

Abstract
Atherosclerosis is the leading cause of death in the United States, and human cytomegalovirus (HCMV), a member of the herpes virus family, may play a role in the development of the disease. We previously showed that HCMV regulated endothelial apoptosis. In this study, we investigated the induction of apoptosis and signal transduction pathways regulating this process in HCMV-infected endothelial cells. As observed previously, HCMV induced a typical cytopathic effect in human aortic endothelial cells (HAECs), ie, the formation of single nucleated or multinucleated giant cells. Although infected HAECs were resistant to apoptosis at earlier stages of infection, they became apoptotic with prolonged infection as demonstrated by positive staining using terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL). This apoptotic process was mediated by the caspase-dependent mitochondrial apoptotic pathway as indicated by increased expression and cleavage of caspases 3 and 9 as well as increased expressions of pro-apoptotic molecules Bax and Bak. Blocking caspases 3 or 9 significantly inhibited the HCMV-induced apoptosis. Further exploration of the upstream pathway demonstrated upregulation of the tumor suppressor p53 gene and activation of the ataxia telangiectasia mutant (ATM) pathway in the infected cells. Blocking p53 inhibited HCMV-stimulated Bax and Bak expression as well as caspase-3 activation and blocking the ATM pathway inhibited HCMV-stimulated p53 activation. Although early infection may render cells antiapoptotic, prolonged infection, however, induced endothelial apoptosis through ATM and p53-dependent activation of the mitochondrial death pathway. This proapoptotic effect may be relevant to endothelial dysfunction and HCMV-associated vascular diseases.
Keywords: cytomegalovirus, endothelium, ATM, p53, apoptosis
Human cytomegalovirus (HCMV) is a member of the Herpesviridae family and infects 50% to 90% of adults in most populations. An increasing body of evidence suggests that HCMV infection may play a role in vascular diseases, such as atherosclerosis and1–4 thrombosis,5,6 and allograft rejection7,8 and restenosis.9,10 Although it is still far from consistent, population studies have generally demonstrated an association between HCMV serum antibody positivity and increased risk of coronary atherosclerosis; HCMV DNA and antigens have been detected in atherosclerotic lesions.11 Furthermore, HCMV infection has been shown to promote atherogenesis in mice.12,13 However, the mechanisms of the pathogenesis are not clear.
HCMV infects many different tissues and organ systems. Endothelium is unique in many ways mediating HCMV-induced pathogenesis. Endothelium is a critical component within the circulatory compartment, which provides a protective barrier and regulates vascular functions in defense against HCMV invasion. Damage to macrovascular and microvascular endothelium is an important event in many forms of vascular injuries. HCMV has an established tropism in a variety of endothelial cells.14,15 Establishment of HCMV propagation within endothelial cells can potentially cause endothelial dysfunction. HCMV infection may initiate endothelial inflammation and vasculitis, which form part of the atherosclerotic or restenotic processes.10,13,16,17 HCMV infection disturbs endothelial integrity by increased expressions of endothelial surface adhesion molecules that are responsible for leukocyte migration and adhesion to vessel wall.13,18,19 Moreover, HCMV infection is capable of changing endothelium from an anticoagulant to a procoagulant status.20,21 Furthermore, endothelium is a reservoir for HCMV latency development.22 Reactivation of the latent HCMV in these host endothelial cells can immediately cause endothelial damage in large and small vessels—an event that preludes to many forms of vascular injuries. Therefore, understanding the molecular events during active and latent infections of endothelium is critical to the understanding of HCMV-induced vascular diseases.
In previous studies, we and others have shown that at earlier stages of HCMV infection (typically the first 72 to 96 hours postinfection, which include the classically defined immediate early, early, and late infection), infected endothelial cells are resistant to apoptotic stimulation such as serum starvation or ultraviolet-induced DNA damage.23–25 Inhibition of p53 function either by cytoplasmic sequestration or by direct inhibition on p53 transactivation has been suggested as a responsible mechanism.23–25 However, this process of acute infection may not accurately reflect the chronic in vivo pathogenesis of vascular disease, which generally develops over extended periods of time. In the present study, we have extended our investigation to explore the molecular changes in endothelial cells after prolonged HCMV infection. We observed that prolonged HCMV infection induced endothelial apoptosis. We found that caspase-3, caspase-9, proapoptotic Bax, and Bak were induced in HCMV-infected cells. Furthermore, the induction of apoptosis involves ataxia tel-angiectasia mutant (ATM) -dependent and p53-dependent activation of the mitochondrial death pathway.
Methods
p53 siRNA, Kinase Inhibitor, and Antibodies
p53 siRNA was purchased from Ambion (Austin, Tex). ATM inhibitor caffeine was purchased from Sigma. Caspase inhibitors (caspase-3 inhibitor II, Z-DEVD-FMK; caspase-8 inhibitor II, Z-IETD-FMK; caspase-9 inhibitor I, Z-LEHD-FMK; the negative control Z-FA-FMK) were purchased from EMD Biosciences (San Diego, Calif). For Western blot analysis and immunofluorescence staining, monoclonal and polyclonal antibodies from Cell Signaling (Beverly, Mass) and Santa Cruz Biotechnology (Santa Cruz, Calif) were used.
Cell Culture
Primary human aortic endothelial cells (HAECs) from Cell Applications (San Diego, Calif) were grown in K12 medium, containing 20% fetal bovine serum, penicillin (100 U/mL), streptomycin (100 μg/mL), sodium pyruvate (1 mmol/L), l-glutamine (4 mmol/L), and heparin (30 μg/mL) and supplemented with endothelial cell growth factor (100 μg/mL). Cells cultured up to 5 passages were used in experiments. In all cases, floating and attached cells in each sample were combined for processing at the end of incubation.
Infection of HAECs With HCMV
Three strains of HCMV (AD169 [ATCC VR-538], Towne [ATCC VR-977], and VHL/E [from W.J.W.]) were used in the study. The virus was propagated as previously described.14 HCMV-infected cells were harvested, and the cell-associated virus was released from cells by freeze/thaw cycle. Subconfluent HAEC monolayers were infected with HCMV at multiplicity of infection (MOI) 1 and incubated at 37°C for 1 hour for virus adsorption. The monolayers were then washed 3 times with Dulbecco phosphate-buffered saline (PBS), fresh complete medium was added, and cells were cultured at 37°C in a CO2 incubator. Supernatant and cell fractions were harvested at various postinfection (pi) times.
p53 siRNAs and Endothelial Cell Transfection
Silencing of p53 gene expression in primary aortic endothelial cells was achieved using the siRNA technique. Transfection of HAEC was performed using LipofectAMINE 2000 (Invitrogen, Carlsbad, Calif) according to the manufacturer’s instruction.
Western Blot Analysis
HAECs were collected from mock-infected and HCMV-infected cultures and washed with ice-cold PBS. Cells were lysed in protein lysis buffer (20 mmol/L Tris [pH 7.4], 150 mmol/L NaCl, 1 mmol/L EDTA, 1 mmol/L EGTA, 1% Triton, 2.5 mmol/L sodium pyrophosphate, 1 mmol/L β-glycerol phosphate, 1 mmol/L Na3VO4, 10 βg/mL of each protease inhibitors [aprotinin, leupeptin and pepstatin], and 1 mmol/L phenylmethylsulfonyl fluoride) for 1 hour on ice. Protein concentration was measured by the Bradford method (Bio-Rad). Fifteen μg of protein per lane was separated by 10% or 12% SDS-polyacrylamide gels and transferred to PVDF membranes. The membrane was blocked in 5% nonfat powdered milk in TBST (50 mmol/L Tris, pH 7.5, 150 mmol/L NaCl, 0.05% Tween 20). The membrane was incubated with the primary antibody in 2% powdered milk in TBST, washed extensively with TBST, and then incubated with secondary antirabbit or antimouse horseradish peroxidase-labeled antibody. Bands were visualized with ECL (Amersham Biosciences, Piscataway, NJ) according to the manufacturer’s instruction.
Immunofluorescent Staining and Microscopy
For immunofluorescence assays, cells were grown on glass cover-slips and infected. After infection, cells were washed with PBS, fixed with 4% paraformaldehyde for 10 minutes, and permeabilized with 0.2% Triton X-100 for 5 minutes. The coverslips were blocked with 1% bovine serum albumin, incubated with the primary antibody, washed extensively with PBS, and then incubated with secondary antirabbit or antimouse fluorescein isothiocyanate (FITC) or Texas Red-labeled antibody. One percent bovine serum albumin in PBS was used for blocking nonspecific binding sites and for dilution of primary and secondary antibodies. The DNA dye 4′6′Diamidino-2-phenylindole dihydrochloride (DAPI) was added at a concentration of 0.1 μg/mL and incubated for 15 minutes to counterstain double-stranded DNA in nuclei. The slides were examined with a Leica DMLS epifluorescence microscope equipped with a Leica DC 100 digital camera and the data were analyzed with Image-Pro Plus V4.5 software (Media Cybernetics, Inc).
Apoptosis TUNEL Assay
Apoptosis was detected by terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick-end labeling (TUNEL) using in situ cell death detection kit (Roche Applied Science, Indianapolis, Ind) according to the manufacturer’s instruction. Briefly, infected and mock-infected cells were harvested, washed with PBS, fixed with 4% paraformaldehyde in PBS for 10 minutes, and permeabilized with 0.2% Triton X-100 for 5 minutes. The fixed cells were incubated with TUNEL reaction mixture containing the terminal deoxynucleotidyl transferase at 37°C for 1 hour. Double-stranded DNA in nuclei was counterstained after TUNEL staining with DAPI (0.1 μg/mL). The cells were air-dried, and coverslips were placed on a drop of antifade solution and sealed to slide with mounting solution. Images of nuclear fluorescence were obtained by fluorescence microscopy as described.
Results
Cytopathic Effect on Endothelial Cells at Prolonged HCMV Infection
To investigate whether HCMV infection changes endothelial cell viability/mortality, we infected HAEC with the laboratory-adapted virus strains AD169, Towne, and VHL/E, a clinical isolate whose natural endothelial cytopathogenicity has been preserved by propagation in endothelial cells. Only VHL/E strain demonstrated a high rate of infection at the same level of MOI and clearly showed the typical pathogenicity phenotype. We therefore focused on the VHL/E strain for the rest of this study. At earlier stage of infection (days 1 to 5 pi), the infected cells remained healthy and viable (data not shown). However, the cell viability declined in infected cells at later stage of infection (after 5 days pi) compared with mock-infected negative controls (Figure 1A). We next examined whether the HCMV-induced endothelial morphological changes were caused by apoptosis. Internucleosomal fragmentation of cellular DNA is a hallmark of apoptosis. Thus, we evaluated the effects of HCMV infection on the fragmentation of chromosomal DNA by incorporation of fluoresce-in-dUTP into the 3′-OH of nicked chromosomal DNA (TUNEL analysis). Microscopic analysis of fluorescein-dUTP–labeled DNA showed that infecting HAECs with HCMV induced apoptosis and the apoptosis was specifically confined in the infected cells (Figure 1B). Kinetic studies showed that apoptotic changes started after 5 days pi and increased with time, indicating that prolonged HCMV infection induced endothelial cell apoptosis (Figure 1C and 1D).
Figure 1.
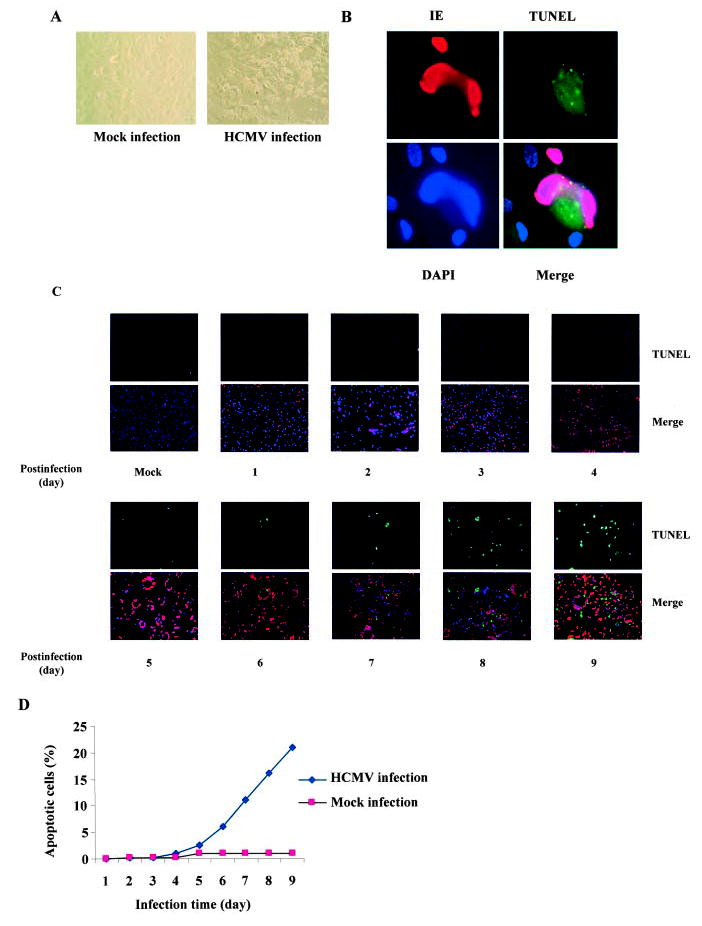
Prolonged HCMV infection induced endothelial apoptosis. A, HCMV infection resulted in substantial cytopathicity in endothelial cells. HAECs were infected with VHL/E strain of HCMV for 9 days and changes in morphology and viability were observed under light microscope. B, Detection of endothelial apoptosis in HCMV-infected HAECs. VHL/E-infected HAECs (day 9 pi) were stained with anti-IE (HCMV immediate early antigen) antibody to indicate infected cells (red), TUNEL (green) for apoptosis, and DAPI (blue) for nuclei. Merged image shows colocalization. C, Time-dependent apoptosis of HCMV-infected HAECs. HAECs were infected with VHL/E for indicated time periods; cells were stained with anti-IE antibody (red), TUNEL (green), and DAPI (blue). Merged images show colocalization. D, Time-dependent death curves of VHL/E-infected HAECs. TUNEL-positive cells and total cells were counted in 5 fields per coverslip. Results were expressed as the average percentage of TUNEL-positive cells over total cells.
Activation of the Caspase Apoptosis Pathway by HCMV Infection
Apoptosis is triggered by caspase-dependent and caspase-independent pathways. In caspase-dependent pathway, apoptotic signals from membrane-associated receptors and mitochondrial sensors converge on a common pathway with the release of cytochrome c from mitochondria and activation of the caspases. Caspase-3 is a terminal caspase effector involved in apoptosis by proteolytically cleaving essential cellular proteins. To investigate whether caspase-3 plays a role in HCMV-induced endothelial apoptosis, we measured caspase-3 expression and activation in HCMV infected cells. As shown in Figure 2A, VHL/E strain-infected cells showed most dramatic changes in expression and activation of caspase-3. Time course showed that caspase-3 (32-kDa) expression noticeably increased 3 days after HCMV infection and was cleaved into 17-kDa to 19-kDa molecules indicating caspase-3 activation. The activation of caspase-3 was specifically in the infected cells as shown in the immunofluorescence stain (Figure 2C). These results indicate that HCMV-induced apoptosis was mediated, at least in part, by caspase-3 activation.
Figure 2.
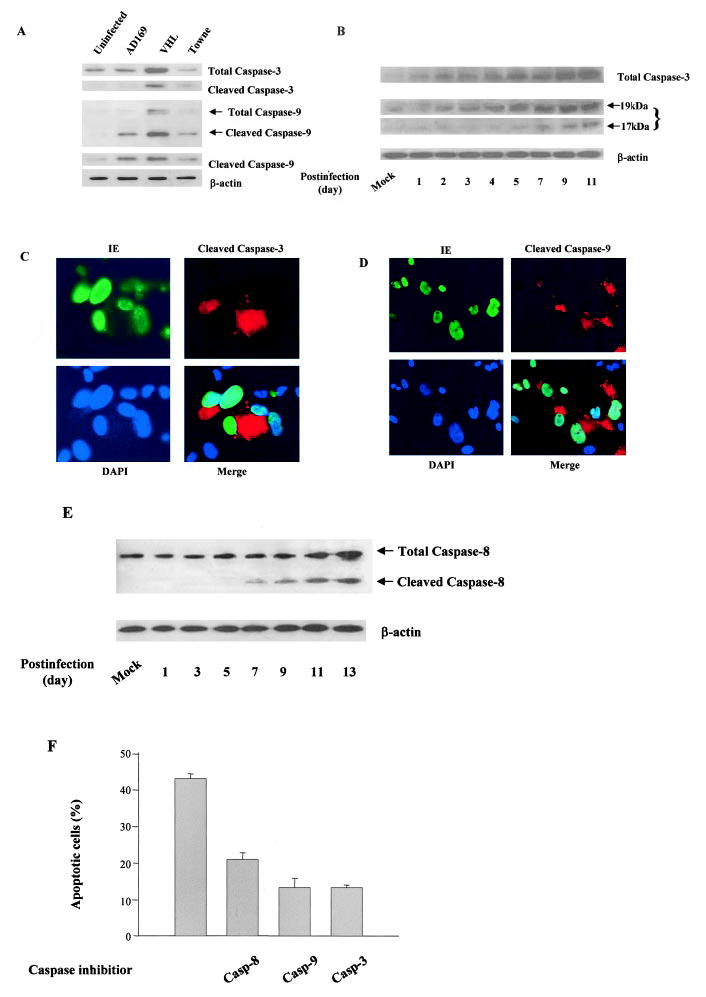
Activation of the mitochondrial apoptosis pathway in HCMV-infected endothelial cells. A, Increased cleavage of caspases-3 and 9 in HCMV-infected endothelial cells. Total protein from mock, AD169, VHL/E, and Towne strains of infected cells (day 9) was extracted and blotted with anticaspase-9 and anticaspase-3 antibodies. β-Actin was used as an internal sample control. B, Time-dependent increases of expression and cleavage of caspase-3 in HCMV-infected cells. After indicated infection time, total protein from mock-infected and VHL/E-infected cells was extracted and blotted with antibodies against total and cleaved caspase-3. C, Cleavage of caspase-3 in HCMV-infected cells. VHL/ E-Infected HAECs (day 9) were stained with anti-IE antibody-FITC (green), anti-cleaved caspase-3 antibody-Texas Red (red), and DAPI (blue). Merged image shows colocalization. D, Cleavage of caspase-9 in HCMV-infected cells. VHL/ E-infected HAECs (day 9) were stained anti-IE antibody-FITC (green), anticleaved caspase-9 antibody-Texas Red (red), and DAPI (blue). Merged image shows colocalization. E, Time-dependent increases in expression and cleavage of caspase-8 in HCMV-infected cells. After indicated infection time, total protein from mock-infected and VHL/E-infected cells was extracted and blotted with antibody against caspase-8. F, The involvement of caspases-3, -9, and -8 in HCMV-induced endothelial apoptosis. HAECs were infected with VHL/E for 8 days and treated with 10 μmol/L of caspase inhibitor for 3 days (fresh inhibitors were added daily). TUNEL-positive cells and total cells were counted in 4 fields per coverslip. Results were expressed as the mean±SEM percentage of TUNEL-positive cells over total cells. Using ANOVA, all 3 caspase inhibitors resulted in significantly less apoptosis (P<0.001); the inhibiting effect by caspase-3 or 9 inhibitors was greater than that by caspase-8 (P=0.024).
Caspase-3 can be activated through specific proteolytic cleavage by upstream components of different apoptotic pathways. Caspase-9, involved in the mitochondrion-dependent pathway, is the upstream caspase that activates caspase-3. To explore the role of the mitochondrion-dependent pathway in HCMV-triggered apoptosis, we measured the expression and cleavage of caspase-9. As shown in Figure 2A, compared with mock-infected cells, the HCMV-infected endothelial cells had increased expression and cleavage of caspase-9, particularly in VHL/E strain-infected cells. Again, the activation of caspase-9 was specifically in the infected cells as shown in the immunofluorescence stain (Figure 2D). Therefore, we concluded that caspase-9 activation was associated with and likely essential for efficient apoptotic response to HCMV infection. We further showed increased expression and activation of caspase-8 in the infected cells (Figure 2E) indicating that the receptor-mediated apoptosis pathway was also activated.
To further examine whether the HCMV-induced activation of caspases was essential for HCMV-induced apoptosis, we blocked caspase activity with specific inhibitors (caspase-3 inhibitor II, Z-DEVD-FMK for caspase-3; Z-LEHD-FMK for caspase-9; Z-IETD-FMK for caspase-8; and Z-FA-FMK as negative control). As shown in Figure 2F, HCMV-induced apoptosis was suppressed by all 3 caspase inhibitors (ANOVA F=84.33, P<0.001). The inhibiting effect was more significant by caspase-3 and caspase-9 inhibitors than that by the caspase-8 inhibitor (P=0.024). These findings confirm that caspase activation mediated by either receptor or mitochondrial pathways plays a central role in HCMV induced apoptosis (Figure 2F).
Expression of Bcl Family Molecules Bax and Bak in HCMV-Infected Endothelial Cells
Activation of caspases is modulated by several mechanisms. Members of the Bcl-2 family, which include antiapoptotic Bcl-2 and Bcl-XL, and the proapoptotic Bax and Bak control the release of cytochrome c from mitochondria and the activation of the caspase pathway. The upregulation of Bax and Bak expression and downregulation of Bcl-2 have been demonstrated during apoptosis. We therefore examined the expression of Bak and Bax in the HCMV-infected endothelial cells. As shown in Figure 3A, the expression of Bax and Bak was low in mock-infected cells. However, there was a dramatic induction in Bax expression by HCMV infection, particularly in VHL/E strain-infected cells. The increase in the expression of both Bax and Bak started at 1 day pi and sustained after 9 days pi (Figure 3B). This increased expression of Bax and Bak was confined to infected cells as indicated by immunofluorescence stains (Figure 3C and 3D). These results indicate the activation of pro-apoptotic Bax and Bak in HCMV-infected endothelial cells and further demonstrate the involvement of the mitochondrial apoptosis pathway in HCMV-induced endothelial apoptosis.
Figure 3.
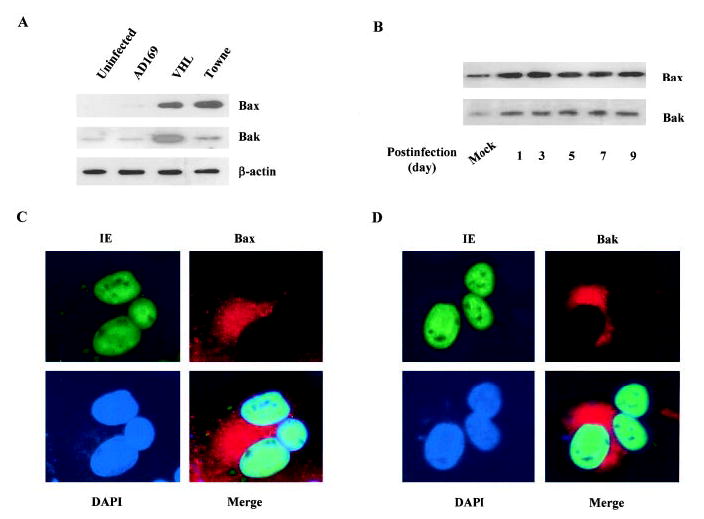
Increased Bax and Bak levels in HCMV-infected endothelial cells. A, Increase of Bax and Bak expression in HCMV-infected endothelial cells. Total protein from uninfected (cultured for the same duration) and infected (by AD169, VHL/E, and Towne strains) HAECs (day 9) was extracted and blotted with anti-Bax and anti-Bak antibodies. B, Time-dependent increase of Bax and Bak expression in HCMV-infected cells. After indicated infection time, total protein from mock-infected and VHL/E-infected HAECs was extracted and blotted with anti-Bax and anti-Bak antibodies. C, Increased Bax level in HCMV-infected HAECs. VHL/E-infected HAECs (day 9) were stained with anti-IE antibody-FITC (green), anti-Bax antibody-Texas Red (red), and DAPI (blue). Merged image shows colocalization. D, Increased Bak level in HCMV-infected HAEC. VHL/E-infected HAECs (day 9) were stained with anti-IE antibody-FITC (green), anti-Bak antibody-Texas Red (red), and DAPI (blue). Merged image shows colocalization.
Activation of the p53 Pathway in HCMV-Infected Cells
An important regulator of apoptosis is p53. p53 triggers apoptosis in response to a variety of stress stimuli and induces apoptosis by targeting gene regulation and transcription-independent signaling. Bax and Bak are p53 targets and are upregulated in a number of systems during p53-mediated apoptosis and are directly involved in mitochondrial apoptosis. We therefore examined whether p53 was involved in the HCMV-induced endothelial apoptosis. We first tested p53 activation in HCMV-infected cells. Serine residue 15 (ser15) is an important site for p53 DNA binding and interaction with other transcriptional factors, whereas serine 20 (ser20) is a site that binds to MDM2 when dephosphorylated and dissociates with MDM2 when phosphorylated. As shown in Figure 4A and 4B, the phosphorylation of ser15 was increased in VHL/E-infected cells indicating the activation of p53. The phosphorylation of ser20 was also increased, suggesting the activation and stabilization of p53. Furthermore, HCMV-infected endothelial cells had higher p53 levels at the late stage of infection. Whether this elevation in p53 is caused by increased expression or decreased degradation requires further investigation. The p53 appeared to be specifically activated in HCMV-infected cells as indicated by immunofluorescence stains (Figure 4C and 4D). These findings suggest that p53 can be activated by HCMV infection in endothelial cells.
Figure 4.
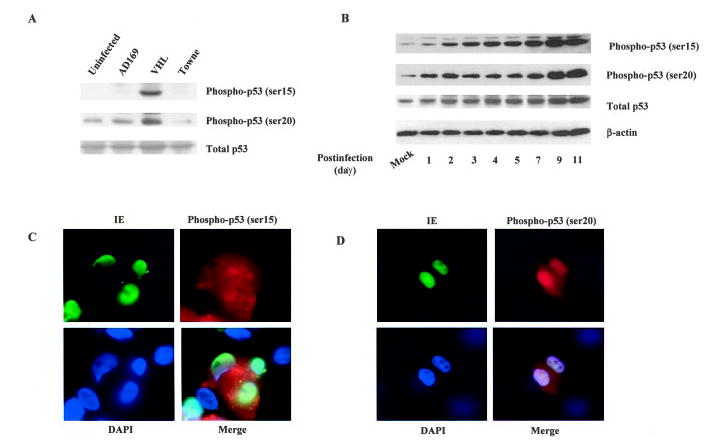
HCMV infection induced p53 phosphorylation in endothelial cells. A, Increase of p53 phosphorylation in HCMV-infected endothelial cells. Total protein from mock-infected and VHL/E-infected HAECs (day 9) was extracted and blotted with anti-phospho p53 (ser15), anti-phospho p53 (ser20), and anti-total p53 antibodies. B, Time-dependent increase in p53 phosphorylation in HCMV-infected cells. HAECs were infected with VHL/E as indicated, and total cell lysates were blotted with antibodies specific for p53 phosphorylated on ser15, ser20, total p53, or β-actin. C, Increased p53 phosphorylation in HCMV-infected HAECs. VHL/ E-infected HAECs (day 9) were stained with anti-IE antibody-FITC (green), anti-phosphor p53 (ser15) antibody-Texas Red (red), and DAPI (blue). Merged image shows colocalization. D, VHL/E-infected HAECs (day 9) were stained with anti-IE (green), anti-phospho p53 (ser 20) (red) antibodies, and DAPI (blue). Merged image shows colocalization.
Involvement of the p53 Pathway in HCMV-Induced Endothelial Apoptosis
To examine whether this p53 activation was involved in HCMV-induced apoptosis, we blocked p53 expression using p53-specific siRNA. As shown in Figure 5, HCMV-induced p53 expression was suppressed in the cells transfected with p53 siRNA in a dose-dependent manner. At the same time, the expressions of Bax and Bak, 2 direct targets of p53, were also reduced with p53 siRNA. Moreover, HCMV-induced expression and cleavage of caspase-3 were inhibited by p53 siRNA. These findings indicate that p53 is directly involved in HCMV-induced endothelial apoptosis.
Figure 5.
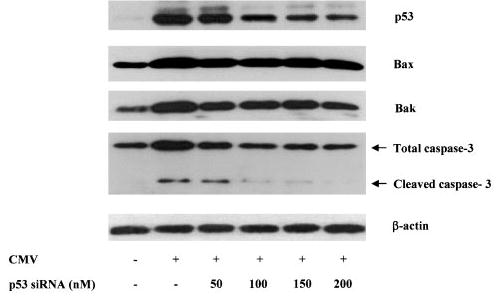
The involvement of p53 pathway in HCMV-induced endothelial apoptosis. HAECs were infected with VHL/E for 5 days before transfected with different amount of p53 siRNA. Twenty-four hours after transfection, total protein lysates from mock-infected and infected cells were prepared and blotted with antibodies against p53, Bax, Bak, caspase-3, and β-actin.
Role of ATM Kinase in HCMV-Induced p53 Phosphorylation
Phosphorylations of p53 on ser15 and ser20 are regulated, in part, by DNA damage-dependent kinases ATM and Chk2.26 To investigate the upstream signals of HCMV-induced p53 phosphorylation and activation, we measured the activation of Chk2/ATM pathway. As shown in Figure 6A, the expression of ATM and phosphorylation of Chk2 were increased in HCMV-infected cells indicating the activation of the ATM pathway. To further investigate the involvement of this pathway in HCMV-induced p53 activation, we treated HCMV-infected cells with caffeine—an inhibitor of ATM.27,28 We found that caffeine limited HCMV-induced Chk2 phosphorylation and p53 phosphorylation in a dose-dependent manner (Figure 6B). Thus, the ATM pathway, at least partially, contributes to HCMV-induced p53 activation.
Figure 6.

Involvement of the ATM/Chk2 pathway in HCMV-induced apoptosis. A, Time-dependent activation of the ATM pathway in HCMV-infected cells. HAECs were infected with VHL/E as indicated, and total cell lysates were blotted with antibodies specific for total ATM, phosphorylated Chk2, and β-actin. B, The involvement of the ATM pathway in HCMV-induced endothelial apoptosis. HAECs were treated with indicated concentrations of caffeine followed by infection with VHL/E. Five days after infection, cell lysates were subjected to immunoblotting with antibodies specific against phosphorylated Chk2, phospho p53 at ser15, and β-actin.
Discussion
HCMV is a widespread opportunistic pathogen that causes acute, latent, and chronic infections. Although the primary infection may be asymptomatic in immunocompetent individuals, the virus can cause a wide variety of severe diseases in immunocompromised hosts. Involvement of HCMV infection has been discovered in atherosclerosis,1–4 thrombosis,5,6 allograft rejection,7,8 and restenosis.9,10 However, the mechanisms of the pathogenesis are not clear. Earlier, we and others showed that HCMV-infected cells were resistant to apoptosis at the earlier stage of infection.23–25 At this stage, the actively proliferating HCMV may increase resistance of endothelial cells to apoptosis, which provides a period long enough for HCMV to use the cellular reproductive machinery for virus propagation. The mechanism of resistance to apoptosis was suggested to be cytoplasmic sequestration of p53.23 We have shown that HCMV can inhibit p53 nuclear localization signal function.24,25 In the present study, we show that HCMV can induce cytopathic effects and apoptotic changes in endothelial cells after a prolonged period of infection (>5 days). This cytopathic effect may occur in vivo during chronic infection or reactivation, which leads to endothelial dysfunction and contributes to the pathogenesis of vascular diseases such as atherosclerosis. The biphasic effect of the HCMV on endothelial apoptotic process was a time-and dose-dependent phenomenon. Although HCMV-infected endothelial cells may be resistant to cell death at the earlier stage, prolonged productive infection will induce cell death. Furthermore, exposure to higher doses of HCMV, ie, by a higher MOI, induces this cytopathic effect earlier in the process (data not shown).
In this study, we further addressed the signal transduction pathways for HCMV-induced cell death as proposed in Figure 7. The activation of caspase-3 in HCMV-infected cells indicates the involvement of the common caspase-3 apoptotic pathway in the process. Caspase-3 is a terminal caspase and is involved in the execution of cell death via cleavage of critical cellular proteins such as poly (ADP-ribose) polymerase, lamins, and gelsolin. Caspase-3 is activated by a variety of different stimuli, including infection with HIV,29 adenovirus,30 Sendai virus,31 and sindbis virus.32 Caspase-3 can be activated by extrinsic and intrinsic pathways. In the current study, we have shown that both pathways can be activated by HCMV infection. The activation of caspase-9 by HCMV infection suggests that this could be an intrinsic mitochondrion-mediated process, for which we have further explored the upstream pathways.
Figure 7.
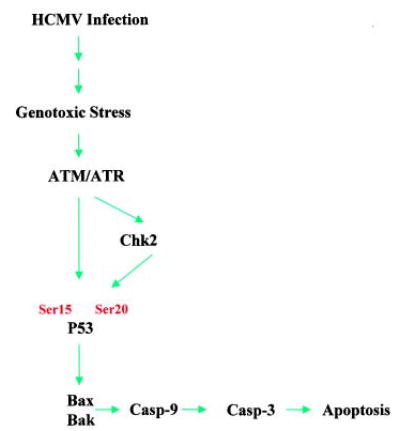
Proposed model of the HCMV-induced apoptotic signaling pathways. HCMV infection causes genotoxic stress, which activates ATM kinase. The activated ATM can activate p53 either directly or through activation of Chk2. The activation of p53 leads to the activation of mitochondrial pathway, ie, Bax and Bak, hence the activation of caspase-9 and caspase-3 resulting in cell apoptosis.
We have demonstrated HCMV-induced expression of pro-apoptotic Bcl-2 members Bax and Bak, which are involved in activation of mitochondrial apoptotic pathway. The activation of Bax and Bak may be a complicated process in HCMV-infected cells. At the earlier stage of infection, Bax and Bak expression increased after 1 day of infection, before the activation of the ATM/Chk2 pathway and p53, which started 5 days postinfection. Whether this Bax and Bak upregulation is induced by HCMV invasion or immediate early genes (genotoxic stress) or both is interesting to examine. Even though the Bax and Bak levels increased at earlier stage of infection, this activation did not lead to apoptosis. It suggests that there may be virus-induced antiapoptotic mechanisms that can antagonize Bax and Bak during this earlier infection stage for which further investigation is needed. However, antiapoptotic Bcl-2 may not be involved because phosphorylation and expression of Bcl-2 remain unchanged in the infected cells (data not shown). At the later stage of infection, the activation of Bax and Bak is partially mediated by p53 because it can be inhibited by p53 siRNA. The activation of Bax and Bak at this stage may be related to virus replication-induced genotoxic stress that induces apoptosis.
p53 Is considered the guardian of the genome and has a number of biological functions including cell cycle arrest, DNA repair, and apoptosis.33 Our study indicates that p53 activation may mediate the apoptotic changes in prolonged HCMV infection. Blocking p53 expression by siRNA resulted in reduced expression of Bax and Bak suggests that the HCMV-induced upregulation of Bax and Bak is p53-dependent. Bax and Bak as p53 targets are upregulated in a number of systems during p53-mediated apoptosis and are directly involved in mitochondrial apoptosis.34 In addition to expression, p53 may also be responsible for direct Bax and Bak activation in the HCMV-infected endothelial cells. However, where the p53-mediated Bax/Bak activation takes place remains to be resolved. As we can see from Figure 4C, there was a large proportion of the activated p53 located in cytoplasm and only a small proportion was in nucleus. Whether this small amount active nuclear p53 would be sufficient or whether p53 could directly trigger the Bax/Bak action in cytoplasm or mitochondria is an interesting question to explore. Indeed, recent studies show that p53 has an extranuclear role in the cytoplasm to directly induce apoptosis.35
The nuclear kinase ATM and its related kinase ATR are critically involved in integrating the initial DNA damage signals to cell cycle checkpoints and apoptosis. Activation of ATM/ATR by DNA damage has been widely demonstrated and ATM is the key regulatory molecule in DNA damage signaling and apoptosis.26 ATM resides predominantly in the nucleus and responds to DNA damage by phosphorylating numerous substrates. p53 is the main target of the ATM pathway, which has been reported to directly phosphorylate p53 on ser15.36,37 Our experiments have shown that ATM/ Chk2 pathway was activated in HCMV-infected cells and blocking ATM activity inhibited p53 phosphorylation on ser15 and ser20 sites. These findings suggest that ATM pathway may directly mediate HCMV-induced p53 phosphorylation on these 2 sites. We have also shown the phosphorylation and activation of Chk2–a checkpoint kinase.38,39 Because Chk2 can directly phosphorylate p53 on ser20,40 the observed phosphorylation on p53 ser20 may also be mediated by Chk2 kinase. However, further kinase assay may be needed to show whether ATM and Chk2 from HCMV-infected cells can directly phosphorylate p53 in vitro. Because ATM kinase is a key regulator of multiple signaling cascades that respond to DNA damage, the involvement of ATM pathway indicates that HCMV-induced endothelium damage may be caused by virus-induced DNA damage or genotoxic stress.
In summary, we have shown that prolonged HCMV infection can induce endothelial apoptosis. Contrary to the antiapoptotic effect during earlier stage of HCMV infection, the induction of apoptosis after prolonged HCMV infection could be responsible for endothelial dysfunction, damage to endothelial integrity, and ensuing vascular diseases in those with chronic or reactivated latent HCMV infection. The elucidation of the molecular mechanisms leading to activation of intrinsic mitochondrial apoptotic pathway through ATM and p53 regulation will offer potential targets to attenuate the HCMV-induced cytopathic effects and, hence, vascular diseases by HCMV infection.
Acknowledgments
This project is supported by a National Institutes of Health/National Heart, Lung and Blood Institute grant R01-HL071608. X.L.W. is an AHA Established Investigator.
References
- 1.Dummer S, Lee A, Breinig MK, Kormos R, Ho M, Griffith B. Investigation of cytomegalovirus infection as a risk factor for coronary atherosclerosis in the explanted hearts of patients undergoing heart transplantation. J Med Virol. 1994;44:305–309. doi: 10.1002/jmv.1890440316. [DOI] [PubMed] [Google Scholar]
- 2.Nieto FJ, Adam E, Sorlie P, Farzadegan H, Melnick JL, Comstock GW, Szklo M. Cohort study of cytomegalovirus infection as a risk factor for carotid intimal-medial thickening, a measure of subclinical atherosclerosis. Circulation. 1996;94:922–927. doi: 10.1161/01.cir.94.5.922. [DOI] [PubMed] [Google Scholar]
- 3.Espinola-Klein C, Rupprecht HJ, Blankenberg S, Bickel C, Kopp H, Rippin G, Victor A, Hafner G, Schlumberger W, Meyer J. Impact of infectious burden on extent and long-term prognosis of atherosclerosis. Circulation. 2002;105:15–21. doi: 10.1161/hc0102.101362. [DOI] [PubMed] [Google Scholar]
- 4.Grahame-Clarke C, Chan NN, Andrew D, Ridgway GL, Betteridge DJ, Emery V, Colhoun HM, Vallance P. Human cytomegalovirus seropositivity is associated with impaired vascular function. Circulation. 2003;108:678–683. doi: 10.1161/01.CIR.0000084505.54603.C7. [DOI] [PubMed] [Google Scholar]
- 5.Abgueguen P, Delbos V, Chennebault JM, Payan C, Pichard E. Vascular thrombosis and acute cytomegalovirus infection in immunocompetent patients: report of 2 cases and literature review. Clin Infect Dis. 2003;36:E134–E139. doi: 10.1086/374664. [DOI] [PubMed] [Google Scholar]
- 6.Vercellotti GM. Effects of viral activation of the vessel wall on inflammation and thrombosis. Blood Coagul Fibrinolysis. 1998;9:S3–S6. [PubMed] [Google Scholar]
- 7.Toyoda M, Galfayan K, Galera OA, Petrosian A, Czer LS, Jordan SC. Cytomegalovirus infection induces anti-endothelial cell antibodies in cardiac and renal allograft recipients. Transpl Immunol. 1997;5:104–111. doi: 10.1016/s0966-3274(97)80050-0. [DOI] [PubMed] [Google Scholar]
- 8.Borchers AT, Perez R, Kaysen G, Ansari AA, Gershwin ME. Role of cytomegalovirus infection in allograft rejection: a review of possible mechanisms. Transpl Immunol. 1999;7:75–82. doi: 10.1016/s0966-3274(99)80023-9. [DOI] [PubMed] [Google Scholar]
- 9.Manegold C, Alwazzeh M, Jablonowski H, Adams O, Medve M, Seidlitz B, Heidland U, Haussinger D, Strauer BE, Heintzen MP. Prior cytomegalovirus infection and the risk of restenosis after percutaneous trans-luminal coronary balloon angioplasty. Circulation. 1999;99:1290–1294. doi: 10.1161/01.cir.99.10.1290. [DOI] [PubMed] [Google Scholar]
- 10.Zhou YF, Leon MB, Waclawiw MA, Popma JJ, Yu ZX, Finkel T, Epstein SE. Association between prior cytomegalovirus infection and the risk of restenosis after coronary atherectomy. N Engl J Med. 1996;335:624–630. doi: 10.1056/NEJM199608293350903. [DOI] [PubMed] [Google Scholar]
- 11.Tanaka S, Toh Y, Mori R, Komori K, Okadome K, Sugimachi K. Possible role of cytomegalovirus in the pathogenesis of inflammatory aortic diseases: a preliminary report. J Vasc Surg. 1992;16:274–279. doi: 10.1067/mva.1992.37474. [DOI] [PubMed] [Google Scholar]
- 12.Hsich E, Zhou YF, Paigen B, Johnson TM, Burnett MS, Epstein SE. Cytomegalovirus infection increases development of atherosclerosis in Apolipoprotein-E knockout mice. Atherosclerosis. 2001;156:23–28. doi: 10.1016/s0021-9150(00)00608-0. [DOI] [PubMed] [Google Scholar]
- 13.Froberg MK, Adams A, Seacotte N, Parker-Thornburg J, Kolattukudy P. Cytomegalovirus infection accelerates inflammation in vascular tissue overexpressing monocyte chemoattractant protein-1. Circ Res. 2001;89:1224–1230. doi: 10.1161/hh2401.100601. [DOI] [PubMed] [Google Scholar]
- 14.Waldman WJ, Sneddon JM, Stephens RE, Roberts WH. Enhanced endothelial cytopathogenicity induced by a cytomegalovirus strain propagated in endothelial cells. J Med Virol. 1989;28:223–230. doi: 10.1002/jmv.1890280405. [DOI] [PubMed] [Google Scholar]
- 15.Sinzger C, Schmidt K, Knapp J, Kahl M, Beck R, Waldman J, Hebart H, Einsele H, Jahn G. Modification of human cytomegalovirus tropism through propagation in vitro is associated with changes in the viral genome. J Gen Virol. 1999;80:2867–2877. doi: 10.1099/0022-1317-80-11-2867. [DOI] [PubMed] [Google Scholar]
- 16.Zhu J, Quyyumi AA, Norman JE, Csako G, Epstein SE. Cytomegalovirus in the pathogenesis of atherosclerosis: the role of inflammation as reflected by elevated C-reactive protein levels. J Am Coll Cardiol. 1999;34:1738–1743. doi: 10.1016/s0735-1097(99)00410-6. [DOI] [PubMed] [Google Scholar]
- 17.Koskinen P, Lemstrom K, Bruggeman C, Lautenschlager I, Hayry P. Acute cytomegalovirus infection induces a subendothelial inflammation (endothelialitis) in the allograft vascular wall. A possible linkage with enhanced allograft arteriosclerosis. Am J Pathol. 1994;144:41–50. [PMC free article] [PubMed] [Google Scholar]
- 18.Sedmak DD, Knight DA, Vook NC, Waldman JW. Divergent patterns of ELAM-1, ICAM-1, and VCAM-1 expression on cytomegalovirus-infected endothelial cells. Transplantation. 1994;58:1379–1385. [PubMed] [Google Scholar]
- 19.Koskinen PK. The association of the induction of vascular cell adhesion molecule-1 with cytomegalovirus antigenemia in human heart allografts. Transplantation. 1993;56:1103–1108. doi: 10.1097/00007890-199311000-00011. [DOI] [PubMed] [Google Scholar]
- 20.Van Dam-Mieras MC, Bruggeman CA, Muller AD, Debie WH, Zwaal RF. Induction of endothelial cell procoagulant activity by cytomegalovirus infection. Thromb Res. 1987;47:69–75. doi: 10.1016/0049-3848(87)90241-6. [DOI] [PubMed] [Google Scholar]
- 21.Neumann FJ, Kastrati A, Miethke T, Pogatsa-Murray G, Seyfarth M, Schomig A. Previous cytomegalovirus infection and risk of coronary thrombotic events after stent placement. Circulation. 2000;101:11–13. doi: 10.1161/01.cir.101.1.11. [DOI] [PubMed] [Google Scholar]
- 22.Jarvis MA, Nelson JA. Human cytomegalovirus persistence and latency in endothelial cells and macrophages. Curr Opin Microbiol. 2002;5:403–407. doi: 10.1016/s1369-5274(02)00334-x. [DOI] [PubMed] [Google Scholar]
- 23.Kovacs A, Weber ML, Burns LJ, Jacob HS, Vercellotti GM. Cytoplasmic sequestration of p53 in cytomegalovirus-infected human endothelial cells. Am J Pathol. 1996;149:1531–1539. [PMC free article] [PubMed] [Google Scholar]
- 24.Wang J, Marker PH, Belcher JD, Wilcken DE, Burns LJ, Vercellotti GM, Wang XL. Human cytomegalovirus immediate early proteins upregulate endothelial p53 function. FEBS Lett. 2000;474:213–216. doi: 10.1016/s0014-5793(00)01604-5. [DOI] [PubMed] [Google Scholar]
- 25.Wang J, Belcher JD, Marker PH, Wilcken DE, Vercellotti GM, Wang XL. Cytomegalovirus inhibits p53 nuclear localization signal function. J Mol Med. 2001;78:642–647. doi: 10.1007/s001090000157. [DOI] [PubMed] [Google Scholar]
- 26.Shiloh Y. ATM and related protein kinases: safeguarding genome integrity. Nat Rev Cancer. 2003;3:155–168. doi: 10.1038/nrc1011. [DOI] [PubMed] [Google Scholar]
- 27.Zhou BB, Chaturvedi P, Spring K, Scott SP, Johanson RA, Mishra R, Mattern MR, Winkler JD, Khanna KK. Caffeine abolishes the mammalian G(2)/M DNA damage checkpoint by inhibiting ataxia-telangiectasia-mutated kinase activity. J Biol Chem. 2000;275:10342–10348. doi: 10.1074/jbc.275.14.10342. [DOI] [PubMed] [Google Scholar]
- 28.Sarkaria JN, Busby EC, Tibbetts RS, Roos P, Taya Y, Karnitz LM, Abraham RT. Inhibition of ATM and ATR kinase activities by the radiosensitizing agent, caffeine. Cancer Res. 1999;59:4375–4382. [PubMed] [Google Scholar]
- 29.Stewart SA, Poon B, Song JY, Chen IS. Human immunodeficiency virus type 1 vpr induces apoptosis through caspase activation. J Virol. 2000;74:3105–3111. doi: 10.1128/jvi.74.7.3105-3111.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schmidt M, Afione S, Kotin RM. Adeno-associated virus type 2 Rep78 induces apoptosis through caspase activation independently of p53. J Virol. 2000;74:9441–9450. doi: 10.1128/jvi.74.20.9441-9450.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bitzer M, Prinz F, Bauer M, Spiegel M, Neubert WJ, Gregor M, Schulze-Osthoff K, Lauer U. Sendai virus infection induces apoptosis through activation of caspase-8 (FLICE) and caspase-3 (CPP32) J Virol. 1999;73:702–708. doi: 10.1128/jvi.73.1.702-708.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nava VE, Rosen A, Veliuona MA, Clem RJ, Levine B, Hardwick JM. Sindbis virus induces apoptosis through a caspase-dependent, CrmA-sensitive pathway. J Virol. 1998;72:452–459. doi: 10.1128/jvi.72.1.452-459.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Burns TF, El-Deiry WS. The p53 pathway and apoptosis. J Cell Physiol. 1999;181:231–239. doi: 10.1002/(SICI)1097-4652(199911)181:2<231::AID-JCP5>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 34.Shen Y, White E. p53-dependent apoptosis pathways. Adv Cancer Res. 2001;82:55–84. doi: 10.1016/s0065-230x(01)82002-9. [DOI] [PubMed] [Google Scholar]
- 35.Chipuk JE, Green DR. p53’s believe it or not: lessons on transcription-independent death. J Clin Immunol. 2003;23:355–361. doi: 10.1023/a:1025365432325. [DOI] [PubMed] [Google Scholar]
- 36.Banin S, Moyal L, Shieh S, Taya Y, Anderson CW, Chessa L, Smorodinsky NI, Prives C, Reiss Y, Shiloh Y, Ziv Y. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science. 1998;281:1674–1677. doi: 10.1126/science.281.5383.1674. [DOI] [PubMed] [Google Scholar]
- 37.Khanna KK, Keating KE, Kozlov S, Scott S, Gatei M, Hobson K, Taya Y, Gabrielli B, Chan D, Lees-Miller SP, Lavin MF. ATM associates with and phosphorylates p53: mapping the region of interaction. Nat Genet. 1998;20:398–400. doi: 10.1038/3882. [DOI] [PubMed] [Google Scholar]
- 38.McGowan CH. Checking in on Cds1 (Chk2): A checkpoint kinase and tumor suppressor. Bioessays. 2002;24:502–511. doi: 10.1002/bies.10101. [DOI] [PubMed] [Google Scholar]
- 39.Herzog CR. Chk2 meets Plk3 in damage control. Cell Cycle. 2002;1:408–409. doi: 10.4161/cc.1.6.268. [DOI] [PubMed] [Google Scholar]
- 40.Shieh SY, Ahn J, Tamai K, Taya Y, Prives C. The human homologs of checkpoint kinases Chk1 and Cds1 (Chk2) phosphorylate p53 at multiple DNA damage-inducible sites. Genes Dev. 2000;14:289–300. [PMC free article] [PubMed] [Google Scholar]