

Abstract
It has been reported that interferons (IFNs) may have antitumor activity in multiple myeloma (MM). The mechanism for their effect on MM, however, remains elusive. This study shows that IFN-α and -β, but not -γ, induce apoptosis characterized by Annexin V positivity, nuclear fragmentation and condensation, and loss of clonogenicity in 3 MM cell lines (U266, RPMI-8266, and NCI-H929), and in plasma cells from 10 patients with MM. Apo2 ligand (Apo2L, also TRAIL) induction was one of the earliest events following IFN administration in U266 cells. Treatment of these cells with TRAIL, but not with Fas agonistic antibodies, induces apoptosis. Cell death induced by IFNs and Apo2L in U266 cells was partially blocked by a dominant-negative Apo2L receptor, DR5, demonstrating the functional significance of Apo2L induction. This study shows that IFNs activate caspases and the mitochondrial-dependent apoptotic pathway, possibly mediated by Apo2L production. Thus, IFN-α and -β induce cytochrome c release from mitochondria starting at 12 hours, with an amplified release seen at 48 hours. Moreover, Bid cleavage precedes the initial cytochrome c release, whereas the late, amplified cytochrome c release coincides with changes in levels of Bcl-2, Bcl-XL, and reduction of mitochondrial membrane potential. These results link the Apo2L induction and modulation of Bcl-2 family proteins to mitochondrial dysfunction. Furthermore, IFNs and Apo2L induce cell death of CD38+/CD45−/dim plasma cells, without significant effect on nonplasma blood cells, in a caspase and Bcl-2 cleavage-dependent manner. These results warrant further clinical studies with IFNs and Apo2L in MM.
Introduction
Multiple myeloma (MM), currently an incurable disease, is the second most common blood cancer. It is characterized by the presence of malignant plasma cells predominantly located in bone marrow.1 Interferons (IFNs), a family of pleiotropic cytokines, have been used for the treatment of MM alone or in combination with other chemotherapeutic drugs.2–4 Despite their clinical effectiveness for antitumor growth, how IFNs act on MM is unclear.5 IFNs, which consist of type I (predominantly α and β) and type II (γ) IFNs, play an essential role in host defense, having both antiviral and antitumor effects. Type I and type II IFNs bind to their specific receptors to phosphorylate and activate the Janus kinases and the signal transducers and activators of transcription (STATs).6 Once activated, STAT proteins are dimerized and translocate to the nucleus, where they bind to distinct DNA motifs to induce a large number of IFN-responsive genes. Type I IFNs primarily activate STAT 1 and 2, which are then translocated to the nucleus to bind to IFN-stimulated regulatory elements to induce gene expression. Type I and type II IFNs elicit distinct signaling pathways; however, they also induce a set of common genes. Of these IFN-induced genes, some are reported to be associated with apoptosis. In spite of the growing knowledge of signaling pathways for IFNs,6 how IFN-induced gene expression is linked to the cell death machinery remains elusive.
Apoptosis is a genetically regulated cell death process. Cells undergo apoptosis by default, and all the critical components for apoptosis are compartmentalized within distinct subcellular organelles. Once committed to death, the cell undergoes the relatively stereotypic execution and degradation phases involving chromatin condensation, phosphatidyl-serine externalization, and selective proteolysis by a family of cysteine proteases, named caspases.7 It is important to identify and characterize the precommitment signals that engage the execution and degradation machinery, because these signals hold promise for identifying novel pharmaceutical targets useful for augmenting tumor cell death in cancer therapy. Mitochondria play a central role in the execution process of apoptosis.8,9 Once the cells are committed to cell death, apoptogenic factors, such as cytochrome c (cyt c)10–13 and Smac/DIABLO,14,15 are released from mitochondria to initiate the caspase cascade and thus may represent irreversible commitment events. Cyt c acts as a cofactor to stimulate the complexing of Apaf-1 (human homolog of Caenorhabditis elegans CED-4) with caspase 9.16,17 This complex then initiates activation of the caspase cascade, which culminates in proteolytic targeting of key intracellular proteins.18 Smac,14 once maturated and released into cytosol, is able to interact with inhibitors of apoptosis proteins to promote caspase activation. One other important apoptotic event in many cell systems is the loss of an electrical potential across the inner mitochondrial membrane,19 manifested by a reduction in mitochondrial membrane potential (Δψm), indicative of mitochondrial permeability transition (MPT). Given the critical role of cyt c in the initiation and execution of apoptosis, it is important to understand how cyt c release is regulated. Little is known regarding IFN-induced apoptosis with respect to the mechanism of action and effect on mitochondria.
The Bcl-2 family of proteins plays a pivotal role in regulating cyt c release and apoptosis.8,20 This expanding family consists of both anti-apoptotic molecules, such as Bcl-2 and Bcl-XL, as well as pro-apoptotic ones, such as Bax and Bid. Bcl-2 can block the release of cyt c from mitochondria11,12 and prevent the activation of caspase 3,21 whereas Bax and Bid can promote cyt c release from mitochondria and thus activate the caspase cascade.22–25 The interactions between pro- and anti-apoptotic molecules seem to be the determining factors for cell survival. Not surprisingly, most death modulators function by acting through Bcl-2 family proteins to regulate cyt c release.8,26 It has been recently reported that Bid could induce cyt c release without MPT, whereas Bax induces cyt c release and MPT.25 How Bcl-2 family proteins regulate MPT and cyt c release is a recent focus of apoptosis research.
Of the many disparate death stimuli, the signaling pathway initiated by tumor necrosis factor and the related Fas ligand (also Apo-1) is best characterized.27 These cytokines are expressed as membrane-bound ligands that can be cleaved to a soluble form. Engagement by these ligands of their respective receptors, followed by recruitment of an adapter molecule, such as FADD (Fas-associated death domain protein28,29) leads to direct activation of the caspase cascade. This activation is accomplished by recruitment of caspase 8, followed by its proteolytic activation. Once activated, caspase 8 can proteolytically cleave Bid, with the truncated Bid targeting mitochondria and inducing cyt c release and further amplification of the caspase cascade.22–24,30 The engagement of mitochondria through truncated Bid in receptor-mediated apoptosis further supports the pivotal role of mitochondria in apoptosis. Interestingly, Bcl-2 is a proteolytic target of caspases and is converted in situ to a pro-apoptotic molecule13,31 to promote cyt c release and apoptosis. We have recently described in MM 2 distinct stages of cyt c release during genotoxic stress, characterized by a limited initial cyt c release, sufficient to activate caspases, followed by a second one, characterized by a positive feedback amplification of cyt c release.13
In this study we examined the apoptotic pathways in MM following treatment with IFNs. We show here that type I but not type II IFNs induce typical apoptosis through activation of the Apo2 ligand (Apo2L, also TRAIL) pathway and modulation of the Bcl-2 family of proteins in 3 MM cell lines and in plasma cells isolated from 10 patients with MM. Our data indicate that Apo2L induction is at least partially necessary for caspase activation and the Bcl-2 and Bid cleavage-dependent pathway. Our data further support the notion that Apo2L could become a useful molecule for management of MM.
Materials and methods
Cell culture, bone marrow plasma cell isolation, and treatments
Human U266, NCI-H929, and RPMI-8226 MM cells were obtained from American Type Culture Collection (ATCC, Rockville, MD) and cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum, 2 mM L-glutamine, and antibiotics.13 Bone marrow biopsies and aspirate smears from consenting patients with established diagnosis of plasma cell myeloma were examined by light microscopy by a hematopathologist (E.D.H.). Plasma cells were further isolated by density centrifugation on Ficoll-Hypaque (Amersham Pharmacia Biotech, Uppsala, Sweden), followed by negative selection with the use of magnetic beads (Stem Cell Technology, Vancouver, BC), or by positive selection (MACSelect; Miltenyi Biotec, CA) by using anti-CD138 antibodies.32 To confirm MM, cells were examined by light microscopy by a hematopathologist and/or assayed by flow cytometry for expression of CD38+ and CD45−/dim, specific for plasma cells. Phycoerythrin-conjugated CD38 and peridinin chlorophyll protein-conjugated CD45 were obtained from Becton Dickinson Biosciences [BD; San Jose, CA]). Samples containing significant proportion of plasma cells were further cultured in RPMI-1640 medium supplemented with 20% fetal calf serum, 100 mM minimal essential medium (MEM), nonessential amino-acids, 1 mM sodium pyruvate, 1 mM glutamine, and 5 mg/mL gentamycin (GIBCO BRL, Gaithersburg, MD). Cells were treated with IFN-α-2a (Roche Molecular Biochemicals, Indianapolis, IN), IFN-β-2a (Biogen, Cambridge, MA), IFN-γ (GIBCO BRL), Apo2L, and soluble receptor to Apo2L (Alexis Biochemicals, San Diego, CA), or with the Fas agonistic monoclonal antibody (mAb; clone CH11; Panvera, Madison, WI). In certain experiments, the eluted nonplasma cell lymphocytes (examined by light microscopy and flow cytometry) were also treated for comparison. Isolated cells were treated with IFN, followed by triple staining for CD38, CD45, and Annexin V (Biosource International, Camarillo, CA) to determine the effects of IFNs and Apo2L on apoptosis.
Clonogenic cell survival assays
Cellular clonogenicity was measured as previously described.26 Briefly, U266 cells were treated and serially diluted to 100 or 50 cells/mL in fresh RPMI 1640 medium supplemented with 10% fetal calf serum containing medium. This cell suspension was then distributed to a U-bottom 96-well plate, with each well containing 100 μL medium. Cell colonies were counted 10 to 14 days after initial plating, and the percentage of cells that were clonogenic calculated by the ratio of the theoretical Poisson density of the number of negative wells observed, against the initial cell plating density (0.5, 1, or 5 cells/well); that is, the % clonogenicity = (Ln × [96/NegWells])/(plate density) × 100, where NegWells is the number of wells that have failed to grow colonies and plate density is the original cell plating density per well.
Apoptosis assays
Apoptosis was either measured by nuclear fragmentation and condensation examined by fluorescence microscopy after Hoechst 33258 staining or by examining phosphatidylserine exposure on cell membranes, using Annexin V, as described.13,33 Cells were stained with fluorescein isothiocyanate (FITC)-Annexin V (25 ng/mL; green fluorescence), simultaneously with dye exclusion (propidium iodide [PI]; negative for red fluorescence). This assay13,34 discriminates between intact (FITC−/PI−), early apoptotic (FITC+/PI−), and late apoptotic cells (FITC+/PI+). Comparative experiments were performed at the same time by bivariate flow cytometry using a FACScan and analyzed with the CellQuest software (BD) on data obtained from the cell population from which debris were gated out.
The caspase protease assays were performed as described,13,33 using the acetyl-Ile-Glu-Thr-Asp-pNA (IETD) and acetyl-Asp-Glu-Val-Asp-pNA (DEVD) p-nitroanilide (pNA)-derived chromogenic substrates for caspase 8 and 3 activity, by enzyme-catalyzed release of pNA monitored at 405 nm in a 96-well microtiter plate reader (Spectramax 340; Molecular Devices, Sunnyvale, CA). Caspase peptide inhibitors (z-VAD-fmk, benyloxycarbonyl-Val-Ala-Asp-fmk), or substrates were from BioMol (Plymouth Meeting, PA); all other chemicals were from Sigma (St Louis, MO), unless otherwise specified.
To determine of the role of the Apo2L pathway, a FLAG epitope-tagged dominant-negative Apo2L receptor, DR5Δ (TRAIL-R2DN) was used as described30 for stable transfection of U266 cells. DR5Δ lacks the death domain and has been shown to function as a dominant-negative inactivating the function of the endogenous DR5.35 Exponentially growing (106) cells were washed and resuspended in a 6-well plate, mixed with 4 μg DMRIE-C reagent (GIBCO-BRL), incubated with 2 μg DNA in OPTI-MEM I, and further cultured overnight. Next day the cells were switched to complete medium containing 1 mg/mL G418 for further selection, and individual clones expressing FLAG.DR5Δ were isolated. One representative clone was chosen for these studies because it abundantly expressed FLAG.DR5Δ, in addition to a vector control-only expressing clone.
Immunoblotting
Western immunoblotting was performed as described.13 Briefly, cells were washed and lysed in buffer containing 150 mM NaCl, 25 mM HEPES pH 7.4, 1% (vol/vol) NP-40, 0.25% (wt/vol) sodium deoxycholate, 1 mM EGTA, EDTA, and phenylmetholsulfonyl fluoride, 10 mg/mL aprotinin, leupeptin, and pepstatin. Cellular proteins from total cell lysates (10 μg/sample) were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred onto a nitrocellulose membrane (Schleicher and Schull, Keene, NH). The membranes were probed using immunoblot analyses with mAb to the human Bcl-2, and polyclonal antibodies (pAbs) to caspase 3 (Santa Cruz, Santa Cruz, CA), Apo2L/TRAIL (Santa Cruz), cyt c (Pharmingen, San Diego, CA), or Bid pAbs,22 followed by incubation with the secondary antibody conjugated to horseradish peroxidase for 1 hour at room temperature. Equal protein loading was confirmed by staining filters with Ponceau S and/or reprobing with actin mAb (Sigma). Immunoreactive bands were detected using enhanced chemiluminescence (Amersham Pharmacia Biotech) and visualized by autoradiography.
Mitochondrial isolation and membrane potential
Cell fractionation was done as previously reported.36 Briefly, cells (1 × 106/sample) were washed twice with ice-cold phosphate-buffered saline then kept on ice for 20 minutes in buffer A, containing 20 mM HEPES-KOH (pH 7.2), 100 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 250 mM sucrose, and protease inhibitors (1 mM phenylmetholsulfonyl fluoride and dithiothreitol, 10 mg/mL aprotinin, leupeptin, and pepstatin). The cells were treated at different time points and harvested at the same time for cell fractionation. The cell suspension was gently homogenized with a Dounce homogenizer (5–8 strokes), and the cell lysis was checked by trypan blue staining. The homogenate was centrifuged at 750g for 5 minutes, and the supernatant subjected to further centrifugation at 10 000g. This pellet, containing mitochondria, was designated P10. The supernatant was subjected to further ultracentrifugation at 100 000g for 45 minutes. The resulting pellet and supernatant, representing the endoplasmic reticulum and cytosolic fractions, were designated as P100 and S100, respectively. The purity of the cellular fractions was examined by Western blotting using α-Calnexin, α-PCNA, and mitochondrial cytochrome oxidase 1 (MTCO1) mAbs, as endoplasmic reticulum (ER) membrane, nuclear, and mitochondrial markers, respectively. Cyt c was not detectable in the P-100 fraction.
The mitochondrial membrane potential indicator DIOC6 (3) (2 μL of 2 mM stock solution in dimethyl sulfoxide) was added to a U266 cell suspension (4 × 105 cells/mL) in fresh RPMI 1640 medium (pH 7.2) and incubated at 37°C for 5 minutes. A change in DIOC6 (3) fluorescence indicates a change in Δψm and/or in mitochondrial mass. Therefore, parallel experiments were performed by using the mitochondrial specific dye nonyl acridine orange (100 nM) to determine mitochondrial mass.37 DIOC6 (3) or nonyl acridine orange fluorescence was examined at 530 ± 30 nm. DIOC6 (3) data were validated by addition of 20 mM m-chlorophenylhydrazone after 5 minutes of DIOC6 (3) loading.
RNA analyses
Total RNA was isolated from U266 cells at various intervals after IFN treatment by using the TRIZOL reagent (GIBCO BRL). To determine the steady-state levels of RNA, the RiboQuant system (Pharmingen) for RNase protection was used, as described.33,38 The hApo-3C template set (Pharmingen), containing 10 complementary DNA probes, including the housekeeping gene product L32 as a control, was used for the T7 polymerase-directed synthesis of high-specific activity [32P]-labeled antisense RNA probes.
Results
Type I but not type II IFNs induce typical apoptosis and loss of clonogenicity
IFNs have been reported to be important regulators of growth and survival.6 Exponentially growing cultures of U266, an interleukin 6-dependent MM cell line resistant to ionizing radiation and Fas, were treated with IFN-α, -β, and -γ and examined for phosphatidyl-serine exposure on the cell membranes, indicative of apoptosis. There was no change in the Annexin V staining at 6 and 24 hours following IFN-α treatment compared with untreated cells. However, 22% of cells showed Annexin V positivity at 48 hours, which reached 37% at 72 hours and 45% at 96 hours following treatment with 2000 U/mL IFN-α (Figure 1A). IFN-α and, more potently, IFN-β induced Annexin V positivity in a dose-dependent manner (data not shown). In contrast, under the same experimental conditions, IFN-γ treatment did not induce Annexin V-positive staining.
Figure 1. IFN-α induces apoptosis in U266 MM cells.
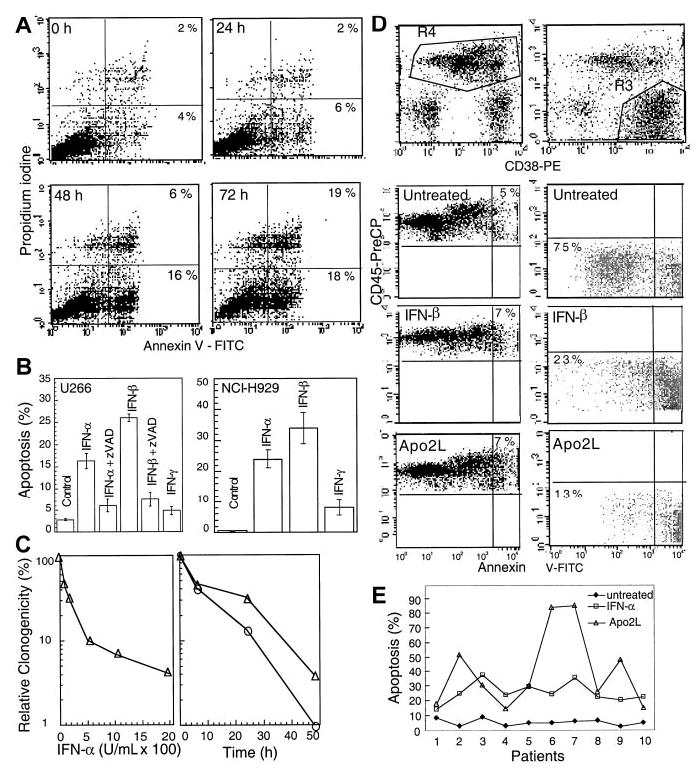
(A) At the indicated times following IFN-α treatment, cells were subjected to bivariate flow cytometry following Annexin V/PI staining. The data were obtained from the cell population from which debris were gated out against forward and side scatter. (B) Cells were treated with IFNs for 72 hours in the presence or absence of 75 μM z-VAD-fmk (added at the time of IFN addition, and every 24 hours thereafter), and apoptosis was determined at 72 hours by Hoechst 33258 staining and expressed as the percentage of the total cells. (C) Clonogenic survival was determined following treatment for 48 hours with IFN-α (100–2000 U/mL; left panel). A time course was determined for cells treated for 6, 24, or 48 hours (right panel) with IFN-α (o) or -β (Δ) (2000 U/mL) and then cultured as described in “Materials and methods.” Data shown represent the mean value of 2 duplicate experiments, normalized against untreated cells. (D) Plasma cells were isolated from bone marrow aspirates of consenting patients with MM as described in “Materials and methods,” treated with IFN-α, -β, -γ, and Apo2L before being examined by triple staining for CD38, CD45, and Annexin V, indicative of apoptosis. R3 and R4 represent CD38+/CD45−/dim and CD45+ cells, respectively, with the gated regions being analyzed for Annexin V staining. (E) The response of patient plasma cells to INF-α and Apo2L was determined by Hoechst staining as above.
To confirm the above results, we further examined nuclear morphologic changes by determining nuclear condensation and fragmentation, another hallmark for apoptosis. Hoechst 33258 staining showed that 16% and 26% of cells displayed the nuclear condensation at 72 hours after IFN-α and -β treatments, respectively. The pancaspase inhibitor z-VAD-fmk13 efficiently prevented IFN-induced apoptosis (Figure 1B) without having any significant effect on cell growth when used alone. In contrast, IFN-γ had no significant effect.
We also examined the response of another typical MM cell line, NCI-H929. IFN-α and -β, but not -γ, also induced typical apoptosis in these cells (Figure 1B, right), which was inhibitable by z-VAD-fmk. In contrast, IM-9, a cell line derived from an MM patient, but later classified as a lymphoblast,39 was not sensitive to IFNs. Clonogenic assays, used to further characterize apoptosis, indicated that IFN-α and -β induced a progressive loss of clonogenic survival in a dose-dependent manner. When treated for 48 hours with IFN-α and -β (1000 or 2000 U/mL), almost all cells lost their clonogenic survival ability. However, a similar treatment for 24 hours, resulted in only 15% or 32% of clonogenic cells compared with untreated cells following IFN-α and -β treatments, respectively (Figure 1C). These data suggest that type I IFNs can induce the loss of clonogenicity before the appearance of apoptotic morphology.
We further asked whether IFNs can induce apoptosis in plasma cells freshly isolated from the bone marrow of MM patients. We found that apoptosis was induced in patient-derived primary MM plasma cells by type I IFNs. For 4 of the patient samples, the nonplasma lymphocytes were also used as control. We found that only the samples containing malignant (CD38+/CD45−/dim) plasma cells underwent apoptosis on treatment with IFNs (Table 1). This finding was further confirmed by triple staining for CD38, CD45, and Annexin V. Almost all CD38+/CD45−/dim cells became Annexin V positive when treated with IFNs and Apo2L for 48 hours. In contrast, the majority of nonplasma cells were not sensitive to Apo2L and IFNs (Figure 1D).
Table 1.
Response to interferons and Apo2L of plasma cells isolated from the bone marrow of MM patients
| Apoptosis (%)
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Patient no. | Age* (y) | % of PCs at Dx† | Previous treatment | % of PCs at selection† | % of PCs treated‡ | Control | α | β | γ | Apo2L |
| 1 | 66 | 80 | CTX × 6 | Clusters | 90 | 8.7 | 15 | 15 | ND | 19 |
| 2 | 77 | 100 | RTX × 4 | 90 | 79 | 3.2 | 26 | 22 | 8 | 53 |
| 3 | 69 | 80 | RTX × 4 | 51 | 80 | 9.4 | 38 | 23 | ND | 31 |
| 4 | 74 | 70 | RTX × 4 | 80 | 81 | 3.2 | 24 | 13 | ND | 15 |
| 5 | 46 | 80 | No | 80 | 100 | 5.2 | 31 | 27 | 5 | 31 |
| 6 | 45 | 8 | RMP, DVD × 8, CTX × 4 | ND | 90 | 5.2 | 25 | 21 | 14 | 85 |
| 7 | 51 | 30 | RTX × 4 | 20 | 90 | 6.3 | 36 | 34 | 17 | 86 |
| 8B | 44 | 60–70 | VAD × 3 | 75 | 9 | 1.9 | 2 | 13 | 4 | 11 |
| CTX × 2 | ||||||||||
| DVD, RTX, De, DT | ||||||||||
| 8C | 90 | 6.5 | 23 | 32 | 26 | 27 | ||||
| 9B | 61 | 55 | No | 55 | 0 | 2.7 | 2 | 4 | 1 | 5 |
| 9C | 85 | 2.7 | 21 | 28 | 13 | 49 | ||||
| 10B | 59 | 50 | No | 10–20 | 6 | 1.5 | 8 | 6 | 6 | 3 |
| 10C | 86 | 5.3 | 23 | 16 | 20 | 15 | ||||
| 5B§ | 46 | 80 | RTX × 2 | ND | 26 | 3.6 | ND | 7 | 10 | 5 |
| 5C§ | 92 | 6.7 | ND | 33 | 5 | 31 | ||||
PC, plasma cells; Dx, diagnosis; CTX, Cytoxan; ND, not detected; RTX, Rituxan; RMP, ritoxin + melphalan + prednisone; DVD, doxorubicin + vincristine + dexamethasone; B, C, nonplasma and plasma cells after CD138 selection; VAD, vincristine + adriamycin + doxorubicin; De, Decadron; DT, DTPASE.
PCs were isolated from the bone marrow of patients with MM by negative (patients 1–4) or positive selection. The plasma B cells (enriched more than 80%) were treated with IFN-α, β, or γ (2000 U/mL) or with Apo2L (400 ng/mL) for 48 hours. The apoptotic cells (> 200/sample) were then stained with Hoechst 33258, scored by fluorescence microscopy, and expressed as the percentage of the total cells.
Age at first diagnosis of multiple myeloma.
Identified from medical files and proved by pathologist.
Percentage of PCs in the samples treated with interferon (α, β, or γ) or with Apo2L.
This patient was seen twice: at diagnosis and 2 months later.
IFNs induce Apo2L/TRAIL in MM
We sought to determine next whether IFNs affect expression of known apoptosis-related genes and chose to examine components of receptor-mediated apoptosis.27 Control and IFN-β–treated (chosen because it had the most potent effect) U266 cells, examined by RNase protection at 4 to 24 hours, a time at which no apoptosis was observed, revealed that Apo2L messenger RNA (mRNA) was induced abundantly by IFN-β (Figure 2A). In contrast, there were no significant changes in expression levels of the Apo2L receptors DR5 and DR4, or of Caspase 8 and Fas. Moreover, there was no detectable Apo2L mRNA expression in untreated cells. Western blotting revealed that Apo2L was induced as early as 6 hours following type I IFN treatment and a more pronounced expression at later times following treatment (Figure 2B). In contrast, there was only a slight change in Apo2L levels following IFN-γ treatment early on, with no additional change at a later time. Similar analyses of patient-derived plasma cells revealed that IFN-β, at a concentration as low as 200 U/mL, was effective, whereas a higher concentration of IFN-α was required for Apo2L induction (Figure 2C). Significantly, Apo2L induced apoptosis in plasma cells derived from MM. Of these treatments, soluble Apo2L was the most effective inducer of apoptosis (Table 1). Interestingly, plasma cells isolated from one patient (No. 6), who received extensive chemotherapy (13 cycles) but had a persistent tumor, were very sensitive to Apo2L treatment. This finding suggests that Apo2L may be useful for treating resistant or relapsing myeloma. Because the number of patients is relatively small, a large-scale study will be needed in the future to define a role for Apo2L in treatment of relapsing or resistant MM. Nevertheless, these findings indicate that IFN-α and -β induce Apo2L/TRAIL expression in the MM cell line U266 and in freshly isolated plasma cells of MM patients.
Figure 2. IFN-α and -β induce Apo2L expression in MM.

(A) U266 cells were treated with IFN-β, and the steady state mRNA expression was analyzed by multiprobe RNase protection assay for caspase 8 (casp. 8), Fas, DR4, DR5, Apo2L, and, as a control, L32, as described33,38 and detailed in “Materials and methods.” Apo2L protein levels were determined for U266 cells (B) and plasma cells of MM patients (C) treated with IFNs for 24 hours and then subjected to Western blotting using antibodies against Apo2L, and as a control, β-actin.
IFN-α and -β induce caspase activation
We next asked whether caspases are activated following IFN treatment that induces Apo2L. Protease activity assays indicate that caspase 8 was activated 6 hours following IFN treatment, followed by caspase 3 (Figure 3A). Moreover, Western blotting showed that caspase 3 was converted to the p17 kD active form as early as 24 hours after IFN-α and -β, but not IFN-γ, treatment of U266 cells (Figure 3B). These data suggest that caspases were activated before the appearance of any morphologic changes associated with apoptosis. Similar to U266 cells, Western blotting showed that type I but not type II IFNs induced caspase 3 cleavage in freshly isolated plasma cells of MM patients (Figure 3C).
Figure 3. IFN-α and -β induce caspase activation in MM cells.
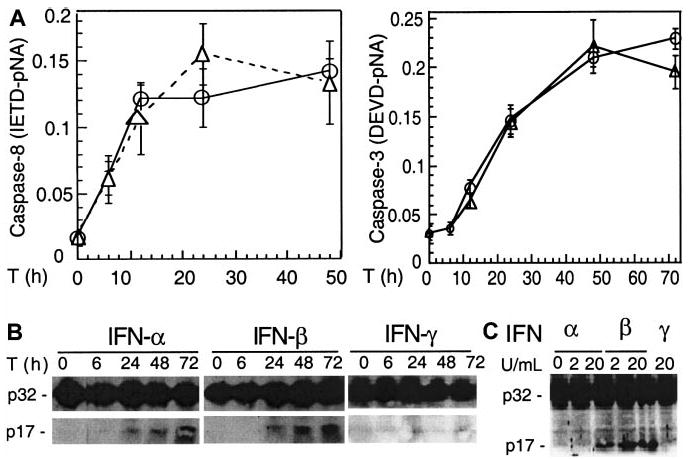
U266 cells were collected at the indicated times (T) after IFN treatment and analyzed by protease activity assays for caspase 8 and caspase 3 by using the specific IETD and DEVD-pNA substrates (A) and Western blotting for caspase 3 cleavage (B). Data were representative of 3 separate experiments. (C) IFN-α and -β, but not -γ, induced caspase 3 cleavage in freshly isolated MM plasma cells (patient No. 2). Primary MM cells were isolated from bone marrow aspirates of patients, as described in “Materials and methods,” treated for 48 hours with IFNs, then subjected to Western blotting for caspase 3. The p32 kD band represents procaspase 3, whereas p17 represents its activated form.
IFNs induce distinct levels of cyt c release from mitochondria
We and others have reported that apoptosis in MM is associated with an increase in cytosolic cyt c levels.13,40 Examination of the distribution of cyt c in cytosolic and mitochondrial cellular fractions following IFN treatment of U266 cells revealed that cyt c was undetectable in cytosol isolated from untreated cells. Small amounts of cyt c were present in the cytosol by Western blotting at 16 and 24 hours following IFN-α and -β treatments, and these amounts increased significantly at 48 hours. In contrast, there were no significant levels of cytosolic cyt c present in the cytosol after IFN-γ treatment even at 48 hours (Figure 4A). These results suggest that type I IFN-induced apoptosis was associated with induction of a differential cyt c release from mitochondria at 24 and 48 hours after IFN treatment. To determine whether this differential cyt c release had a functional significance, we examined next the Δψm, a critical mitochondrial event implicated in apoptosis.19 The Δψm-sensitive lipophilic fluorochrome DIOC6 (3) indicated that there was no change in its intensity and bimodal distribution at 24 hours. A subpopulation of these cells showed decreased DIOC6 (3) staining, indicating that these cells had a lower Δψm. The proportion of cells displaying a lower Δψm significantly increased in IFN-β–treated cells at 48 hours, reaching 76% at 72 hours (Figure 4B). Importantly, the proportion of apoptotic cells at 72 hours following IFN-β treatment was only 26% (Figure 1). IFN-α treatment was similar, whereas IFN-γ had no effect on the DIOC6 (3) staining (data not shown). Altogether, these results indicate that the early cyt c release from mitochondria took place before the reduction of Δψm, with the later, augmented cyt c release coinciding with the reduction of Δψm.
Figure 4. IFN-α and -β induce cyt c release and Δψm reduction.
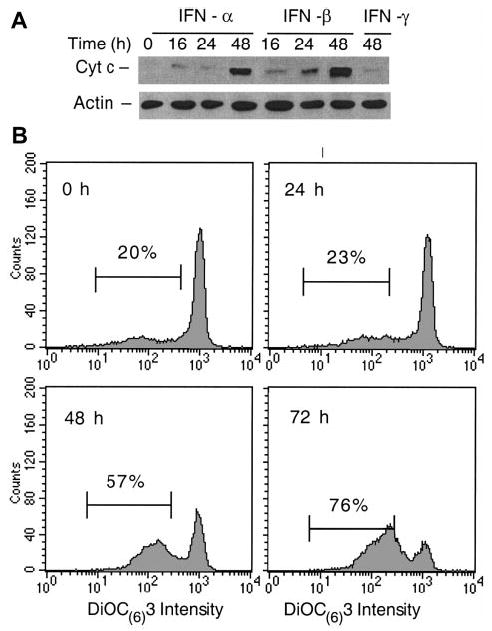
(A) Cyt c levels were determined in cellular fractions isolated by differential centrifugation from control or IFN-treated U266 cells as described in “Materials and methods.” Immuno-blotting with anti–cyt c mAb and β-actin as a control were used to determine the levels of cyt c in the S100 fractions, representing the cytosolic cyt c in 20 μg of cellular protein. (B) The Δψm was determined by using 10 nM DIOC6 (3) as described in “Materials and methods.” The percentage of Δψm (low) after the cell debris were excluded was determined, and comparative experiments were performed on the same day. The data shown are representative of 3 separate duplicate experiments.
IFN-α and -β induce Bid and Bcl-2 cleavage and down-regulation of Bcl-xL
Next, we further examined whether IFNs induced cyt c release through activation of Bcl-2 family proteins. We found that IFNs induced cleavage of Bid and Bcl-2. Thus, Western blot analyses revealed 2 Bid cleavage products, of p13 and p15 kD, which appeared at 6 to 24 hours after type I IFN treatment (Figure 5A). The appearance of Bid cleavage preceded the initial release of cyt c from mitochondria into the cytosol and the activation of caspase 8. Western analyses also revealed that Bcl-2 was cleaved at 48 hours after type I IFN treatment (Figure 5B, left panel). Cleavage of both Bid and Bcl-2 could be inhibited by z-VAD-fmk (Figure 5A,B). Similar observations were extended to freshly isolated plasma cells from MM patients, in which a concentration of IFN-β as low as 200 U/mL induced Bcl-2 cleavage (Figure 5B, right panel). In addition, IFN-α but not -γ induced down-regulation of Bcl-xL. In contrast, IFNs did not affect levels of any other Bcl-2 family proteins, such as Bad, Bak, and Bax (Figure 5C). These results indicate that some of these Bcl-2 family proteins are modulated in IFN-induced apoptosis, with Bid and Bcl-2 cleavage corresponding to the early and late stages of cyt c release.
Figure 5. IFN-α and -β induce Bid and Bcl-2 cleavage.
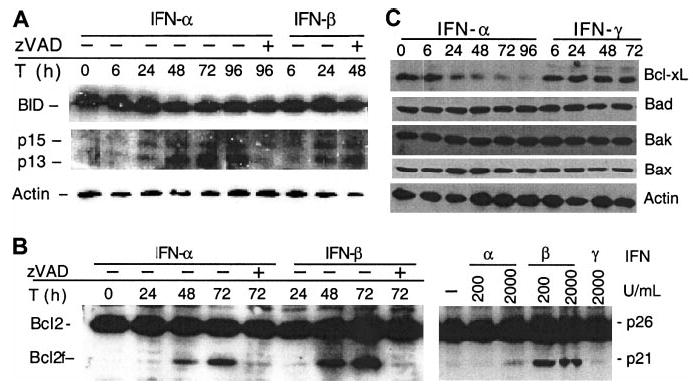
U266 cells were treated and harvested at the indicated times (T) in the presence or absence of z-VAD-fmk. (A) Bid and Bcl-2 were analyzed by Western blotting using a Bid pAb (a kind gift from X. Wang, UT Southwestern22) and Bcl-2 mAb. z-VAD-fmk was added every 24 hours following IFN treatment. The data shown are representative of 3 different experiments. (B) IFN-α and -β, but not -γ, induce Bcl-2 cleavage in U266 (left) and freshly isolated MM cells (right). Plasma cells were isolated from patients and treated for 48 hours, before being subjected to Western blotting for Bcl-2. (C) U266 cells were treated as indicated and then subjected to Western blotting with the appropriate antibodies for Bcl-xL, Bad, Bak, and Bax, and, as a control, β-actin.
IFNs induce apoptosis in MM through the Apo2L/TRAIL pathway
The above results prompted us to further examine whether IFN-induced Apo2L expression might be directly involved in enhanced cytotoxicity against MM. Examination of the cellular response to Fas or Apo2L showed that, consistent with previous reports, U266 was insensitive to anti–Fas agonistic antibodies (Figure 6A). Strikingly, U266 cells were sensitive instead to Apo2L, which induced apoptosis at a concentration as low as 100 ng/mL (Figure 6A). RPMI-8226 cells were sensitive to both Apo2L and anti–Fas agonistic antibodies. Induction of apoptosis by Apo2L was specific because soluble Fc Apo2L receptor (Fc-DR5) was able to prevent Apo2L-induced apoptosis (data not shown). Furthermore, Apo2L but not Fas mAb, induced Bcl-2 and caspase 3 cleavage in U266 (Figure 6B). Freshly isolated plasma cells from a MM patient were also responsive to Apo2L, whereas the non-plasma cells from the same patient did not undergo apoptosis following Apo2L treatment (Table 1). Apo2L was also able to induce caspase 3 and Bcl-2 cleavage (Figure 6C).
Figure 6. Apo2L and Fas induce apoptosis.

(A). U266 (left) and RPMI-8266 (right) were treated with Apo2L and anti–Fas mAb (CH 11) and examined for induction of apoptosis by Hoechst staining. U266 (B) and primary MM plasma cells (C) were treated with Apo2L for 48 hours and then subjected to Western blotting for caspase 3 and Bcl-2. The concentration of Apo2L and Fas is given as ×100 ng/mL.
To examine the Apo2L contribution to cell death, we stably expressed a FLAG epitope-tagged dominant-negative Apo2L receptor, DR5Δ (TRAIL-R2DN), into U266 cells. As shown in Figure 7A, treatment with IFN-α, -β, or Apo2L killed 15% to 35% of U266 cells, as measured by Hoechst 33258 staining. In contrast, DR5Δ-expressing cells were partially resistant to the cytotoxic effect of all agents, with only 5% to 8% of cells being killed by IFN-α, -β, or Apo2L treatments. In addition, Bcl-2 cleavage induced by IFN-α, -β, and Apo2L was also prevented (Figure 7B). These results indicate that Apo2L induction is required for IFN-induced apoptosis and that Apo2L could become useful for treating MM.
Figure 7. DR5Δ blocks IFN-induced apoptosis.
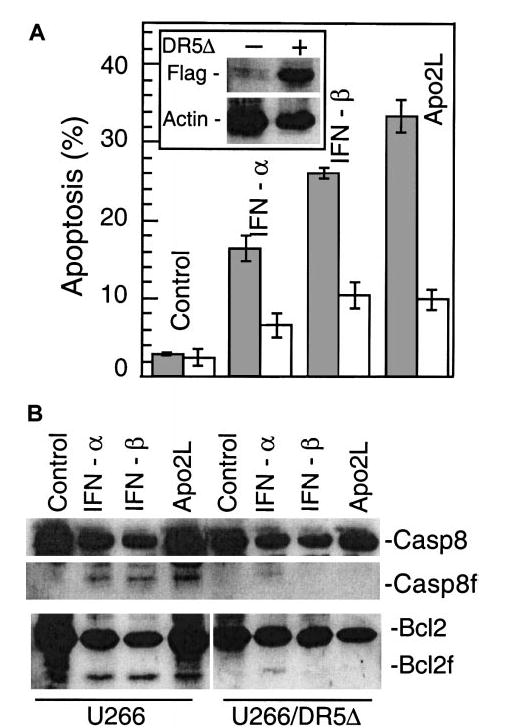
U266 cells were transfected with pcDNA3-DR5Δ (residues 1-268). DR5Δ (also called TRAIL-R2) lacks the death domain and has been shown to function as a dominant-negative molecule inactivating the function of the endogenous DR5 (alsoTRAIL-R2).35 DR5Δ also contains a FLAG epitope-tag that facilitates examination of its expression levels (A). Parental and DR5Δ-FLAG–containing U266 cells were treated with Apo2L (100 ng/mL), IFN-α, and -β (200 U/mL), and apoptosis was determined at 72 hours by Hoechst 33258 staining and expressed as the percentage of the total cells. At least 200 cells were counted for each sample. Expression of DR5Δ was determined by Western blotting using an anti-FLAG antibody and β-actin as a protein loading control (inset). In a parallel experiment (B), the cells were also lysed and subjected to Western blotting for caspase 8 and Bcl-2, upper and lower panel, respectively.
Discussion
In an effort to characterize the molecule(s) responsible for IFN-induced apoptosis of MM, we identified Apo2L as a critical determinant. Our data show that IFNs induce up-regulation of Apo2L, which is responsible, at least partially, for apoptosis. Apo2L mRNA was induced dramatically as early as 4 hours after treatment of U266 cells. We previously reported that the promoter region of Apo2L contains 2 IFN-stimulated regulatory elements, and IFNs can significantly induce the promoter activity of this gene.41 Western blotting confirmed that the Apo2L protein was induced as early as 6 hours and continued to accumulate during IFN-induced apoptosis. The induction of Apo2L is of functional significance because U266 cells were sensitive to Apo2L but not Fas agonistic antibodies. Moreover, a dominant-negative Apo2L receptor, DR5Δ, was able to partially block IFN and Apo2L-induced Bcl-2 cleavage and apoptosis.
We delineate here, for the first time, the signaling pathway for IFN-induced apoptosis in MM (Figure 8). Apo2L induction is followed by caspase 8 activation, Bid cleavage, cyt c release, caspase 3 activation, and the subsequent mitochondrial dysfunction. Bid was partially converted to the p15 and p13 fragments at a stage prior to the appearance of apoptotic morphology but coinciding with the initial cyt c release and caspase 3 activation. In contrast, Bcl-2 cleavage was detected only at 48 hours, a time corresponding to the amplification of cyt c release. It seems that Bid and Bcl-2 cleavage are functionally linked to the initial and augmented cyt c release, respectively. It was recently demonstrated that Apo2L could activate caspase 8 and downstream events of apoptosis.28,29 It has been established that Bid is cleaved during receptor-mediated caspase 8 activation, with the truncated product being targeted to mitochondria and causing cyt c release.22,23 Using isolated mitochondria from mouse liver, we confirmed25 that the truncated Bid induces cyt c release without jeopardizing the mitochondrial functions, as indicated by maintenance of Δψm and preservation of integrity of mitochondrial membrane transition pore. (T. Xia and Q. Chen, unpublished data, January 2001). We and others have reported that Bcl-2 cleavage is an important component of the positive feedback loop that provides the link between caspases and mitochondrial dysfunction.13,42 Consistent with previous observations, our data here suggest that Bcl-2 is cleaved downstream of caspase activation, indicating a feedback amplification of caspase-induced mitochondrial dysfunction in IFN-induced apoptosis. This is reminiscent of the distinct stages of cyt c release we recently described during genotoxic stress-induced apoptosis.13 To support this observation, we and others have observed that transfection of truncated Bcl-2 without the BH-4 domain indeed induces cyt C release and subsequent apoptosis43 (Q. Chen and A. Almasan, unpublished data, February 2001). In both cases, there are distinct initial and amplification stages of cyt c release, with the amplification step being accomplished through a positive feedback loop involving caspase-induced Bcl-2 cleavage. This 2-stage cyt c release model reconciles the dual function of mitochondria as a powerhouse for the diverse cellular metabolic needs and as a source for critical factors required for initiation of apoptosis, such as cyt c and caspase 9. Together, our data further imply that Apo2L uses mitochondria to activate the apoptosis process. The exact contribution of Apo2L-induced cyt c release by caspases to this positive feedback amplification remains to be defined.
Figure 8. Model for activation of apoptosis in MM by IFNs.
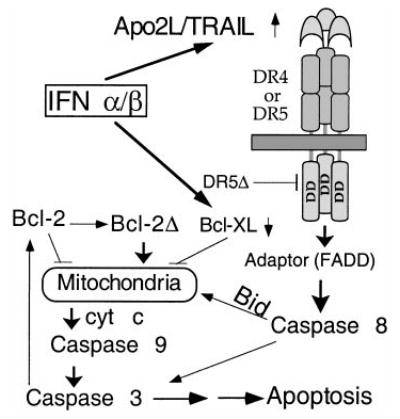
Following transcriptional induction by IFNs, Apo2L binds to its receptor DR5 (or DR4) and, through an adaptor intermediate (FADD), recruits caspase 8 to the cell membrane. Following caspase 8 activation by proteolysis, Bid is cleaved and translocates to mitochondria, causing release of low levels of cyt c into the cytosol, leading to caspase 9 and 3 activation. This results in attack of the anti-apoptotic protein Bcl-2 on the mitochondrial membranes, producing a truncated Bcl-2Δ protein that causes release of more cyt c, caspase activation, and apoptosis. Bcl-xL transcriptional down-regulation is an additional mechanism by which IFNs may decrease levels of anti-apoptotic proteins, shifting the balance toward a pro-apoptotic state.
Apo2L and FasL could use distinct pathways to induce apoptosis, because Bid44 and caspase 845 knock-out mice are resistant to Fas-induced apoptosis in hepatocytes but sensitive to other types of death stimuli, including Apo2L. Our results show that U266 cells are sensitive to Apo2L but not to Fas agonistic mAb, further suggesting that there are distinct apoptotic pathways activated by FasL and Apo2L in our system. It was reported that, even though the Fas receptor was also induced by IFNs, U266 cells were insensitive to FasL, probably because of their intrinsic high level of Bcl-XL.46 Our findings are consistent with reports that, although U266 cells are resistant to Fas, they are sensitive to IFN47 or Apo2L.48 In fact, it has been reported that Fas-mediated apoptosis of MM cell lines required cotreatment with IFN-α.47 This approach could not distinguish the apoptotic effects of IFNs and Fas, because IFNs induce apoptosis in U266 cells. Our data imply that the lack of sensitivity of U266 cells to Fas is due to their deficiency in Fas signaling components toward apoptosis rather than the deregulated Bcl-XL levels. We observed that IFN-α but not -γ induced down-regulation of Bcl-XL, an inhibitor of apoptosis. This observation may explain, in part, why IFN-γ cannot induce apoptosis, even though Apo2L is slightly induced at an earlier time point. It is not known precisely how down-regulation of Bcl-XL contributes to IFN-induced cyt c release and mitochondrial dysfunction, but it is likely to shift the balance in favor of apoptosis. IFNs affect the expression of a number of genes, which are not only associated with apoptosis but also with cell cycle progression.49
IFNs play an important role in the immune system. One mechanism by which IFNs may achieve their role in immune surveillance could be through regulation of other cytokines, which, depending on the cell type, may induce cell cycle arrest or activate apoptosis. There are reports indicating that IFNs could also cause cell cycle arrest and activate other signaling pathways, such as mitogen-activated protein kinase50 or protein kinase C.49 Our data provide evidence that IFNs exert their profound effect by inducing apoptosis in MM and suggest that Apo2L may be an important mediator for these effects, although it may not be the sole molecule responsible for IFN-induced apoptosis. It has been reported that interleukin 6 (IL-6)51 and RNA-dependent protein kinase (PKR) may also be involved in regulation of IFN-induced apoptosis in MM. It remains to be determined whether PKR,52 IL-6 down-regulation,51 or other IFN-induced genes also have a role in apoptosis in our system, as our results do not exclude their possible involvement.
There is a heterogeneous IFN response in different cell types, or even within the same type of cells. We also examined other cell lines for their sensitivity to IFNs, Apo2L, and Fas agonistic antibody. RPMI-8226 and NCI-H929, other typical MM cell lines, were sensitive to IFN-α and -β but not -γ. In contrast, Molt-4, Jurkat, and IM-9 cells did not undergo apoptosis on treatment with IFNs. Interestingly, we found that IFNs induced Apo2L gene expression in IM-9, Molt-4, and Jurkat cells, although these cells do not die on treatment with IFNs. It is possible that Apo2L induction is necessary but not sufficient to induce apoptosis. Also it is likely that the responsiveness to IFNs is cell-type dependent. Indeed, the tumor responsiveness to IFN therapy may vary with cell types and from patient to patient. IFNs induce multiple events that, in concert with Apo2L, may kill these cells. It is reported that, although many human tumor cell lines were reported to be sensitive to Apo2L, most normal cells were not. This apparent protection of normal cells from the cytotoxic effect of Apo2L is believed to be based on a unique set of decoy receptors (DcRs); these cells either lack the DcR1 (also TRAIL-R3/LIT35,53–55) or have a truncated DcR2 (also TRAIL-R4/TRUNDD56,57), so they are unable to signal, but they compete instead for receptor binding to Apo2L. There is a clear need to understand the mechanism of IFN sensitivity in different systems and to examine Apo2L expression in other tumor cell types. Further studies are required to examine whether other signaling components of the Apo2L pathway, such as its DcRs, or inhibitors are regulated by IFNs.
In summary, we delineated the signaling pathways of IFN-induced apoptosis in MM and identified Apo2L as an important mediator, with mitochondria, caspases, and Bcl-2 family proteins playing an important role in this process. Importantly, we show that IFNs induce Apo2L in plasma cells of MM patients. We also confirmed a previous report58 that MM (CD38+/CD45−/dim), but not nonplasma blood cells, are sensitive to Apo2L. It is reported that Apo2L can induce apoptosis of an intermediate subtype of erythroblasts, but not of immature and mature erythroblast, an effect which could be protected by erythropoietin.59 It remains to be determined whether IFNs can induce Apo2L during MM therapy or whether Apo2L can induce MM cell death in vivo. Given the lack of cytotoxicity of Apo2L toward most other type of blood cells, as well as other cell types in mice60 and nonhuman primates,61 it would be of interest to examine whether Apo2L could be used for treating MM patients. The molecular mechanisms of cell sensitivity to IFNs, Apo2L, and Fas and their application to clinical treatment of MM warrant further investigation. Clearly, further delineation of the molecular signaling pathway to IFN-induced apoptosis holds promises to address these issues. Our findings support a rationale to explore Apo2L in the management of MM, a novel alternative for this currently incurable disease.
Acknowledgments
We thank Drs X. Wang (UT Southwestern), E. S. Alnemri, and S. M. Srinivasula (T. Jefferson) for the Bid antibodies and pCDNA3-DR5Δ construct; P. Rayman and T. Bloom (laboratory of Dr J. Finke, Department of Immunology, Cleveland Clinic) for advice and help with isolation and examination of MM cells; and A. Raber, C. Stanko, and S. Stahl (Flow Cytometry Core facility of Cleveland Clinic) for flow cytometry.
Footnotes
Supported in part by research grants CA81504 and CA82858 from the National Cancer Institute and by the Radiological Society of North America (A.A).
References
- 1.Hallek M, Leif Bergsagel P, Anderson KC. Multiple myeloma: increasing evidence for a multistep transformation process. Blood. 1998;91:3–21. [PMC free article] [PubMed] [Google Scholar]
- 2.Mellstedt H, Aahre A, Bjorkholm M, et al. Interferon therapy in myelomatosis. Lancet. 1979;2:697. doi: 10.1016/s0140-6736(79)92099-3. [DOI] [PubMed] [Google Scholar]
- 3.Kyle RA. Maintenance therapy and supportive care for patients with multiple myeloma. Semin Oncol. 1999;26:35–42. [PubMed] [Google Scholar]
- 4.Oken MM, Leong T, Lenhard RE, Jr, et al. The addition of interferon or high dose cyclophosphamide to standard chemotherapy in the treatment of patients with multiple myeloma: phase III Eastern Cooperative Oncology Group Clinical Trial EST 9486. Cancer. 1999;86:957–968. [PubMed] [Google Scholar]
- 5.Grander D. How does interferon-alpha exert its antitumour activity in multiple myeloma? Acta Oncol. 2000;39:801–805. doi: 10.1080/028418600750063532. [DOI] [PubMed] [Google Scholar]
- 6.Stark GR, Kerr IM, Williams BR, Silverman RH, Schreiber RD. How cells respond to interferons. Annu Rev Biochem. 1998;67:227–264. doi: 10.1146/annurev.biochem.67.1.227. [DOI] [PubMed] [Google Scholar]
- 7.Alnemri ES, Livingston DJ, Nicholson DW, et al. Human ICE/CED-3 protease nomenclature. Cell. 1996;87:171. doi: 10.1016/s0092-8674(00)81334-3. [DOI] [PubMed] [Google Scholar]
- 8.Gross A, McDonnell JM, Korsmeyer SJ. BCL-2 family members and the mitochondria in apoptosis. Genes Dev. 1999;13:1899–1911. doi: 10.1101/gad.13.15.1899. [DOI] [PubMed] [Google Scholar]
- 9.Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281:1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- 10.Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- 11.Kluck R, Bossy-Wetzel E, Green D, Newmeyer D. The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science. 1997;275:1132–1136. doi: 10.1126/science.275.5303.1132. [DOI] [PubMed] [Google Scholar]
- 12.Yang J, Liu X, Bhalla K, et al. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science. 1997;275:1129–1132. doi: 10.1126/science.275.5303.1129. [DOI] [PubMed] [Google Scholar]
- 13.Chen Q, Gong B, Almasan A. Distinct stages of cytochrome c release from mitochondria: evidence for a feedback amplification loop linking caspase activation to mitochondrial dysfunction in genotoxic stress induced apoptosis. Cell Death Differ. 2000;7:227–233. doi: 10.1038/sj.cdd.4400629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell. 2000;102:33–42. doi: 10.1016/s0092-8674(00)00008-8. [DOI] [PubMed] [Google Scholar]
- 15.Verhagen AM, Ekert PG, Pakusch M, et al. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell. 2000;102:43–53. doi: 10.1016/s0092-8674(00)00009-x. [DOI] [PubMed] [Google Scholar]
- 16.Li P, Nijhawan D, Budihardjo I, et al. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- 17.Zou H, Henzel WJ, Liu X, Lutschg A, Wang X. Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell. 1997;90:405–413. doi: 10.1016/s0092-8674(00)80501-2. [DOI] [PubMed] [Google Scholar]
- 18.Cryns V, Yuan J. Proteases to die for. Genes Dev. 1998;12:1551–1570. doi: 10.1101/gad.12.11.1551. [DOI] [PubMed] [Google Scholar]
- 19.Zamzami N, Marchetti P, Castedo M, et al. Sequential reduction of mitochondrial transmembrane potential and generation of reactive oxygen species in early programmed cell death. J Exp Med. 1995;182:367–377. doi: 10.1084/jem.182.2.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Adams JM, Cory S. The Bcl-2 protein family: arbiters of cell survival. Science. 1998;281:1322–1326. doi: 10.1126/science.281.5381.1322. [DOI] [PubMed] [Google Scholar]
- 21.Chinnaiyan A, Orth K, O’Rourke K, et al. Molecular ordering of the cell death pathway. Bcl-2 and Bcl-xL function upstream of the CED-3-like apoptotic proteases. J Biol Chem. 1996;271:4573–4576. doi: 10.1074/jbc.271.9.4573. [DOI] [PubMed] [Google Scholar]
- 22.Luo X, Budihardjo I, Zou H, Slaughter C, Wang X. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell. 1998;94:481–490. doi: 10.1016/s0092-8674(00)81589-5. [DOI] [PubMed] [Google Scholar]
- 23.Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94:491–501. doi: 10.1016/s0092-8674(00)81590-1. [DOI] [PubMed] [Google Scholar]
- 24.Gross A, Yin XM, Wang K, et al. Caspase cleaved BID targets mitochondria and is required for cytochrome c release, while BCL-XL prevents this release but not tumor necrosis factor-R1/Fas death. J Biol Chem. 1999;274:1156–1163. doi: 10.1074/jbc.274.2.1156. [DOI] [PubMed] [Google Scholar]
- 25.Shimizu S, Tsujimoto Y. Proapoptotic BH3-only Bcl-2 family members induce cytochrome c release, but not mitochondrial membrane potential loss, and do not directly modulate voltage-dependent anion channel activity. Proc Natl Acad Sci U S A. 2000;97:577–582. doi: 10.1073/pnas.97.2.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen Q, Takeyama N, Brady G, Watson AJM, Dive C. Blood cells with reduced mitochondrial membrane potential and cytosolic cytochrome c can survive and maintain clonogenicity given appropriate signals to suppress apoptosis. Blood. 1998;92:4545–4553. [PubMed] [Google Scholar]
- 27.Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science. 1998;281:1305–1308. doi: 10.1126/science.281.5381.1305. [DOI] [PubMed] [Google Scholar]
- 28.Bodmer JL, Holler N, Reynard S, et al. TRAIL receptor-2 signals apoptosis through FADD and caspase-8. Nat Cell Biol. 2000;2:241–243. doi: 10.1038/35008667. [DOI] [PubMed] [Google Scholar]
- 29.Kischkel FC, Lawrence DA, Chuntharapai A, et al. Apo2L/TRAIL-dependent recruitment of endogenous FADD and caspase-8 to death receptors 4 and 5. Immunity. 2000;12:611–620. doi: 10.1016/s1074-7613(00)80212-5. [DOI] [PubMed] [Google Scholar]
- 30.Gong B, Almasan A. Apo2 ligand/TNF-related apoptosis-inducing ligand and death receptor 5 mediate the apoptotic signaling induced by ionizing radiation in leukemic cells. Cancer Res. 2000;60:5754–5760. [PubMed] [Google Scholar]
- 31.Cheng EH, Levine B, Boise LH, Thompson CB, Hardwick JM. Bax-independent inhibition of apoptosis by Bcl-XL. Nature. 1996;379:554–556. doi: 10.1038/379554a0. [DOI] [PubMed] [Google Scholar]
- 32.Borset M, Hjertner O, Yaccoby S, Epstein J, Sanderson RD. Syndecan-1 is targeted to the uropods of polarized myeloma cells where it promotes adhesion and sequesters heparin-binding proteins. Blood. 2000;96:2528–2536. [PubMed] [Google Scholar]
- 33.Gong B, Chen Q, Endlich B, Mazumder S, Almasan A. Ionizing radiation-induced, Bax-mediated cell death is dependent on activation of serine and cysteine proteases. Cell Growth Diff. 1999;10:491–502. [PubMed] [Google Scholar]
- 34.Martin SJ, Reutelingsperger CP, McGahon AJ, et al. Early redistribution of plasma membrane phosphatidylserine is a general feature of apoptosis regardless of the initiating stimulus: inhibition by overexpression of Bcl-2 and Abl. J Exp Med. 1995;182:1545–1556. doi: 10.1084/jem.182.5.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.MacFarlane M, Ahmad M, Srinivasula SM, et al. Identification and molecular cloning of two novel receptors for the cytotoxic ligand TRAIL. J Biol Chem. 1997;272:25417–25420. doi: 10.1074/jbc.272.41.25417. [DOI] [PubMed] [Google Scholar]
- 36.Vander Heiden MG, Chandel NS, Williamson EK, Schumacker PT, Thompson CB. Bcl-xL regulates the membrane potential and volume homeostasis of mitochondria. Cell. 1997;91:627–637. doi: 10.1016/s0092-8674(00)80450-x. [DOI] [PubMed] [Google Scholar]
- 37.Gong B, Chen Q, Almasan A. Ionizing radiation stimulates mitochondrial gene expression and activity. Radiat Res. 1998;150:505–512. [PubMed] [Google Scholar]
- 38.Gong B, Almasan A. Differential upregulation of p53-responsive genes by genotoxic stress in hematopoietic cells containing wild-type and mutant p53. Gene Expr. 1999;8:197–206. [PMC free article] [PubMed] [Google Scholar]
- 39.Pellat-Deceunynk C, Amiot M, Bataille R, et al. Human myeloma cell lines as a tool for studying the biology of multiple myeloma: a reappraisal 18 years after. Blood. 1995;86:4001–4002. [PubMed] [Google Scholar]
- 40.Chauhan D, Pandey P, Ogata A, et al. Cytochrome c-dependent and -independent induction of apoptosis in multiple myeloma cells. J Biol Chem. 1997;272:29995–29997. doi: 10.1074/jbc.272.48.29995. [DOI] [PubMed] [Google Scholar]
- 41.Gong B, Almasan A. Genomic organization and transcriptional regulation of the human Apo2L/TRAIL gene. Biochem Biophys Res Commun. 2000;278:747–752. doi: 10.1006/bbrc.2000.3872. [DOI] [PubMed] [Google Scholar]
- 42.Cheng EHY, Kirsch DG, Clem RJ, et al. Conversion of Bcl-2 to a Bax-like death effector by caspases. Science. 1997;278:1966–1968. doi: 10.1126/science.278.5345.1966. [DOI] [PubMed] [Google Scholar]
- 43.Kirsch DG, Doseff A, Chau BN, et al. Caspase-3-dependent cleavage of Bcl-2 promotes release of cytochrome c. J Biol Chem. 1999;274:21155–21161. doi: 10.1074/jbc.274.30.21155. [DOI] [PubMed] [Google Scholar]
- 44.Yin XM, Wang K, Gross A, et al. Bid-deficient mice are resistant to Fas-induced hepatocellular apoptosis. Nature. 1999;400:886–891. doi: 10.1038/23730. [DOI] [PubMed] [Google Scholar]
- 45.Varfolomeev EE, Schuchmann M, Luria V, et al. Targeted disruption of the mouse Caspase 8 gene ablates cell death induction by the TNF receptors, Fas/Apo1, and DR3 and is lethal prenatally. Immunity. 1998;9:267–276. doi: 10.1016/s1074-7613(00)80609-3. [DOI] [PubMed] [Google Scholar]
- 46.Catlett-Falcone R, Landowski TH, Oshiro MM, et al. Constitutive activation of Stat3 signaling confers resistance to apoptosis in human U266 myeloma cells. Immunity. 1999;10:105–115. doi: 10.1016/s1074-7613(00)80011-4. [DOI] [PubMed] [Google Scholar]
- 47.Spets H, Georgii-Hemming P, Siljason J, Nilsson K, Jernberg-Wiklund H. Fas/APO-1 (CD95)-mediated apoptosis is activated by interferon-gamma and interferon- in interleukin-6 (IL-6)-dependent and IL-6-independent multiple myeloma cell lines. Blood. 1998;92:2914–2923. [PubMed] [Google Scholar]
- 48.Mariani SM, Matiba B, Armandola EA, Krammer PH. Interleukin 1 beta-converting enzyme related proteases/caspases are involved in TRAIL-induced apoptosis of myeloma and leukemia cells. J Cell Biol. 1997;137:221–229. doi: 10.1083/jcb.137.1.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sangfelt O, Erickson S, Castro J, et al. Induction of apoptosis and inhibition of cell growth are independent responses to interferon-alpha in hematopoietic cell lines. Cell Growth Differ. 1997;8:343–352. [PubMed] [Google Scholar]
- 50.Goh KC, Haque SJ, Williams BR. p38 MAP kinase is required for STAT1 serine phosphorylation and transcriptional activation induced by interferons. EMBO J. 1999;18:5601–5608. doi: 10.1093/emboj/18.20.5601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Frassanito MA, Cusmai A, Iodice G, Dammacco F. Autocrine interleukin-6 production and highly malignant multiple myeloma: relation with resistance to drug-induced apoptosis. Blood. 2001;97:483–489. doi: 10.1182/blood.v97.2.483. [DOI] [PubMed] [Google Scholar]
- 52.Williams BR. PKR; a sentinel kinase for cellular stress. Oncogene. 1999;18:6112–6120. doi: 10.1038/sj.onc.1203127. [DOI] [PubMed] [Google Scholar]
- 53.Degli-Esposti MA, Smolak PJ, Walczak H, et al. Cloning and characterization of TRAIL-R3, a novel member of the emerging TRAIL receptor family. J Exp Med. 1997;186:1165–1170. doi: 10.1084/jem.186.7.1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sheridan JP, Marsters SA, Pitti RM, et al. Control of TRAIL-induced apoptosis by a family of signaling and decoy receptors. Science. 1997;277:818–821. doi: 10.1126/science.277.5327.818. [DOI] [PubMed] [Google Scholar]
- 55.Pan G, Ni J, Wei YF, et al. An antagonist decoy receptor and a death domain-containing receptor for TRAIL. Science. 1997;277:815–818. doi: 10.1126/science.277.5327.815. [DOI] [PubMed] [Google Scholar]
- 56.Degli-Esposti MA, Dougall WC, Smolak PJ, et al. The novel receptor TRAIL-R4 induces NF-kappaB and protects against TRAIL-mediated apoptosis, yet retains an incomplete death domain. Immunity. 1997;7:813–820. doi: 10.1016/s1074-7613(00)80399-4. [DOI] [PubMed] [Google Scholar]
- 57.Marsters SA, Sheridan JP, Pitti RM, et al. A novel receptor for Apo2L/TRAIL contains a truncated death domain. Curr Biol. 1997;7:1003–1006. doi: 10.1016/s0960-9822(06)00422-2. [DOI] [PubMed] [Google Scholar]
- 58.Gazitt Y. TRAIL is a potent inducer of apoptosis in myeloma cells derived from multiple myeloma patients and is not cytotoxic to hematopoietic stem cells. Leukemia. 1999;13:1817–1824. doi: 10.1038/sj.leu.2401501. [DOI] [PubMed] [Google Scholar]
- 59.Griffith TS, Wiley SR, Kubin MZ, et al. Monocyte-mediated tumoricidal activity via the tumor necrosis factor-related cytokine, TRAIL. J Exp Med. 1999;189:1343–1354. doi: 10.1084/jem.189.8.1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Walczak H, Miller RE, Ariail K, et al. Tumoricidal activity of tumor necrosis factor-related apoptosis-inducing ligand in vivo. Nat Med. 1999;5:157–163. doi: 10.1038/5517. [DOI] [PubMed] [Google Scholar]
- 61.Ashkenazi A, Pai RC, Fong S, et al. Safety and antitumor activity of recombinant soluble Apo2 ligand. J Clin Invest. 1999;104:155–162. doi: 10.1172/JCI6926. [DOI] [PMC free article] [PubMed] [Google Scholar]