

Introduction
The identification of the molecular mechanisms involved in the regulation of synaptic receptors is essential to understanding how synaptic plasticity is regulated. Our understanding of AMPA receptor regulation has grown in recent years as key interacting proteins have been identified and mechanisms controlling these interactions have been elucidated (Malinow and Malenka, 2002). Significantly less is known about the regulation of synaptic NMDA receptors. Although the NMDAR is a relatively stable resident of the synapse (Ehlers et al., 2000; Lin et al., 2000; Luscher et al., 1999), a number of studies have shown that both the number of receptors and their subunit compositions can be altered. These changes appear to be mediated in a number of ways including changes in synthesis and degradation (Ehlers, 2003; Follesa and Ticku, 1996; Rao and Craig, 1997), lateral diffusion within the membrane (Tovar and Westbrook, 2002), and alterations in receptor trafficking (Crump et al., 2001; Rao and Craig, 1997; Snyder et al., 2001; Watt et al., 2000, reviewed in Choquet and Triller, 2003). Recent studies have also shown that both synaptic and extrasynaptic NMDARs can be regulated by clathrin-mediated internalization (Li et al., 2002; Montgomery and Madison, 2002; Nong et al., 2003; Nong et al., 2004; Roche et al., 2001; Snyder et al., 2001; Vissel et al., 2001). This was first suggested by Vissel et al., 2001, who identified a motif in the proximal region of the carboxy terminus of NR2A that regulated use-dependent internalization of the receptor. Nong et al. (2003) showed that glycine could prime NMDARs for internalization, and Li et al. (2003) suggested that endocytosis was involved in rundown of extrasynaptic NMDARs.
Previous work in our laboratory identified an AP-2 binding site that functions as an internalization motif, YEKL (Figure 1A), which is important in regulating surface expression of constructs containing the distal C-terminus of the NR2B subunit (Roche et al., 2001). However, this study did not address whether or not internalization involving the YEKL motif played a role in controlling synaptic receptors or if only extrasynaptic receptors were regulated in this fashion. The age-dependent decrease in the internalization of NMDARs in cultured neurons was consistent with internalization restricted to extrasynaptic receptors. In the present study, we investigated the molecular basis of NMDAR internalization and the role of PDZ proteins in regulating synaptic NMDARs. We find that three processes, PDZ anchoring, AP-2 mediated internalization, and phosphorylation of Y1472 together play a key role in the regulation of NR2B-containing synaptic NMDARs.
Figure 1. Expression of an NR2B subunit with a mutation at Y1472 causes a specific increase in NMDA-mEPSC amplitude.
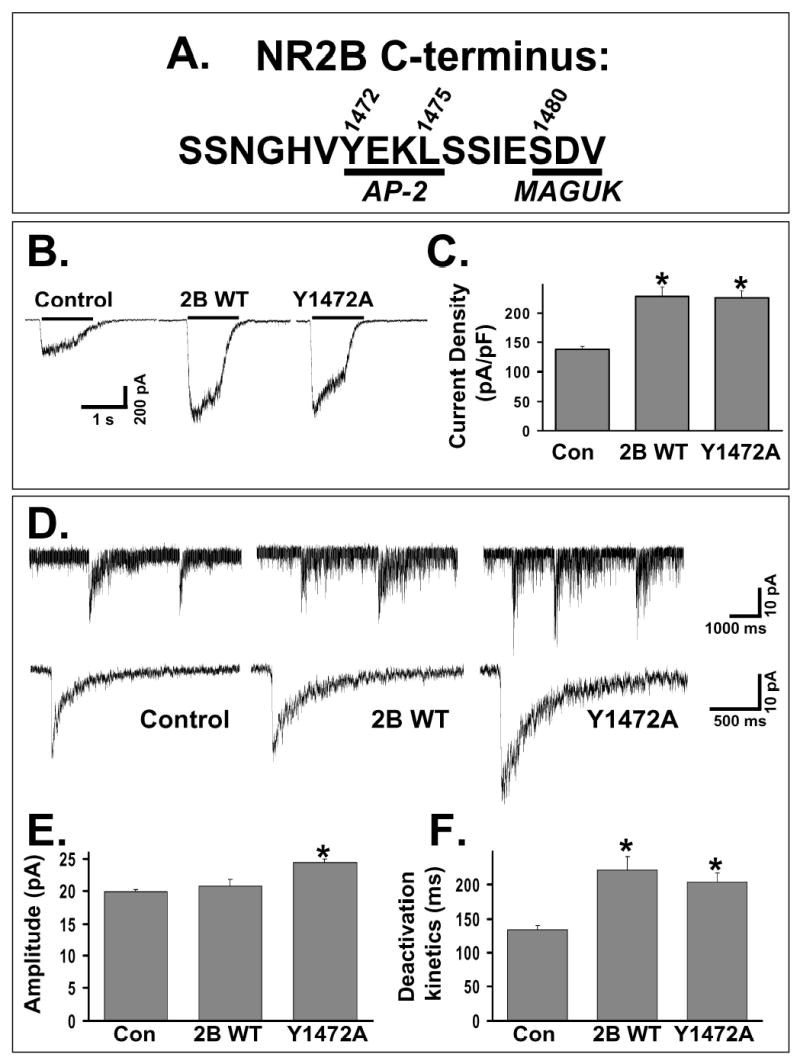
A: The amino acid sequence of the distal C-terminus of the NR2B subunit. Two motifs are underlined, a consensus internalization motif for AP-2 binding (YEKL, 1472-1475) and the PDZ-binding domain that mediates MAGUK binding (SDV, 1480-1482). Amino acids 1472, 1475, and 1480 are sites where mutations were made.
B: Representative traces from control CGC or CGC transfected with cDNAs for wildtype or mutant subunits of the NMDAR. Whole-cell recordings were done 48 hours after transfection with application of 200 μM NMDA and 20 μM D-serine with 1 μM TTX in Mg2+-free solution.
C: Summary of current density responses from multiple CGCs (control, n = 10; NR2B WT, n = 12; Y1472A, n= 15). Transfection with either NR2B WT or Y1472A caused a similar, significant increase in the current density of response (p < 0.01, ANOVA).
D: Representative NMDA-mEPSCs recorded and averaged from individual control or transfected (with NR2B WT or Y1472A) CGCs. Recordings were made in Mg2+-free extracellular solution with 50 μM bicuculline, 1 μM TTX, and 10 μM NBQX.
E: Average amplitude of NMDA-mEPSCs from multiple CGCs (control, n = 25; NR2B WT, n = 15; Y1472A, n= 20). Expression of NR2B WT caused no change in amplitude compared to untransfected CGCs, but expression of Y1472A caused a significant increase (ANOVA, * = p<0.01).
F: Average of the weighted time constant of decay (τw) from multiple CGCs. Expression of either NR2B WT or Y1472A caused a significant slowing of the deactivation kinetics of the NMDA-mEPSCs (ANOVA, * = p<0.01).
Results
Mutation at Y1472 of NR2B increases the number of synaptic NMDARs
We used cerebellar granule cells (CGCs) in culture, a system that allows precise analysis of functional NMDARs that form when transfected NR2 subunits coassemble with the native NR1 subunit population (Prybylowski et al., 2002). Expression of NR2B WT caused an increase in the current density in response to applied NMDA (200 μM NMDA/20 μM D-serine), as previously shown (Prybylowski et al., 2002). Expression of Y1472A caused a significant increase in the current density, which was not different from NR2B WT expression (Figure 1A–C). Experiments in HEK cells also indicated similar surface expression levels and cell death (data not shown). Therefore, a similar number of total surface NMDARs are present with expression of either Y1472A or NR2B WT.
We then analyzed NMDA miniature excitatory postsynaptic currents (NMDA-mEPSCs) in transfected CGCs. Expression of NR2B WT caused a slowing of the deactivation kinetics of the NMDA-mEPSC (due to the slow kinetics of NMDARs containing the NR2B subunit {Cull-Candy et al., 2001}) without a change in the amplitude of response (Figure 1D–F). This indicates that NR2B-containing receptors entered the synapse, which was previously confirmed by an increase in ability of an NR2B-selective antagonist to block NMDA-mEPSCs (Prybylowski et al., 2002). In contrast to NR2B WT, expression of Y1472A caused both an increase in the amplitude as well as a slowing of the deactivation kinetics of the NMDA-mEPSC (Figure 1D–F). Statistical analyses indicate that NMDA-mEPSCs from Y1472A transfected CGCs were larger than both control and NR2B WT transfected CGCs (ANOVA, p<0.01). There was no significant change in the frequency of NMDA-mEPSCs among the different groups (control, 0.07 ± 0.01; 2B WT, 0.07 ± 0.01; Y1472A, 0.07 ± 0.01 Hz). Because of the low background noise, single channel currents could be directly measured from the tails of NMDA-mEPSCs. Single channel currents measured in untransfected and Y1472A transfected CGCs (4.0 ± 0.2 and 4.0 ± 0.3 pA, respectively, from at least 10 cells) were not different. Although we measured conductances of NMDARs that are presumably synaptic, it has been previously shown that synaptic and extrasynaptic NMDARs have similar conductances in cerebellar granule cells (Clark et al., 1997). There was also no difference in agonist affinity between NR2B WT and Y1472A, and similar results were obtained with mutation of Y1472G compared to Y1472A (data not shown). Although synaptic NMDARs may not be saturated by glutamate release by a single vesicle, a larger receptor pool would increase the amplitude of response independently of whether the receptor pool is saturated. In addition, a change in the open probability of the receptor would cause a change in the response to agonist, which is the same for 2B WT and Y1472A. The increased NMDA-mEPSC amplitude without a change in single channel conductance, therefore, suggests that NR2B-containing NMDARs with mutation at Y1472 are increased in number at the synapse compared to those containing NR2B WT subunits.
Relationship of the PDZ-binding domain and AP-2-dependent internalization
The increased NMDA-mEPSC amplitude of Y1472A without a corresponding change in the current density of response to NMDA (which measures both synaptic and extrasynaptic NMDARs) suggests that the change was limited to the synapse. Deletion of the PDZ-binding domain causes a decreased synaptic localization of NR2B-containing NMDARs (Barria et al., 2002; Prybylowski et al., 2002; Steigerwald et al., 2000), so we tested the impact of Y1472A mutation on the trafficking of NMDARs in conjunction with mutations that disrupt PDZ binding. S1480A mutation dramatically decreases the affinity of the NR2B C-terminus for the PDZ domains of MAGUKs (Lim et al., 2002). As expected, receptors containing the mutant S1480A NR2B subunit did not localize to the synapse, as shown by their inability to slow the deactivation kinetics of the NMDA-mEPSC, in contrast to NR2B WT (Figure 2A1–3). S1480A had normal surface expression in CGCs (current densities of response to NMDA application were 173 ± 8 and 188 ± 20 pA/pF for NR2B WT {n=12} and S1480A {n=6} transfected CGCs, respectively), indicating that the PDZ-binding domain is not required for extrasynaptic receptor delivery (Prybylowski et al., 2002; Steigerwald et al., 2000).
Figure 2. Normal PDZ binding is not necessary for the synaptic localization of NR2B-containing NMDARs if AP-2 binding is blocked.
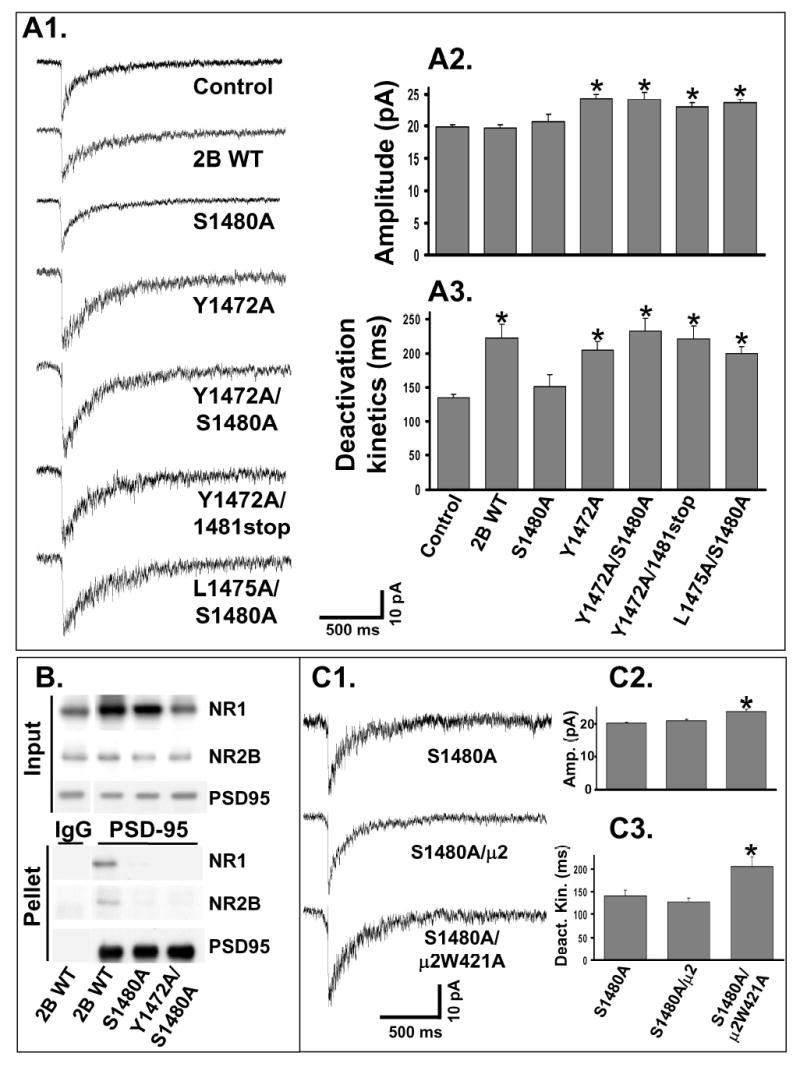
A1: Representative NMDA-mEPSCs were recorded from individual control or transfected CGCs. Y1472A/S1480A, L1475A/S1480A and Y1472A/1481stop (lacking the last amino acid of NR2B) represent double mutations in NR2B.
A2: Average of the NMDA-mEPSC amplitude from multiple CGCs. Expression of NR2B WT (n = 15) or S1480A (n = 16) did not alter NMDA-mEPSCs amplitude compared to control (n = 25). Expression of Y1472A (n = 20), Y1472A/S1480A (n = 15), Y1472A/1481stop (n = 12) or L1475A/S1480A (n = 19) significantly increased NMDA-mEPSCs amplitude compared to control (ANOVA, *=p<0.05).
A3: Average of the weighted time constants of decay (τw). Expression of NR2B WT, Y1472A, Y1472A/S1480A, L1475A/S1480A or Y1472A/1481stop significantly slowed the NMDA-mEPSC deactivation kinetics compared to control (ANOVA, * = p<0.01). Expression of S1480A did not alter NMDA-mEPSC deactivation kinetics compared to control.
B: Deoxycholate solubilized samples from HEK 293 cells were incubated with anti-PSD-95 antibody or non-specific IgG coupled to Protein A agarose. Input and pellet samples were used for Western blot. Images are representative of 3 independent experiments.
C1: CGCs were transfected either with S1480A alone or together with the μ2 WT or μ2W421A AP-2 subunits. Recordings were made at least 3 days after transfection, and representative NMDA-mEPSCs are shown.
C2: Averaged amplitude of NMDA-mEPSCs from multiple CGCs. Coexpression of μ2W421A with S1480A (n = 18) significantly increased NMDA-mEPSC amplitude (p <0.05, ANOVA). Coexpression of μ2 WT with S1480A (n = 10) caused no change in amplitude compared to S1480A expression alone (n = 16).
C3: Average of the weighted time constant of decay (τw) from multiple CGCs. Coexpression of μ2W421A with S1480A significantly slowed the NMDA-mEPSC deactivation kinetics compared to S1480A alone (p <0.05, ANOVA). Coexpression of μ2 WT with S1480A caused no change.
We then tested an NR2B construct with a double mutation at both Y1472A and S1480A (Y1472A/S1480A), disrupting both the AP-2 and the PDZ-binding motifs. Agonist (200μM NMDA/20 μM D-serine) response from cells expressing Y1472A/S1480A was not different from NR2B WT (173 ± 8 and 165 ± 4 pA/pF for NR2B WT {n=12} and Y1472A/S1480A {n=6}, respectively), indicating normal total surface expression and no change in functional properties of the NR2B subunit. Y1472A/S1480A caused a significant slowing of the deactivation kinetics as well as an increase in the amplitude of the NMDA-mEPSC (Figure 2A1–3). As shown in Figure 2A1, NMDA-mEPSCs from Y1472A/S1480A transfected CGCs closely resembled Y1472A transfected CGCs. This result was reproduced with another double mutant, Y1472A/1481stop, which lacks the last amino acid of the NR2B subunit (V1482) that is critical for PDZ binding (Hung and Sheng, 2002). Y1472A/1481stop also caused both a slowing of the deactivation kinetics as well as an increased amplitude of the NMDA-mEPSC (Figure 2A1–3).
Since Y1472 of NR2B may be associated with functions in addition to mediating the interaction with AP-2, we studied the other critical amino acid of the YXXφ consensus site, L1475 (Bonifacino and Traub, 2003). Mutation of this site to a non-permissive amino acid should have an effect similar to that of Y1472A mutation if the results obtained from the latter mutation were due to the disruption of AP-2 binding. L1475A/S1480A also slowed the deactivation kinetics and caused an amplitude increase of the NMDA-mEPSC (Figure 2A). These results showed that mutation of the AP-2 binding site of NR2B, without altering Y1472, could compensate for the loss of the PDZ-binding domain and allow localization of NR2B-containing NMDARs at the synapse.
It is possible that mutation of the AP-2 binding site created a novel interaction between PDZ proteins and the NR2B subunit. To address this, we expressed PSD-95-GFP and NR1-1a with WT and mutated NR2B subunits in HEK 293 cells and analyzed their co-immunoprecipitation. As seen in Figure 2B, NMDAR subunits containing the S1480A mutation or the double mutation Y1472A/S1480A in the NR2B subunit showed no significant interaction with PSD-95. Although the mutation Y1472A/S1480A does not bind well to PSD-95, we cannot rule out that this construct has novel protein-protein interactions. We therefore tested the effect of altering interactions with AP-2 more directly using a dominant-negative construct of the μ2 subunit.
The μ2 subunit of the AP-2 complex has been shown to interact with the YXXφ consensus motif (Nakatsu and Ohno, 2003). We verified that the mutations in the AP-2 binding motif (either Y1472A or L1475A), with or without mutation of S1480A, blocked the interaction with the μ2 subunit of AP-2 using a yeast two-hybrid assay (data not shown). We cotransfected S1480A with μ2 WT and μ2 W421A subunits into CGCs to confirm the role of the AP-2 complex in regulating NR2B localization. μ2 W421A has been previously shown to block internalization of proteins with tyrosine-based internalization motifs through the AP-2 complex (Nesterov et al., 1999; Vissel et al., 2001). Therefore, μ2 W421A can be used to characterize the role of AP-2 dependent internalization due to tyrosine motifs. Coexpression of μ2 WT had no effect in S1480A transfected CGCs (Figure 2C). However, coexpression of μ2 W421A caused a significant increase in the deactivation kinetics and amplitude of NMDA-mEPSCs in S1480A transfected CGCs (Figure 2C). Therefore, NR2B-containing NMDARs that lack normal PDZ binding are able to localize to the synapse if AP-2 dependent internalization is blocked. Expression of μ2 W421A also caused a smaller but significant increase in the NMDA-mEPSC amplitude in CGCs which were not transfected with exogenous NMDAR subunits (22.0±0.4 versus 19.9±0.2 pA, for μ2 W421A {n=10} and control {n=23}, respectively), suggesting that endogenous synaptic NMDARs are also regulated by internalization.
A peptide including the YEKL motif of NR2B increases synaptic NMDA receptors
We then explored the possibility that AP-2 binding to NR2B could be competed by a peptide that contains the distal C-terminus of NR2B including the YEKL motif but without the PDZ-binding domain (NGHVYEKLSSIE, the YEKL peptide). In particular, this study could measure changes in endogenous receptors without expression of exogenous subunits. Our control was the identical peptide with a substitution of alanine at the tyrosine residue (NGHVAEKLSSIE, the AEKL peptide), which would not compete for AP-2 binding to NR2B. Inclusion of the AEKL or YEKL peptide did not cause any change in the response to NMDA over 20 minutes (5 second application of 200 μM NMDA/20 μM D-serine given every minute, Figure 3A). In recordings of NMDA-mEPSCs made with the YEKL peptide, there was an increase in amplitude in events recorded from 10–15 minutes compared to 0–5 minutes after the start of the whole cell recording (Figure 3B–C, p<0.05). In contrast, there was no change in the amplitude of NMDA-mEPSCs recorded with the AEKL peptide over 20 minutes. The amplitude of NMDA-mEPSCs recorded at 10–15 minutes also was significantly larger for cells where the YEKL peptide was included compared to the AEKL peptide. These data suggest that competition of binding of AP-2 to the NR2B tail can regulate synaptic NMDA receptors on a relatively rapid time scale.
Figure 3. Domains in the distal C-terminus of NR2B control its localization.
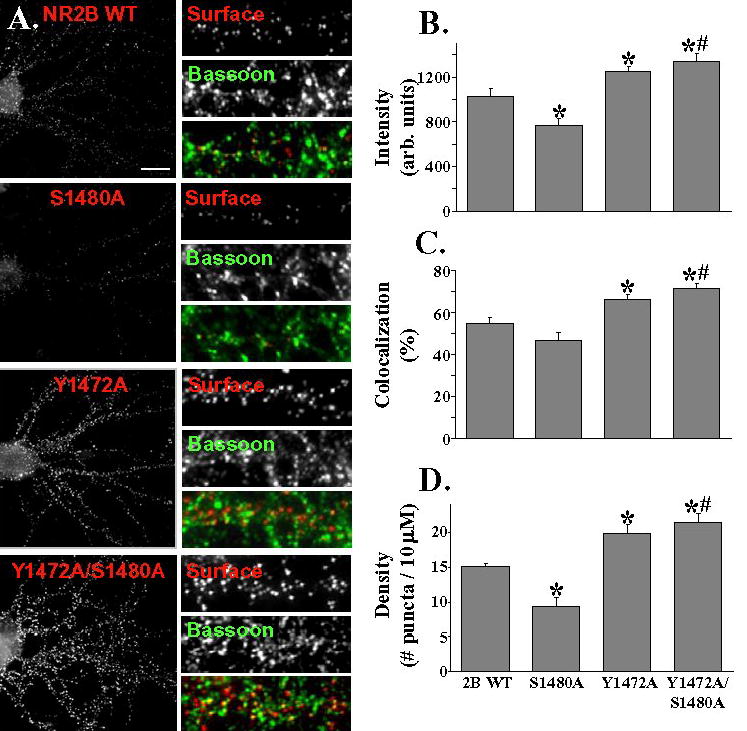
A: Hippocampal neurons were transfected with NR2B constructs containing an extracellular Flag epitope (the same constructs that were used for electrophysiology experiments). Surface staining was done with Flag antibody (in red), followed by fixation and permeabilization, and then staining was done for the presynaptic marker protein, Bassoon (in green). Scale bar = 10 μm.
B: Dendritic puncta were analysed after transfection with different constructs. 5 dendritic sections were measured and averaged to give a value for each cell included. At least 11 cells were counted from 2 separate transfections for each group. In comparison to NR2B WT, the S1480A mutant showed a decrease in intensity while Y1472A and Y1472A/S1480A showed an increase in intensity (* = p<0.05). Y1472A/S1480A shows a significantly greater puncta intensity compared to S1480A (# = p<0.01, Student's t-test)
C: Co-localization of Flag puncta to Bassoon puncta was measured. Value is given in percent co-localization (defined as the number of Flag puncta overlapping or adjacent to Bassoon puncta, divided by the total number of Flag puncta). In comparison to 2B WT, Y1472A and Y1472A/S1480A show increased co-localization with Bassoon, while the S1480A mutant does not show a significant difference. There was a significant difference in percent co-localization with Bassoon in comparing S1480A and Y1472A/S1480A (Student's t-test, # = <0.01).
D: The Y1472A and Y1472A/S1480A mutants show increased density (# of puncta/10μm) of puncta compared to 2B WT, while the S1480A mutant shows decreased density of puncta. In comparison to the S1480A mutant, the Y1472A/S1480A showed significantly higher density of puncta (Student's t-test, # = <0.01).
Regulation of NMDARs by Fyn kinase
Phosphorylation of the tyrosine residue of the YXXφ motif has been shown to block binding of AP-2 (Bonifacino and Traub, 2003; Owen and Evans, 1998). As Y1472 of NR2B is phosphorylated by Fyn kinase (Nakazawa et al., 2001), we hypothesized that phosphorylation of this residue would affect the synaptic localization of NMDARs. In HEK 293 cells, co-expression of Fyn Y531F, a constitutively active Fyn, with NR1 and NR2B caused a significant increase in tyrosine phosphorylation of the NR2B subunit (using immunoprecipitation of NR2B and immunoblotting with the anti-phosphotyrosine antibody, 4G10 {data not shown}).
Expression of Fyn Y531F increased the amplitude of NMDA-mEPSCs with no change in the deactivation kinetics of the response, suggesting that a change occurred at synaptic NMDARs (Figure 4). There was no change in the single channel current of cells expressing Fyn Y531F (4.1 ± 0.2 and 4.2 ± 0.2 pA for control and Fyn Y531F, respectively; data from at least 7 cells). Previous studies in heterologous systems indicated that recombinant NR1/NR2B receptors are not regulated by Fyn (Kohr and Seeburg, 1996), and expression of Fyn Y531F did not cause a change in the response to agonist (current density of 107±9 pA/pF and 104±15 pA/pF for control {n=15} and Fyn Y531F {n=8} transfected, respectively), supporting the idea that there is no change in functional properties of the total NMDAR pool. Therefore, phosphorylation of NMDARs by Fyn increases the number of synaptic NMDARs.
Figure 4. A peptide containing the distal tyrosine motif of NR2B increases amplitude of NMDA-mEPSCs.
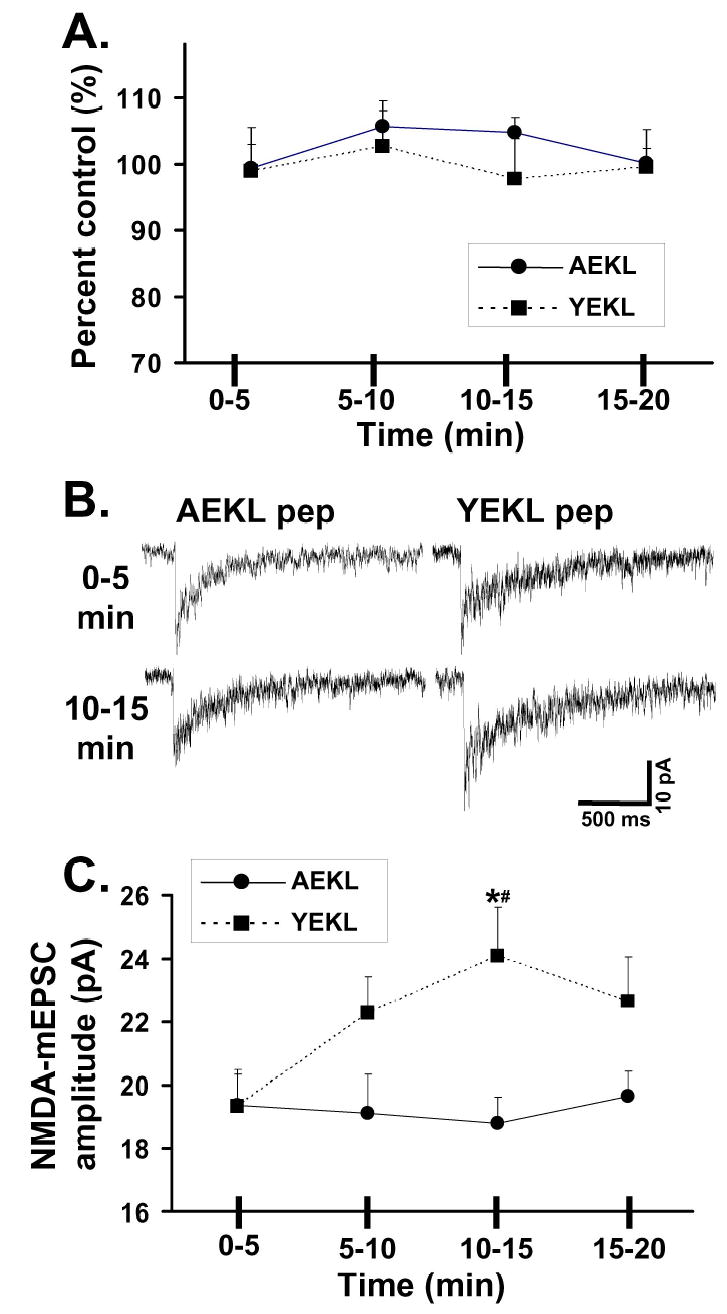
A: Responses were recorded with inclusion of peptide in the recording solution. YEKL peptide (NGHVYEKLSSIE) contains the distal tyrosine motif of NR2B, while AEKL peptide (NGHVAEKLSSIE) lacks the critical tyrosine for binding to AP-2. Responses were measured every minute to a 1 second application of 200 μM NMDA/20 μM D-serine (control equals value of first response). Responses within the time periods were averaged for each cell (n=7 for YEKL; n=9 for AEKL).
B. Representative NMDA-mEPSCs were recorded and averaged from CGCs during different times of recording.
C. Average of the NMDA-mEPSC amplitude (from data pooled during time periods) from multiple CGCs. While there was no statistical change in the amplitude of recordings over time with the AEKL peptide (n=8), inclusion of the YEKL peptide (n=7) in the recording pipette caused an increase in the amplitude of NMDA-mEPSCs in the time frame of 10–15 minutes compared to YEKL 0-5 or to AEKL 10-15 (p<0.05).
The ability of Fyn Y531F to increase the number of synaptic NMDARs suggested that it might also promote the synaptic localization of NMDARs lacking a PDZ-binding domain. Coexpression of S1480A with Fyn Y531F caused an increase in the amplitude and deactivation kinetics of the NMDA-mEPSC (Figure 4A–C), indicating that tyrosine phosphorylation is sufficient to allow the synaptic localization of NR2B subunits lacking PDZ-binding domains. Coexpression of Fyn Y531F with NR2B WT also caused an increase in NMDA-mEPSC amplitude that was not present with expression of NR2B WT. In contrast, there was no evidence of increased amplitude of NMDA-mEPSCs in CGCs expressing Y1472A together with Fyn Y531F compared to those without Fyn. Therefore, mutation at Y1472A and increased tyrosine phosphorylation of the NR2B subunit do not have an additive effect on NMDA-mEPSC amplitude.
Synaptic localization of NR2A-containing NMDARs is not controlled by the PDZ-binding domain or the distal tyrosine motif
There are multiple reports that the synaptic trafficking and localization of NR2A-and NR2B-containing NMDARs receptors are different and that these subunit differences are regulated by development and activity (Barria and Malinow, 2002; Rumbaugh and Vicini, 1999; Tovar and Westbrook, 1999; for review of the topic, Prybylowski and Wenthold, 2004). We thus assessed whether the AP-2 binding that regulates the NR2B subunit also controls the NR2A subunit. Because receptors containing the NR2A subunit are characterized by relatively fast deactivation kinetics compared to other NR2 subunits (Cull-Candy et al., 2001), entry of NR2A WT into the synapse is accompanied by a speeding of the deactivation kinetics of the NMDA-mEPSC, as shown in Figure 5B. Thus, faster deactivation kinetics of NMDA-mEPSCs are a marker of the synaptic localization of the NR2A subunit. An S1462A mutant of NR2A was studied with mutation of the serine in the PDZ-binding domain. S1462A mutation of NR2A corresponds to S1480A mutation of NR2B, as the NR2A and NR2B subunits have identical PDZ-binding domains, -SDV, at their distal C-termini (see Figure 5A). S1462A expression produces NMDA-mEPSCs with significantly faster deactivation kinetics than control. The deactivation kinetics of S1462A were indistinguishable from those obtained with NR2A WT (Figure 5B,D). Thus, unlike the exclusion of NR2B subunits lacking PDZ-binding domains (S1480A) from the synapse (Figure 2), NR2A subunits lacking a PDZ-binding domain still localize to the synapse.
Figure 5. Increased tyrosine phosphorylation promotes the synaptic localization of NMDARs.
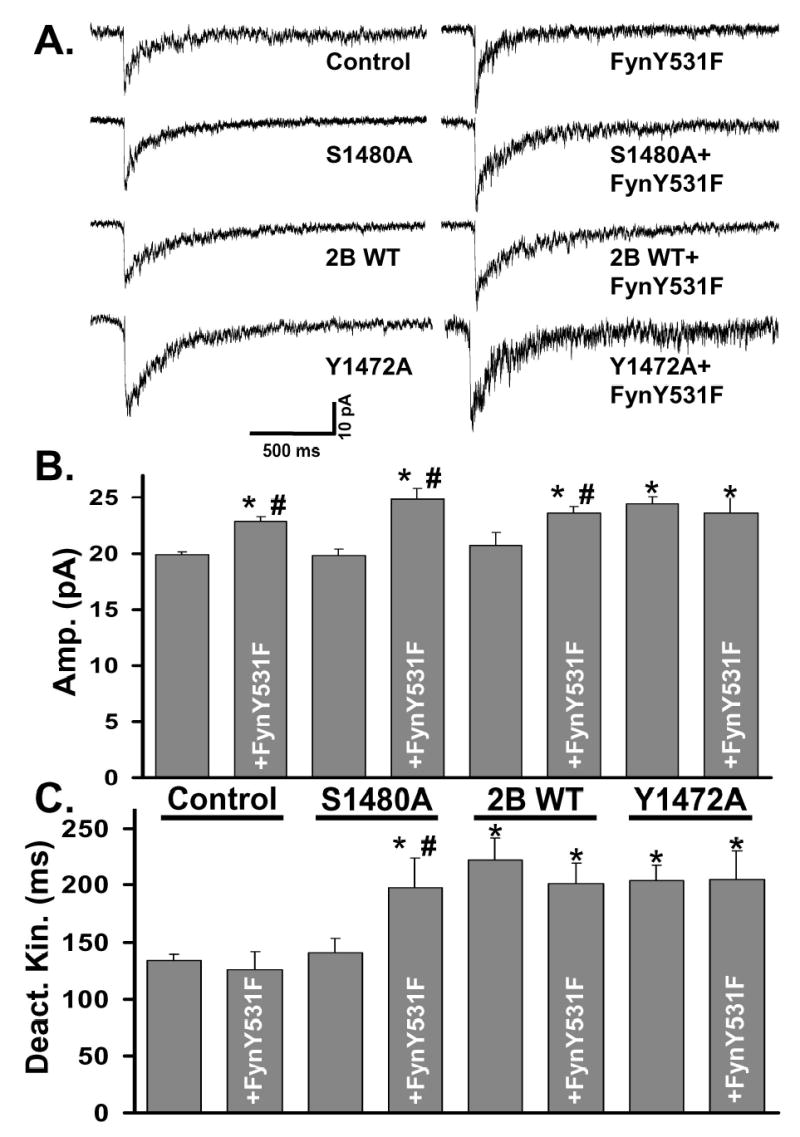
A: Representative NMDA-mEPSCs were recorded and averaged from individual control or transfected CGCs as described in Figure 1.
B: Average of the NMDA-mEPSC amplitude from multiple CGCs. Expression of S1480A (n = 16) caused no change in NMDA-mEPSC amplitude. Expression of Fyn Y531F alone (n = 17) or with any of the NR2B constructs significantly increased NMDA-mEPSC amplitude compared to control (ANOVA, *=p<0.05). Coexpression of Fyn Y531F together with S1480A (n = 19) or NR2B WT (n = 10) caused a significant increase from the NR2B subunit alone (ANOVA, #=p<0.05). Coexpression of Fyn Y531F with Y1472A (n=6) did not cause any change from Y1472A alone (n=20).
C: Average of the weighted time constants of decay (τw). Expression of S1480A or Fyn Y531F alone did not alter NMDA-mEPSC deactivation kinetics. Expression of NR2B WT or Y1472A significantly slowed the deactivation kinetics of the response (*=p<0.01) with no change with coexpression with Fyn Y531F. Coexpression of Fyn Y531F with S1480A significantly slowed the deactivation kinetics of NMDA-mEPSCs compared to control (*=p<0.01) or S1480A alone (#=p < 0.01).
Since the synaptic exclusion of NR2B subunits lacking PDZ-binding domains can be reversed by disruption of AP-2 binding to YEKL (Figure 2), we hypothesized that NR2A subunits lacking a PDZ-binding domain localize to the synapse because NR2A-containing NMDARs are not controlled by AP-2 binding to a distal consensus tyrosine motif YKKM containing Y1454 (which has been shown to poorly bind the μ2 subunit of AP-2 {Lavezzari et al., 2004}). We found that expression of an NR2A mutant at Y1454A (the distal tyrosine corresponding to Y1472 in NR2B) produced fast NMDA-mEPSCs like NR2A WT with no change in the amplitude from control or NR2A WT (Figure 5). Thus, mutation of the YKKM motif of the NR2A subunit does not promote the synaptic localization of NMDARs, suggesting that the distal tyrosine motif of NR2B is a subunit-specific regulator of NMDAR localization.
Since normal PDZ binding is necessary for localizing NR2B, but not NR2A, to the synapse, this difference could be due to the amino acid differences between NR2B and NR2A in the distal C-terminus (Fig. 5A). We generated a construct, 2AMut5, which is NR2A with 5 amino acid mutations to create a sequence identical to that of NR2B in its last 16 amino acids (five amino acids were mutated, R1451G, R1452H, K1455E, M1457L, and P1458S, Figure 5A). 2AMut5 entered the synapse as measured by the fast deactivation kinetics of NMDA-mEPSCs in CGCs transfected with this construct compared to control. These deactivation kinetics with 2AMut5 were not statistically different from those of NR2A WT (Figure 5D). Thus, NR2A can enter the synapse with the distal amino acids of NR2B.
To see if 2AMut5 is dependent upon PDZ binding to localize at the synapse as is NR2B, we generated 2AMut6, which has the 5 mutations present in 2AMut5 as well as mutation of the serine of the PDZ-binding domain (S1462A). We find that expression of any of the NR2A constructs caused a similar increase in the whole cell response, indicating similar surface expression levels {current density to application of 200 μM NMDA/20 μM D-serine of 108±8 pA/pF for control (n=15); 176±17 for 2A WT (n=10); 178±28 for S1462A (n=6); 178±20 for Y1454A (n=7); 173±20 for 2AMut5 (n=11); 175±21 for 2AMut6 (n=6)}. The deactivation kinetics of NMDA-mEPSCs from 2AMut6 transfected CGCs were significantly slower than those of NR2A WT (Figure 5D). In 2AMut6 transfected CGCs, there was no change in the deactivation kinetics of NMDA-mEPSCS in CGCs compared to control, indicating that this 2AMut6 subunit does not localize at the synapse, unlike NR2A WT (Figure 5). Therefore, amino acids differences between NR2A and NR2B in their distal C-terminus dictate their trafficking properties.
Discussion
Our results show that stabilization through a PDZ protein and internalization through an interaction with AP-2 are important determinants of the number and composition of synaptic NR2B-containing NMDARs. Y1472 of NR2B, in addition to being a critical residue of the AP-2 binding motif, is also a substrate for Fyn kinase dependent phosphorylation, providing a mechanism for rapid modulation of synaptic receptors. These findings suggest a model for regulation of NR2B-containing NMDARs, which links phosphorylation, internalization, and PDZ protein interactions (Figure 6). Since receptors containing the NR2A subunit are not regulated in a similar manner, these events may play a key role in subunit-specific trafficking of NMDARs.
Figure 6. Localization of NR2A at the synapse is independent of the mechanisms regulating NR2B.
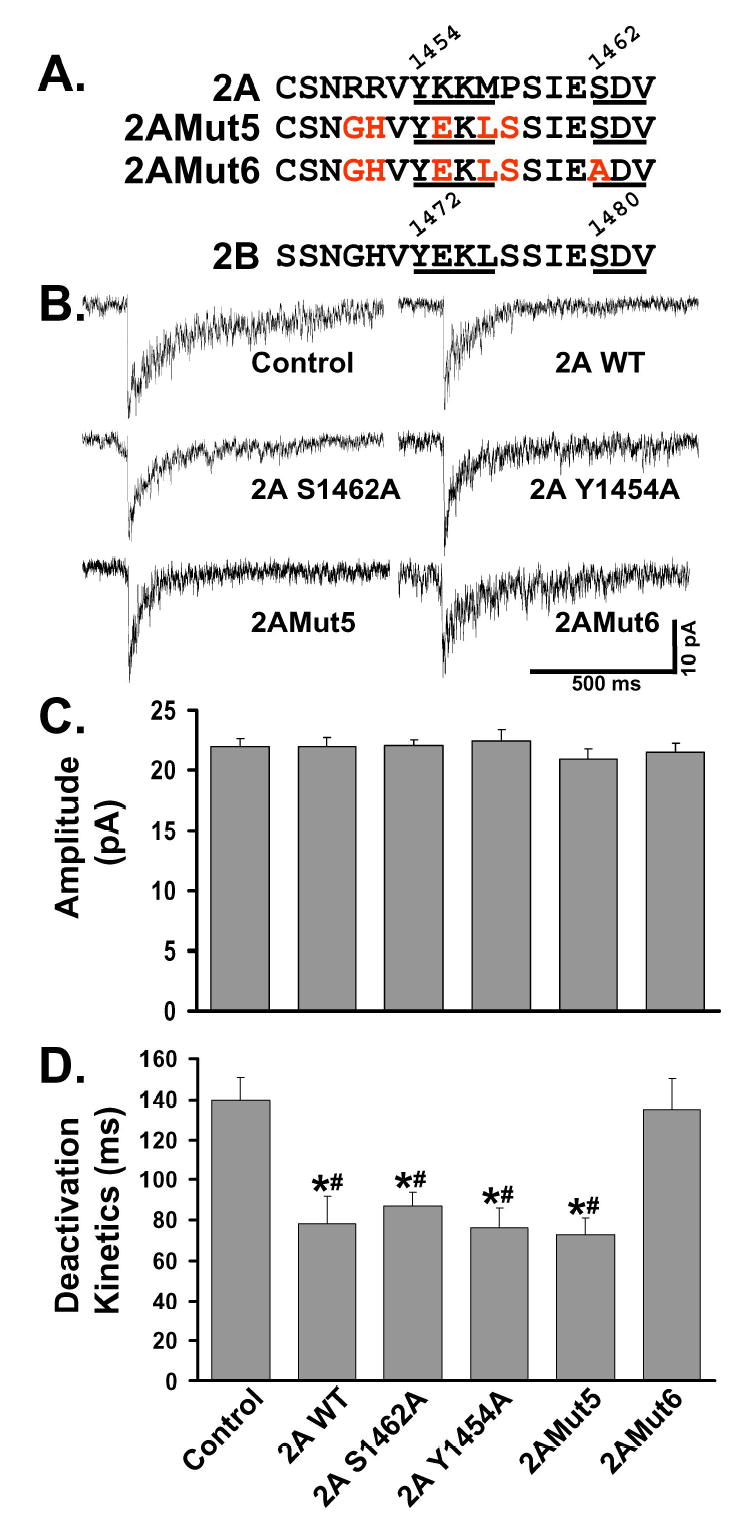
A: Comparison of the amino acid sequences of the distal C-termini of the NR2A and NR2B subunits. Both contain a tyrosine based motif that fits the consensus for binding of the μ2 subunit of AP-2 (YEKL, 1472-1475 for NR2A; YKKM, 1454-1457 for NR2B). NR2A and NR2B have identical PDZ-binding domains, SDV, as their distal 3 amino acids. From NR2A WT, S1462A (within PDZ-binding domain) and Y1454A (within consensus internalization motif) were generated at the corresponding amino acids to NR2B S1480A and Y1472A. NR2AMut5 is a mutation of NR2A, in which the 16 distal amino acids in NR2A are converted to the corresponding amino acids in NR2B (five amino acids were mutated: R1451G, R1452H, K1455E, M1457L, and P1458S). NR2AMut6 has the five mutations present in NR2AMut5, plus a mutation of the serine of the PDZ-binding domain (S1462A).
B: Representative NMDA-mEPSCs were recorded and averaged from individual control or transfected CGCs as described in Figure 1. CGCs transfected with various NR2A constructs were compared to control.
C: Average of the NMDA-mEPSC amplitude from multiple CGCs. Expression of any of these NR2A constructs did not alter the amplitude of the NMDA-mEPSC compared to those of control, untransfected CGCs (n’s given below). Bar labels are given in Panel D.
D: Average deactivation kinetics of NMDA-mEPSCs from multiple CGCs. Expression of NR2A WT (n=8), S1462A (n=12), Y1454A (n=9), or NR2Mut5 (n=8) produced NMDA-mEPSCs that were significantly faster than control (*=p<0.05). There was no statistical difference between the deactivation kinetics of NR2A WT, S1462A, Y1454A, and NR2AMut5. NR2AMut6 (n=13) expression produced NMDA-mEPSCs that were not significantly different from control (n=13) CGCs, but which were significantly slower than other NR2A constructs.
Regulation of NR2 subunits by PDZ interactions
Although not directly proven, it is generally thought that synaptic NMDARs are retained at the synapse by an attachment to PSD-95 (or another PSD-95 family member) through a PDZ interaction with the NR2 subunits (Scannevin et al., 2000). Removal of the receptor would require either breaking the PDZ interaction or removal of the PDZ protein (which may include associated proteins) along with the NMDAR. Recent work has shown that the affinity of the PDZ interaction can be altered by activity-dependent phosphorylation of Ser1480 by casein kinase II, suggesting that the binding of the NMDAR to PDZ proteins is a regulated process (Chung et al., 2004). In addition, cotransfection with PSD-95 can slow the internalization rate of chimeras containing the tail of the C-terminus of the NR2B subunit (Roche et al., 2001). These data support the idea that maintenance of the PDZ interaction is a critical step in keeping NMDA receptors at the synapse, and that disrupting this interaction may be the first step in the removal of the receptor. Our data show that an interaction of the NR2B subunit with AP-2 is also required for removal, indicating that internalization is a dominant regulator of NR2B-containing receptors at the synapse. Our results, however, also suggest that the PDZ interaction with NR2 subunits may not be required for synaptic localization. NR2B-containing NMDARs lacking PDZ-binding domains are retained at the synapse if the AP-2 internalization motif of NR2B is mutated or AP-2 binding to tyrosine motifs is blocked (Figure 2). In addition, a PDZ-binding domain mutant of NR2A localizes to the synapse irrespective of mutation in the internalization motif (Figure 5). Retention at the synapse may, therefore, depend upon other proteins, such as the EphB receptor (Takasu et al., 2002), which has been shown to interact with NMDARs through extracellular domains.
Regulation of NR2B subunit by Fyn phosphorylation
Regardless of their role as anchoring molecules, PSD-95 and other MAGUKs may play a critical role in recruiting Fyn, which has been shown to be the predominant kinase responsible for the phosphorylation of Y1472 of the NR2B subunit (Nakazawa et al., 2001). PSD-95 associates with Fyn and brings the kinase into close proximity with the NMDAR (Tezuka et al., 1999; Ali and Salter, 2001). Therefore, NR2B-containing receptors at the synapse may require phosphorylation of Y1472 for retention, which may occur if the receptor is in close proximity to a Fyn/PSD-95 complex. If this is the case, the PDZ interaction may not directly anchor NR2B-containing receptors at the synapse, but rather indirectly anchor them by keeping PSD-95 and Fyn in close proximity and, consequently, keeping Y1472 phosphorylated and unable to interact with AP-2. In support of a role of tyrosine phosphorylation in NMDAR trafficking, tyrosine phosphorylated NMDARs in the striatum are enriched in the synaptic membrane and decreased in intracellular membranes (Dunah et al., 2001). Blocking tyrosine phosphorylation also causes relatively rapid internalization of extrasynaptic NMDARs (Li et al., 2002). Furthermore, there is increased phosphorylation of NR2B after LTP induction (Rostas et al., 1996, Rosenblum et al., 1996), and Fyn mutant mice have impaired long term potentiation, which can be rescued by postnatal expression of Fyn (Kojima et al., 1997). Our studies identify a novel function of Fyn in regulating internalization and synaptic localization of NR2B-containing NMDARs. This has critical functional consequences since tyrosine phosphorylation of NR2B can be altered in pain paradigms (Guo et al., 2002), with lithium treatment (Hashimoto et al., 2002) and with ethanol treatment (Alvestad et al., 2003).
Regulation of synaptic and extrasynaptic NMDARs
Although NMDARs are concentrated at synapses, functional NMDARs are known to localize at extrasynaptic sites, with potential differences in the role of synaptic versus extrasynaptic NMDARs. In particular, synaptic NMDARs have been proposed to mediate signaling pathways that promote neuronal survival, while extrasynaptic NMDARs play a role in excitotoxicity (Hardingham and Bading, 2003). We have shown previously (Prybylowski et al., 2002) that expression of NR2A WT or NR2B WT subunits in CGCs causes a significant increase in the amplitude of response to agonist application (which activates all surface receptors) with no change in the amplitude or frequency of NMDA-mEPSCs. This provided evidence that while the extrasynaptic receptor population can be regulated by the total amount of assembled complexes, the synaptic NMDAR population is regulated independently of availability of subunit complexes. We found in the present study that mutations in either the PDZ-binding domain or the YEKL motif of the NR2B subunit similarly only affect the synaptic NMDAR population, without significant effect on the whole cell response. It has been previously shown that a peptide that blocks AP-2 binding to the GluR2 subunit of the AMPAR does not alter basal synaptic AMPA transmission or affect total surface expression of AMPARs, but these peptides block both NMDA-induced internalization of AMPARs and long-term depression of synaptic responses (Lee et al., 2002). Therefore, synaptic NMDA and AMPA receptors may be specifically controlled by internalization.
Subunit-specific regulation of NMDARs
Previous studies have shown that receptors containing the NR2B subunit are regulated differently from those containing the NR2A subunit (Cull-Candy et al., 2001). NR2B-containing receptors are abundant at the synapse early in development (Rumbaugh et al., 1999; Tovar et al., 1999; Townsend et al., 2003), while with activity NR2A-containing receptors can replace NR2B-containing receptors (Barria et al., 2002). In particular, introduction of light stimuli allows rapid replacement of synaptic NR2B subunits with NR2A subunits in the visual cortex (Philpot et al., 2001). Distinct subunit composition of synaptic NMDARs has been suggested to underlie differences in synaptic plasticity (Liu et al., 2004). Our results suggest that the differential regulation of NR2A and NR2B containing receptors at the synapse may depend upon specific interactions of NR2B with PDZ proteins and AP-2. Unlike NR2B, we find that NR2A does not require a PDZ-binding domain for synaptic localization and that NR2A subunits are not affected by mutations in their distal YKKM motif (Figure 5). It has been shown previously that the internalization motifs of NR2A and NR2B differ: NR2A has a motif (YKKM) that is similar to the YEKL motif of NR2B, but it is not a substrate for AP-2 binding (Lavezzari et al., 2003; Lavezzari et al., 2004). Rather, internalization of the NR2A subunit appears to involve a more proximal dileucine motif (Lavezzari et al., 2004). We directly studied the differences in NR2A and NR2B by generating a construct, NR2AMut5, which is identical to NR2A except in its last 16 amino acids, which are identical to those of NR2B. NR2AMut5 entered the synapse, but with mutation of the serine of the PDZ-binding domain (NR2AMut6) this construct did not localize to the synapse. Thus, mutation of the C-terminus of NR2A to the amino acids of NR2B generates a subunit that is dependent upon PDZ binding to localize at the synapse.
Since the PDZ-binding domains of NR2B and NR2A are identical, our data on the ability of NR2A and the inability of NR2B to traffic to the synapse in the absence of PDZ binding are surprising. However, other findings in other studies could be attributable to differences in PDZ interactions of these subunits. NR2A knockout mice show a developmental loss of miniature NMDA currents (evoked in the presence of tetrodotoxin to allow release of glutamate from single vesicles), which likely corresponds to loss of NMDARs that are spatially closest to release sites (Towsend et al., 2003). In contrast, electrically evoked currents are maintained in these NR2A knockout mice, suggesting that the remaining NMDARs (predominantly NR2B-containing) are spatially more distant from release sites but can respond to glutamate diffusion produced by release of multiple vesicles. In addition, these authors found a decreased binding of PSD-95 to NR2B in the absence of NR2A. This led to a model suggesting that activity-dependent insertion of PSD-95 into the postsynaptic density of NR2A knockout mice leads to a NMDAR-free “hole” in the synapse due to loss of NR2B (Towsend et al., 2003). Our data provide mechanistic support for these findings and indicate that the NR2B subunit is under internalization pressure in the absence of PDZ binding. Since the NR2A subunit can localize to the synapse in the absence of PDZ binding, it suggests that this subunit may be relatively resistant to changes in the post-synaptic density because it does not have the internalization behavior of NR2B.
Evidence for dynamic regulation of synaptic NMDARs by internalization
Our studies on the interaction between the NR2B subunit and AP-2 using a peptide corresponding to the distal portion of the NR2B subunit, including the YEKL motif but excluding the PDZ-binding domain, revealed a surprisingly rapid change in synaptic NMDA receptors. Infusion of the peptide led to a significant increase in the amplitude of the NMDA-mEPSC within 10 minutes (Figure 3), suggesting a dynamic basal regulation of endogenous NMDARs by internalization. This time frame is similar to that reported for AMPA receptors after infusion of a peptide that interferes with the interaction of AP-2 with GluR2 (Lee et al., 2002). While NMDARs at the synapse have been thought to be relatively static compared to AMPARs (Malinow and Malenka, 2002), there is evidence of changes in synaptic NMDARs within a similar time frame. For example, glycine priming has been shown to produce relatively rapid internalization of NMDARs (within 10 minutes), with a decline in synaptic NMDA response that can be blocked with a peptide that inhibits clathrin-dependent endocytosis (Nong et al., 2003). In addition, when synaptic NMDARs are irreversibly blocked with MK-801, there appears to be rapid lateral diffusion of functional NMDA receptors into the synapse, suggesting that a large portion of synaptic NMDARs could be exchanged in under 10 minutes (Tovar and Westbrook, 2002). Lateral diffusion is very likely to also play a role in changes in synaptic NMDARs to allow the receptor to move from the synapse to an extrasynaptic site (Choquet and Triller, 2003), as clathrin and endocytic zones have been shown to be concentrated outside the postsynaptic density (Blanpied et al., 2002; Petralia et al., 2003; Racz et al., 2004).
While the AP-2 complex is widely distributed (and immunolabeling can be found at the postsynaptic membrane {Petralia et al., 2003; Racz et al., 2004}), phosphorylation of the μ2 subunit and association with PI[4,5]P2 is required for clathrin-dependent internalization of proteins containing the YXXφ motif (Nakatsu et al., 2003). Thus, two events could regulate the binding of AP-2 to NR2B: 1) the availability and phosphorylation state of the site on the receptor subunit, such as YEKL for NR2B, and 2) the availability and phosphorylation state of the proper adaptor subunit. Electron microscopy studies indicate that AP-2 resides significantly closer to the post-synaptic density (PSD) than clathrin or dynamin (Racz et al., 2004). Therefore, binding of adaptor proteins could occur within the PSD and mark receptor complexes for internalization at lateral endocytic zones (Racz et al., 2004). Thus, reductions in NMDA responses observed following changes in synaptic activity may be linked to regulated endocytosis (Montgomery and Madison, 2002).
Experimental Procedures
DNA constructs
NR2B-Flag (NR2B subunit tagged at the N-terminus with Flag epitope, a gift of Dr. F. Anne Stephenson, UCL) has been previously described (Hawkins et al., 1999). The cDNA was isolated from mouse brain and has a sequence identical to that of GenBank # D10651. The NR2A was a gift of Dr. Peter Seeburg (Max Planck Institute), and a myc tag was placed immediately after its leader sequence at amino acid position 23. All constructs were generated with site-directed mutagenesis (QuikChange Site-Directed Mutagenesis Kit, Stratagene) using NR2B-Flag or NR2A-myc as the template. All constructs were verified by DNA sequencing. μ2 WT and μ2 W421A subunits of the AP-2 complex were gifts from Dr. Alexander Sorkin (UCHSC). Fyn Y531F was a gift of Dr. Dorit Ron (UCSF).
Cerebellar granule cell culture
Primary cultures of rat cerebellar granule cells (CGCs) were prepared from postnatal day 7 (P7) Sprague Dawley rat cerebella (Prybylowski et al., 2002). Cells were cultured in basal Eagle’s medium supplemented with 10% fetal bovine serum (FBS), 2 mM glutamine, and 100 μg/ml gentamicin, with a final concentration of 25 mM KCl (Invitrogen). To achieve functional synapse formation, on day in vitro 5 (DIV5), media was replaced with low (5 mM) potassium media (MEM supplemented with 10 μM cytosine arabinofuranoside, 5 mg/ml glucose, 0.1 mg/ml transferrin, 0.025 mg/ml insulin, 2 mM glutamine and 20 μg/ml gentamicin; Invitrogen). CGCs were transfected on DIV5, using a modification of the calcium phosphate precipitation technique (previously described in Prybylowski et al., 2002). EGFP plasmid (Clontech) was co-transfected to allow visualization of successfully transfected cells.
Immunoprecipitations
HEK 293 cells were transfected overnight using calcium phosphate (Invitrogen). Cells were transfected with NR1-1a (previously described in Prybylowski et al., 2002), PSD-95-GFP (gift of Dr. David Bredt, UCSF) and NR2B cDNAs (at a 5NR1:10NR2B:1PSD-95-GFP ratio). Transfected cells were maintained with NMDAR antagonists (500 μM ketamine and 2 mM kynurenic acid; Sigma) for 48 hours after transfection. Cells were collected and solubilized with 1% DOC (pH 7.5) as previously outlined (Sans et al., 2000). Samples were incubated overnight with Protein A agarose (Pierce), which was pre-coupled (for 3 hours at 4°C with 10 μl rabbit serum) either with anti-PSD-95 (T60, previously described {Sans et al., 2000}) or with pre-immune IgG. Samples were then centrifuged and washed, and the pellet was collected for standard Western blotting. Resulting blots were incubated with monoclonal antibodies, including anti-NR2B (Transduction Labs, 1:250), anti-GFP (Chemicon, 1:2000), or anti-NR1 (clone 54.2, 1:4000). Goat anti-mouse secondary antibody was used (Amersham), and blots were processed using the Amersham ECL plus kit.
Solutions and drugs for electrophysiology
The recording chamber was continuously perfused at 5 ml/min with an extracellular media composed of (in mM): NaCl (145), KCl (5), MgCl2 (1), CaCl2 (1), HEPES (5), glucose (5), sucrose (25), phenol red (0.25 mg/l) and D-serine (20 μM) (all from Sigma). Successfully transfected cells (2–4 days after transfection) were visualized using GFP fluorescence. CGCs were voltage clamped at −60 mV, and whole-cell recordings were performed (using Axopatch 200; Axon Instrument). CGCs transfected with μ2 constructs were recorded at least 3–4 days after transfection to allow assembly into AP-2 complexes. A potassium gluconate recording solution was used containing (in mM): potassium gluconate (145), HEPES (10), ATP.Mg (5), GTP.Na (0.2), and BAPTA (10), adjusted to pH 7.2 with KOH. Drug application was done using a multibarrel pipette controlled by a stepper motor (SF-77b, World Precision Instruments). To measure NMDA miniature excitatory postsynaptic currents (NMDA-mEPSCs), Mg2+-free extracellular solution with 1 μM tetrodotoxin (TTX), 5 μM NBQX, and 50 μM bicuculline (Sigma) was applied to the cell being recorded. All experiments were performed at room temperature (24–26°C). Data analysis was done as previously described in Prybylowski et al., 2002 using Clampfit 9 (Axon Instruments) and Minianalysis (Synaptosoft). Single channel current measurements were made from the tails of NMDA-mEPSCs. Fitting of the decay phase of averaged currents was performed using a simplex algorithm for least squares exponential fitting routines, and we used a weighted mean decay time constant τw = [If/(If+Is)] * tf + [Is/(If+Is)] * ts. For all groups, at least 7 CGCs were used. Data values are expressed as mean ± SEM unless otherwise indicated. p values represent the results of ANOVA analysis, with p<0.05 being defined as the level for significance.
Peptide studies
Peptides of the NR2B tail were generated by Princeton BioMolecules. One contained the YEKL motif (NGHVYEKLSSIE, YEKL peptide) while the control peptide lacked the internalization motif present in NR2B (NGHVAEKLSSIE, AEKL peptide). To allow normal internalization in these experiments, these experiments were done with recording solution that lacked BAPTA. Recordings were done with the peptide (100 μM) included in recording solution containing protease inhibitors (1 μg/ml each of aprotonin and leupeptin).
Yeast two-hybrid protein interaction assay
The interaction between NR2B C-terminal tails (from amino acid position 1315 to 1482) with various mutations at the YEKL wmotif (amino acids 1472-1475) and the AP-2 μ2 subunit was investigated with a yeast two-hybrid interaction assay. NR2B C-terminal sequences were subcloned into the binding domain containing pGBKT7 vector. The AP-2 μ2 subunit was subcloned into the activation domain containing pACTII vector and the positive control TGN38 into the binding domain containing pGBT9 vector (original μ2 and TGN38 constructs were gifts from Dr. Juan Bonifacino, NIH). AH109 yeast cells (BD Clontech) were co-transformed with NR2B containing vectors (or TGN38 as a control) and μ2 vector. After incubation at 30°C for 2–5 days, the presence of blue colonies was an indication of an interaction between the NR2B C-terminus and the μ2 subunit.
Model summarizing the role of PDZ proteins, AP-2 mediated internalization, and phosphorylation by Fyn kinase in the regulation of NR2B-containing receptors at the synapse.
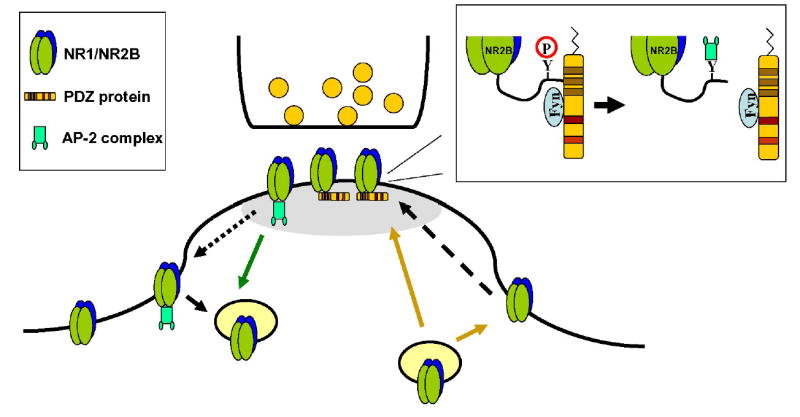
NMDARs enter the post-synaptic density (gray shading) either through direct delivery or by lateral diffusion from extrasynaptic sites (arrows). Fyn is recruited by the PDZ protein. Fyn facilitates the phosphorylation of Y1472 of NR2B and prevents AP-2 binding (inset). Removal of the receptor from the synapse requires AP-2 binding. While AP-2 binding may occur at the synapse, internalization likely occurs at lateral endocytic zones.
Acknowledgments
We are grateful to Drs. Juan Bonifacino, David Bredt, Dorit Ron, Peter Seeburg, Alexander Sorkin, and F. Anne Stephenson for cDNA constructs. We thank Dr. Jeffrey Diamond for helpful comments. This work was supported by the Pharmacology Research Associate (PRAT) program (K.P.), NIDCD Intramural Program (K.P., K.C., L.K., R.J.W.) and NINDS R01 NS047700-06 (S.V.). This research was performed while K.P. held a National Research Council Research Associateship Award at NIDCD.
References
- Ali DW, Salter MW. NMDA receptor regulation by Src kinase signalling in excitatory synaptic transmission and plasticity. Curr Opin Neurobiol. 2001;11:336–42. doi: 10.1016/s0959-4388(00)00216-6. [DOI] [PubMed] [Google Scholar]
- Alvestad RM, Grosshans DR, Coultrap SJ, Nakazawa T, Yamamoto T, Browning MD. Tyrosine dephosphorylation and ethanol inhibition of N-Methyl-D-aspartate receptor function. J Biol Chem. 2003;278:11020–5. doi: 10.1074/jbc.M210167200. [DOI] [PubMed] [Google Scholar]
- Barria A, Malinow R. Subunit-specific NMDA receptor trafficking to synapses. Neuron. 2002;35:345–53. doi: 10.1016/s0896-6273(02)00776-6. [DOI] [PubMed] [Google Scholar]
- Blanpied TA, Scott DB, Ehlers MD. Dynamics and regulation of clathrin coats at specialized endocytic zones of dendrites and spines. Neuron. 2002;36:435–49. doi: 10.1016/s0896-6273(02)00979-0. [DOI] [PubMed] [Google Scholar]
- Bonifacino JS, Traub LM. Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu Rev Biochem. 2003;72:395–447. doi: 10.1146/annurev.biochem.72.121801.161800. [DOI] [PubMed] [Google Scholar]
- Choquet D, Triller A. The role of receptor diffusion in the organization of the postsynaptic membrane. Nat Rev Neurosci. 2003;4:251–65. doi: 10.1038/nrn1077. [DOI] [PubMed] [Google Scholar]
- Chung HJ, Huang YH, Lau LF, Huganir RL. Regulation of the NMDA receptor complex and trafficking by activity-dependent phosphorylation of the NR2B subunit PDZ ligand. J Neurosci. 2004;24:10248–59. doi: 10.1523/JNEUROSCI.0546-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark BA, Farrant M, Cull-Candy SG. A direct comparison of the single-channel properties of synaptic and extrasynaptic NMDA receptors. J Neurosci. 1997;17:107–16. doi: 10.1523/JNEUROSCI.17-01-00107.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crump FT, Dillman KS, Craig AM. cAMP-dependent protein kinase mediates activity-regulated synaptic targeting of NMDA receptors. J Neurosci. 2001;21:5079–88. doi: 10.1523/JNEUROSCI.21-14-05079.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cull-Candy S, Brickley S, Farrant M. NMDA receptor subunits: diversity, development and disease. Curr Opin Neurobiol. 2001;11:327–35. doi: 10.1016/s0959-4388(00)00215-4. [DOI] [PubMed] [Google Scholar]
- Dunah AW, Standaert DG. Dopamine D1 receptor-dependent trafficking of striatal NMDA glutamate receptors to the postsynaptic membrane. J Neurosci. 2001;21:5546–58. doi: 10.1523/JNEUROSCI.21-15-05546.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers MD. Related reinsertion or degradation of AMPA receptors determined by activity-dependent endocytic sorting. Neuron. 2000;28:511–25. doi: 10.1016/s0896-6273(00)00129-x. [DOI] [PubMed] [Google Scholar]
- Ehlers MD. Activity level controls postsynaptic composition and signaling via the ubiquitin-proteasome system. Nat Neurosci. 2003;6:231–42. doi: 10.1038/nn1013. [DOI] [PubMed] [Google Scholar]
- Follesa P, Ticku MK. Chronic ethanol-mediated up-regulation of the N-methyl-D-aspartate receptor polypeptide subunits in mouse cortical neurons in culture. J Biol Chem. 1996;271:13297–9. doi: 10.1074/jbc.271.23.13297. [DOI] [PubMed] [Google Scholar]
- Guo W, Zou S, Guan Y, Ikeda T, Tal M, Dubner R, Ren K. Tyrosine phosphorylation of the NR2B subunit of the NMDA receptor in the spinal cord during the development and maintenance of inflammatory hyperalgesia. J Neurosci. 2002;22:6208–17. doi: 10.1523/JNEUROSCI.22-14-06208.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE, Bading H. The Yin and Yang of NMDA receptor signalling. Trends Neurosci. 2003;26:81–9. doi: 10.1016/S0166-2236(02)00040-1. [DOI] [PubMed] [Google Scholar]
- Hashimoto R, Hough C, Nakazawa T, Yamamoto T, Chuang DM. Lithium protection against glutamate excitotoxicity in rat cerebral cortical neurons: involvement of NMDA receptor inhibition possibly by decreasing NR2B tyrosine phosphorylation. J Neurochem. 2002;80:589–97. doi: 10.1046/j.0022-3042.2001.00728.x. [DOI] [PubMed] [Google Scholar]
- Hawkins LM, Chazot PL, Stephenson FA. Biochemical evidence for the co-association of three N-methyl-D-aspartate (NMDA) R2 subunits in recombinant NMDA receptors. J Biol Chem. 1999;274:27211–8. doi: 10.1074/jbc.274.38.27211. [DOI] [PubMed] [Google Scholar]
- Hung AY, Sheng M. PDZ domains: structural modules for protein complex assembly. J Biol Chem. 2002;277:5699–702. doi: 10.1074/jbc.R100065200. [DOI] [PubMed] [Google Scholar]
- Kohr G, Seeburg PH. Subtype-specific regulation of recombinant NMDA receptor-channels by protein tyrosine kinases of the src family. J Physiol. 1996;492:445–52. doi: 10.1113/jphysiol.1996.sp021320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima N, Wang J, Mansuy IM, Grant SG, Mayford M, Kandel ER. Rescuing impairment of long-term potentiation in fyn-deficient mice by introducing Fyn transgene. Proc Natl Acad Sci. 1997;94:4761–5. doi: 10.1073/pnas.94.9.4761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavezzari G, McCallum J, Lee R, Roche KW. Differential binding of the AP-2 adaptor complex and PSD-95 to the C-terminus of the NMDA receptor subunit NR2B regulates surface expression. Neuropharmacology. 2003;45:729–37. doi: 10.1016/s0028-3908(03)00308-3. [DOI] [PubMed] [Google Scholar]
- Lavezzari G, McCallum J, Dewey CM, Roche KW. Subunit-specific regulation of NMDA receptor endocytosis. J Neurosci. 2004;24:6383–91. doi: 10.1523/JNEUROSCI.1890-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Liu L, Wang YT, Sheng M. Clathrin adaptor AP2 and NSF interact with overlapping sites of GluR2 and play distinct roles in AMPA receptor trafficking and hippocampal LTD. Neuron. 2002;36:661–74. doi: 10.1016/s0896-6273(02)01024-3. [DOI] [PubMed] [Google Scholar]
- Li B, Chen N, Luo T, Otsu Y, Murphy TH, Raymond LA. Differential regulation of synaptic and extra-synaptic NMDA receptors. Nat Neurosci. 2002;5:833–4. doi: 10.1038/nn912. [DOI] [PubMed] [Google Scholar]
- Lim IA, Hall DD, Hell JW. Selectivity and promiscuity of the first and second PDZ domains of PSD-95 and synapse-associated protein 102. J Biol Chem. 2002;277:21697–711. doi: 10.1074/jbc.M112339200. [DOI] [PubMed] [Google Scholar]
- Lin JW, Ju W, Foster K, Lee SH, Ahmadian G, Wyszynski M, Wang YT, Sheng M. Distinct molecular mechanisms and divergent endocytotic pathways of AMPA receptor internalization. Nat Neurosci. 2000;3:1282–90. doi: 10.1038/81814. [DOI] [PubMed] [Google Scholar]
- Liu L, Wong TP, Pozza MF, Lingenhoehl K, Wang Y, Sheng M, Auberson YP, Wang YT. Role of NMDA receptor subtypes in governing the direction of hippocampal synaptic plasticity. Science. 2004;304:1021–4. doi: 10.1126/science.1096615. [DOI] [PubMed] [Google Scholar]
- Luscher C, Xia H, Beattie EC, Carroll RC, von Zastrow M, Malenka RC, Nicoll RA. Role of AMPA receptor cycling in synaptic transmission and plasticity. Neuron. 1999;24:649–58. doi: 10.1016/s0896-6273(00)81119-8. [DOI] [PubMed] [Google Scholar]
- Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 2002;25:103–26. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- Montgomery JM, Madison DV. State-dependent heterogeneity in synaptic depression between pyramidal cell pairs. Neuron. 2002;33:765–77. doi: 10.1016/s0896-6273(02)00606-2. [DOI] [PubMed] [Google Scholar]
- Nakatsu F, Ohno H. Adaptor protein complexes as the key regulators of protein sorting in the post-Golgi network. Cell Struct Funct. 2003;28:419–29. doi: 10.1247/csf.28.419. [DOI] [PubMed] [Google Scholar]
- Nakazawa T, et al. Characterization of Fyn-mediated tyrosine phosphorylation sites on GluR epsilon 2 (NR2B) subunit of the N-methyl-D-aspartate receptor. J Biol Chem. 2001;276:693–9. doi: 10.1074/jbc.M008085200. [DOI] [PubMed] [Google Scholar]
- Nesterov A, Carter RE, Sorkina T, Gill GN, Sorkin A. Inhibition of the receptor-binding function of clathrin adaptor protein AP-2 by dominant-negative mutant mu2 subunit and its effects on endocytosis. EMBO J. 1999;18:2489–99. doi: 10.1093/emboj/18.9.2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nong Y, et al. Glycine binding primes NMDA receptor internalization. Nature. 2003;422:302–7. doi: 10.1038/nature01497. [DOI] [PubMed] [Google Scholar]
- Nong Y, Huang YQ, Salter MW. NMDA receptors are movin’ in. Curr Opin Neurobiol. 2004;14:353–61. doi: 10.1016/j.conb.2004.05.001. [DOI] [PubMed] [Google Scholar]
- Owen DJ, Evans PR. A structural explanation for the recognition of tyrosine-based endocytotic signals. Science. 1998;282:1327–32. doi: 10.1126/science.282.5392.1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petralia RS, Wang YX, Wenthold RJ. Internalization at glutamatergic synapses during development. Eur J Neurosci. 2003;18:3207–17. doi: 10.1111/j.1460-9568.2003.03074.x. [DOI] [PubMed] [Google Scholar]
- Philpot BD, Sekhar AK, Shouval HZ, Bear MF. Visual experience and deprivation bidirectionally modify the composition and function of NMDA receptors in visual cortex. Neuron. 2001;29:157–69. doi: 10.1016/s0896-6273(01)00187-8. [DOI] [PubMed] [Google Scholar]
- Prybylowski K, et al. Relationship between availability of NMDA receptor subunits and their expression at the synapse. J Neurosci. 2002;22:8902–10. doi: 10.1523/JNEUROSCI.22-20-08902.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prybylowski K, Wenthold RJ. N-Methyl-D-aspartate receptors: subunit assembly and trafficking to the synapse. J Biol Chem. 2004;279:9673–6. doi: 10.1074/jbc.R300029200. [DOI] [PubMed] [Google Scholar]
- Racz B, Blanpied TA, Ehlers MD, Weinberg RJ. Lateral organization of endocytic machinery in dendritic spines. Nat Neurosci. 2004;7:917–8. doi: 10.1038/nn1303. [DOI] [PubMed] [Google Scholar]
- Rao A, Craig AM. Activity regulates the synaptic localization of the NMDA receptor in hippocampal neurons. Neuron. 1997;19:801–12. doi: 10.1016/s0896-6273(00)80962-9. [DOI] [PubMed] [Google Scholar]
- Roche KW, et al. Molecular determinants of NMDA receptor internalization. Nat Neurosci. 2001;4:794–802. doi: 10.1038/90498. [DOI] [PubMed] [Google Scholar]
- Rosenblum K, Dudai Y, Richter-Levin G. Long-term potentiation increases tyrosine phosphorylation of the N-methyl-D-aspartate receptor subunit 2B in rat dentate gyrus in vivo. Proc Natl Acad Sci. 1996;93:10457–60. doi: 10.1073/pnas.93.19.10457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rostas JA, Brent VA, Voss K, Errington ML, Bliss TV, Gurd JW. Enhanced tyrosine phosphorylation of the 2B subunit of the N-methyl-D-aspartate receptor in long-term potentiation. Proc Natl Acad Sci. 1996;93:10452–6. doi: 10.1073/pnas.93.19.10452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumbaugh G, Vicini S. Distinct synaptic and extrasynaptic NMDA receptors in developing cerebellar granule neurons. J Neurosci. 1999;19:10603–10. doi: 10.1523/JNEUROSCI.19-24-10603.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sans N, et al. A developmental change in NMDA receptor-associated proteins at hippocampal synapses. J Neurosci. 2000;20:1260–71. doi: 10.1523/JNEUROSCI.20-03-01260.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scannevin RH, Huganir RL. Postsynaptic organization and regulation of excitatory synapses. Nat Rev Neurosci. 2000;1:133–41. doi: 10.1038/35039075. [DOI] [PubMed] [Google Scholar]
- Snyder EM, et al. Internalization of ionotropic glutamate receptors in response to mGluR activation. Nat Neurosci. 2001;4:1079–85. doi: 10.1038/nn746. [DOI] [PubMed] [Google Scholar]
- Steigerwald F, et al. C-Terminal truncation of NR2A subunits impairs synaptic but not extrasynaptic localization of NMDA receptors. J Neurosci. 2000;20:4573–81. doi: 10.1523/JNEUROSCI.20-12-04573.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takasu MA, Dalva MB, Zigmond RE, Greenberg ME. Modulation of NMDA receptor-dependent calcium influx and gene expression through EphB receptors. Science. 2002;295:491–5. doi: 10.1126/science.1065983. [DOI] [PubMed] [Google Scholar]
- Tezuka T, Umemori H, Akiyama T, Nakanishi S, Yamamoto T. PSD-95 promotes Fyn-mediated tyrosine phosphorylation of the N-methyl-D-aspartate receptor subunit NR2A. Proc. Natl Acad Sci U S A. 1999;96:435–40. doi: 10.1073/pnas.96.2.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tovar KR, Westbrook GL. The incorporation of NMDA receptors with a distinct subunit composition at nascent hippocampal synapses in vitro. J Neurosci. 1999;19:4180–8. doi: 10.1523/JNEUROSCI.19-10-04180.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tovar KR, Westbrook GL. Mobile NMDA receptors at hippocampal synapses. Neuron. 2002;34:255–64. doi: 10.1016/s0896-6273(02)00658-x. [DOI] [PubMed] [Google Scholar]
- Townsend M, Yoshii A, Mishina M, Constantine-Paton M. Developmental loss of miniature N-methyl-D-aspartate receptor currents in NR2A knockout mice. Proc Natl Acad Sci U S A. 2003;100:1340–5. doi: 10.1073/pnas.0335786100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vissel B, Krupp JJ, Heinemann SF, Westbrook GL. A use-dependent tyrosine dephosphorylation of NMDA receptors is independent of ion flux. Nat Neurosci. 2001;4:587–96. doi: 10.1038/88404. [DOI] [PubMed] [Google Scholar]
- Watt AJ, van Rossum MC, MacLeod KM, Nelson SB, Turrigiano GG. Activity coregulates quantal AMPA and NMDA currents at neocortical synapses. Neuron. 2000;26:659–70. doi: 10.1016/s0896-6273(00)81202-7. [DOI] [PubMed] [Google Scholar]