

Abstract
Virion infectivity factor (Vif) is essential for the replication of human immunodeficiency virus type 1 (HIV-1) in vivo, but its function remains uncertain. Recently, we have shown that Vif proteins are able to form multimers, including dimers, trimers, or tetramers. Because the multimerization of Vif proteins is required for Vif function in the viral life cycle, we propose that it could be a novel target for anti-HIV-1 therapeutics. Through a phage peptide display method, we have identified a set of 12-mer peptides containing a PXP motif that binds to HIV-1 Vif protein. These proline-enriched peptides potently inhibited the Vif-Vif interaction in vitro. We have also screened a set of synthesized Vif peptides (15-mer), which covers all the amino acids of the HIV-1 Vif protein sequence, for their ability to inhibit the Vif-Vif interaction in vitro. We demonstrated that Vif-derived proline-enriched peptides that contain the 161PPLP164 domain are able to inhibit the Vif-Vif interaction. Conversely, the deletion of the 161PPLP164 domain of Vif protein will significantly impair the capability of Vif proteins to interact with each other, indicating that the 161PPLP164 domain plays a key role in Vif multimerization. All these results demonstrate that the proline-enriched peptides block the multimerization of Vif through interfering with the polyproline interfaces of Vif formed by 161PPLP164 domain. Moreover, these peptides which inhibit the Vif-Vif interaction in vitro potently inhibit HIV-1 replication in the “nonpermissive” T-cells. We propose that this study starts a novel strategy to develop structural diverse inhibitors of Vif such as peptidomimetics or small organic molecules.
Virion infectivity factor (Vif)1 protein of HIV-1 is required for viral replication in vivo (1, 2). In cell culture systems, HIV-1Δvif viruses are incapable of establishing infection in certain cells, such as H9 T-cells, peripheral blood lymphocytes, and monocyte-derived macrophages (3–6). HIV-1 viruses with a defective vif gene are not able to complete intracellular reverse transcription and endogenous reverse transcription in cell-free virions when mild detergent is utilized to make the viral envelope permeable (7–10). Most studies indicated that the expression of viral components, including viral proteins and nucleic acids, is not altered in the virions produced from nonpermissive cells (3, 10, 11). However, the deletion of the vif gene will result in alterations of virion morphology (12–14). Various hypotheses have been proposed regarding the molecular mechanisms of Vif protein. It has been reported that defect of vif could affect the maturation of Gag precursor (15). Furthermore, Vif could directly bind to the protease domain of pol precursor and prevent the improper cleavage of Gag precursors before viral assembly (16). It was also proposed that Vif protein is required to counteract an unknown endogenous inhibitor(s) in the virus-producing cells (17, 18). Recent studies further indicated this endogenous inhibitor is CEM15, which is only expressed in the nonpermissive cells. Introduction of CEM15 into the permissive cells will generate a nonpermissive phenotype (19). However, the function of CEM15 remains unknown. Because its sequence is similar with APOBEC-1(apoB mRNA-editing catalytic subunit 1), a cytidine deaminase that can change cytidine into uridine in the mRNA of apolipoprotein B, CEM15 could affect the genomic RNA of HIV-1. Interestingly, we and others show that Vif is an RNA-binding protein and is an integral component of a messenger ribonucleotide protein complex of viral RNA (20, 21). The Vif protein in this ribonucleoprotein complex may protect viral RNA from various endogenous inhibitors and could mediate viral RNA engagement with HIV-1 Gag precursors. As such, Vif could play a key role in the proper trafficking of the viral genetic substance (genomic RNA) in the lentivirus-producing cells.
Because Vif is essential for HIV-1 replication, it is an important target for anti-HIV therapeutics. However, because its molecular mechanism in viral life cycle remains to be further determined, it is quite difficult to generate a small molecule inhibitor(s) to block Vif function at the present time. Recently, we have found that Vif proteins are able to form multimer (22). It is well known that multimerization is critical to the biological activity of many prokaryotic and eukaryotic proteins and is a common mechanism for the functional activation/inactivation of proteins. Therefore, multimerization has been an ideal target for the development of inhibitors of various proteins (23–25).
In this report, we demonstrate that Vif multimerization could be a promising intervention target for anti-HIV-1 agent development. We have found that a set of proline-enriched peptides is able to bind to Vif protein, inhibit the Vif-Vif interaction, and inhibit viral replication in cell culture. Our data demonstrates that, although the function and structure of Vif remains uncertain, we have still successfully developed the potent Vif antagonists, based upon the biochemical characteristics of Vif protein.
MATERIALS AND METHODS
Plasmid Constructions, Expression of GST Fusion Proteins, and Synthesis of 35S-Labeled Proteins by in Vitro Translation
The construction of pGEX-Vif, pCITE-Vif, pCITE-Vif-(Δ151–192), and pCITE-Vif-(Δ151–164) were described previously (20, 22). Vif(ΔPPLP) genes were generated by PCR-mediated mutagenesis and then inserted into pGEX vector. The vif(ΔPPLP) gene was also inserted into pCITE-4a vector (Novagen, Madison, WI) for in vitro translation. 35S-Labeled Vif or its mutant proteins were synthesized by in vitro transcription and translation utilizing SPT3 kits (Novagen) in the presence of [35S]methionine (1,000 Ci/mmol; Amersham Biosciences), as described previously (20). The GST, GST-Vif, and other GST fusion Vif mutant proteins were produced according to the previously described methods (20, 22). The tyrosine kinase Hck genes were generated by PCR amplification and then inserted into the pGEX vector. GST-Hck fusion protein was expressed and purified with the same procedure as for GST-Vif.
Phage Display Peptide Screening
Vif-binding peptides displayed on M13 phages were selected using the Ph.D.-12 phage display peptide library kit (New England Biolabs, Beverly, MA). Phage panning procedures were performed according to the manufacturer’s protocol with some modifications. Briefly, GST-Vif fusion protein attached to glutathione-conjugated agarose beads was used as a target for phage panning. For each round of panning, 1011 phages were first absorbed with GST followed by mixing with 3 ml of GST-Vif attached to glutathione-agarose beads. After binding at room temperature for 1 h, the GST-Vif binding phages were then eluted by 5 mm reduced glutathione. The eluted phages were amplified by mixing the elution with 20 ml of Escherichia coli ER2738 culture (optical density at 0.6). After incubation at 37 °C with vigorous shaking for 4 h, the bacterial cells were pelleted, and the phages in the supernatant were precipitated by polyethylene glycol (20%)/NaCl (2.5 m). After resuspension in Tris-buffered saline and reprecipitation by polyethylene glycol, the phages were suspended in 200 μl of Tris-buffered saline, 0.02%NaN3. The titration of the eluted or amplified phages was determined by infecting the E. coli ER2738 mixed in the conditioned medium-agar plates, as described in the kit protocol. After three rounds of panning, individual phage plaques from the GST or GST-Vif elution were selected for amplification, respectively. Phage DNA was then purified and sequenced.
Determination of Binding Affinity by Enzyme-linked Immunosorbent Assay (ELISA)
An ELISA was performed to measure the relative binding affinity of phages to GST, GST-Vif, or GST-Vif (Δ151–192). The protocol supplied by the manufacturer was followed. Briefly, 150 μl of 100 μg/ml GST and GST-Vif in 0.1 m NaHCO3, pH 8.6, was coated on 96-well microtiter plates, respectively, and incubated at 4 °C overnight. The plates were blocked with blocking buffer (0.1 m NaHCO3, pH 8.6, 5 mg/ml bovine serum albumin) for 2 h at room temperature. The individual phage clones were 4-fold-serially diluted (from 1011 to 105), added to the wells coated with GST, GST-Vif, or GST-Vif-(Δ151–192), and incubated for 2 h at room temperature. After washing, horseradish peroxide-conjugated anti-M13 antibody was added to bind the phages. After incubation at room temperature for 1 h, the excess antibody was washed, the substrate was added, and color development was allowed to proceed. The phages captured by Vif were therefore semi-quantitated. Optical density at 405 nm equal to or greater than 0.15 was considered as positive.
Peptide Synthesis
HIV-1 consensus B Vif (15-mer) peptides were provided by the National Institutes of Health AIDS Research and Reference Reagent Program. All the other peptides were synthesized by solid-phase techniques using a Symphony Multiplex synthesizer (Protein Technologies, Inc., Tucson, AZ) and a 9050 Pepsynthesizer Plus automated peptide synthesizer (Perseptive Biosystems, Cambridge, MA) with Nα-Fmoc (N-(9-fluorenyl)methoxycarbonyl)/tert-butyl chemistry. Biotin peptides were biotinylated by adding biotin (Sigma) at the N terminus. The peptides were characterized by analytical HPLC and matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. All the peptides were at least 95% pure, as determined by HPLC.
In Vitro Vif-Vif/Vif-Hck Interactions and Their Inhibition by Peptides
A GST pull-down assay was used to study the in vitro protein-protein interactions. The GST fusion proteins on agarose beads are first generated as described previously except without elution with 5 mm glutathione (20). The 35S-labeled, in vitro translated Vif proteins were then mixed with GST fusion protein-conjugated agarose beads in washing/binding buffer (150 mm NaCl, 10 mm Tris-HCl, pH 8.0, and 0.1% Triton-X-100). Binding is allowed to proceed at 23 °C for 20 min and then at 4 °C for 1 h. For the inhibition of Vif-Vif/Vif-Hck binding by peptides, 35S-labeled Vif proteins were added to GST-Vif/GST-Hck-conjugated agarose beads and incubated with peptides at different concentrations in binding buffer at 4 °C for 1 h. The beads were then washed with washing/binding buffer 3 times, and the bead-bound 35S-labeled Vif proteins were fractionated by SDS-PAGE followed by autoradiography and quantitated using a PhosphorImager (Molecular Dynamics, Sunnyview, CA).
Peptide Internalization Experiment
H9 cells were suspended in serum-free RPMI 1640 supplemented with 4 mm l-glutamine and incubated with the peptides for 30 min. After washing 3 times with phosphate-buffered saline (PBS), the cells were fixed with 4% formaldehyde in PBS for 10 min at room temperature. Cells were then washed twice with PBS and treated with 0.1% Triton X-100 in PBS for 10 min. After an additional 2 washes with PBS, cells were incubated with blocking buffer (3% bovine serum albumin in PBS) for 1 h at room temperature followed by incubation with streptavidin-fluorescein isothiocyanate (Sigma) at 2 μg/ml in blocking buffer for 5–10 min in the dark. Cells were then washed with PBS, and cell suspensions were smeared on glass microscope slides for fluorescence microscopy using an Olympus BX60 fluorescence microscope.
Viral Infectivity Assay
H9 cells (1 × 106) were mixed with HIV-1NL4–3 viruses at a multiplicity of infection of 0.01. After incubation at 37 °C for 5 h, the excess viruses were removed, and the cells were cultured in the presence of RPMI 1640 medium plus 10% fetal bovine serum with or without peptides at a concentration of 50 μm. Every 3–4 days, the supernatants were harvested and refreshed. The effects of these peptides upon viral infectivity were monitored by detecting the HIV-1 p24 antigen level in the cell culture supernatant via ELISA, as described previously (20, 26, 27).
RESULTS
Identification of PXP Motif-containing Peptides Binding to Vif Protein
To search for the peptides that bind with HIV-1 Vif protein, the phage peptide display method was employed. The procedures described in the manual supplied by manufacturer were followed. After three rounds of panning, the phage-displayed peptides that bind with GST-Vif were identified by sequencing DNA in the knot region of the phages. Through theses methods we have identified a set of 12-mer peptides containing a PXP motif that bind to the Vif protein (Table I).
Table I.
PXP motif containing peptides identified by phage display peptide screening
| Sequence ID | Peptides |
|---|---|
| VMI1 | SNFASITTPRPH |
| VMI2 | WPTNPTTVPVPS |
| VMI3 | LTSDTYFLPVPA |
| VMI4 | SLHWPVSHPPPP |
| VMI5 | SVSVGMKPSPRP |
| VMI6 | WHSQRLSPVPPA |
| VMI7 | SNQGGSPLPRSV |
| VMI8 | SEPHLPFPVLPH |
| VMI9 | LPLPAPSFHRTT |
| VMI10 | YPLPHPMWSMLP |
| VMI11 | TMTPPPTSVRGT |
| VMI12 | TPLPTIRGDTGT |
| VMI13 | GPPPHHRDYHGP |
| VMI14 | YPAPIKVLLPNS |
| VMI15 | SPYPMALFPLHN |
| VMI16 | SPYPSWSTPAGR |
To determine the binding affinity of various PXP motif-containing peptides to Vif protein, a simple assay based upon an ELISA was used to determine the relative affinity. The phages at various concentrations captured by Vif were semi-quantitated. Fig. 1A demonstrates that among PXP motif containing peptides, VMI 5, VMI 7, VMI 9, and VMI 16 bind to Vif at the highest affinities. The C terminus-deleted Vif protein binds with PXP motif-containing peptides at low affinity, indicating that PXP motif-containing peptides bind to Vif protein through the C terminus of the Vif protein.
Fig. 1.
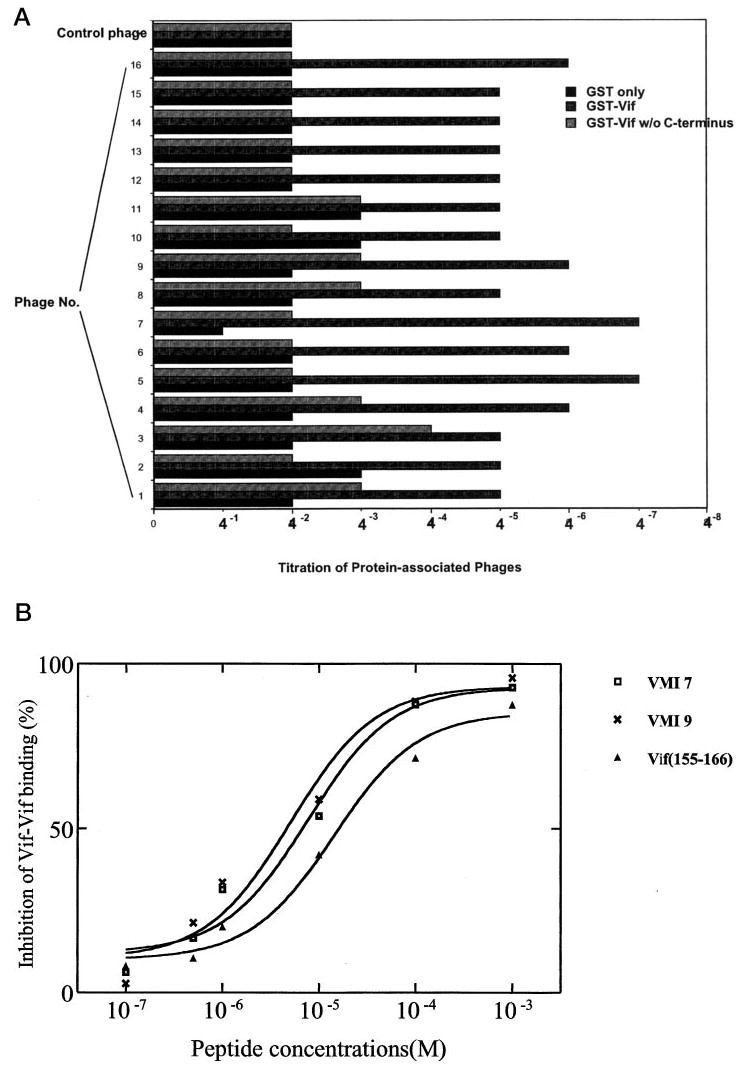
A, relative affinity comparisons between PXP motif-containing peptides. GST fusion protein (100 μg/ml) of Vif, Vif-(Δ151–192), and GST only were coated onto the 96-well plate. The phages clones isolated through the GST-Vif-containing column were serially diluted and added. After incubation to allow phage-Vif binding, excess phages were washed off. Anti-M13 phage antibody conjugated with horseradish peroxide was added to bind the phages that were captured by GST-Vif. After washing, the substrate was added, and color development was allowed to proceed. The phages captured by GST-Vif were, therefore, semi-quantitated. Optical density at 405 nm equal to or greater than 0.15 was considered to be positive. The phage sample number (VMI) is the same as shown in Table I. B, in vitro binding affinity of peptides to Vif. Various peptides (10−7, 5 × 10−7, 10−6, 10−5, 10−4, 10−3 m) were added to the mixture of 35S-labeled Vif and GST-Vif-conjugated agarose beads. The 35S-labeled Vif binding to GST-Vif was dissociated from beads by adding 2% SDS loading buffer and then analyzed by SDS-PAGE followed by autoradiography and quantitation using PhosphorImager. IC50 is the concentration of the peptides inhibiting 50% 35S-labeled Vif binding to GST-Vif in GST pull-down assays.
PXP Motif-containing Peptides Inhibit Vif-Vif Interaction in Vitro
We noted that Vif proteins of various HIV-1 strains all contain proline-rich sequence at their C terminus (161PPLP164 in NL4–3 strain), and we have demonstrated that the proline-enriched domain (151AALIKPKQIKPPLP164) is required for Vif multimerization (22). Therefore, it is interesting to examine whether PXP motif-containing peptides inhibit Vif-Vif interaction. To this end, some of these peptides containing the PXP domain, identified from phage display libraries, were chemically synthesized and examined for their ability to inhibit Vif-Vif binding. Fig. 1B indicates that peptides containing the PXP motif, such as SNQGGSPLPRSV (VMI 7) or LPLPAPSFHRTT (VMI 9), could significantly inhibit Vif-Vif interaction. The IC50 for the inhibition of Vif multimerization is 7.43 μm for VMI 7 and 4.84 μm for VMI 9 (Fig. 1B). A Vif-derived 12-mer peptide, 155KPKQIKPPLPSV166 (Vif-(155–166)), which is originated from the proline-enriched C terminus of Vif, also has the similar inhibition activity upon Vif-Vif interaction (IC50 = 17.39 μm).
We have also screened a set of synthesized Vif peptides (15-mer), which includes all the amino acids of HIV-1 Vif protein, for their ability to block the Vif-Vif interaction in vitro. We demonstrated that proline-enriched Vif peptides, such as 153LITPKKIKPPLPSVT167 and 157KKIKPPLPSVTKLTE171, which contain the 161PPLP164 domain, are able to inhibit the Vif-Vif interaction significantly, further supporting that PXP motif-containing peptides inhibit Vif multimerization (Fig. 1B and Fig. 2). Conversely, this result also suggests that the 151–165 region of Vif is responsible for Vif-Vif binding. The peptides, derived from region of 145–163, which is upstream of the 161PPLP164 domain, are also able to moderately inhibit Vif-Vif interaction, suggesting that the amino acid residues at this region could also participate in the Vif-Vif interaction.
Fig. 2. The inhibition of HIV-1 Vif (15-mer) peptides upon Vif-Vif binding.

HIV-1 consensus B Vif (15-mer) peptides (100 μm) were added to the mixture of 35S-labeled Vif- and GST-Vif-conjugated agarose beads. The 35S-labeled Vif binding to GST-Vif was dissociated from beads by adding 2% SDS loading buffer and then analyzed by SDS-PAGE followed by autoradiography and quantitation using a PhosphorImager
The 161PPLP164 Domain Is Required for Vif Multimerization
Because the PXP motif is also shared by Vif in the 161PPLP164 domain that is located within the putative Vif multimerization domain and the PXP motif-containing peptides are able to inhibit Vif-Vif interaction, it is interesting to investigate whether the 161PPLP164 domain is required for Vif-Vif interaction. To this end, site-directed mutagenesis was performed to delete 161PPLP164, 151AALIKPKQIKPPLP164, and the Vif C terminus (151–192). The mutants were expressed with an in vitro translation system in the presence of [35S]methionine or expressed as GST fusion proteins. The 35S-labeled Vif or Vif mutants were then bound with GST-Vif or GST-Vif(ΔPPLP) that were conjugated with glutathione-coated agarose beads. As described previously, Vif mutant proteins deleted at the C terminus (151–192) or 151AALIKPKKIKPPLP164 have decreased binding to Vif (Fig. 3) (22). Vif mutant protein just deleted at the 161PPLP164 domain also showed a decrease in its binding to Vif. Interestingly, the protein-protein interactions between the Vif mutants deleted at 161PPLP164 domain were significantly decreased (Fig. 3). These data indicated that the 161PPLP164 domain is required for Vif-Vif multimerization.
Fig. 3. Deletion of PPLP eliminates Vif-Vif interaction.

GST-Vif- or GST-Vif(ΔPPLP)-conjugated agarose beads were mixed with 35S-labeled Vif or its mutants in binding buffer and incubated at 4 °C for 1 h. The 35S-labeled Vif or its mutants remaining on beads were dissociated from beads by adding 2%SDS loading buffer and then analyzed by SDS-PAGE followed by autoradiography and quantitation using a Phosphor-Imager. A, GST-Vif/35S-Vif; B, GST-Vif/35S-Vif-(Δ151–192); C, GST-Vif/35S-Vif-(Δ151–164); D, GST-Vif/35S-Vif(ΔPPLP); E, GST-Vif(ΔPPLP)/35S-Vif; F, GST-Vif(ΔPPLP)/35S-Vif(ΔPPLP).
PXP Motif-containing Peptides Inhibit Vif-Hck Binding
It has been demonstrated that Hck kinase can also bind with Vif through the 161PPLP164 domain (28). It is possible that PXP motif-containing peptides are able to block the interaction between Vif and Hck and other protein kinases. To this end, 35S-labeled Vif was allowed to bind with GST-Hck in the presence or absence of various peptides. As described by others, Vif is able to bind with Hck. In the presence of VMI 7, VMI 9, and Vif (155–166), the binding between Vif and Hck is significantly decreased (Fig. 4).
Fig. 4. The in vitro inhibition by the peptides on Vif-Vif or Vif-Hck binding.
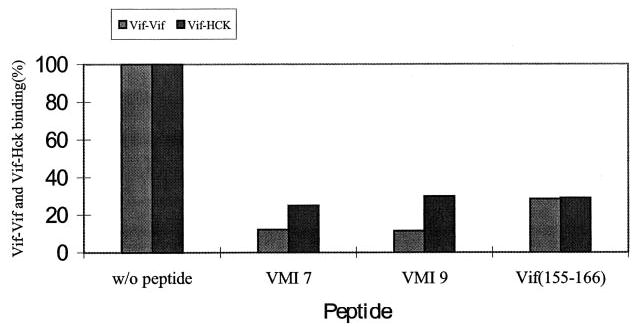
Various peptides (100 μm) were added to the mixtures of 35S-labeled Vif and GST-Vif- or GST-Hck-conjugated agarose beads. The 35S-labeled Vif binding to GST-Vif or GST-Hck was dissociated from beads by adding 2% SDS containing loading buffer and then analyzed by SDS-PAGE followed by autoradiography and quantitation using a PhosphorImager.
PXP Motif-containing Peptides Inhibit HIV-1 Replication
To examine the inhibitory effects of PXP motif-containing peptides upon viral infectivity in cell culture, the peptides must be introduced into the virally infected cells by a reliable method. As antennapedia homeodomain (Ant, RQIKIWFQNRRMKWKK) has been widely used to effectively carry peptides into various living cells (29–34), Ant fusion peptides, Ant-VMI 7, Ant-VMI 9, and Ant-Vif (155–166), were synthesized, and their activity for in vivo inhibition of HIV-1 replication were investigated. These peptides did not show any toxicity to H9 cells at the concentration of 50 μm (data not shown). To examine whether these fusion peptides are able to enter the H9 cells, the cell permeability of biotin Ant-VMI 9 was determined. Fig. 5A indicates that the Ant fusion peptide can efficiently enter the H9 cells and locate in the cytoplasm. Because Vif mainly locates in the cytoplasm of virus-infected cells, the fusion peptide should physically interact with Vif protein.
Fig. 5.

A, internalization of peptides. H9 cells were incubated with biotinylated peptides VMI 9 and Ant-VMI 9 for 30 min. The excess peptides were then washed off. After fixing, the internalized peptides were detected with streptavidin-fluorescein isothiocyanate followed by visualization with fluorescence microscopy. A, Ant-VMI 9, fluorescence; B, Ant-VMI 9, phase-contrast; C, VMI 9, fluorescence; D, VMI 9, phase contrast. B, Ant fusion peptides inhibit HIV-1 replication. H9 cells were infected by virions at 37 °C HIV-1NL4–3 for 4 h. The infected H9 cells (1 × 106) were then cultured in duplicate in 2 ml of RPMI 1640 medium plus 10% fetal bovine serum without or with peptides (50 μm). Portions of the supernatants (0.5 ml) were collected every 3–4 days. The HIV-1 p24 antigen levels were determined by ELISA. This data represent three independent experiments.
The fusion peptides were then added into the cell culture to examine their capability to inhibit HIV-1 replication. H9 cells, a nonpermissive cell line that requires Vif to counteract the endogenous inhibitor, were infected with HIV-1 viruses in the presence or absence of various fusion peptides. At the concentration of 50 μm, the fusion peptides, Ant-VMI 7, Ant-VMI 9, and Ant-Vif (155–166) are able to effectively inhibit HIV-1 replication. As a control, the Ant peptide itself does not have any anti-HIV-1 activity (Fig. 5B).
DISCUSSION
We have demonstrated that the 151AALIKPKQIKPPLP164 domain of HIV-1 Vif is critical for Vif multimerization, which is required for Vif function (22). In this report, we have further demonstrated that the 161PPLP164 domain plays a key role in Vif-Vif interaction. Our current results suggest that Vif-Vif binding occurs, at least in part, through the direct interaction between 161PPLP164 domains in each Vif molecule. Because the function of Vif remains unknown, it is difficult to investigate the molecular mechanism regarding how Vif multimerization is required for Vif function. However, recent studies indicate that Vif is required to counteract the endogenous inhibitor CEM15, which is a putative cytidine deaminase (19, 35). Because Vif binds to HIV-1 RNA, it is reasonable to assume that Vif-RNA binding could protect the HIV-RNA from RNA editing (20, 21). If so, Vif-RNA binding could be the major mechanism for Vif function. It is therefore quite important to study the correlation between Vif-RNA binding and Vif-Vif interaction. Because Vif binds to RNA through its N terminus, whereas Vif-Vif interaction takes place at the C terminus, Vif-Vif interaction could be correlated with Vif-RNA binding. Conversely, Vif is able to bind with Gag protein through the positive-charged amino acids in the 151–164 region at the C terminus, and Vif binds to Hck also through the 161PPLP164 domain. Therefore, the Vif-Vif interaction could be reversibly correlated with Vif-Gag binding or Vif-Hck binding (13, 28). These hypotheses remain to be fully tested.
Through screening phage display peptide libraries, a set of proline-enriched peptides binding to Vif was identified and is able to block the Vif-Vif interaction. The proline-enriched sequence is a hydrophobic region and usually binds to the hydrophobic interface of SH3/WW domains in protein-protein interactions (36). Vif-Vif interaction could occur between the two 161PPLP164 domains or the 161PPLP164 domains and other regions in the Vif protein. It seems that the PXP motif-containing peptides mimic the hydrophobic structure of the 161PPLP164 domain and bind to the hydrophobic interface of Vif, which is quite critical for Vif multimerization. Among these proline-enriched peptides, the peptides containing the PXPXP motif have the higher binding affinity to Vif protein. We have also tested the synthesized peptides derived from the Vif protein upon Vif-Vif interaction. Our data demonstrated that the peptides containing the 161PPLP164 domain are able to inhibit Vif-Vif interaction, indicating that the 161PPLP164 domain plays a key role in Vif-Vif interaction.
In this report, we demonstrated that proline-enriched PXP motif-containing peptides not only inhibit Vif-Vif interaction but also the binding between Vif and Hck. It is notable that the PXP motif-containing peptides have been shown to inhibit the activation of various SH3 domain-contained protein kinases (36, 37). Because the peptides identified in this report do not have any toxicity to the cultured cells at concentrations used to inhibit HIV-1 replication, they should have certain specificity in blocking Vif-Vif or Vif-Hck interactions rather than inhibiting the activation of other protein kinases used in maintaining the normal functions of the cells.
Because Vif is required for HIV-1 replication and Vif multimerization is important for the function of Vif, the inhibitor(s) that blocks the formation of Vif multimer should inhibit HIV-1 replication. A reliable method was used to allow the peptides that inhibit Vif-Vif interaction to effectively enter HIV-1-infected cells. Indeed, the peptides that effectively inhibit Vif-Vif interaction potently inhibit HIV-1 replication in cell culture (Fig. 5B).
In this report, we have shown that the 161PPLP164 domain of Vif is a valuable target for developing Vif inhibitors. Because the PXP motif-containing peptides potently inhibit Vif-Vif interaction and inhibit HIV-1 replication in nonpermissive cells, it is interesting to further investigate the structural mechanisms of these peptide inhibitors and develop more potent nonpeptide Vif inhibitors, such as peptidomimetic or small organic molecular inhibitors. Because of the essential role of Vif in HIV-1 replication, we believe that the development of these Vif inhibitors may represent a new strategy for anti-AIDS therapy (38, 39).
Acknowledgments
We thank the National Institutes of Health AIDS Research and Reference Reagent Program for providing HIV-1 consensus B Vif (15-mer) peptides.
Footnotes
This work was supported by National Institutes of Health Grant AI47720 (to H. Z.).
The abbreviations used are: Vif, virion infectivity factor; HIV-1, human immunodeficiency virus type 1; GST, glutathione S-transferase; ELISA, enzyme-linked immunosorbent assay; HPLC, high performance liquid chromatography; Ant, antennapedia; PBS, phosphate-buffered saline.
References
- 1.Wieland U, Hartmann J, Suhr H, Salzberger B, Eggers HJ, Kuhn JE. Virology. 1994;203:43–51. doi: 10.1006/viro.1994.1453. [DOI] [PubMed] [Google Scholar]
- 2.Sova P, van Ranst M, Gupta P, Balachandran R, Chao W, Itescu S, McKinley G, Volsky DJ. J Virol. 1995;69:2557–2564. doi: 10.1128/jvi.69.4.2557-2564.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gabuzda DH, Lawrence K, Langhoff E, Terwilliger E, Dorfman T, Haseltine WA, Sodroski J. J Virol. 1992;66:6489–6495. doi: 10.1128/jvi.66.11.6489-6495.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Strebel K, Daugherty D, Clouse K, Cohen D, Folks T, Martin MA. Nature. 1987;328:728–730. doi: 10.1038/328728a0. [DOI] [PubMed] [Google Scholar]
- 5.Gabuzda DH, Li H, Lawrence K, Vasir BS, Crawford K, Langhoff E. J Acquired Immune Defic Syndr. 1994;7:908–915. [PubMed] [Google Scholar]
- 6.Dornadula G, Yang S, Pomerantz RJ, Zhang H. J Virol. 2000;74:2594–2602. doi: 10.1128/jvi.74.6.2594-2602.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Courcoul M, Patience C, Rey F, Blanc D, Harmache A, Sire J, Vigne R, Spire B. J Virol. 1995;69:2068–2074. doi: 10.1128/jvi.69.4.2068-2074.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goncalves J, Korin Y, Zack J, Gabuzda D. J Virol. 1996;70:8701–8709. doi: 10.1128/jvi.70.12.8701-8709.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sova P, Volsky DJ. J Virol. 1993;67:6322–6326. doi: 10.1128/jvi.67.10.6322-6326.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.von Schwedler U, Song J, Aiken C, Trono D. J Virol. 1993;67:4945–4955. doi: 10.1128/jvi.67.8.4945-4955.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fouchier RA, Simon JH, Jaffe AB, Malim MH. J Virol. 1996;70:8263–8269. doi: 10.1128/jvi.70.12.8263-8269.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Borman AM, Quillent C, Charneau P, Dauguet C, Clavel F. J Virol. 1995;69:2058–2067. doi: 10.1128/jvi.69.4.2058-2067.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bouyac M, Courcoul M, Bertoia G, Baudat Y, Gabuzda D, Blanc D, Chazal N, Boulanger P, Sire J, Vigne R, Spire B. J Virol. 1997;71:9358–9365. doi: 10.1128/jvi.71.12.9358-9365.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hoglund S, Ohagen A, Lawrence K, Gabuzda D. Virology. 1994;201:349–355. doi: 10.1006/viro.1994.1300. [DOI] [PubMed] [Google Scholar]
- 15.Simm M, Shahabuddin M, Chao W, Allan JS, Volsky DJ. J Virol. 1995;69:4582–4586. doi: 10.1128/jvi.69.7.4582-4586.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Potash MJ, Bentsman G, Muir T, Krachmarov C, Sova P, Volsky DJ. Proc Natl Acad Sci U S A. 1998;95:13865–13868. doi: 10.1073/pnas.95.23.13865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Madani N, Kabat D. J Virol. 1998;72:10251–10255. doi: 10.1128/jvi.72.12.10251-10255.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Simon JH, Gaddis NC, Fouchier RA, Malim MH. Nat Med. 1998;4:1397–1400. doi: 10.1038/3987. [DOI] [PubMed] [Google Scholar]
- 19.Sheehy AM, Gaddis NC, Choi JD, Malim MH. Nature. 2002;418:646–650. doi: 10.1038/nature00939. [DOI] [PubMed] [Google Scholar]
- 20.Zhang H, Pomerantz RJ, Dornadula G, Sun Y. J Virol. 2000;74:8252–8261. doi: 10.1128/jvi.74.18.8252-8261.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dettenhofer M, Cen S, Carlson BA, Kleiman L, Yu XF. J Virol. 2000;74:8938–8945. doi: 10.1128/jvi.74.19.8938-8945.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang S, Sun Y, Zhang H. J Biol Chem. 2001;276:4889–4893. doi: 10.1074/jbc.M004895200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Angers S, Salahpour A, Bouvier M. Annu Rev Pharmacol Toxicol. 2002;42:409–435. doi: 10.1146/annurev.pharmtox.42.091701.082314. [DOI] [PubMed] [Google Scholar]
- 24.Cochran AG. Chem Biol. 2000;7:85–94. doi: 10.1016/s1074-5521(00)00106-x. [DOI] [PubMed] [Google Scholar]
- 25.Huang Z. Pharmacol Ther. 2000;86:201–215. doi: 10.1016/s0163-7258(00)00052-8. [DOI] [PubMed] [Google Scholar]
- 26.Zhang H, Dornadula G, Pomerantz RJ. J Virol. 1996;70:2809–2824. doi: 10.1128/jvi.70.5.2809-2824.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang H, Dornadula G, Beumont M, Livornese L, Jr, Van Uitert B, Henning K, Pomerantz RJ. N Engl J Med. 1998;339:1803–1809. doi: 10.1056/NEJM199812173392502. [DOI] [PubMed] [Google Scholar]
- 28.Hassaine G, Courcoul M, Bessou G, Barthalay Y, Picard C, Olive D, Collette Y, Vigne R, Decroly E. J Biol Chem. 2001;276:16885–16893. doi: 10.1074/jbc.M009076200. [DOI] [PubMed] [Google Scholar]
- 29.Dostmann WR, Taylor MS, Nickl CK, Brayden JE, Frank R, Tegge WJ. Proc Natl Acad Sci U S A. 2000;97:14772–14777. doi: 10.1073/pnas.97.26.14772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bonfanti M, Taverna S, Salmona M, D’Incalci M, Broggini M. Cancer Res. 1997;57:1442–1446. [PubMed] [Google Scholar]
- 31.Derossi D, Calvet S, Trembleau A, Brunissen A, Chassaing G, Prochiantz A. J Biol Chem. 1996;271:18188–18193. doi: 10.1074/jbc.271.30.18188. [DOI] [PubMed] [Google Scholar]
- 32.Schwartz JJ, Zhang S. Curr Opin Mol Ther. 2000;2:162–167. [PubMed] [Google Scholar]
- 33.Yuan J, Kramer A, Eckerdt F, Kaufmann M, Strebhardt K. Cancer Res. 2002;62:4186–4190. [PubMed] [Google Scholar]
- 34.Chikh GG, Kong S, Bally MB, Meunier JC, Schutze-Redelmeier MP. J Immunol. 2001;167:6462–6470. doi: 10.4049/jimmunol.167.11.6462. [DOI] [PubMed] [Google Scholar]
- 35.Pomerantz RJ. Nature. 2002;418:594–595. doi: 10.1038/418594a. [DOI] [PubMed] [Google Scholar]
- 36.Kay BK, Williamson MP, Sudol M. FASEB J. 2000;14:231–241. [PubMed] [Google Scholar]
- 37.Sparks AB, Rider JE, Hoffman NG, Fowlkes DM, Quillam LA, Kay BK. Proc Natl Acad Sci U S A. 1996;93:1540–1544. doi: 10.1073/pnas.93.4.1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kieber-Emmons T, Murali R, Greene MI. Curr Opin Biotechnol. 1997;8:435–441. doi: 10.1016/s0958-1669(97)80065-1. [DOI] [PubMed] [Google Scholar]
- 39.Moore GJ. Proc West Pharmacol Soc. 1997;40:115–119. [PubMed] [Google Scholar]