobic groove when positioned within the groove. However, the ANB moiety is relatively polar and may reduce the hydrophobicity of the groove somewhat. Nevertheless, in most locations, it appears that the ANB-lysine does not inhibit Bcl-2 dimer formation (see Fig. 2) (data not shown). Furthermore, previous mutagenesis experiments have failed to identify any single point mutations that disrupt Bcl-2 homodimers, suggesting that the homodimer interface may be extensive enough that many single site alterations can be tolerated individually. Consistent with the prediction from our modeling study and previous mutagenesis data, most of the single lysine Bcl-2 mutants that we generated retained as least some antiapoptotic activity when assayed in the MCF-7 cells (data not shown). Since these mutants containing a charged lysine residue instead of the neutral ANB-lysine continue to inhibit apoptosis, the reduction in hydrophobicity due to the lysine does not interfere with Bcl-2 interactions important for function.
Fig. 2. Mapping Bcl-2 homodimerization interface using site-specific photocross-linking.
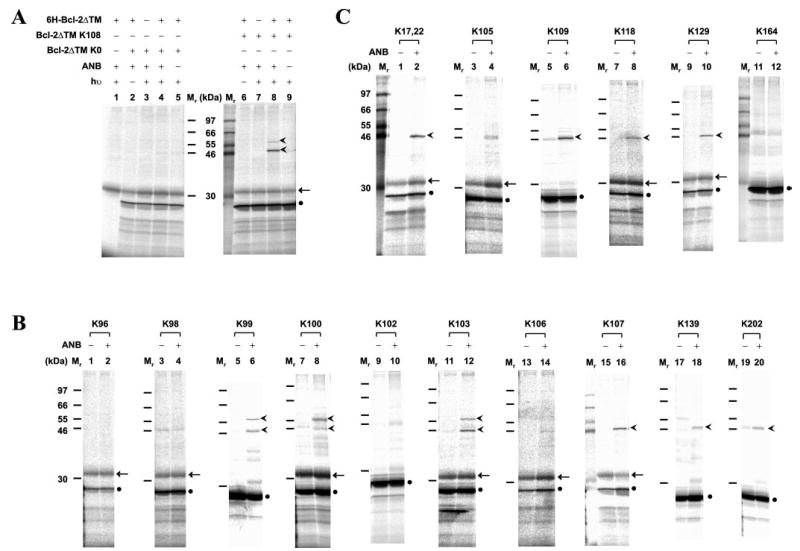
A, photocross-linking of in vitro synthesized Bcl-2ΔTM with a single ANB probe attached to Lys108 to His-tagged Bcl-2ΔTM. After cross-linking, the fraction that bound to Ni2+-chelating resin was eluted, and photoadducts were separated by SDS-PAGE and detected by phosphorimaging (indicated by the arrowheads adjacent to lane 8). The adducts were not detected in control reactions lacking RNA encoding of Bcl-2ΔTM K108, UV irradiation (hν), 6H-Bcl-2ΔTM protein, or εANB-Lys-tRNALys (ANB), as shown in lanes 1, 6, 7, and 9, respectively. The adducts were also absent when RNA encoding Bcl-2ΔTM K0 was used (lanes 2–5). Background detected in some or all of the control reactions included in vitro synthesized Bcl-2ΔTM and an unidentified reticulocyte protein, indicated by a filled circle and arrow, respectively. The Mr of standards is indicated between the two panels. B and C, photocross-linking of His-tagged Bcl-2ΔTM proteins to in vitro synthesized Bcl-2ΔTM mutant proteins, each with a single ANB probe attached to a Lys residue within the acceptor surface or the donor surface, respectively (defined in Fig. 3). The symbols used to indicate Bcl-2ΔTM photoadducts, background proteins and Mr of standards are defined in A.
Mapping the Bcl-2 Homodimer Interface by Site-specific Photocross-linking
Several previous studies suggest that the helix 2 of Bcl-2 family proteins containing the BH3 sequence is likely to be involved in homo- and heterodimerization (6, 14, 44). To examine the possibility that this region is in the homodimer interface Bcl-2ΔTM K108, a mutant with a single ANB-Lys replacing the last residue of helix 2 (Tyr108) was used for photocross-linking to purified His-tagged Bcl-2ΔTM. Two specific photoadducts were detected between the proteins (Fig. 2A, lane 8, arrowheads). In control experiments (Fig. 2A), neither product was formed when UV irradiation (lane 6), εANB-Lys-tRNALys (lane 9), RNA encoding Bcl-2ΔTM K108 (lane 1), or His-tagged Bcl-2ΔTM (lane 7) was omitted. Both adducts were preferentially bound to Ni2+-chelating beads compared with other products seen in the total photocross-linking reaction (data not shown). The formation of both photoadducts was via the ANB probe attached to Lys108, since neither was detected when RNA encoding the Lys-null Bcl-2ΔTM K0 was used (Fig. 2A, lanes 2–5). The relative molecular mass (Mr) of both photoadducts is close to 50 kDa, the Mr predicted for the Bcl-2ΔTM homodimer. The difference in the apparent Mr between the two photoadducts most likely resulted from the ANB probe at Lys108 of in vitro synthesized Bcl-2ΔTM reacting with two different sites in the His-tagged Bcl-2ΔTM, since precedents of this phenomenon have been reported previously (45, 46). Among the other radioactive proteins detected, the most abundant one was the in vitro synthesized Bcl-2ΔTM protein (Fig. 2A, filled circle). The other one was a translation product from a reticulocyte mRNA, since it also appeared when εANB-Lys-tRNALys, UV light, or both Bcl-2ΔTM K108 and K0 RNAs were omitted (Fig. 2A, arrow).
Three lines of evidence from this experiment suggest that adduct formation is due to position 108 being in the interface of a Bcl-2 homodimer. (i) The ANB probe at Lys108 of one Bcl-2 molecule was able to react specifically with the other Bcl-2 molecule in solutions containing ~2.5 μm (Fig. 2A) to ~0.25 μm (data not shown) total Bcl-2. (ii) Cross-linking between the two Bcl-2 molecules was observed in reactions containing a high concentration of reticulocyte lysate proteins, including ~150 μm globin. (iii) The cross-linking occurred via the ANB-derived reactive nitrene which has a very short lifetime (nanoseconds) and covalently links to the side chain of Lys108.
To map the hydrophobic groove of Bcl-2 previously predicted to be involved in Bcl-2 dimerization (parts of helices 2, 3, 4, and 8 as well as the loops between helices 2 and 3 and helices 4 and 5 including the BH1–3 domain sequences), individual mutants were generated with the ANB probe at each of the residues 96, 98, 99, 100, 102, 103, 106, 107, 139, and 202. These proteins were used to perform photocross-linking experiments with His-tagged Bcl-2ΔTM protein as above. Although a complete set of controls was included in each experiment, for clarity only reactions with and without εANB-Lys-tRNALys are presented in Fig. 2B. Specific photoadducts were detected when the probe was located at position 99, 100, 103, 107, 139, or 202 but not for other positions (Fig. 2B). These results provide direct physical evidence that the hydrophobic groove of Bcl-2 is involved in Bcl-2 homodimerization. They also demonstrate that the outcome of the photocross-linking experiments is very sensitive to the location of the ANB probe, as expected.
Additional mutants were assayed to determine if other parts of the Bcl-2 surface including the BH4 domain sequence or helix 1 are involved in homodimerization. We found that positions 17 or 22, 109, 118, and 129 are near the dimer interface, since ANB at these positions generated specific photoadducts containing the two Bcl-2ΔTM proteins, whereas ANB at positions 105 and 164 did not result in significant adduct formation compared with the controls (Fig. 2C) (data not shown). Thus, the homodimerization interface is not limited to the hydrophobic groove.
Modeling the Bcl-2 Homodimer Interface
The photocross-linking data in Fig. 2 were used to model which residues would be in the Bcl-2 homodimer interface, based on the NMR structure of the Bcl-2 core (6). Mapping these residues on the NMR structure suggests that there are two distinct binding surfaces in the homodimer that we refer to as acceptor and donor surfaces (Fig. 3, A and B, respectively). As shown in Fig. 3A, the acceptor surface is defined by photoadducts obtained when the cross-linker was positioned at the residues that are colored red. This surface overlaps extensively with the hydrophobic groove, including the BH1–3 domain sequences that was previously implicated in Bcl-2 family protein dimerization. The glycine at position 145 in the middle of the acceptor surface abolishes hetero- but not homodimerization when mutated to alanine (Fig. 3A, green). The donor surface is oriented ~90° to the acceptor surface, and the positions resulting in photoadducts are colored blue. This surface begins on one side of helix 2, a result consistent with the assignment of this helix as the correspondent to the ligand helix that binds to the hydrophobic acceptor groove on Bcl-xL (14–16). However, our map suggests that the donor surface extends beyond helix 2 into helices 1, 3, and 4, overlapping the BH3 and −4 domain sequences. Unlike previous models, the donor surface we identified is exposed in the NMR structure of Bcl-2 core and does not require a structural rearrangement proposed previously to expose helix 2 such that it can form the entire binding surface. Position 145 is not part of the donor surface identified here.
Fig. 3. Modeling Bcl-2 homodimerization interface.
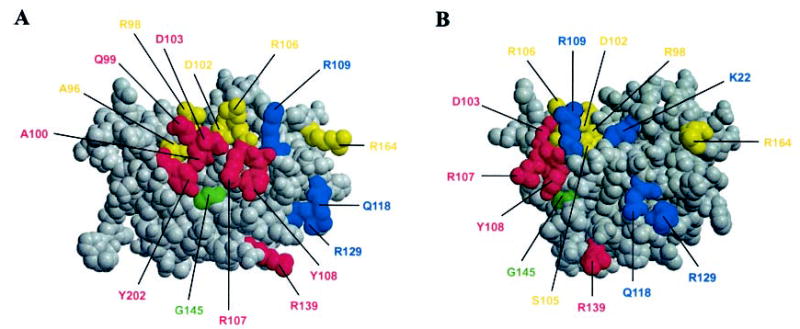
The Bcl-2 dimerization interface identified from the photocross-linking data shown in Fig. 2 modeled as two distinct surfaces, an acceptor surface (A) and a donor surface (B). The model is based on the previously determined Bcl-2 core structure (6). Residues close to the acceptor and donor surfaces are colored in red and blue, respectively. Residues at positions that did not result in photoadducts are yellow. The G145 in the middle of the acceptor surface is green. The flexible loop consisting of residues 25–91 is omitted in the model, since its structure is elusive (6).
Bcl-2 Can Form Oligomers
One prediction of our model is that Bcl-2 may form homo-oligomers, since each Bcl-2 protein has more than one binding surface, and utilization of each need not be mutually exclusive. Consistent with this prediction, gel filtration chromatography demonstrated that although His-tagged Bcl-2ΔTM proteins eluted from a Superdex 200 column mainly as monomers, dimers and oligomers were also evident (Fig. 4). The actual ratio of dimer and oligomer to monomer in the sample is probably higher than that shown by the chromatogram (Fig. 4A), since some dimers and oligomers could dissociate into monomers during chromatography. The elution profile was not changed when the Bcl-2ΔTM proteins were incubated in a solution containing reticulocyte lysate and ATP (i.e. the same solution used for in vitro translation except for the radioactive amino acid and aminoacyl tRNA) prior to gel filtration (data not shown), indicating that at least a fraction of Bcl-2 remains in complexes even in the presence of chaperones and ATP.
Fig. 4. Oligomerization of Bcl-2.
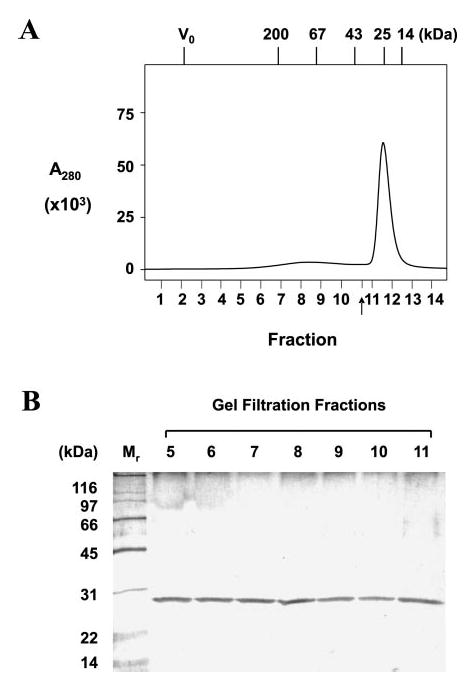
A, oligomerization of purified His-tagged Bcl-2ΔTM protein examined by gel filtration chromatography. The A280 was monitored for each eluted fraction and plotted. The arrow indicates a fraction that was not collected for analysis. The void volume for the column and elution positions for protein standards are indicated at the top of the plot by V0 and Mr, respectively. The predicted Mr for the Bcl-2 monomer, dimer, trimer, and tetramer are about 25, 50, 75, and 100 kDa, respectively. B, the proteins in fractions 5–11 (shown in A) examined by SDS-PAGE and silver staining. His-tagged Bcl-2ΔTM protein was detected in all fractions. 15-Fold less sample was loaded for fraction 11 than for other fractions.
Activated Bax Interacts with Both Bcl-2 Monomers and Dimers
Taken together, the results above suggest that Bcl-2 proteins can form both dimers and oligomers via homotypic interactions that use two distinct binding surfaces. Given these interactions, how would a proapoptotic Bcl-2 family member, such as Bax, interact with Bcl-2? To determine which form(s) of Bcl-2 (monomer, dimer, or both) can be bound by Bax, we used in vitro synthesized Bcl-2ΔTM that was ANB-labeled at Lys107 and purified recombinant Bax protein to perform additional photocross-linking experiments. The Bax protein was a full-length protein expressed in bacteria and purified under conditions that kept the protein in a native conformation similar to that of Bax in healthy cells (35). In healthy cells, Bax proteins reside in the cytosol and do not dimerize with each other or with other Bcl-2 family members. Upon induction of apoptosis, Bax proteins are switched to an active conformation and translocate to mitochondrial and ER membranes, where they form homo-oligomers or heterodimerize with other Bcl-2 family members. Active full-length Bax is likely to interact with the cytosolic domain of membrane-bound Bcl-2 to form a heterodimer, since the C-terminal tail of Bcl-2 is buried in the lipid bilayer and is therefore not available for the initial interaction.
As shown in Fig. 5A, photoadducts between the Bcl-2ΔTM protein and Bax protein were detected. The size of the adducts is most consistent with the formation of a heterodimer. Furthermore, the cross-linked proteins were recognized by the antibody specific for Bax (Fig. 5A, lane 4, arrowheads) and were formed only when Bax protein, UV light, and εANB-Lys-tRNALys were all present (Fig. 5A, lane 4 versus lanes 1–3) and Bcl-2ΔTM RNA was used to program the in vitro translation reaction (data not shown). Triton X-100 (0.25%) was required for the formation of the cross-linked Bcl-2/Bax heterodimer (Fig. 5A, lane 4 versus lane 5), consistent with previous findings indicating that certain nonionic detergents can switch Bax to a dimerization-competent conformation (47).
Fig. 5. Interaction of Bax with both Bcl-2 monomer and dimer.

A, photocross-linking of in vitro synthesized Bcl-2ΔTM protein with a single ANB attached at Lys107 to Bax protein detected by immunoprecipitation using antibodies specific for Bax. The photoadducts were detected in the reaction shown in lane 4 (indicated by arrowheads) but not in control reactions in which εANB-Lys-tRNALys, UV irradiation, Bax protein, or Triton X-100 was omitted, shown in lane 1, 2, 3, or 5, respectively. The in vitro synthesized Bcl-2ΔTM protein was indicated by a filled circle. B, BMH cross-linking of Bcl-2ΔTM proteins with single Cys at position 168 detected by SDS-PAGE and Coomassie staining. The BMH cross-linked Bcl-2ΔTM dimer and monomer are indicated by an arrowhead and a filled circle, respectively. C, photocross-linking of BMH cross-linked His-tagged Bcl-2ΔTM proteins to in vitro synthesized, ANB-labeled Bax protein. The three major photoadducts detected in Ni2+-bound fractions after SDS-PAGE by phosphorimaging were identified as follows: adducts of Bax with Bcl-2ΔTM monomer (bracket), BMH cross-linked Bcl-2ΔTM dimer (arrow), and BMH cross-linked Bcl-2ΔTM/reticulocyte protein (oval). These adducts were not detected in the control reaction in which the BMH cross-linked Bcl-2ΔTM protein, UV irradiation, or εANB-Lys-tRNALys was omitted, shown in lane 1, 2, or 3, respectively. The in vitro synthesized Bax protein is indicated by a filled circle.
Thus, activated Bax can interact directly with Bcl-2 monomers to form Bax/Bcl-2 heterodimers. Therefore, it is important to determine whether or not the activated Bax protein can also interact with Bcl-2 homodimers, since most of the Bcl-2 proteins isolated from mammalian cells appear to be in homodimer or oligomer state (48, 49). To determine whether Bax can interact with the Bcl-2 homodimer directly, we used the thio-specific chemical cross-linker BMH to cross-link two His-tagged Bcl-2ΔTM proteins, each containing a single Cys at position 168. BMH will covalently link two Bcl-2ΔTM molecules together if the two Cys residues are about 16 Å away, a distance possible for these residues in a Bcl-2 homodimer formed via the two different binding surfaces we identified above. We detected BMH-cross-linked Bcl-2ΔTM dimer only in the presence of BMH, not the solvent control (− BMH) (Fig. 5B, lanes 3 and 4, arrowhead). When a Cys-null Bcl-2ΔTM protein (C0) was used in parallel experiments, no cross-linked Bcl-2ΔTM proteins were detected, verifying that the BMH cross-linking reaction is specific (Fig. 5B, lanes 1 and 2).
To test whether Bax can cross-link to the Bcl-2 homodimers, we incubated the sample from the BMH cross-linking reaction with in vitro synthesized Bax protein with ANB probes attached to the endogenous Lys residues. Based on the migration position, one of the photoadducts detected in the Ni2+-bound fraction was due to cross-linking between ANB-labeled Bax and the BMH-linked 6H-Bcl-2ΔTM dimer (Fig. 5C, lane 4, arrow). Photoadducts between Bax and Bcl-2ΔTM monomers were also detected (Fig. 5C, lane 4, bracket). These bands are not Bax homodimers because Bax homodimers do not bind to the Ni2+ resin (data not shown), similar to the Bax monomers (Fig. 5C, lane 1, filled circle). The photoadduct indicated by an oval is probably a cross-linking product of Bax oligomers with 6H-Bcl-2ΔTM. It is also possible that the complex includes one or more proteins from the reticulocyte lysate. Together, these results demonstrate that Bcl-2 monomers, dimers, and possibly larger oligomers bind activated Bax, suggesting that any of these may neutralize Bax apoptotic activity.
Bax Binds to the Acceptor Face of Bcl-2
Our data support a model in which a Bcl-2 homodimer is formed by interaction of the acceptor surface of one molecule with the donor surface of another molecule (Fig. 3). In principle, Bcl-2/Bax heterodimer interface can be mapped in the same way. However, of the various ANB-Bcl-2ΔTM mutants that we have tested, only ANB-Bcl-2ΔTM K107 cross-linked to recombinant Bax (Fig. 5A) (data not shown). Since position 107 is in the acceptor surface of Bcl-2 (Fig. 3A), this result strongly suggests that Bax binds the acceptor surface of Bcl-2. Consistent with Bax binding to only the acceptor surface, a Bcl-2 mutation (G145A) known to abrogate the interaction between Bcl-2 and Bax (17) is located in the middle of the acceptor surface (Fig. 3A). To verify that an interaction between Bcl-2 G145A and Bax is not detected by photocross-linking, we assayed cross-linking between in vitro synthesized Bcl-2ΔTM G145A with ANB attached to Lys107 and Bax. As shown in Fig. 6A, cross-linking was eliminated by the mutation, substantiating the notion that the acceptor surface in Bcl-2 serves as the binding site for Bax. In addition, since the G145A mutation did not affect the cross-linking of two Bcl-2ΔTM proteins via ANB-Lys107 (Fig. 6B, lanes 1–3), even when the mutation was created in both Bcl-2ΔTM proteins (Fig. 6B, lanes 4 –7), the mutation did not abolish homodimerization. Consistent with our finding, Bcl-2 G145A was shown to retain homodimerization and certain antiapoptotic functions in other systems (17, 20). Therefore, the Bcl-2 homodimer interface must be different than that of Bcl-2/Bax heterodimer.
Fig. 6. Effect of Bcl-2 G145A mutation on photocross-linking of Bcl-2/Bax heterodimer and Bcl-2 homodimer.
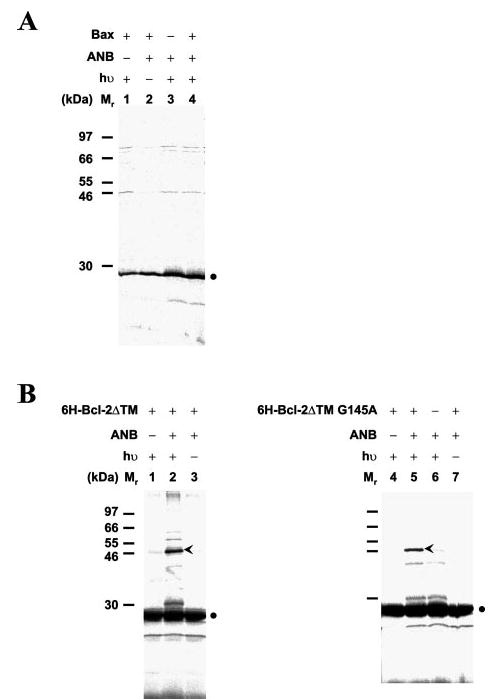
A, photocross-linking of in vitro synthesized Bcl-2ΔTM protein with the G145A mutation and a single ANB attached to Lys107 to Bax protein. When the Bcl-2ΔTM protein contained a G145A mutation, the Bcl-2/Bax photoadduct was not detected after immunoprecipitation using antibodies specific for Bax, although the mutant Bcl-2 protein was produced (filled circle). B, photocross-linking of the same protein with His-tagged Bcl-2ΔTM with (right panel) or without (left panel) the G145A mutation. The Bcl-2/Bcl-2 photoadducts were detected in the Ni2+-bound fraction from both reactions (lanes 2 and 5, arrowheads). These adducts were absent in control reactions shown in other lanes.
Inhibition of Bcl-2 Homodimerization by Activated Bax
The fact that Bax can bind to a Bcl-2 homodimer that is held together with a covalent cross-link demonstrates that the homodimer contains an accessible binding site for Bax, as predicted from our model. However, if binding of Bax to Bcl-2 impacts Bcl-2 homodimerization, as predicted by some models (50), such an effect would not be revealed by this assay. To determine whether or not Bax binding to Bcl-2 inhibits Bcl-2 homodimerization, photocross-linking experiments were performed with in vitro synthesized, ANB-labeled Bcl-2ΔTM K107 and His-tagged Bcl-2ΔTM in the presence of increasing amounts of detergent-activated Bax. As shown in Fig. 7A, lanes 1–4, the cross-linking of two Bcl-2ΔTM proteins was inhibited by the Bax in a concentration-dependent manner. Moreover, the amount of photoadduct between Bax and Bcl-2ΔTM correlated positively with the relative amount of Bax added to the reaction (Fig. 7B, lanes 1–4). Taken together with the data presented in Fig. 5, this result suggests that Bax/Bcl-2 interaction reduces the amount of Bcl-2 homodimer by either binding to the Bcl-2 monomer or binding to and disrupting the Bcl-2 dimer or both.
Fig. 7. Inhibition of Bcl-2 homodimerization by Bax.
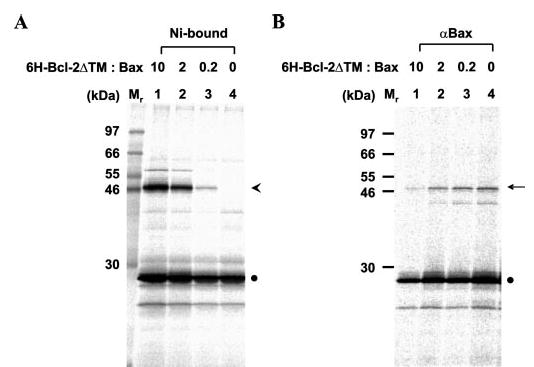
A, photocross-linking of in vitro synthesized Bcl-2ΔTM protein with a single ANB attached to Lys107 to His-tagged Bcl-2ΔTM protein at various Bcl-2ΔTM/Bax ratios. The Bcl-2ΔTM homodimer photoadduct was detected in the Ni2+-bound fractions as shown in Fig. 2. The amount of the photoadduct was reduced as the molar ratio of His-tagged Bcl-2ΔTM to Bax in the reaction (indicated at the top of the gel) was reduced. The arrowhead and filled circle indicate the cross-linked Bcl-2ΔTM dimer and monomer, respectively. B, photocross-linking of in vitro synthesized Bcl-2ΔTM protein with a single ANB attached to Lys107 to Bax protein at various Bcl-2ΔTM/Bax ratios. Aliquots of the samples used for A were examined for the Bcl-2ΔTM/Bax photocross-linking by immunoprecipitation using antibodies specific for Bax. The arrow indicates the photoadduct of the Bcl-2ΔTM and Bax, whose amount was increased when the molar ratio of Bcl-2ΔTM to Bax was reduced.
DISCUSSION
Our photocross-linking studies reveal that the interface of the Bcl-2 homodimer is much more extensive than predicted from structural studies using peptides. The binding interface can be divided into acceptor and donor surfaces and includes at least part of all four conserved BH domain sequences. The acceptor surface, consisting of helix 2, the loop between helix 4 and 5, and helix 8, includes the BH1–3 domain sequences. The donor surface that consists of helices 1–4, including the BH3 and −4 domain sequences, is oriented ~90° from the acceptor surface. Because of the geometry of the two surfaces, binding at one surface does not occlude the other. Therefore, one Bcl-2 molecule can bind either Bax or Bcl-2 via its acceptor surface and simultaneously another Bcl-2 monomer via its donor surface. This aspect of our model explains the formation of both homo- and hetero-oligomers. Moreover, our competition data suggest that binding of Bax to Bcl-2 may initially take place on homodimeric Bcl-2 molecules but may reduce the affinity of the homodimer, leading it to dissociate releasing another monomer. If the monomeric Bcl-2 has a higher affinity for Bax than the homodimer, the Bcl-2 monomers generated in this way may effectively sequester more activated Bax proteins.
Implications of the Two-binding Surface Model for Bcl-2 Dimerization
Bcl-2 homodimerization has been studied using a yeast two-hybrid system and various Bcl-2 deletion mutants (30, 31). Some mutant Bcl-2 proteins were still able to interact although they contained very large deletions. For example, a Bcl-2 mutant with the BH4 domain deleted interacted with Bcl-2 mutants with either the BH1 or BH2 domain deleted. This result is consistent with our two-binding surface model, because the BH4 deletion mutant still has an intact acceptor surface, and deletion of either BH1 or BH2 is not predicted to affect the donor surface of the other Bcl-2. In contrast, if both Bcl-2 mutants lacked either intact acceptor or donor surfaces they did not interact in the two-hybrid assay. For example, deletion of the BH1 domain sequence from both bait and target proteins analyzed in yeast abolished the interaction, since the deletion destroyed the same surface from both proteins. Similarly, deletion of the BH1 sequence from one protein and the BH2 sequence from the other also abolished the interaction between the two proteins, because both deleted sequences belong to the acceptor surface. This result also confirms our prediction that Bcl-2 homodimers consist of donor-acceptor pairs.
The binding affinity (EC50) measured in semisolid binding assays for the Bcl-2 and Bcl-xL homodimers (66.2 and 109.7 nm, respectively) (51) is much lower than the Kd measured in fluorescence equilibrium binding assays for Bcl-xL BH3 peptide binding to Bcl-xL (325 μm) (14). These quantitative data strongly suggest that the dimer interface is much more extensive than that between the BH3 peptide and the Bcl-xL hydrophobic groove, revealed by the NMR structures (14–16).
The Role of the Acceptor and Donor Surfaces in Protein Binding by Bcl-2
Our data suggest that the Bcl-2 acceptor surface binds either Bcl-2 or Bax, whereas the donor surface binds only another Bcl-2. The NMR structure of Bcl-xL in complex with BH3 peptides from different BH3-only proteins implies that the acceptor surface of Bcl-2 may also bind BH3-only proteins. Thus, the hydrophobic groove in the acceptor surface may be able to accommodate a variety of ligands.
Bcl-2 family proteins have been reported to bind to a variety of proteins, including scaffold proteins Apaf-1, CED-4, and Bag-1, mitochondrial proteins VDAC and FKBP38 (an inherent calcineurin inhibitor), kinase Raf-1 (an effector of H-Ras), phosphatase calcineurin, GTPases R-Ras and H-Ras, prenylated Rab acceptor PRA1, and transcription regulators p53 (and p53-binding protein p53-BP2) and Nur77/TR3 (for a review, see Refs. 52 and 53) (54–61). The detailed interface of most of these interactions has not yet been elucidated, and some of these interactions may be the result of low specificity interactions with the acceptor surface of Bcl-2. However, consistent with our data showing that the BH4 domain is part of the interface for Bcl-2 homotypic interaction, there is evidence that Bcl-2 BH4 domain sequences are involved in most of these Bcl-2 heterotypic interactions. Our model suggests that such interactions can occur with Bcl-2 homodimers, since there is always one of each binding surface that is unused in the dimer. Thus, our model clearly explains how, although the vast majority of the Bcl-2 in the cell is homodimeric, the same sequences identified as important for homodimerization can engage in a variety of other protein-protein interactions.
Acknowledgments
We thank Kristine Kim, Kam Zhang, and David Hockenbery (Fred Hutchinson Cancer Research Center) for providing plasmid and Escherichia coli strain for recombinant Bcl-2 protein expression and Trinity Loveless, Tian Xia, and Olga Nickolaeva (Lin laboratory), and Domina Falcone (Andrews Laboratory) for technical support.
Footnotes
This work was supported by National Institutes of Health Grant GM062964 (to J. L.) and Canadian Institutes of Health Research Grant FRN 12517 (to D. W. A.).
The abbreviations used are: BH, Bcl-2 homology; BMH, bis-male-imidohexane; εANB, Nε-(5-azido-2-nitrobenzoyl); FAK, focal adhesion kinase; PARP, poly(ADP-ribose) polymerase; WT, wild type; ANB, 5-azido-2-nitrobenzoyl.
References
- 1.Annis MG, Yethon JA, Leber B, Andrews DW. Biochim Biophys Acta. 2004;1644:115–123. doi: 10.1016/j.bbamcr.2003.07.001. [DOI] [PubMed] [Google Scholar]
- 2.Petros AM, Olejnicizak ET, Fesik SW. Biochim Biophys Acta. 2004;1644:83–94. doi: 10.1016/j.bbamcr.2003.08.012. [DOI] [PubMed] [Google Scholar]
- 3.Scorrano L, Korsmeyer SJ. Biochem Biophys Res Commun. 2003;304:437–444. doi: 10.1016/s0006-291x(03)00615-6. [DOI] [PubMed] [Google Scholar]
- 4.Sharpe JC, Arnoult D, Youle RJ. Biochim Biophys Acta. 2004;1644:107–113. doi: 10.1016/j.bbamcr.2003.10.016. [DOI] [PubMed] [Google Scholar]
- 5.Muchmore SW, Sattler M, Liang H, Meadows RP, Harlan JE, Yoon HS, Nettesheim D, Chang BS, Thompson CB, Wong SL, Ng SL, Fesik SW. Nature. 1996;381:335–341. doi: 10.1038/381335a0. [DOI] [PubMed] [Google Scholar]
- 6.Petros AM, Medek A, Nettesheim DG, Kim DH, Yoon HS, Swift K, Matayoshi ED, Oltersdorf T, Fesik SW. Proc Natl Acad Sci U S A. 2001;98:3012–3017. doi: 10.1073/pnas.041619798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hinds MG, Lackmann M, Skea GL, Harrison PJ, Huang DCS, Day CL. EMBO J. 2003;22:1497–1507. doi: 10.1093/emboj/cdg144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Denisov AY, Madiraju MSR, Chen G, Khadir A, Beauparlant P, Attardo G, Shore GC, Gehring K. J Biol Chem. 2003;278:21124–21128. doi: 10.1074/jbc.M301798200. [DOI] [PubMed] [Google Scholar]
- 9.Woo JS, Jung JS, Ha NC, Shin J, Kim KH, Lee W, Oh BH. Cell Death Differ. 2003;10:1310–1319. doi: 10.1038/sj.cdd.4401303. [DOI] [PubMed] [Google Scholar]
- 10.Suzuki M, Youle RJ, Tjandra N. Cell. 2000;103:645–654. doi: 10.1016/s0092-8674(00)00167-7. [DOI] [PubMed] [Google Scholar]
- 11.Chou JJ, Li H, Salvesen GS, Yuan J, Wagner G. Cell. 1999;96:615–624. doi: 10.1016/s0092-8674(00)80572-3. [DOI] [PubMed] [Google Scholar]
- 12.McDonnell JM, Fushman D, Milliman CL, Korsmeyer SJ, Coburn D. Cell. 1999;96:625–634. doi: 10.1016/s0092-8674(00)80573-5. [DOI] [PubMed] [Google Scholar]
- 13.Mathura VS, Soman KV, Varma TK, Braun W. J Mol Model. 2003;9:298–303. doi: 10.1007/s00894-003-0152-y. [DOI] [PubMed] [Google Scholar]
- 14.Sattler M, Liang H, Nettesheim D, Meadows RP, Harlan JE, Eberstadt M, Yoon HS, Shuker SB, Chang BS, Minn AJ, Thompson CB, Fesik SW. Science. 1997;275:983–986. doi: 10.1126/science.275.5302.983. [DOI] [PubMed] [Google Scholar]
- 15.Petros AM, Nettesheim DG, Wang Y, Olejnicizak ET, Meadows RP, Mack J, Swift K, Matayoshi ED, Zhang H, Thompson CB, Fesik SW. Protein Sci. 2000;9:2528–2534. doi: 10.1110/ps.9.12.2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu X, Dai S, Zhu Y, Marrack P, Kappler JW. Immunity. 2003;19:341–352. doi: 10.1016/s1074-7613(03)00234-6. [DOI] [PubMed] [Google Scholar]
- 17.Yin XM, Oltvai ZN, Korsmeyer SJ. Nature. 1994;369:321–323. doi: 10.1038/369321a0. [DOI] [PubMed] [Google Scholar]
- 18.Sedlak TW, Oltvai ZN, Yang E, Wang K, Boise LH, Thompson CB, Korsmeyer SJ. Proc Natl Acad Sci U S A. 1995;92:7834–7838. doi: 10.1073/pnas.92.17.7834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheng EHY, Levine B, Boise LH, Thompson CB, Hardwick JM. Nature. 1996;379:554–556. doi: 10.1038/379554a0. [DOI] [PubMed] [Google Scholar]
- 20.St Clair EG, Anderson SJ, Oltvai ZN. J Biol Chem. 1997;272:29347–29356. doi: 10.1074/jbc.272.46.29347. [DOI] [PubMed] [Google Scholar]
- 21.Jeong SY, Gaume B, Lee YJ, Hsu YT, Ryu SW, Yoon SH, Youle RJ. EMBO J. 2004;23:2146–2155. doi: 10.1038/sj.emboj.7600225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang E, Zha J, Jockel J, Boise LH, Thompson CB, Korsmeyer SJ. Cell. 1995;80:285–291. doi: 10.1016/0092-8674(95)90411-5. [DOI] [PubMed] [Google Scholar]
- 23.Wang K, Yin XM, Chao DT, Milliman CL, Korsmeyer SJ. Genes Dev. 1996;10:2859–2869. doi: 10.1101/gad.10.22.2859. [DOI] [PubMed] [Google Scholar]
- 24.Zha H, Aime-Sempe C, Sato T, Reed JC. J Biol Chem. 1996;271:7440–7444. doi: 10.1074/jbc.271.13.7440. [DOI] [PubMed] [Google Scholar]
- 25.Zha J, Harada H, Osipov K, Jockel J, Waksman G, Korsmeyer SJ. J Biol Chem. 1997;272:24101–24104. doi: 10.1074/jbc.272.39.24101. [DOI] [PubMed] [Google Scholar]
- 26.Wang K, Gross A, Waksman G, Korsmeyer SJ. Mol Cell Biol. 1998;18:6083–6089. doi: 10.1128/mcb.18.10.6083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.O’Connor L, Strasser A, O’Reilly LA, Hausmann G, Adams JM, Cory S, Huang DCS. EMBO J. 1998;17:384–395. doi: 10.1093/emboj/17.2.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wei MC, Lindsten T, Mootha VK, Weiler S, Gross A, Ashiya M, Thompson CB, Korsmeyer SJ. Genes Dev. 2000;14:2060–2071. [PMC free article] [PubMed] [Google Scholar]
- 29.Kim PK, Annis MG, Dlugosz PJ, Leber B, Andrews DW. Mol Cell. 2004;14:523–529. doi: 10.1016/s1097-2765(04)00263-1. [DOI] [PubMed] [Google Scholar]
- 30.Sato T, Hanada M, Bodrug S, Irie S, Iwama N, Boise LH, Thompson CB, Golemis E, Fong L, Wang HK, Reed JC. Proc Natl Acad Sci U S A. 1994;91:9238–9242. doi: 10.1073/pnas.91.20.9238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hanada M, Aime-Sempe C, Sato T, Reed JC. J Biol Chem. 1995;270:11962–11969. doi: 10.1074/jbc.270.20.11962. [DOI] [PubMed] [Google Scholar]
- 32.Falcone D, Andrews DW. Mol Cell Biol. 1991;11:2656–2664. doi: 10.1128/mcb.11.5.2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. Gene (Amst) 1989;77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- 34.Kim KM, Giedt CD, Basanez G, O’Neil JW, Hill JJ, Han YH, Tzung SP, Zimmerberg J, Hockenbery DM, Zhang KYJ. Biochemistry. 2001;40:4911–4922. doi: 10.1021/bi002368e. [DOI] [PubMed] [Google Scholar]
- 35.Yethon JA, Epand RF, Leber B, Epand RM, Andrews DW. J Biol Chem. 2003;278:48935–48941. doi: 10.1074/jbc.M306289200. [DOI] [PubMed] [Google Scholar]
- 36.Zhu W, Cowie A, Wasfy GW, Penn LZ, Leber B, Andrews DW. EMBO J. 1996;15:4130–4141. [PMC free article] [PubMed] [Google Scholar]
- 37.Zhu W, Leber B, Andrews DW. EMBO J. 2001;20:5999–6007. doi: 10.1093/emboj/20.21.5999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Crowley KS, Reinhart GD, Johnson AE. Cell. 1993;73:1101–1115. doi: 10.1016/0092-8674(93)90640-c. [DOI] [PubMed] [Google Scholar]
- 39.Krieg UC, Johnson AE, Walter P. Proc Natl Acad Sci U S A. 1986;83:8604–8608. doi: 10.1073/pnas.83.22.8604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lin J, Liang Z, Zhang Z, Li G. J Biol Chem. 2001;276:41733–41741. doi: 10.1074/jbc.M103475200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McCallum CD, Do H, Johnson AE, Frydman J. J Cell Biol. 2000;149:591–601. doi: 10.1083/jcb.149.3.591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Do H, Falcone D, Lin J, Andrews DW, Johnson AE. Cell. 1996;85:369–378. doi: 10.1016/s0092-8674(00)81115-0. [DOI] [PubMed] [Google Scholar]
- 43.Brunner J. Annu Rev Biochem. 1993;62:483–514. doi: 10.1146/annurev.bi.62.070193.002411. [DOI] [PubMed] [Google Scholar]
- 44.Minn AJ, Kettlun CS, Liang H, Kelekar A, Vander Heiden MG, Chang BS, Fesik SW, Fill M, Thompson CB. EMBO J. 1999;18:632–643. doi: 10.1093/emboj/18.3.632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Plath K, Mothes W, Wilkinson BM, Stirling CJ, Rapoport TA. Cell. 1998;94:795–807. doi: 10.1016/s0092-8674(00)81738-9. [DOI] [PubMed] [Google Scholar]
- 46.McCormick PJ, Miao Y, Shao Y, Lin J, Johnson AE. Mol Cell. 2003;12:329–341. doi: 10.1016/s1097-2765(03)00304-6. [DOI] [PubMed] [Google Scholar]
- 47.Hsu YT, Youle RJ. J Biol Chem. 1997;272:13829–13834. doi: 10.1074/jbc.272.21.13829. [DOI] [PubMed] [Google Scholar]
- 48.Antonsson B, Montessuit S, Sanchez B, Martinou JC. J Biol Chem. 2001;276:11615–11623. doi: 10.1074/jbc.M010810200. [DOI] [PubMed] [Google Scholar]
- 49.Mikhailov V, Mikhailova M, Pulkrabek DJ, Dong Z, Venkatachalam MA, Saikumar P. J Biol Chem. 2001;276:18361–18374. doi: 10.1074/jbc.M100655200. [DOI] [PubMed] [Google Scholar]
- 50.Oltvai ZN, Milliman CL, Korsmeyer SJ. Cell. 1993;74:609–619. doi: 10.1016/0092-8674(93)90509-o. [DOI] [PubMed] [Google Scholar]
- 51.Diaz JL, Oltersdorf T, Horne W, McConnell M, Wilson G, Weeks S, Garcia T, Fritz LC. J Biol Chem. 1997;272:11350–11355. doi: 10.1074/jbc.272.17.11350. [DOI] [PubMed] [Google Scholar]
- 52.Hacker G, Vaux DL. Curr Biol. 1995;5:622–624. doi: 10.1016/s0960-9822(95)00126-6. [DOI] [PubMed] [Google Scholar]
- 53.Reed JC. Nature. 1997;387:773–776. doi: 10.1038/42867. [DOI] [PubMed] [Google Scholar]
- 54.Hu Y, Benedict MA, Wu D, Inohara N, Nunez G. Proc Natl Acad Sci U S A. 1998;95:4386–4391. doi: 10.1073/pnas.95.8.4386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Huang DCS, Adams JM, Cory S. EMBO J. 1998;17:1029–1039. doi: 10.1093/emboj/17.4.1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shimizu S, Konishi A, Kodama T, Tsujimoto Y. Proc Natl Acad Sci U S A. 2000;97:3100–3105. doi: 10.1073/pnas.97.7.3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li LY, Shih HM, Liu MY, Chen JY. J Biol Chem. 2001;276:27354–27362. doi: 10.1074/jbc.M103821200. [DOI] [PubMed] [Google Scholar]
- 58.Shirane M, Nakayama KI. Nat Cell Biol. 2003;5:28–37. doi: 10.1038/ncb894. [DOI] [PubMed] [Google Scholar]
- 59.Denis GV, Yu Q, Ma P, Deeds L, Faller DV, Chen CY. J Biol Chem. 2003;278:5775–5785. doi: 10.1074/jbc.M210202200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mihara M, Erster S, Zaika A, Petrenko O, Chittenden T, Pancoska P, Moll UM. Mol Cell. 2003;11:577–590. doi: 10.1016/s1097-2765(03)00050-9. [DOI] [PubMed] [Google Scholar]
- 61.Lin B, Kolluri SK, Lin F, Liu W, Han YH, Cao X, Dawson MI, Reed JC, Zhang XK. Cell. 2004;116:527–540. doi: 10.1016/s0092-8674(04)00162-x. [DOI] [PubMed] [Google Scholar]