

SUMMARY
We previously noted strong induction of genes related to intestinal copper homeostasis (Menkes Copper ATPase [Atp7a] and metallothionein) in the duodenal epithelium of iron-deficient rats across several stages of postnatal development (Collins, JF et al [2005] Am J Physiol Gastrointest Liver Physiol, 288: G964–G971). We now report significant copper loading in the livers and intestines of iron-deficient rats. These findings are consistent with the hypothesis that there is increased intestinal copper transport during iron-deficiency. We additionally found that hepatic Atp7b gene expression does not change with iron-deficiency, suggesting that liver copper excretion is not altered. We developed polyclonal antibodies against rat Atp7a and we first demonstrate the specificity of the immunogenic reaction. We then show that the Atp7a protein is present on apical domains of duodenal enterocytes in control rats, and on brush-border (BB) and basolateral (BL) membrane domains in iron-deprived rats. This localization is surprising, as previous in vitro studies have suggested that Atp7a traffics between the trans-golgi network and the basolateral membrane. We further demonstrate that Atp7a protein levels are dramatically increased in BB and BL membrane vesicles isolated from iron-deficient rats. Other experiments show that iron re-feeding corrects the hematological abnormalities seen in iron-deficient rats, but that it does not ameliorate Atp7a protein induction, suggesting that Atp7a does not respond to intracellular iron levels. We conclude that Atp7a is involved in copper loading observed during iron-deficiency, and that increased intestinal copper transport is of physiological relevance, as copper plays important roles in overall body iron homeostasis.
INTRODUCTION
Iron is a component of many proteins which are involved in cellular growth and metabolism. Complex homeostatic mechanisms exist to ensure that adequate iron is available for normal physiological functions, while simultaneously avoiding excessive iron accumulation in body tissues. Free iron is highly reactive and is prone to catalyze the formation of toxic oxygen radicals via the Fenton reaction (1). Iron-overload resulting from genetic diseases, leads to hepatic fibrosis and cancer, diabetes mellitus, neurodegenerative disorders and cardiomyopathy (2). Furthermore, iron-deficiency is major clinical issue, likely afflicting billions of people worldwide including a large number of people in developed countries such as the United States (3–5). Iron-deficiency anemia, due to low dietary Fe intake, results in impaired neurological development and decreased metabolic rate. To avoid iron-deficiency and iron-overload, iron balance is maintained via precise regulation of intestinal iron transport, which is the site for hormonal regulation of overall body iron homeostasis (6,7). This is especially critical for this important trace mineral, as iron excretion is an unregulated, inefficient process.
Within the past few years, several proteins involved in vectorial iron transport across the intestinal epithelium have been isolated and studied. Dietary iron is predominantly in the oxidized, ferric form (Fe3+), which is insoluble and has very low bioavailability. Thus, dietary iron is reduced by the BBM ferrireductase, Duodenal Cytochrome B (Dcytb) (8) to Fe2+ and then transported across the BBM by Divalent Metal Transporter 1 (Dmt1) (9). Once iron has been transported into the cell, it becomes part of the cytosolic iron pool, where it is available for metabolic processes, or is stored in the cytoplasm within ferritin complexes. According to the body’s needs, various amounts of free iron traverse the cytoplasm and are transported across the BLM by the iron-exporter ferroportin (10,11). In addition to ferroportin, hephaestin (Hp) is required for iron transport across the BLM of enterocytes, and it is believed to oxidize Fe2+ to Fe3+ as iron is exported from the cell. Subsequently, oxidized iron can then interact with Transferrin (Tf) in the interstitial fluid, an interaction that mediates iron distribution to cells of the body via the circulatory system.
Intestinal iron transport is known to be the “checkpoint” of body iron homeostasis, and as such several physiological effectors are known to modulate this important process. These include erythroid, hypoxia, inflammatory and stores regulators of intestinal iron transport (1). We now have some insight into how these various stimuli lead to concerted changes in iron homeostasis and affect intestinal iron transport. Current evidence suggests that all of these physiological cues may modulate the production of hepcidin, a circulating, peptide hormone produced by the liver, which has pronounced effects on intestinal iron transport (12,13). Hepcidin expression may be altered dramatically in response to several known stimuli that alter iron absorption. Conditions that lead to increased iron transport (e.g. iron-deficiency, anemia, hypoxia) signal to decrease hepcidin production, whereas conditions that lead to decreased intestinal iron transport (e.g. iron-overload, inflammation) induce its expression (14–16). Recent in vitro observations have shown that hepcidin binds to the ferroportin protein on the plasma membrane of target cells to induce endocytosis and lysosomal degradation (17), although other iron-transport related proteins may also be regulated by hepcidin. The iron-overload phenotypes of animals and humans deficient in hepcidin further strengthen the supposition that this peptide hormone is a negative regulator of intestinal iron absorption (18,19). Furthermore, laboratory rodents over-expressing hepcidin develop a severe anemic phenotype with systemic iron-deficiency (20). Thus, current evidence strongly suggests that hepcidin is a key modulator of intestinal iron transport, a finding that further links intestinal transport with regulation of overall body iron homeostasis.
Copper, like iron, is an essential micronutrient, as demonstrated by nutritional deficiencies and rare genetic disorders (21,22). Copper is essential for many physiological processes, including pigmentation, neurotransmitter synthesis, free-radical detoxification, mitochondrial oxidative phosphorylation, hemoglobin synthesis, iron metabolism and intestinal iron transport (23). Recently, a homolog of the yeast Copper Transporter 1 (Ctr1) has been identified in mammalian cells (24), and it may play a role in the uptake of dietary copper across the apical membrane of enterocytes. However, Ctr1 inactivation in mice leads to embryonic lethality (49, 52), so this possibility remains unproven. There is also in vitro experimental evidence that Dmt1 may play a role in brush-border copper transport (25). Additionally, recent studies suggest that intestinal copper transport is mediated by an unidentified copper ATPase expressed in the apical membrane of intestinal epithelial cells (26). Once copper enters enterocytes, it may be sequestered in metallothionein (27–29), or it may traverse the cytoplasm bound to chaperone proteins and be pumped across the basolateral membrane by the copper ATPase (Atp7a), the mutant gene in Menke’s disease (30). Once copper exits the basal membrane of enterocytes, it is bound to ceruloplasmin (Cp), which presumably oxidizes toxic ferrous iron to the ferric form and then functions to distribute copper to the rest of the body via the circulatory system. Cp is thought to play a role in iron release from some tissues including the liver (31,32) and recent studies suggest it may also be important for intestinal iron transport (33).
The interplay of copper and iron homeostasis has been recognized for more than 150 years and both trace minerals are required to prevent anemia (34). This observation has now been confirmed at the molecular level with the discovery of ceruloplasmin (Cp) and hephaestin (Hp), which are multi-copper ferroxidases that play roles in iron export from some tissue types (12). Cp is a circulating ferroxidase of hepatic origin and Hp is a membrane-anchored ferroxidase that is important for iron release from duodenal enterocytes. The exact role of Cp in intestinal iron absorption has not been resolved conclusively (34), although some experimental results have shown a positive effect of Cp on iron release from enterocytes (35). Recently, the Cp gene was disrupted in mice in an effort to produce an animal model of aceruloplasminemia, which is the lack of Cp (17). The authors of this study concluded that Cp loss did not affect intestinal iron absorption, but the low number of mice used and their mixed genetic backgrounds could not rule out an effect of Cp on this important physiological process (36). However, very recent evidence suggests that Cp may be important for intestinal iron transport under conditions of increased erythropoeisis (33). Furthermore, in sex-linked anemic (sla) mice, which have inactivating mutations in the Hp gene, anemia is seen in younger animals, but the anemia is resolved as the animals age, suggesting that another gene becomes active and compensates for the loss of Hp (37).
Our previous studies lead us to the discovery that the Menkes copper ATPase is a novel, metal-responsive gene that is strongly induced at the mRNA level by iron-deprivation in the duodenum and jejunum of rats at different stages of postnatal development (38,39). This finding was of particular interest as copper is known to have the following profound effects on body iron homeostasis: 1) copper is necessary for intestinal iron transport via Hp and possibly Cp, 2) copper is necessary for iron release from body iron stores in the liver, spleen and reticuloendothelial macrophages, and 3) copper is necessary for hemoglobin production. Thus, in the current manuscript, we have expanded upon these previous observations to demonstrate that iron-deficient rats have significantly increased intestinal and liver copper levels, suggesting that they absorb increased amounts of dietary copper. Importantly, we also show that Atp7a protein expression is also strongly induced by iron-deficiency and that the Atp7a protein is present on both brush-border and basolateral domains of duodenal enterocytes. We further provide experimental evidence that Atp7a induction during iron-deficiency is not ameliorated by iron re-feeding and correction of hematological parameters.
EXPERIMENTAL PROCEDURES
Animal Studies
Sprague Dawley rats at different stages of postnatal developmental were placed on modified AIN-93G rodent diets which contained either 198 ppm Fe (control diet with the same Fe content as normal rodent chow) or 3 ppm Fe (low Fe diet) for various periods of time (as described in detail previously) (38). The diets were identical except for the addition of pure ferric citrate to the control Fe diet, and they both had standard copper levels. Suckling (8 days), weanling (21 days) and adolescent (6 weeks) rats were made iron-deficient by feeding dams the low Fe diet throughout part or all of gestation and lactation. Soon after birth, litter sizes were normalized by cross-fostering the pups to different dams to maintain a constant dam:pup ratio. For the production of 12-week-old iron deficient rats, weaned rats at 3-weeks-of-age were placed on the control or low diet for 9 weeks, and for 36-week-old rats, 8 week-old rats were placed on the respective diets for 28 weeks. Some rats were also placed on the diets for one year beginning at 8-weeks-of-age.
Livers and intestines were removed from control and iron-deficient rats, and copper and iron analysis was performed by flame atomic absorption spectrometry (AAS).
Western Blot Analyses
Brush-border membrane vesicles were prepared by the Mg++ precipitation method (40,41) and basolateral membrane vesicles were prepared by the percoll density gradient method (42,43). The purity of these preps has been addressed extensively in previous publications by us (44–53) and moreover, these well-established techniques have been utilized by numerous investigators over the past two decades (54–60). Membrane proteins were solubilized in Laemmli buffer plus βME, boiled for 5 minutes and then separated by SDS/PAGE, followed by electroblotting onto nitrocellulose membranes. Western blots were processed as described previously (41,61,62), with a commercially available antiserum against Dmt1 (Alpha Diagnostics) or Atp7a (made by us as described below).
We sought to develop a useful antiserum against the rat Atp7a protein to perform quantitative immunoblot and immunolocalization studies in rat intestine. We were unable to find any published reports of Atp7a antiserum for rat or any commercially available reagents, and surprisingly, we discovered that this protein has never been definitively localized in the mammalian intestine. We thus identified two hydrophilic, potentially antigenic sites in the rat Menkes protein and had two peptides synthesized and co-injected into two rabbits. One site was the C-terminus and the other was from within the N-terminal region. The peptide sequences were as follows: NH2-KHSLLVGDFREDDDTTL-COOH, and NH2-KKDRSANHLDHKRE-COOH. Rabbits were immunized three times and a 10-week bleed was utilized for the experiments that are described in this manuscript. ELISA data showed that there was a strong immunogenic response to the C-terminal peptide in both rabbits, with little immune response generated against the N-terminal peptide (data not shown). For some experiments, pre-immune serum was utilized and in others, peptide-blocked antiserum was utilized (see details in the legend of Figure 5).
Figure 5. Western Blot Analysis of Atp7a Protein Expression in BBM Vesicles Isolated From 12-Week-Old Rats.
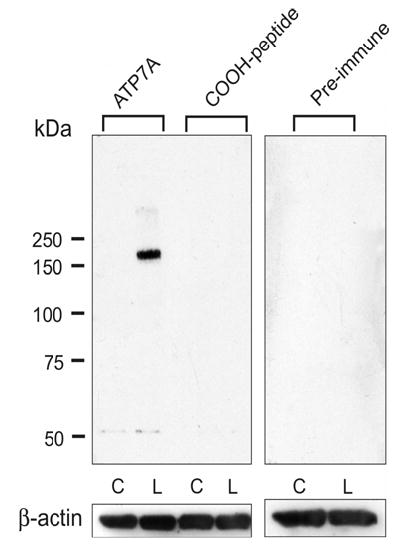
Western blots were processed with Atp7a immune serum, pre-immune serum or antigen-blocked immune serum. A single band at ~180 kDa was detected only in the low Fe group and it was blockable by pre-incubation of the antiserum with the immunogenic peptide (1 mg peptide/ml serum, 2 hours at 15° C). Pre-immune serum also did not detect this protein band. As seen, very little immunoreactive protein was detected in the control groups, even with over-exposure of the blots (not shown). C is control and L is low Fe.
Immunohistochemical Analysis of Rat Duodenum
Transverse sections (2–4 mm in width) were taken from mid-duodenum of iron-deficient and control rats, and were placed in 4% paraformaldehyde in PBS overnight at 4° C. Tissue samples were then transferred to 70% ethanol and embedded in paraffin. 6 μm sections were subsequently cut and affixed to microscope slides. Tissue sections were deparrafinized, blocked and processed with Atp7a antiserum at a 1:100 dilution, by standard methods (41,63). A fluorescent-tagged secondary antibody (Alexa Fluor 568 goat anti-rabbit IgG [H + L] conjugate; Molecular Probes, Eugene, OR) was utilized at a 1:400 dilution and staining was visualized by laser scanning confocal microscopy (Biorad MRC 1024), with a 568 nm excitation wavelength and a HQ 598 40 emission filter. Pre-immune serum was also utilized for some experiments.
RESULTS
Iron-deficient rats at the three younger ages were smaller than controls, however 12-week-old, 36-week-old and one-year-old rats were not (data not shown). Phenotype observation and blood analysis of the rats revealed that iron-deprived rats at 8-days, 21-days, 6- weeks and 12-weeks-of-age showed signs of hypochromic, microcytic anemia (38) (data not shown). However, the 36-week-old and one-year-old rats, despite being deprived of dietary iron for 28 weeks and one year respectively, showed no differences between control and iron-deprived groups in any parameters measured with the CBC blood test (although there was a non-statistical trend in that direction). Furthermore, we also noted that iron-deficient rats at different developmental stages had significantly increased liver copper levels (Figure 1), while liver iron levels were decreased as expected. The liver iron content was as follows (in ppm): for 21-day-old, control rats 171.2±14.85 and iron-deficient rats 19.7±0.96 (n=10 per group, p<0.001), for one-year-old rats, control rats 265.2±35 and iron-deprived rats 58.5±16 for (n=5 per group, p<0.001). We also measured duodenal and jejunal copper levels by flame AAS and found that the intestines of control rats had much lower copper levels than those harvested from iron-deficient rats (in ppm; duodenum- 0.21±0.014 for controls versus 0.82±0.01 for iron-deficient rats, n=3 per group, p<0.004; jejunum-0.53±0.007 for controls versus 0.89±0.08 for iron-deficient rats, n=2–3 per group, p=0.007).
Figure 1. Analysis of Liver Copper Levels In Rats at Different Stages of Post-Natal Development.
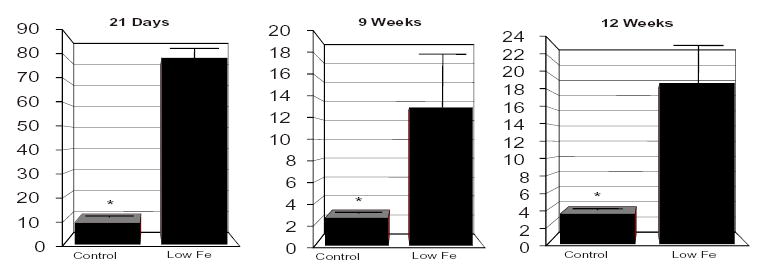
Liver copper levels in iron-deficient rats were significantly increased as compared to controls. The number of livers studied is as follows: for 21-day old rats, 10 controls and 10 iron-deficient; for 9-week-old rats, 9 controls and 9 iron-deficient; for 12-week-old rats, 20 controls and 20 iron-deficient. * p<0.05 between groups.
Furthermore, as body copper levels are known to be principally regulated at the level of excretion in the bile, as mediated by the Wilson’s copper ATPase (Atp7b), we performed real-time PCR experiments for Atp7b expression in control and iron-deficient rats at different ages. Our studies showed that there are no alterations in Atp7b mRNA expression between groups (data not shown), suggesting that liver copper excretion was not altered with iron-deficiency.
We next performed western blot analysis of Dmt1 protein expression in iron-deficient rats at 6- and 12-weeks-of-age (Figure 2). These experiments showed strong induction of Dmt1 expression by iron-deficiency in duodenum with little expression in jejunum, as reported by others previously (64,65). Interestingly, 4 transcript splice variants of Dmt1 have been described in the literature; two 5’ and two 3’ variants. Each of these variants presumably encode slightly different sized Dmt1 proteins. Our data thus suggest that more than one of these variants is likely induced by our dietary feeding strategy. Furthermore, post-translational modification of DMT1 likely occurs and many investigators have detected multiple DMT1 bands on immunoblots. These studies also showed that rats were indeed iron-deficient, as rats with normal iron status have little detectable Dmt1 protein. Furthermore, these immunoblots demonstrate the fact that our brush-border membrane preparation method is indeed highly enriched for brush-border membranes, as this is where Dmt1 is expressed within enterocytes. Importantly, this same vesicle purification method was utilized for western blot analysis of Atp7a protein expression, which led us to the novel observation that Atp7a is expressed on the apical membranes of duodenal entrocytes, and that Atp7a protein on the brush-border is greatly increased by dietary iron-deprivation (as described in detail below).
Figure 2. Western Blot Analysis of Dmt1 Protein Expression in the Duodenum and Jejunum of Iron-Deficient Rats.
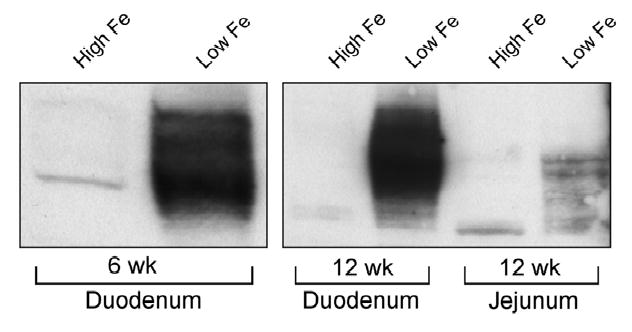
Intestinal mucosa was harvested from groups of rats and brush-border membrane vesicle proteins were purified by the well-established Mg++ precipitation method. SDS/PAGE followed by immunoblotting was subsequently performed with a 1:4000 dilution of the Dmt1 antiserum. As shown, significant induction of DMT1 was seen in 6-week and 12-week-old iron-deficient rats, and a modest induction was also detected in the jejunum of a group of 12-week-old iron-deficient rats.
A preliminary experiment demonstrated that there was strong induction of Atp7a protein expression in crude membrane proteins isolated from a group of 12-week-old iron-deficient rats versus normal controls (data not shown). This finding led us to believe that our antiserum would be useful to study Atp7a protein expression levels in our experimental model of iron-deficiency. We thus preformed a series of immunohistochemistry (IHC) experiments with Atp7a and Na+/K+ ATPase antisera in weanling and adult iron-deficient rats (Figures 3 and 4).These experiments clearly demonstrated that the Atp7a protein was predominantly localized to the apical membrane of enterocytes in control rats and was present on both brush-border and basolateral membranes in iron-deficient rats at two different ages. The localization to the apical membrane of enterocytes was unexpected, as previous in vitro studies have suggested that Atp7a traffics between an intracellular compartment (e.g. the trans-golgi network) and the basolateral membrane upon copper loading, when expressed in polarized renal epithelial cells (66)(66) and in other cell types (67–69)(67–69). Furthermore, Na+/K+ ATPase protein was localized exclusively to basolateral membrane domains as expected and Atp7a pre-immune serum did not detect any immunoreactive proteins in the intestinal villus from either control or iron-deficient rats. These observations confirmed the validity of these studies.
Figure 3. Immunohistochemical (IHC) Analysis of 12-Week-Old Rat Duodenum.
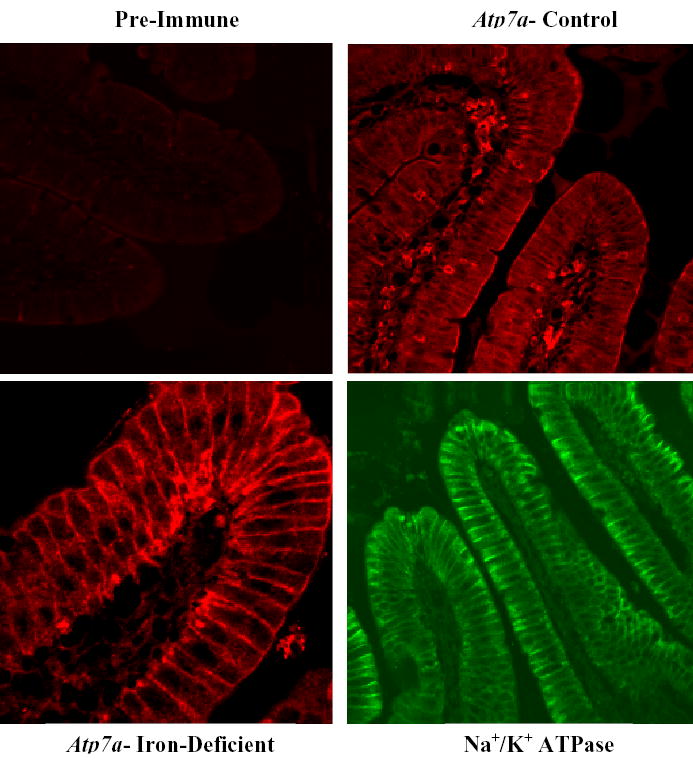
Tissue sections were processed for IHC and reacted with a 1:100 dilution of pre-immune serum, serum from a 10-week immune bleed against Atp7a or a commercially available antiserum against the rat Na+/K+ ATPase. A fluorescent tagged secondary antibody was used and slides were imaged with a BioRad MRC 1024 laser scanning confocal microscope utilizing identical settings for imaging of each slide within each individual experiment. Pre-immune serum showed minimal staining, while the Atp7a immune serum showed strong staining of brush-border membranes (in control and iron-deficient rats) and basolateral membranes (stronger in iron-deficient rats). In some experiments, basolateral staining was also apparent in control rats. As expected, Na+/K+ ATPase antiserum showed only basal and lateral staining. This presented experiment is representative of several that were performed, with very similar results.
Figure 4. Immunohistochemical (IHC) Analysis of Atp7a Expression in 21-Day-Old Rat Duodenum.
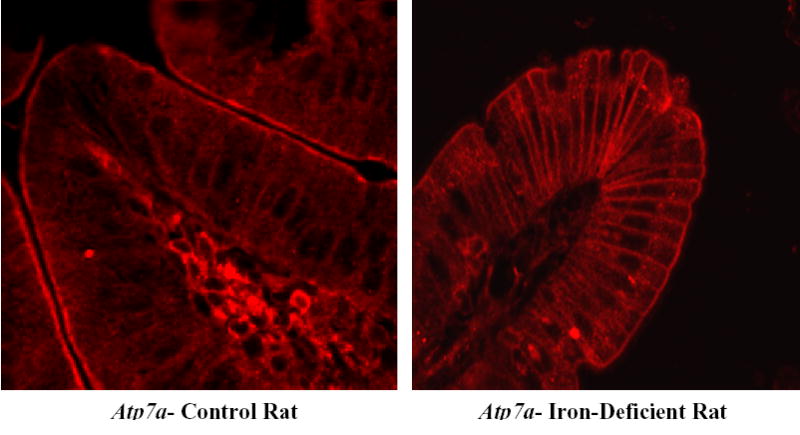
Tissues were processed and images were captured as described in the legend to Figure 3. Identical results were obtained as those seen in 12-week-old rats, in which brush-border staining was present in both control and iron-deficient samples, while basolateral staining was much stronger in the iron-deficient rats. One representative experiment is shown.
We next sought to perform immunoblots to accurately quantitate Atp7a protein expression levels in iron-deficient rats at different postnatal ages, as IHC is semi-quantitative at best. We thus performed experiments to define the specificity of this immunogenic reaction and to confirm observations made by IHC experiments. First, we purified BBMV from iron-deficient and control rats and performed western blots with the 10-week immune bleed from one of the immunized rabbits. These experiments showed detection of a strong band at ~180 kDa that was only seen in the iron-deficient group. We also determined that the reaction could be blocked by pre-treatment of the antiserum with the C-terminal, antigenic peptide (since this was the one that ELISA data showed had generated the immune response) and that the pre-immune serum did not detect any protein bands in either group (Figure 5).
Our next series of experiments was designed to determine if Atp7a could also be detected in a pure prep of basolateral membranes. We thus harvested duodenal mucosa from control and iron-deficient rats, and utilized a well-established basolateral membrane prep method, which involves removing enterocytes by palpation of gut segments filled with a citrate buffer followed by percoll density centrifugation. The resulting membrane vesicles are highly enriched for basolateral membrane protein markers and have little contamination with brush-border membrane vesicles (42, 43). Western blot analysis of these basolateral membrane vesicle proteins was performed and strong induction of Atp7a protein expression was also detected in iron-deficient rats (Figure 6). For this experiment, antiserum against the Na+/K+ ATPase was used as a constitutive control, as it is known to be localized to only basolateral domains in the intestinal epithelium. Pre-immune serum again did not detect any proteins in basolateral membrane vesicle proteins.
Figure 6. Western Blot Analysis of Atp7a Protein Expression in Basolateral Membrane Vesicles (BLMV) Isolated From 12-Week-Old Rats.
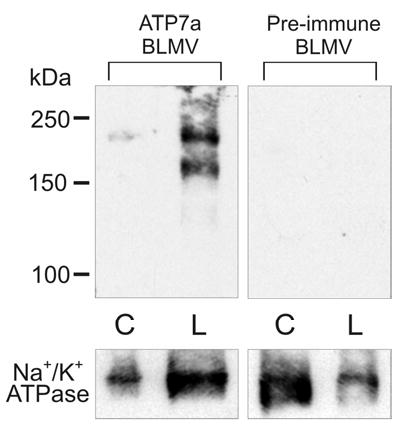
Western blots were processed with Atp7a immune serum and pre-immune serum. In this experiment, two large molecular weight protein bands were detected only in the iron-deficient sample (2 bands were observed in some experiments). Both bands are absent when the pre-immune serum was used and they were blockable by the antigenic peptide (not shown). They may thus represent different molecular forms of the Atp7a protein. These results confirmed the observations made with IHC experiments (shown above), in which little Atp7a protein is present on the basolateral membrane in control rats and that there is a strong induction with iron-deficiency. One representative experiment is shown, out of 3 that were performed with identical results. Na+/K+ ATPase antiserum was utilized as a constitutively expressed basolateral-specific control. In some experiments, we also used β-actin as a control (not shown). C is control and L is low Fe.
We next sought to quantitate Atp7a protein in expression in the brush-border membrane of iron-deficient rats at different postnatal ages, as studies looking at Atp7a mRNA expression had shown regulation throughout postnatal development. We thus produced iron-deficient rats at different stages of postnatal development and performed western blot analyses with BBMV (Figure 7). Results showed that Atp7a protein expression was strongly induced by iron-deprivation in 21-day-, 6-week-, 12-week- and 60-week-old rats, and that immunoreactive protein was barely detectable in control rats at all ages.
Figure 7. Western Blot Analysis of Atp7a Protein Expression in Brush-Border Membrane Vesicles (BBMVs) Isolated From Rat Duodenum at Different Stages of Postnatal Development.
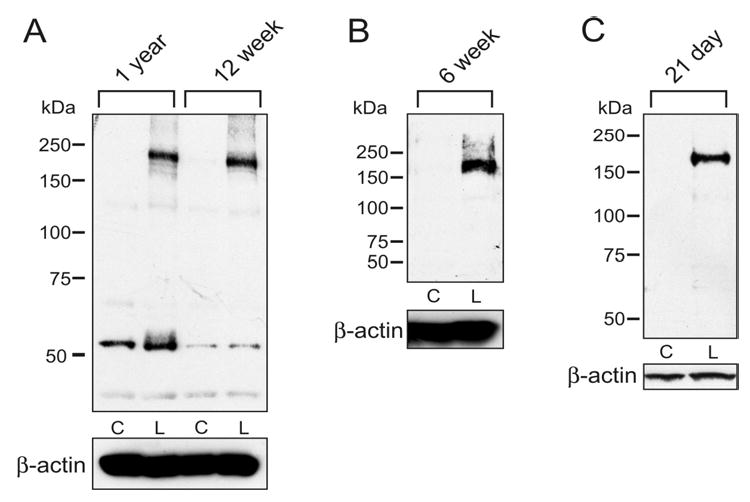
Rats were made iron-deficient by dietary iron deprivation. BBMVs were prepared from duodenal scrapes by a standard method and western blots were processed with Atp7a 10-week immune serum and pre-immune serum. A strong immunoreactive band at ~180 kDa was detected in all iron-deficient groups, but was absent in controls. Pre-immune serum did not detect any protein bands in any samples (not shown). In panel A, smaller immunoreactive bands were detected, however, they were similar in intensity in control and iron-deficient samples, and furthermore, they were absent in most experiments. Each age and treatment group consists of a group of 3-6 rats. C is control and L is low Fe.
Our final experiment was designed to determine the effect that iron-re-feeding would have on expression of Atp7a protein expression in iron-deficient rats. We thus produced a group of iron-deficient rats and fed half the group the control iron diet for 5 days and left the remaining rats on the iron-deficient diet. We then purified BBMV and performed western blot analysis. Interestingly, we found that iron-re-feeding corrected the hematological abnormalities seen in the rats, but had no effect on Atp7a protein expression levels.
DISCUSSION
In order to identify novel genes associated with the important physiological process of intestinal iron transport, we initially induced iron-deficiency in groups of rats at various stages of postnatal development and performed microarray experiments (38,39). This is a logical approach, as many genes involved in iron transport and homeostasis are known to be induced by iron-deprivation. We found that in addition to many known iron-responsive genes, several novel genes were induced by dietary iron-deprivation, including genes related to intestinal copper homeostasis (Menkes Copper ATPase [Atp7a] and metallothionein). Induction of Atp7a at different ages was confirmed by quantitative, real-time PCR (13). We thus hypothesized that upregulation of Atp7a was of potential physiologically relevance, as there is a well-known link between iron and copper in body iron homeostasis. Furthermore, we were intrigued by the fact that Atp7a was coordinately regulated by iron-deficiency, along with several known iron transport-related genes including Dmt1, Dctyb, transferrin receptor 1 and heme oxygenase 1 (38).
We next measured intestinal and liver copper levels in iron-deficient rats at different ages and noted that there were significantly increased copper levels in these two tissues. This finding suggested that iron-deprived rats were absorbing increased amounts of dietary copper, which is consistent with the observation that Atp7a was significantly induced, given its’ known role in intestinal copper transport. However, the possibility remained that liver copper excretion were also altered, as body copper homeostasis is controlled at the level of excretion into the bile. This process is mediated by the Atp7b copper ATPase, the mutant gene in Wilson’s disease. We thus measured Atp7b mRNA expression levels by quantitative, real-time PCR and found in multiple experiments at different ages that Atp7b gene expression was not altered during iron-deficiency. Additionally, as expected, iron-deprived rats had decreased liver iron levels.
Our next objective was to determine if increases in Atp7a mRNA were paralleled by increases in protein expression. We first determined that there were no published reports of Atp7a antiserum against the rat protein and no commercial reagents were available. Thus, we sought to develop our own rat, Atp7a-specific antiserum by immunizing two rabbits with Atp7a-specific peptides. We chose sites of the rat Atp7a protein that were highly hydrophilic and potentially antigenic, based upon several computer prediction programs. Elisa data from the rabbit serum showed that one rabbit in particular generated a very strong immune response to the C-terminal peptide by the second immunization, and we thus chose to perform initial experiments with a 10-week bleed from this rabbit. Utilizing this serum, we demonstrate that Atp7a protein expression is strongly induced in protein samples isolated from both brush-border and basolateral membrane vesicles of iron-deficient rats. In BBM vesicles, we demonstrated that the protein is strongly induced across several stages of postnatal development, which is consistent with previous observations made by microarray and real-time PCR analyses (38). These antibodies have also been used in IHC studies to demonstrate that the Atp7a protein is expressed on both apical and basolateral membrane domains in both control and iron-deficient rats, suggesting that it may play an important role in intestinal copper transport during normal physiological circumstances. We have also conclusively demonstrated that the antiserum is specific for Atp7a, as in control experiments the immunodetection was blocked with the antigenic peptide and there was no protein detection with pre-immune serum by western blot or immunohistochemical analysis in multiple experiments.
This series of IHC and western blotting experiments strongly suggested that Atp7a is a metal-responsive gene that is coordinately regulated along with known iron-responsive genes such as Dmt1 and Dcytb. We also felt that these observations justified further analysis of Atp7a expression and activity to decipher the physiological role of this protein in intestinal metal ion homeostasis under normal and iron-deficient conditions. To this point, several novel observations have been documented: 1) Atp7a is present on the brush-border membrane of duodenal enterocytes, a localization that is congruent with it being involved in copper and/or iron transport, 2) Atp7a gene expression is induced by iron-deficiency, an observation that is intriguing, as regulation of Atp7a gene expression has not been previously described in the scientific literature (only protein trafficking in response to copper loading), 3) Atp7a protein expression is also strongly induced during iron-deprivation and the immunoreactive protein is localized on both apical and basolateral membrane domains. We further noted that other novel genes are coordinately regulated along with Atp7a and known iron-responsive proteins (Dmt1, Dcytb, TfR1), suggesting that this group of genes is likely playing an important physiological role related to intestinal metal ion homeostasis.
Many apically expressed nutrient transporters are known to be acutely regulated by membrane recycling whereby they are physically removed from the plasma membrane of cells under certain physiological circumstances. The same phenomenon has been described for some iron transport-related proteins. For instance, DMT1 is rapidly internalized from the brush-border membrane by feeding iron-deficient rats a bolus of iron (70)(70), and DMT1 gene expression is also rapidly down-regulated by iron re-feeding. These observations suggest that DMT1 responds to intracellular iron levels, rather than to systemic iron status. Thus, we sought to determine whether induction of Atp7a expression on the apical membrane of enterocytes by iron-deficiency could be reversed by re-feeding iron to iron-deficient rats. So, we placed a group of weanling rats on low iron-diet for 9–10 weeks, and then took half the group and put them back on the control diet (with 198 ppm Fe) for 5 days, while the other half remained on the low Fe diet. Interestingly, iron re-feeding did not ameliorate the induction of Atp7a protein expresson on the BBM of duodenal enterocytes. These data suggested that Atp7a expression was not responding to intracellular iron levels like DMT1, but rather to systemic cues most likely related to body iron levels. Or alternatively, Atp7a induction could be related to increased intracellular copper levels, although copper-mediated regulation of Atp7a gene expression has never been described.
Furthermore, as copper and iron homeostasis are intimately intertwined, it seemed very likely that increased copper absorption during iron-deficiency was a specific response intended to somehow compensate for decreased body iron levels. In fact, copper is known to be related to the following physiological functions: 1) intestinal iron transport as mediated by two multi-copper ferroxidases (ceruloplasmin and hephaestin), 2) iron-release from body iron stores in the liver and reticuloendothelial macophages and 3) iron-loading into hemoglobin. It is also conceivable that copper could substitute for iron in other iron-containing protein such as mitochondrial cytochromes involved in respiration. We thus hypothesized that the sustained induction of Atp7a after correction of the hematological abnormalities from iron-deprivation was clear evidence that this phenomenon was not a secondary effect of iron-deficiency, but rather a primary physiological response.
In summary, our previous studies led to the observation that changes in the expression of intestinal metal ion transporters (e.g. iron, copper) occur with iron-deprivation. Based upon these initial observations, we sought to test the hypothesis that several of these coordinately regulated, metal-responsive genes, including Dmt1, Dcytb and Atp7a play key roles in intestinal metal ion homeostasis. Our original observations at the genetic level have been confirmed by several complementary experimental approaches. These include measurement of tissue metal levels and hematological analyses of blood parameters related to iron status, real-time PCR studies, and immunohistochemical and western blot analyses. We have demonstrated that copper levels are significantly increased in the livers and intestines of iron-deficient rats and that expression of the principal liver copper exporter, Atp7b, is not altered during iron-deficiency. Thus, increased copper transport likely occurs during iron-deficiency. We have further developed antibodies against the Menkes copper ATPase, which demonstrated strong induction of protein expression in BB and BLM vesicles across several stages of postnatal development and immnuolocalization of Atp7a protein to apical and basolateral membrane domains in duodenal enterocytes. So overall, these findings demonstrate that many aspects of intestinal metal ion homeostasis are still not clearly defined. Future studies will thus be intended to further increase our understanding of the complex regulatory mechanisms involved in intestinal iron and copper transport and of how these processes contribute to overall body metal ion homeostasis.
Figure 8. Iron Re-Feeding Corrects Hematological Abnormalities But Does Not Effect Atp7a Protein Expression in Iron-Deficient Rats.
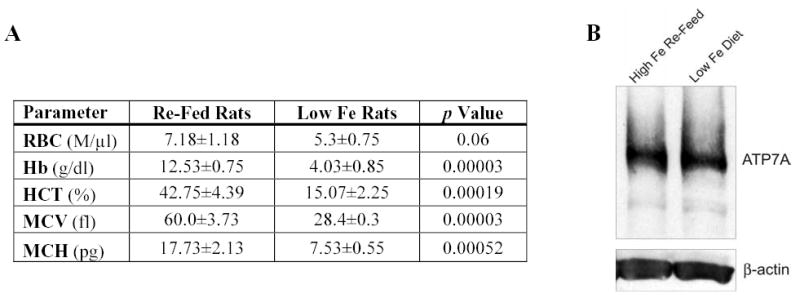
A group of 10 rats was deprived of dietary iron beginning at 3-weeks-of-age for 6 weeks, and then half the rats were re-fed iron for 5 days while the other half were not. Rats were sacrificed and BBMV were purified for western blot analyses. Panel A shows data from CBC analysis of rat blood taken upon sacrifice. Iron re-fed rats had normalization of hematological parameters, while iron-deficient rats showed clear signs of microcytic, hypochromic anemia. In panel A; RBC- red blood count, Hb- hemoglobin, HCT- hematocrit, MCV- mean corpuscular volume, MCH- mean corpuscular hemoglobin. Panel B shows that the Atp7a protein at ~180 kDa was still strongly expressed after iron re-feeding for 5 days. In fact, no changes in expression levels were apparent between the iron-replete and iron-deficient group. β-actin was utilized as a constitutively expressed control.
References
- 1.Hentze MW, Muckenthaler MU, Andrews NC. Cell. 2004;117(3):285–297. doi: 10.1016/s0092-8674(04)00343-5. [DOI] [PubMed] [Google Scholar]
- 2.Griffiths WJ, Kelly AL, Cox TM. Mol Med Today. 1999;5(10):431–438. doi: 10.1016/s1357-4310(99)01541-5. [DOI] [PubMed] [Google Scholar]
- 3.Baltussen R, Knai C, Sharan M. J Nutr. 2004;134(10):2678–2684. doi: 10.1093/jn/134.10.2678. [DOI] [PubMed] [Google Scholar]
- 4.Tapiero H, Gate L, Tew KD. Biomed Pharmacother. 2001;55(6):324–332. doi: 10.1016/s0753-3322(01)00067-1. [DOI] [PubMed] [Google Scholar]
- 5.Baynes RD, Bothwell TH. Annu Rev Nutr. 1990;10:133–148. doi: 10.1146/annurev.nu.10.070190.001025. [DOI] [PubMed] [Google Scholar]
- 6.Frazer DM, Wilkins SJ, Becker EM, Vulpe CD, McKie AT, Trinder D, Anderson GJ. Gastroenterology. 2002;123(3):835–844. doi: 10.1053/gast.2002.35353. [DOI] [PubMed] [Google Scholar]
- 7.Ludwiczek S, Theurl I, Artner-Dworzak E, Chorney M, Weiss G. J Mol Med. 2004;82(6):373–382. doi: 10.1007/s00109-004-0542-3. [DOI] [PubMed] [Google Scholar]
- 8.McKie AT, Barrow D, Latunde-Dada GO, Rolfs A, Sager G, Mudaly E, Mudaly M, Richardson C, Barlow D, Bomford A, Peters TJ, Raja KB, Shirali S, Hediger MA, Farzaneh F, Simpson RJ. Science. 2001;291(5509):1755–1759. doi: 10.1126/science.1057206. [DOI] [PubMed] [Google Scholar]
- 9.Gunshin H, Mackenzie B, Berger UV, Gunshin Y, Romero MF, Boron WF, Nussberger S, Gollan JL, Hediger MA. Nature. 1997;388(6641):482–488. doi: 10.1038/41343. [DOI] [PubMed] [Google Scholar]
- 10.Abboud S, Haile DJ. J Biol Chem. 2000;275(26):19906–19912. doi: 10.1074/jbc.M000713200. [DOI] [PubMed] [Google Scholar]
- 11.McKie AT, Marciani P, Rolfs A, Brennan K, Wehr K, Barrow D, Miret S, Bomford A, Peters TJ, Farzaneh F, Hediger MA, Hentze MW, Simpson RJ. Mol Cell. 2000;5(2):299–309. doi: 10.1016/s1097-2765(00)80425-6. [DOI] [PubMed] [Google Scholar]
- 12.Park CH, Valore EV, Waring AJ, Ganz T. J Biol Chem. 2001;276(11):7806–7810. doi: 10.1074/jbc.M008922200. [DOI] [PubMed] [Google Scholar]
- 13.Pigeon C, Ilyin G, Courselaud B, Leroyer P, Turlin B, Brissot P, Loreal O. J Biol Chem. 2001;276(11):7811–7819. doi: 10.1074/jbc.M008923200. [DOI] [PubMed] [Google Scholar]
- 14.Nicolas G, Chauvet C, Viatte L, Danan JL, Bigard X, Devaux I, Beaumont C, Kahn A, Vaulont S. J Clin Invest. 2002;110(7):1037–1044. doi: 10.1172/JCI15686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weinstein DA, Roy CN, Fleming MD, Loda MF, Wolfsdorf JI, Andrews NC. Blood. 2002;100(10):3776–3781. doi: 10.1182/blood-2002-04-1260. [DOI] [PubMed] [Google Scholar]
- 16.Muckenthaler M, Roy CN, Custodio AO, Minana B, deGraaf J, Montross LK, Andrews NC, Hentze MW. Nat Genet. 2003;34(1):102–107. doi: 10.1038/ng1152. [DOI] [PubMed] [Google Scholar]
- 17.Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, Ganz T, Kaplan J. Science. 2004;306(5704):2090–2093. doi: 10.1126/science.1104742. [DOI] [PubMed] [Google Scholar]
- 18.Nicolas G, Bennoun M, Devaux I, Beaumont C, Grandchamp B, Kahn A, Vaulont S. Proc Natl Acad Sci U S A. 2001;98(15):8780–8785. doi: 10.1073/pnas.151179498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roetto A, Papanikolaou G, Politou M, Alberti F, Girelli D, Christakis J, Loukopoulos D, Camaschella C. Nat Genet. 2003;33(1):21–22. doi: 10.1038/ng1053. [DOI] [PubMed] [Google Scholar]
- 20.Nicolas G, Bennoun M, Porteu A, Mativet S, Beaumont C, Grandchamp B, Sirito M, Sawadogo M, Kahn A, Vaulont S. Proc Natl Acad Sci U S A. 2002;99(7):4596–4601. doi: 10.1073/pnas.072632499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harris ZL, Klomp LW, Gitlin JD. Am J Clin Nutr. 1998;67(5 Suppl):972S–977S. doi: 10.1093/ajcn/67.5.972S. [DOI] [PubMed] [Google Scholar]
- 22.Bull PC, Cox DW. Trends Genet. 1994;10(7):246–252. doi: 10.1016/0168-9525(94)90172-4. [DOI] [PubMed] [Google Scholar]
- 23.Harris ED. Annu Rev Nutr. 2000;20:291–310. doi: 10.1146/annurev.nutr.20.1.291. [DOI] [PubMed] [Google Scholar]
- 24.Zhou B, Gitschier J. Proc Natl Acad Sci U S A. 1997;94(14):7481–7486. doi: 10.1073/pnas.94.14.7481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arredondo M, Munoz P, Mura CV, Nunez MT. Am J Physiol Cell Physiol. 2003;284(6):C1525–1530. doi: 10.1152/ajpcell.00480.2002. [DOI] [PubMed] [Google Scholar]
- 26.Knopfel M, Smith C, Solioz M. Biochem Biophys Res Commun. 2005;330(3):645–652. doi: 10.1016/j.bbrc.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 27.Williams LM, Cunningham H, Ghaffar A, Riddoch GI, Bremner I. Toxicology. 1989;55(3):307–316. doi: 10.1016/0300-483x(89)90020-6. [DOI] [PubMed] [Google Scholar]
- 28.Dameron CT, Harrison MD. Am J Clin Nutr. 1998;67(5 Suppl):1091S–1097S. doi: 10.1093/ajcn/67.5.1091S. [DOI] [PubMed] [Google Scholar]
- 29.Kelly EJ, Palmiter RD. Nat Genet. 1996;13(2):219–222. doi: 10.1038/ng0696-219. [DOI] [PubMed] [Google Scholar]
- 30.Vulpe CD, Packman S. Annu Rev Nutr. 1995;15:293–322. doi: 10.1146/annurev.nu.15.070195.001453. [DOI] [PubMed] [Google Scholar]
- 31.Mukhopadhyay CK, Attieh ZK, Fox PL. Science. 1998;279(5351):714–717. doi: 10.1126/science.279.5351.714. [DOI] [PubMed] [Google Scholar]
- 32.Young SP, Fahmy M, Golding S. FEBS Lett. 1997;411(1):93–96. doi: 10.1016/s0014-5793(97)00478-x. [DOI] [PubMed] [Google Scholar]
- 33.Srujana Cherukuri, R. P., Saul Nurko, Paul L. Fox. (2005) Unexpected role of ceruloplasmin in intestinal iron absorption. In. Book of Abstracts- First International Congress of the International BioIron Society, International BioIron Society, Prague, Czech Republic
- 34.Fox PL. Biometals. 2003;16(1):9–40. doi: 10.1023/a:1020799512190. [DOI] [PubMed] [Google Scholar]
- 35.Wollenberg P, Mahlberg R, Rummel W. Biol Met. 1990;3(1):1–7. doi: 10.1007/BF01141169. [DOI] [PubMed] [Google Scholar]
- 36.Harris ZL, Durley AP, Man TK, Gitlin JD. Proc Natl Acad Sci U S A. 1999;96(19):10812–10817. doi: 10.1073/pnas.96.19.10812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vulpe CD, Kuo YM, Murphy TL, Cowley L, Askwith C, Libina N, Gitschier J, Anderson GJ. Nat Genet. 1999;21(2):195–199. doi: 10.1038/5979. [DOI] [PubMed] [Google Scholar]
- 38.Collins JF, Franck CA, Kowdley KV, Ghishan FK. Am J Physiol Gastrointest Liver Physiol. 2005 doi: 10.1152/ajpgi.00489.2004. [DOI] [PubMed] [Google Scholar]
- 39.Collins, J. F. (2005) Biological ResearchIn Press
- 40.Acra S, Ghishan FK. Gastroenterology. 1991;101(2):430–436. doi: 10.1016/0016-5085(91)90022-d. [DOI] [PubMed] [Google Scholar]
- 41.Collins JF, Xu H, Kiela PR, Zeng J, Ghishan FK. Am J Physiol. 1997;273(6 Pt 1):C1937–1946. doi: 10.1152/ajpcell.1997.273.6.C1937. [DOI] [PubMed] [Google Scholar]
- 42.Acra S, Dykes W, Nylander W, Ghishan FK. Am J Physiol. 1993;264(1 Pt 1):G45–50. doi: 10.1152/ajpgi.1993.264.1.G45. [DOI] [PubMed] [Google Scholar]
- 43.Collins JF, Xu H, Kiela PR, Zeng J, Ghishan FK. Biochim Biophys Acta. 1998;1369(2):247–258. doi: 10.1016/s0005-2736(97)00226-5. [DOI] [PubMed] [Google Scholar]
- 44.Kikuchi K, Abumrad NN, Ghishan FK. Gastroenterology. 1988;95(2):388–393. doi: 10.1016/0016-5085(88)90495-7. [DOI] [PubMed] [Google Scholar]
- 45.Said HM, Ghishan FK, Redha R. Am J Physiol. 1987;252(2 Pt 1):G229–236. doi: 10.1152/ajpgi.1987.252.2.G229. [DOI] [PubMed] [Google Scholar]
- 46.Borowitz SM, Ghishan FK. Pediatr Res. 1985;19(12):1308–1312. doi: 10.1203/00006450-198512000-00021. [DOI] [PubMed] [Google Scholar]
- 47.Ghishan FK, Borowitz S, Mulberg A. Diabetes. 1985;34(8):723–727. doi: 10.2337/diab.34.8.723. [DOI] [PubMed] [Google Scholar]
- 48.Barnard JA, Ghishan FK, Wilson FA. J Clin Invest. 1985;75(3):869–873. doi: 10.1172/JCI111785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ghishan FK, Wilson FA. Am J Physiol. 1985;248(1 Pt 1):G87–92. doi: 10.1152/ajpgi.1985.248.1.G87. [DOI] [PubMed] [Google Scholar]
- 50.Daher M, Acra S, Dykes W, Ghishan FK. Proc Soc Exp Biol Med. 1992;201(3):254–260. doi: 10.3181/00379727-201-43504. [DOI] [PubMed] [Google Scholar]
- 51.Ghishan FK, Sutter W, Said H, Leonard D, Pietsch J, Abumrad N. Biochim Biophys Acta. 1989;979(1):77–81. doi: 10.1016/0005-2736(89)90525-7. [DOI] [PubMed] [Google Scholar]
- 52.Kikuchi K, Ghishan FK. Gastroenterology. 1987;93(1):106–113. doi: 10.1016/0016-5085(87)90321-0. [DOI] [PubMed] [Google Scholar]
- 53.Ghishan FK, Kikuchi K, Arab N. Biochem J. 1987;243(3):641–646. doi: 10.1042/bj2430641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bjorkman DJ, Brigham EJ. Biochem Biophys Res Commun. 1990;170(2):433–440. doi: 10.1016/0006-291x(90)92110-l. [DOI] [PubMed] [Google Scholar]
- 55.Moe AJ, Pocius PA, Polan CE. J Nutr. 1985;115(9):1173–1179. doi: 10.1093/jn/115.9.1173. [DOI] [PubMed] [Google Scholar]
- 56.Stieger B, Murer H. Eur J Biochem. 1983;135(1):95–101. doi: 10.1111/j.1432-1033.1983.tb07622.x. [DOI] [PubMed] [Google Scholar]
- 57.Hauser H, Howell K, Dawson RM, Bowyer DE. Biochim Biophys Acta. 1980;602(3):567–577. doi: 10.1016/0005-2736(80)90335-1. [DOI] [PubMed] [Google Scholar]
- 58.Dudeja PK, Tyagi S, Gill R, Said HM. Dig Dis Sci. 2003;48(1):109–115. doi: 10.1023/a:1021794600864. [DOI] [PubMed] [Google Scholar]
- 59.Tyagi S, Joshi V, Alrefai WA, Gill RK, Ramaswamy K, Dudeja PK. Dig Dis Sci. 2000;45 (12):2282–2289. doi: 10.1023/a:1005670404456. [DOI] [PubMed] [Google Scholar]
- 60.Faelli A, Tosco M, Orsenigo MN, Esposito G, Capraro V. Arch Int Physiol Biochim. 1983;91 (5):423–432. doi: 10.3109/13813458309067990. [DOI] [PubMed] [Google Scholar]
- 61.Collins JF, Bulus N, Ghishan FK. Am J Physiol. 1995;268(6 Pt 1):G917–924. doi: 10.1152/ajpgi.1995.268.6.G917. [DOI] [PubMed] [Google Scholar]
- 62.Xu H, Collins JF, Bai L, Kiela PR, Ghishan FK. Am J Physiol Cell Physiol. 2001;280(3):C628–636. doi: 10.1152/ajpcell.2001.280.3.C628. [DOI] [PubMed] [Google Scholar]
- 63.Xu H, Uno JK, Inouye M, Xu L, Drees JB, Collins JF, Ghishan FK. Am J Physiol Gastrointest Liver Physiol. 2003;285(6):G1317–1324. doi: 10.1152/ajpgi.00172.2003. [DOI] [PubMed] [Google Scholar]
- 64.Trinder D, Oates PS, Thomas C, Sadleir J, Morgan EH. Gut. 2000;46(2):270–276. doi: 10.1136/gut.46.2.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zoller H, Koch RO, Theurl I, Obrist P, Pietrangelo A, Montosi G, Haile DJ, Vogel W, Weiss G. Gastroenterology. 2001;120(6):1412–1419. doi: 10.1053/gast.2001.24033. [DOI] [PubMed] [Google Scholar]
- 66.Greenough M, Pase L, Voskoboinik I, Petris MJ, O'Brien AW, Camakaris J. Am J Physiol Cell Physiol. 2004;287(5):C1463–1471. doi: 10.1152/ajpcell.00179.2004. [DOI] [PubMed] [Google Scholar]
- 67.Mercer JF, Barnes N, Stevenson J, Strausak D, Llanos RM. Biometals. 2003;16(1):175–184. doi: 10.1023/a:1020719016675. [DOI] [PubMed] [Google Scholar]
- 68.Pase L, Voskoboinik I, Greenough M, Camakaris J. Biochem J. 2004;378(Pt 3):1031–1037. doi: 10.1042/BJ20031181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Petris MJ, Voskoboinik I, Cater M, Smith K, Kim BE, Llanos RM, Strausak D, Camakaris J, Mercer JF. J Biol Chem. 2002;277(48):46736–46742. doi: 10.1074/jbc.M208864200. [DOI] [PubMed] [Google Scholar]
- 70.Yeh KY, Yeh M, Watkins JA, Rodriguez-Paris J, Glass J. Am J Physiol Gastrointest Liver Physiol. 2000;279(5):G1070–1079. doi: 10.1152/ajpgi.2000.279.5.G1070. [DOI] [PubMed] [Google Scholar]