

Abstract
Subacute sclerosing panencephalitis (SSPE) is a progressive fatal neurodegenerative disease associated with persistent infection of the central nervous system (CNS) by measles virus (MV), biased hypermutations of the viral genome affecting primarily the matrix (M) gene with the conversion of U to C and A to G bases, high titers of antibodies to MV, and infiltration of B cells and T cells into the CNS. Neither the precipitating event nor biology underlying the MV infection is understood, nor is their any satisfactory treatment. We report the creation of a transgenic mouse model that mimics the cardinal features of SSPE. This was achieved by initially infecting mice expressing the MV receptor with lymphocytic choriomeningitis virus Cl 13, a virus that transiently suppressed their immune system. Infection by MV 10 days later resulted in persistent MV infection of neurons. Analysis of brains from infected mice showed the biased U to C hypermutations in the MV M gene and T and B lymphocyte infiltration. These sera contained high titers of antibodies to MV. Thus, a small animal model is now available to both molecularly probe the pathogenesis of SSPE and to test a variety of therapies to treat the disease.
Measles virus (MV) is a negative-stranded RNA virus whose genome contains eight genes and is among the most contagious of infectious agents for humans (1, 2). Although controlled by vaccination in most industrial countries, MV infects >30,000,000 persons, and ∼530,000 die each year (1–3). Of these, from ∼8.5 (4) to 22 (5) per 1,000,000 MV infections develop a persistent infection of the central nervous system (CNS) called subacute sclerosing panencephalitis (SSPE), a chronic fatal neurodegenerative disease for which there is no established treatment. SSPE is identifiable from six clear fingerprints (1, 6): (a) progressive fatal CNS disease; (b) replication of MV in neurons; (c) defective virus that can be recovered by cocultivation with permissive cells; (d) dramatic biased hypermutation in the MV genome primarily affecting one out of the eight genes, the matrix (M) gene, with the conversion of U to C and A to G bases; (e) high titers of antibodies to MV; and (f) infiltration of B cells and T cells into the CNS. Studies of identical human twins infected simultaneously with MV (presumably the same strain) show a discordance, because SSPE developed in only one twin (7–9). Additionally, unimmunized patients who receive intensive immunosuppressive therapy are more vulnerable to SSPE than immunocompetent individuals infected with MV (10, 11), and the majority of SSPE cases occur in children infected before the age of two (1, 6), when the immune system is still immature. These observations suggest that SSPE likely arises after MV infects an immunosuppressed subject. We have tested this hypothesis by humanized mice using a transgenic (tg) approach (12) with one of the MV receptors, CD46. CD46 (13–15) and signal lymphocyte activation molecule (SLAM) (16) are receptors for MV. Of these two, CD46 but not SLAM is present on human neurons (17). Mice that lack CD46 or SLAM are resistant to MV infection. Using such CD46 tg mice, we ask whether continuous and/or transient depletion of the host's immune system is sufficient and essential to allow MV infection a window of opportunity to persistently infect neurons and cause an SSPE-like chronic degenerative CNS disease. We show here that transient suppression of immune response preceeding MV infection mimics the cardinal features of SSPE.
Results AND Discussion
CD46 tg mice on the C57BL/6 background were crossed on to the Rag1−/− background (Rag1−/− C57BL/6 mice) to delete their B and T lymphocytes and, thus, remove their adaptive immune system. When infected with a cloned Edmonston virus (18, 19), all such mice developed a progressive fatal CNS disease documented by heavy MV infection of their neurons (Fig. 1, A, C, and E–H). The mean time to death ± SD was 47 ± 4 d after MV inoculation (range = 28–65 d) for 30 mice. The clinical course consisted of weight loss, ruffled fur, and lethargy, followed by wobbled gait and kyphosis beginning 3–4 d before the mice were killed or died. The importance of the immune system was clear when such CD46 × Rag1−/− mice remained free from infected neurons and CNS disease after reconstitution with 5 × 107 or 107 splenic lymphocytes but not with 5 × 106 cells 3 d before or 3 d after injection of MV (Fig. 1, B and D). Virus could not be isolated directly from primary neuronal cultures obtained from these MV-infected CD46 × Rag1−/− mice, despite the presence of viral RNA and protein. MV was isolated when brain material was cocultivated on permissive Vero cells. Electron microscopy of brains from MV-infected mice showed viral nucleoprotein (N) structures (Fig. 1, I and J) accumulated in neurons but the absence of budding virus particles (not depicted).
Figure 1.
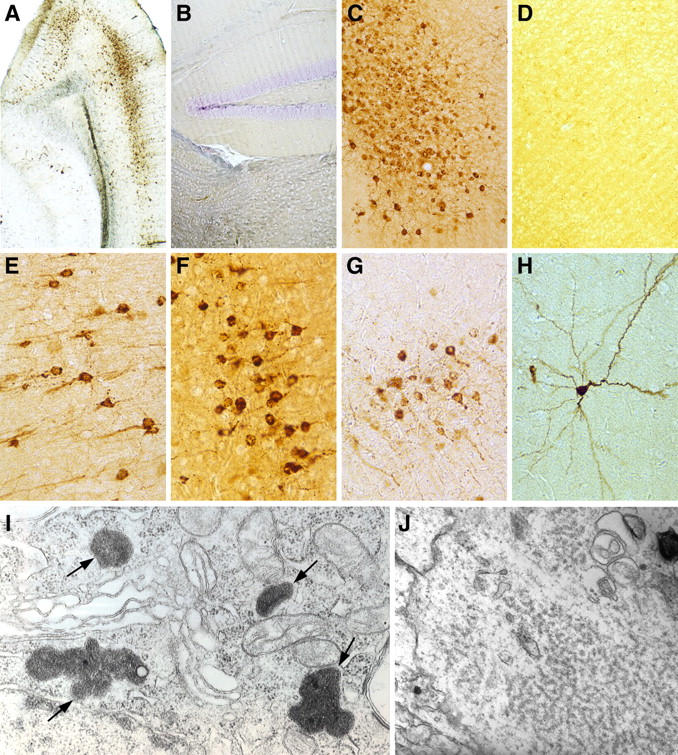
Persistent MV infection in the transgene mice. The SSPE tg mouse model was generated by first creating tg mice expressing the MV receptor CD46 (reference 12) and then breeding these mice on a Rag1−/− background (references 19, 30). (A, C, and E–H) MV replication in individual 6–8-wk-old mice given MV strain Edmonston i.c. All mice died by day 65. Mean time to death ± SD of 47 ± 4 d after viral inoculation. A and C, 50×; E–H, 100×. Equivalent virus replication was seen in all such MV-infected CD46 × Rag1−/− mice throughout the CNS. However, when mice were given 5 × 107 or 107, but not 5 × 106, syngeneic splenocytes either 3 d before or 3 d after MV challenge, 10 out of 10 mice in each group survived >200 d. (B and D) Representative image from the hippocampus (B) and cortex (D) of 10 out of 10 mice receiving 107 or 5 × 107 splenic lymphocytes 3 d before MV challenge, respectively (50×). When five Rag1−/− or five CD46 mice alone were inoculated i.c. with 105 MV, all failed to express MV RNA or protein in the CNS. (I and J) Photomicrographs from an electron microscopic study of an MV-infected neuron in the cortex (I) and hippocampus (J) from a CD46 × Rag1−/− tg mouse. The arrows in I indicate four collections of viral nucleocapsid inclusions (44,800×), whereas J shows cytoplasmic inclusions (78,000×) of loosely coiled viral nucleocapsid helices. In the CNS, only neurons were shown to contain MV antigens.
To determine the genotype of MV within the CNS, total RNA was isolated from the brains of five separate CD46 × Rag1−/− mice infected with MV that developed the SSPE-like disease. Genomic-sense viral RNA was amplified by RT-PCR for the entire M gene sequence, as well as for the complete sequences of the N and fusion (F) protein genes. The full-length cDNAs were cloned into pBS-SK(-) vector, and five M cDNAs and four cDNAs for each N and F gene per mouse were subjected to sequence analysis and compared with sequences from cloned Edmonston virus (18, 19) used to initiate infection.
The mean mutation rates for the N, F, and M genes were 1.75, 1.96, and >10 mutations per 1,000 nucleotides, respectively, or, when normalized to each gene's size, 2.80, 3.28, and >10 point mutations per gene, respectively. These mutation rates are unusually high, because wild-type MVs exhibit a considerably lower mutation rate (20). Important was the preference for A to G and U to C point mutations; these transition mutations accounted for 69, 73, and >95% of all mutations for the N, F, and M genes, respectively. Out of the twelve theoretically possible types of point mutations, this high percentage of A to G and U to C mutations represents a striking mutational bias and suggests the superimposition of mutations induced by a host enzyme, perhaps an adenosine deaminase (6, 21, 22), on the much lower mutation rates associated with MV replication machinery. Sequence analysis comparing M and F genes from cloned Edmonston virus (18, 19) with M and F genes cloned from our virally infected CD46 × Rag1−/− tg mice revealed long stretches of A to G hypermutations in the MV M gene (Fig. 2) and the COOH−-terminal end of the F gene. This was observed with five independent mice that also showed clonal expansion of M genes with biased hypermutations. >50% of all A residues changed to G in the M open reading frame. All mice analyzed possessed Alu I− hypermutated M genes ranging in amount from 5 to ∼40% of the entire M gene population (Fig. 2).
Figure 2.

Anatomical location and frequency of 69 A to G hypermutations. The hypermutations were recorded in the MV M gene open reading frame from one representative mouse out of five studied. 12 cDNA clones were isolated, and each position of A to G changes in the MV M is marked with an asterisk. Data showing the A to G changes from base 215 to 340 are displayed. For our initial set of five MV M sequences from this mouse, one MV M clone was isolated that possessed 20 A to G changes and also ablated a unique Alu I site within the MV M sequence. This observation allowed us to screen for additional Alu I− hypermutated M sequences from this as well as other mice. 11 additional hypermutated MV M sequences were uncovered using the Alu I screen, strongly suggesting that an MV with 20 A to G M gene changes arose, replicated, and underwent further mutation leading to a total number of 69 A to G changes. Quantitation of Alu I− versus Alu I+ MV M clones from this mouse revealed that ∼40% of the MV M clones were hypermutated to A to G changes. The Alu I screen also revealed the presence and clonal expansion of hypermutated M genes in the other mice analyzed. These four other mice had 62, 91, 113, and 135 unique A to G changes, respectively. The clonal expansion of one MV M with a total of 69 A to G mutations is indicated. (bottom) Several of these mutations are shown and compared with the authentic sequence of the inoculated MV. a.a., amino acid.
Thus, several major characteristics of humans with SSPE were mimicked by CD46 × Rag1−/− tg mice infected with MV. Both groups undergo a chronic progressive fatal CNS disease with replication of MV in neurons, and both manifest an electron microscopic profile of “SSPE virus” that shows absence of MV budding and formation of nucleocapsids in a random disorder in the cytosol and nuclei of neurons, as well as the biased hypermutations of U to C and A to G primarily in the MV M gene but also in the F gene (unpublished data). Past attempts to create a rodent model of SSPE by inoculation of MV into newborn or suckling animals instead yielded models of acute or subacute encephalitis (for review see reference 23). Other experiments suggest that the high degree of susceptibility to MV and a variety of other viruses may relate to the immaturity of neurons in young mice and their heightened degree of apoptosis (24).
Because patients with clinical manifestations of SSPE when their disease emerges are not severely immunosuppressed, as Rag1−/− mice are, and also make high titers of antibodies to MV (1, 6–11), we reasoned that a transient immunosuppressive event occurring at the time of or shortly before MV infection might allow the virus a narrow window of opportunity to infect neurons in the CNS. Once infection becomes established, a continuous supply of MV antigens could prime and generate a hyperimmune response to MV. To test this hypothesis, we used lymphocytic choriomeningitis virus (LCMV) Cl 13, which belongs to a family (the arenaviridae) that differs from MV. When LCMV Cl 13 is administered i.v. at a single dose of 2 × 106 PFU into adult immunocompetent mice, it causes a generalized suppression of the immune system because of its ability to infect >75% of DCs (25). Although there is an initial but low generation of antiviral cytotoxic T lymphocytes by day 5 after infection that causes immunopathologic attack on DCs, most infected DCs survive, remain infected, and fail to present antigen and to activate T cells or B cells adequately, as their MHC and costimulatory molecules are down-regulated (25, 26). Suppression of both the humoral and cellular arm of the immune system occurs beginning after day 3 of infection, reaching maximal effect at days 10–14 and waning after day 20; by day 60 after infection, LCMV is usually cleared from the mouse, which is then generally able to generate immune responses. Fig. 3 illustrates that a single hit by MV does not cause CNS disease, nor does a single hit by LCMV Cl 13 (not depicted).
Figure 3.

Data implicating a dual viral hit mechanism for causing persistent MV SSPE-like infection. The first hit is immunosuppressive LCMV Cl 13 given i.v. at a dose of 2 × 106. (A–C) When 105 PFU of MV is administered i.c. 10 d later, the neurons of all inoculated mice become persistently infected with MV. LCMV Cl 13 infects DCs (references 25, 26) and impairs their antigen-presenting capacity so they cannot activate naive T cells or B cells. The result is transient immunosuppression beginning at days 4–5 after LCMV inoculation; peak suppression occurs at days 10–15 and slowly diminishes thereafter, lasting for ∼60 d. A–C are from three individual mice given MV 10 d after LCMV Cl 13 inoculation and killed at 150 (A), 200 (B), and 240 (C) d, respectively, after MV inoculation. Sections are peroxidase stained with antibody to MV, and abundant brown staining neurons are indicated by arrows. (D) Presence of CD4 T cells in the leptomeninges. (E) B cells around blood vessels in the brain parenchyma. (F) CD8 T cells in the brain parenchyma by themselves. Arrowheads in D–F indicate several of these lymphocytes. CD4 and CD8 T lymphocytes and B cells were found in all three locations in four out of four mice studied 200 d after receiving MV and 210 d after receiving LCMV Cl 13.
On the contrary, when LCMV Cl 13 was given initially and MV was given 10 d later, eight out of eight CD46 tg mice (100%) developed a persistent MV infection of neurons (Fig. 3, A–C). Sera harvested from such mice showed heightened titers of antibodies to MV being 10–30-fold higher than immunocompetent mice inoculated with MV. Thus, by ELISA, antibody to MV titers recorded as the reciprocal dilution of sera required to reach a 50% end point were 322 ± 30 versus 14 ± 8, and the neutralization titers were 110 ± 20 versus <10 for MV persistently infected mice compared with noninfected immunized mice, respectively. Results were similar when four additional 8-wk-old CD46 tg mice were given LCMV Cl 13 at day 0 and MV 10 d later. All four developed persistent MV infection of their CNS. Like patients with SSPE, such mice displayed infiltration of predominantly CD4 T cells and B cells, as well as CD8 T cells in their brains (Fig. 3, D–F). In contrast, if MV was given 3 d after LCMV administration, at the time of no or minimal virus-induced immunosuppression (25), none out of the six mice (0%) developed MV-infected neurons. When MV was given 30 d after the initial LCMV infection, only 2 out of 10 mice (20%) develop MV-infected neurons. At this time, the antigen-presenting capacity of DCs as judged in a mixed leukocyte reaction assay was about one-third or one-fourth less suppressive than a mixed leukocyte reaction at day 10 after LCMV Cl 13 infection. Thus, the picture of high MV antibodies, persistent MV infection of the CNS, and B cells and T cells infiltrating the CNS is realized in the dual-hit model. Presumably the continuous replication of MV drives the high antibody response.
The longer-term persistent infection seen with the dual viral hits probably results from two factors. The first is the ability of anti-MV antibodies to prolong the lifespan of MV-infected cells (27), a phenomenon known as antibody-induced antigenic modulation that is believed to exert a similar influence on other viral infections (27). The second is the enhanced U to C, A to G mutations in the M gene occurring in humans with SSPE (1, 6, 21, 22). In support of that later conclusion are observations when reverse genetics was used to swap the hypermutated M gene of the Biken strain from an SSPE patient for the normal nonmutated M gene in Edmonston MV (19). When this Biken M SSPE recombinant MV was inoculated into CD46 × Rag1−/− mice, a protracted progressive infection resulted and death occurred >50 d later than expected (19). Likewise, when we inoculated several additional CD46 × Rag1−/− mice with LCMV Cl 13 first and then with MV 10 d later, the recipients survived up to 220 d after MV infection despite considerable virus replication in their neurons. It has been postulated (6, 19, 21, 22) that the MV sequence variation of U to C and A to G may result from RNA editing by the host enzyme double-stranded RNA adenosine deaminase (ADAR). The opportunity to test this hypothesis is now possible using our CD46 × Rag1−/− SSPE tg mice in which one of the ADAR enzymes (ADAR1a) has been deleted. Such studies are currently underway.
Finally, treatments for SSPE patients remain controversial. Drugs like Amatidine, steroids, Ribavirin, and type I and II IFNs have been tried alone or in combination with either negative or conflicting results (28). Now that an animal model of SSPE is available, testing of various therapies is possible, and a vehicle is at hand to study generation, selection, and molecular bases of mutations in the M gene.
MATERIALS AND METHODS
Mice and viruses.
Generation and genomic characterization of YAC-CD46 and Rag1−/− mice have been described previously, as has breeding of YAC-CD46 tg mice onto the Rag1−/− background (12, 19). All mice used were on the C57BL/6 (H-2b) background. Description, profiling, and quantitation of the MV Edmonston strain and LCMV Cl 13 have been published previously (12, 19, 25). Both viruses used were plaque purified, cloned, and sequenced (18, 29). For MV, 105 PFU of virus was administered intracerebrally (i.c.), and LCMV Cl 13 was given i.v. at a dose of 2 × 106 PFU.
MV was recovered from the brains only after cocultivation on permissive cells like Vero. In brief, suspension of brain tissue was placed on a Ficoll-Hypaque gradient as previously described (12) and cocultivated with Vero cells. Amounts of infectious virus were determined by plaquing log dilutions of supernatant fluids. Cultures from infected brains formed syncytia, and identity of MV was determined on infected Vero cells by immunofluorescence using a specific antibody to MV (12).
Immunochemical, electron microscopic, and antibody detection assays.
Immunochemistry was done on tissue frozen in CME and cut into 40-μM sections with a vibratome (10002; Leica). Sections were placed on roundbottom 96-well plates, fixed for 1 min in 1% paraformaldehyde, and washed. To detect MV antigens, sections were stained with a primary MV-specific polyclonal antibody. To detect infiltrating lymphocytes, the sections were stained with primary antibodies to CD8 (anti–Ly-2 and Ly-3), to CD4 (anti-L3T4), and to B cells (anti-B220). Secondary antibody was biotinylated goat anti–human IgG, followed by avidin–PD conjugate. The color reaction was developed with diaminobenizine in the presence of hydrogen peroxide. Specific details as to antibody specificities, reagents, controls, and the microscope used have been previously published (12, 19).
Antibodies to MV were detected by both ELISA and neutralization assays and followed our previously published reports (12). To detect neutralizing antibody, stock MV was diluted to 600 PFU and mixed with sera obtained before MV inoculation or sera obtained at the time of death from MV-inoculated mice. Sera was heat inactivated at 56°C for 30 min and then diluted at ratios of 1:10, 1:20, 1:50, 1:100, 1:200, and 1:500 and mixed with 600 PFU of input virus. After a 30-min incubation at 37°C, the mixture was placed on ice for 10 min before being added to six-well Petri dishes containing adherent Vero cells. After a 30-min incubation period, 1% agar in RPMI 1640 media containing 7% heat-inactivated FCS was added, and plates were placed in a CO2 incubator and followed daily for the formation of syncytia. Plates were fixed and plaques were counted as described previously (12, 19).
For electron microscopy, the infected mouse brain was divided sagitally, and one half was processed for examination by light microscopic immunocytology to localize sites of MV expression. The other half was fixed in 2% glutaraldehyde in phosphate-buffered saline, postfixed in 1% OSO4, dehydrated, and embedded in Eri 812 resin. Thin sections were stained with uranyl and lead salts and examined in an electron microscope (100CX; JEOL) at 80 kV.
Genotyping MV within the CNS.
Total RNA was isolated from the CNS of MV-infected brains or MV-infected HeLa cells and genomic-sense viral RNA amplified by RT-PCR for the complete sequences of M, N, and F genes. Full-length cDNAs were cloned into pBS-SK(-) vector.
Alu I screen for hypermutated sequences.
Hypermutation of the M gene commonly knocked out a unique Alu I site, allowing an enrichment for additional hypermutated M gene sequences. The specific clonability of M inserts was lower than those for the N and F clones, suggesting that hypermutated sequences are inherently unstable in DH5α, the bacteria used initially for cloning. Subsequent M cDNAs were cloned with Stbl2 competent bacteria (Invitrogen). M gene cDNAs derived from four randomly chosen MV-infected mice were subjected to an Alu I digest, and full-length Alu I− cDNAs were gel purified, inserted into pBS-SK(-), and amplified in the Stbl2 competent bacteria for sequence determination. Because all Alu I− cDNAs analyzed possessed hypermutations and exhibited similar specific clonability as the N and F genes in Stbl2 bacteria, the Alu I digest was also used to approximate the proportion of hypermutated M genes within the M gene population of the five individual mouse brains. M gene cDNAs were generated by RT-PCR and digested with Alu I, after which the products were separated by agarose gel electrophoresis. Each of the resultant DNA bands was gel purified and quantified in duplicate spectroscopically to determine the proportion of Alu I− to Alu I+ M genes within the original population.
Acknowledgments
S. Dales wishes to acknowledge the general hospitality of Günter Blobel, and L. Martin acknowledges the technical assistance of Megan Welch.
This is publication 17330-NP from the Department of Molecular and Integrative Neurosciences and the Department of Infectology of the Scripps Research Institute. This work was supported in part by National Institutes of Health (NIH) grant AI036222. L. Martin was supported by NIH training grants AG00080 and NS41219.
The authors have no conflicting financial interests.
References
- 1.Griffin, D. 2001. Measles virus. Fields Virology. B. Fields, D. Knipe, and P. Howley, editors. Lippincott-Raven, Philadelphia. 1401–1442.
- 2.Oldstone, M.B.A. 1998. Viruses, Plagues, and History. Oxford University Press, New York, NY. 211 pp.
- 3.Centers for Disease Control and Prevention. 2005. Progress in reducing measles mortality—worldwide, 1999-2003. Morb. Mortal. Wkly. Rep. 54:200–203. [PubMed] [Google Scholar]
- 4.Centers for Disease Control and Prevention. 1982. Subacute sclerosing panencephalitis surveillance—United States. Morb. Mortal. Wkly. Rep. 31:585–588. [PubMed] [Google Scholar]
- 5.Bellini, W.J., J.S. Rota, L.E. Lowe, R.S. Katz, P.R. Dyken, S.R. Zaki, W.-J. Shieh, and P.A. Rota. 2005. Subacute sclerosing panencephalitis: more cases of this fatal disease are prevented by measles immunization than previously recognized. J. Infect. Dis. In press. [DOI] [PubMed] [Google Scholar]
- 6.Cattaneo, R., A. Schmid, D. Eschle, K. Baczko, V. ter Meulen, and M.A. Billeter. 1988. Biased hypermutation and other genetic changes in defective measles viruses in human brain infections. Cell. 55:255–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dhib-Jalbut, S., and F.S. Haddad. 1984. Subacute sclerosing panencephalitis in one member of identical twins. Neuropediatrics. 15:49–51. [DOI] [PubMed] [Google Scholar]
- 8.Cianchetti, C., M.G. Marrosu, P.E. Manconi, M. Loi, and A. Cao. 1983. Subacute sclerosing panencephalitis in only one of identical twins. Case report with study of cell-mediated immunity. Eur. Neurol. 22:428–432. [DOI] [PubMed] [Google Scholar]
- 9.Houff, S.A., D.L. Madden, and J.L. Sever. 1979. Subacute sclerosing panencephalitis in only one of identical twins. A seven-year follow-up. Arch. Neurol. 36:854–856. [DOI] [PubMed] [Google Scholar]
- 10.Fukuya, H., M. Ohfu, Y. Tomoda, K. Nibu, S. Ichiki, and A. Midsudome. 1992. A case of subacute sclerosing panencephalitis developing 8 years after immunosuppressive treatment for acute lymphocytic leukemia. No. To. Hattatsu. 24:60–64. [PubMed] [Google Scholar]
- 11.Coulter, J.B., N. Balch, and P.V. Best. 1979. Subacute sclerosing panencephalitis after drug-induced immunosuppression. Arch. Dis. Child. 54:640–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oldstone, M.B.A., H. Lewicki, D. Thomas, A. Tishon, S. Dales, J. Patterson, M. Manchester, D. Homann, and A. Holz. 1999. Measles virus infection in a transgenic model: virus-induced central nervous system disease and immunosuppression. Cell. 98:629–640. [DOI] [PubMed] [Google Scholar]
- 13.Naniche, D., G. Varior-Krishnan, F. Cervoni, T.F. Wild, B. Rossi, C. Rabourdin-Combe, and D. Gerlier. 1993. Human membrane cofactor protein (CD46) acts as a cellular receptor for measles virus. J. Virol. 67:6025–6032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dorig, R., A. Marcel, A. Chopra, and C.D. Richardson. 1993. The human CD46 molecule is a receptor for measles virus (Edmonston strain). Cell. 75:295–305. [DOI] [PubMed] [Google Scholar]
- 15.Manchester, M., M.K. Liszewski, J.P. Atkinson, and M.B.A. Oldstone. 1994. Multiple isoforms of CD46 (membrane cofactor protein) serve as receptors for measles virus. Proc. Natl. Acad. Sci. USA. 91:2161–2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tatsuo, H., N. Ono, K. Tanaka, and Y. Yanagi. 2000. SLAM (CDw150) is a cellular receptor for measles virus. Nature. 406:893–897. [DOI] [PubMed] [Google Scholar]
- 17.Oldstone, M.B., D. Homann, H. Lewicki, and D. Stevenson. 2002. One, two, or three step: measles virus receptor dance. Virology. 299:162–163. [DOI] [PubMed] [Google Scholar]
- 18.Radecke, F., P. Spielhofer, H. Schneider, K. Kaelin, M. Huber, C. Dotsch, G. Christiansen, and M.A. Billeter. 1995. Rescue of measles viruses from cloned DNA. EMBO J. 14:5773–5784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Patterson, J.B., T.I. Cornu, J. Redwine, S. Dales, H. Lewicki, A. Holz, D. Thomas, M.A. Billeter, and M.B.A. Oldstone. 2001. Evidence that the hypermutated M protein of a subacute sclerosing panencephalitis measles virus actively contributes to the chronic progressive CNS disease. Virology. 291:215–225. [DOI] [PubMed] [Google Scholar]
- 20.Parks, C.L., R.A. Lerch, P. Walpita, H.-P. Wang, M.S. Sidhu, and S.A. Udem. 2001. Comparison of predicted amino acid sequences of measles virus strains in the Edmonston vaccine lineage. J. Virol. 75:910–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bass, B.L., H. Weintraub, R. Cattaneo, and M.A. Billeter. 1989. Biased hypermutation of viral RNA genomes could be due to unwinding/modification of double-stranded RNA. Cell. 56:331. [DOI] [PubMed] [Google Scholar]
- 22.Patterson, J.B., and C.E. Samuel. 1995. Expression and regulation by interferon of a double-stranded RNA-specific adenosine deaminase from human cells: evidence for two forms of the deaminase. Mol. Cell. Biol. 15:5376–5388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liebert, U.G., and D. Finke. 1995. Measles virus infections in rodents. Curr. Top. Microbiol. Immunol. 191:149–166. [DOI] [PubMed] [Google Scholar]
- 24.Oliver, K.R., M.F. Scallan, H. Dyson, and J.K. Fazakerley. 1997. Susceptibility to a neurotropic virus and its changing distribution in the developing brain is a function of CNS maturity. J. Neurovirol. 3:38–48. [DOI] [PubMed] [Google Scholar]
- 25.Sevilla, N., S. Kunz, A. Holz, H. Lewicki, D. Homann, H. Yamada, K.P. Campbell, J.C. de la Torre, and M.B.A. Oldstone. 2000. Immunosuppression and resultant viral persistence by specific viral targeting of dendritic cells. J. Exp. Med. 192:1249–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hahm, B., M.J. Trifilo, E.I. Zuniga, and M.B.A. Oldstone. 2005. Viruses evade the immune system through type I interferon-mediated STAT2-dependent, but STAT1-independent, signaling. Immunity. 22:247–257. [DOI] [PubMed] [Google Scholar]
- 27.Oldstone, M.B.A., R.S. Fujinami, and P.W. Lampert. 1980. Membrane and cytoplasmic changes in virus-infected cells induced by interactions of antiviral antibody with surface viral antigen. Prog. Med. Virol. 26:45–93. [PubMed] [Google Scholar]
- 28.Gascon, G.G. 2003. Randomized treatment study of inosiplex versus combined inosiplex and intraventricular interferon-alpha in subacute sclerosing panencephalitis (SSPE): international multicenter study. J. Child Neurol. 18:819–827. [DOI] [PubMed] [Google Scholar]
- 29.Salvato, M., E. Shimomaye, P. Southern, and M.B.A. Oldstone. 1988. Virus-lymphocyte interactions. IV. Molecular characterization of LCMV Armstrong (CTL+) and that of its variant, Clone 13 (CTL−). Virology. 164:517–522. [DOI] [PubMed] [Google Scholar]
- 30.Lawrence, D.M.P., M.M. Vaughn, A.R. Belman, J.S. Cole, and G.F. Rall. 1999. Immune response-mediated protection of adult but not neonatal mice from neuron-restricted measles virus infection and central nervous system disease. J. Virol. 73:1795–1801. [DOI] [PMC free article] [PubMed] [Google Scholar]