

Abstract
The Vif (virion infectivity factor protein of human immunodeficiency virus type I (HIV-1) is essential for viral replication in vivo and productive infection of peripheral blood mononuclear cells, macrophages, and H9 T-cells. However, the molecular mechanism(s) of Vif remains unknown and needs to be further determined. In this report, we show that, like many other proteins encoded by HIV-1, Vif proteins possess a strong tendency toward self-association. In relatively native conditions, Vif proteins formed multimers in vitro, including dimers, trimers, or tetramers. Through in vivo binding assays such as coimmunoprecipitation and the mammalian two-hybrid system, we also demonstrated that Vif proteins could interact with each other within a cell, indicating that the multimerization of Vif proteins is not simply due to fortuitous aggregation. Further studies indicated that the domain affecting Vif self-association is located at the C terminus of this protein, especially the proline-enriched 151–164 region. Moreover, we found that a Vif mutant with deletion at amino acid 151–164 was unable to rescue the infectivity of vif-defective viruses generated from H9 T-cells, suggesting that the multimerization of Vif proteins could be important for Vif function in the viral life cycle. Our studies identified a new feature of Vif and should accelerate our understanding of its role in HIV-1 pathogenesis.
The accessory genes of HIV-1,1 including vif, vpr, nef, and vpu, have been shown to play important roles during HIV-1 infection (1). It has been demonstrated that Vif affects the late stages of the viral life cycle, possibly through the assembly of viral particles (2–4). The vif-defective (vif–) viruses are able to penetrate into target cells but not accomplish reverse transcription (4–7). The requirement for Vif, however, is cell type-specific. The vif– viruses exhibit a negative phenotype only when produced from primary T-lymphocytes, terminally differentiated macrophages, or a few T-lymphoid cell lines, such as H9. These cells were entitled as “nonpermissive” cells. In some T-cell lines such as SupT1, C8166, and other non-T-cells such as HelaCD4 cells, however, productive replication of vif– HIV-1 viruses can be achieved. These cell lines therefore were named as “permissive” cells (2, 4, 8). There are two possibilities for Vif function in the nonpermissive cells; Vif may counteract an endogenous inhibitor existing in the nonpermissive cells or alternatively, substitute a Vif homologue that exists in the permissive cells but not nonpermissive cells (9). A recent study showed that the permissive HelaCD4 cells expressing the HIV-1F12 Vif were resistant to the replication of wild-type HIV-1, suggesting that there may be a Vif homologue in the permissive cells that was inhibited by HIV-1F12 Vif (10). Conversely, the progeny viruses generated from the heterokayons that were formed between permissive and nonpermissive cells showed a phenotype similar to that generated from the nonpermissive cells. This result suggested that nonpermissive cells, most likely the natural targets of HIV-1, contain a potent endogenous inhibitor of HIV-1 replication that is counteracted by Vif (11, 12). However, the nature of endogenous inhibitor and the molecular mechanism(s) regarding how Vif interacts with it remain unknown.
Recently, it has been shown that Vif is associated with a complex in the virus-producing cells (13). Although it has been demonstrated that Vif of HIV-1 interacts with the NCp7 domain of p55 Gag precursor in vitro through its positively charged amino acid-enriched C terminus and colocalizes with Gag precursors in a cell, no direct interaction was observed between Vif and Gag precursors (13–16). We further demonstrated that Vif is an RNA binding protein and able to form an RNase-sensitive messenger ribonucleoprotein complex with viral unspliced RNA in the cytoplasm of HIV-1-infected cells. As Vif-RNA binding could be displaced by Gag-RNA binding, Vif may mediate viral RNA engagement with HIV-1 Gag precursors and thus could be involved in genomic RNA folding and packaging (31). In this study, we demonstrate a new biochemical characteristic of Vif protein; Vif proteins have a strong tendency to form multimers, which could play an important role for the Vif function in HIV-1 life cycle.
EXPERIMENTAL PROCEDURES
Plasmid Constructions
With infectious clone pNL4–3 as template, deletion mutants of HIV-1 Vif were generated by polymerase chain reaction (PCR)-mediated and site-directed mutagenesis (17). The PCR-generated wild-type vif gene and its mutants were then inserted into pCITE-4a vector (Novagen, Madison, WI) for in vitro translation. The vif gene was also inserted into pGEX vector for in vitro expression and isolation of GST-Vif fusion protein. For studying intracellular Vif-Vif interaction, vif genes were tagged with FLAG (DYKDDDDK) or c-Myc (EQKLISEEDL) epitope-encoding sequences at the 3′ terminus, respectively, via PCR. These tagged vif genes were then inserted into the vector pCI-Neo, which contains a chimeric intron just downstream of the cytomegalovirus enhancer and immediate early promoter (Promega, Madison, WI). The resulting plasmids were named pCI-vif-c-Myc or pCI-vif-FLAG, respectively. For mammalian two-hybrid analysis, pGal-Vif or pGal-VifΔ151–164 were constructed by replacing the HindIII-BamHI fragment (containing vp gene) of pSG5GalVP with a PCR-amplified complete vif gene or its mutant Δ151–164. The pVif-VP or pVifΔ151–164-VP were constructed by replacing the EcoRI-BglII fragment (containing gal4 gene) of pSG5GalVP with a PCR-amplified complete vif gene or its mutant Δ151–164, respectively (18). The integrity of all the constructs was confirmed by DNA sequencing.
Protein Expression and in Vitro Binding Assays
The vector pGEX, with or without the vif gene, was transformed into BL21 competent cells (Novagen, Madison, WI). After growth at 37 °C to ~0.6 optical density, The expression of GST or GST-Vif proteins were induced by 0.4 mm isopropylthio-β-d-galactoside. The bacterial cells were lysed by adding lysing buffer (1% Triton X-100, 0.1 mg/ml lysozyme, 2 mm EDTA, 1 mm phenylmethylsulfonyl fluoride, 2 μg/ml leupeptin, 1 μg/ml aprotinin), followed by sonication. The sample was pelleted at 12,000 × g for 10 min at 4 °C, and the supernatant was applied to a glutathione-conjugated agarose bead (Sigma) column. After batch binding, the matrix was washed by the addition of 10 bed volumes of phosphate-buffered saline 3 times. The GST or GST-Vif-conjugated agarose beads were then aliquoted and stored at −20 °C. Conversely, 35S-labeled Vif or its mutant proteins were synthesized utilizing SPT3 kits (Novagen, Madison, WI). The protocol supplied by manufacturer was followed. After in vitro translation, RNase A (0.2 mg/ml) was added to stop the reaction and remove tRNAs and the in vitro transcribed-mRNA. The trichloroacetic acid-insoluble radioactive amino acids were quantitated in the presence of a scintillantion mixture.
For GST pull-down assays, a GST- or GST-Vif-conjugated bead slurry was mixed with 35S-labeled Vif or its mutants (50,000 cpm) in a binding buffer (150 mm NaCl, 20 mm Tris-HCl (pH 7.5), 0.1% Triton X-100). After binding at 4 °C for 1 h, the mixture were centrifuged at 3,000 × g for 1 min, and the beads were washed with binding buffer three times. The 35S-labeled Vif proteins were dissociated from beads by adding SDS-containing loading buffer, and heating at 95 °C for 5 min. The samples were then electrophoresed in SDS-PAGE gels (15% Tris-HCl ready gel made by Bio-Rad, Hercules, CA). After treatment with the fixing buffer (10% acetic acid, 10% methanol) and then Amplify (Amersham Pharmacia Biotech), the gels were dried and exposed to x-ray film or quantitatively analyzed utilizing a PhosphorImager (Molecular Dynamics, Sunnyview, CA).
Furthermore, in vitro-translated, 35S-labeled Vif (50,000 cpm) was also directly loaded into a 4–20% Tris/glycine gel (SDS-free) via 10% glycerol-containing loading buffer, with SDS at various concentrations, and electrophoresed with an SDS-free Tris/glycine running buffer. After fixing and drying, the gel was directly subjected to autoradiography.
Western Blotting and Coimmunoprecipitation
The COS-1 or 293T cells were transfected with 5 μg of pCI-vif-c-Myc and pCI-vif-FLAG using a calcium phosphate precipitation method (17, 19). After 48 h, the cells were lysed in a cell lysing buffer (150 mm NaCl, 50 mm Tris-HCl, (pH 8.0), 5 mm EDTA, 1% Triton X-100, 10% glycerol, 1 mm phenylmethylsulfonyl fluoride, 2 μg/ml aprotinin, 2 μg/ml leupeptin, 2 μg/ml pepstatin A). For direct Western blotting, the whole-cell lysates were mixed with acetone (1:3). The mixture was incubated on ice for 20 min, followed by centrifugation at 12,000 × g for 10 min. The pellets were air-dried and resuspended in SDS-containing sample buffer. The samples were electrophoresed in SDS-PAGE gels and then electronically transferred onto a nylon/nitrocellulose membrane. The primary antibodies, goat anti-c-Myc antibody (A14) (Research Antibodies, Santa Cruz, CA), or mouse anti-FLAG antibody (M2) (Stratagene, La Jolla, CA) were used to bind the samples, respectively. The horseradish peroxidase-conjugated anti-goat IgG antibody or anti-mouse IgG antibody (Research Antibodies, Santa Cruz, CA) was used as the secondary antibody. A Chemiluminescence-based system (ECL; Amersham Pharmacia Biotech) was used to visualize the antigen-antibody binding.
For coimmunoprecipitation, cell lysates from COS-1 or 293T cells expressing Vif-FLAG and/or Vif-c-Myc were incubated with A14 antic-Myc antibody (Santa Cruz) (1 μg/ml) by mixing 12 h at 4 °C, followed by incubation with protein A-conjugated Sepharose CL-4B (Amersham Pharmacia Biotech) for an additional 2 h. The pellet was washed three times with cell lysing buffer. The pellet was then resuspended in SDS-containing buffer, heated at 95 °C, and centrifuged at 12,000 × g. The supernatant was then subjected to SDS-PAGE. After transfer onto a nylon/nitrocellulose membrane, the samples were detected with a mouse M2 anti-FLAG antibody. An horseradish peroxidase-conjugated anti-mouse IgG (Research Antibodies, Santa Cruz, CA) was used as a secondary antibody.
Mammalian Two-hybrid System Assay
A mammalian two hybrid system, which was modified from the GAL4-based yeast two-hybrid assay, was used to study the self-association of HIV-1 Vif proteins in vivo (18, 20). The procedure was as described previously, with some modifications (18, 20). Briefly, 5 μg of pGal-Vif and pVif-VP were cotransfected with pG5BCAT into COS-1, using the Superfect transfection reagent (Qiagen, Valencia, CA). 48 h post-transfection, the cells were lysed in reporter lysing buffer (Promega, Madison, WI) and subjected to a chloramphenicol acetyltransferase (CAT) assay, as described previously (19).
Single-round Viral Infectivity Assays
The biological activity of Vif mutants was evaluated by using a single-round viral infectivity assay, with some modifications (7). To generate recombinant HIV-1 viruses, H9 cells were transfected with 5 μg of pNL4–3ΔvifΔenv, pMD.G (containing vesicular stomatitis virus (VSV) envelope), and wild-type vif gene or its mutants (in pCI-Neo construct) by electroporation (7, 21). The electroporation (350 V, 250 microfarad, 5.1–6.3 ms) was performed by a gene pulser apparatus and capacitance (Bio-Rad, Hercules, CA). Thereafter, conditioned medium (RPMI 1640 plus 10% fetal bovine serum) was used to maintain the transfected H9 cells. Two days after transfection, the viral particles in supernatant were collected and pelleted via ultracentrifugation (7). After normalization by the HIV-1 p24 antigen level, which was detected via enzyme-linked immunosorbent assays (kits from DuPont), the viruses were used to infect 5 × 105 HeLaCD4-CAT cells (22). 48 h post-infection, the cells were lysed in reporter lysing buffer (Promega) and subjected to CAT assays.
RESULTS
Vif Proteins Can Form Multimers in Vitro
To examine whether Vif proteins have a tendency toward self-association, GST-Vif was expressed in BL 21 bacterial cells and isolated onto glutathione-conjugated agarose beads. In vitro-translated, 35S-labeled Vif proteins were allowed to incubate with the GST-Vif-conjugated beads. The bead-associated 35S-labled Vif was then analyzed by SDS-PAGE, followed by direct autoradiography. Fig. 1A illustrates that GST-Vif (lane 2), but not GST (lane 3), can strongly bind to 35S-labeled, in vitro-translated Vif protein, indicating a Vif-Vif interaction. Further, 35S-labeled, in vitro-translated HIV-1 Vif protein was directly loaded onto a Tris/glycine-native gel (SDS-free) for electrophoresis, with loading buffers containing 10% glycerol only or SDS at various concentrations. At the native or relatively native conditions, the 35S-labeled Vif proteins migrated in the 4–15% Tris/glycine gels as monomers (23 kD), dimers (46 kD), and trimers (69 kD) or tetramers (92 kD) (Fig. 1B). With the increment of concentrations of SDS in the loading buffer, the major forms of Vif eventually became a monomer (23 kD). When the sample was heated at 95 °C for 5 min, all the multimers of Vif proteins disappeared, suggesting that the Vif-Vif binding is unlikely to be covalent. It is notable that, prior to the sample loading, 35S-labeled, in vitro-translated HIV-1 Vif protein was treated with RNase A to remove possible RNA contamination. Therefore, the Vif-Vif binding should be RNA-independent.
Fig. 1. Vif self-association in a cell-free system.
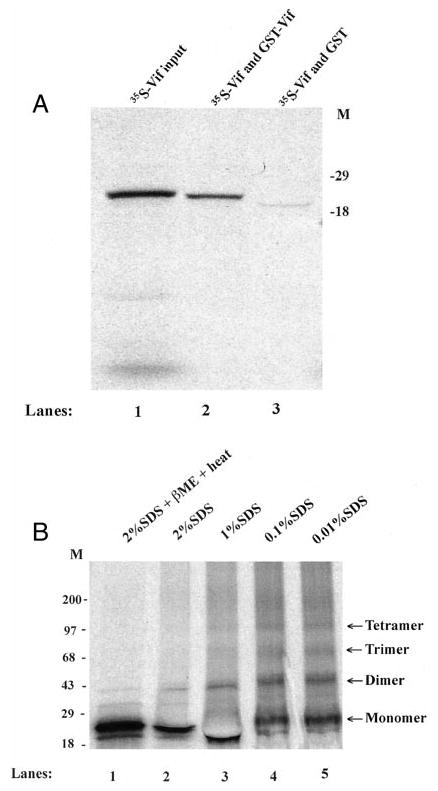
A, in vitro translated, 35S-labeled HIV-1NL4–3 Vif proteins were allowed to bind with GST-Vif conjugated on beads. After binding, the bead-associated 35S-labeled Vif was analyzed via SDS-PAGE and direct autoradiography. B, Vif proteins form dimers and multimers in native or mild-denatured loading buffer. In vitro-translated 35S-labeled HIV-1NL4–3 Vif proteins were loaded directly onto a 4–20% Tris-HCl gel (SDS-free) with native loading buffer (62.5 mm Tris-HCl (pH 6.8), 20% glycerol) plus SDS at different concentrations. Electrophoresis was performed with a Tris/glycine running buffer containing 0.05% SDS, followed by autoradiography. βME, β-mercaptoethanol.
The Binding Site for Vif Multimerization Is Located in the C Terminus
To determine the binding sites for Vif multimerization, a series of deletions in Vif protein have been generated through PCR-based mutagenesis, followed by in vitro translation in the presence of [35S]methionine. These Vif mutants were then allowed to bind to GST-Vif fusion protein conjugated to agarose beads. Fig. 2A indicates that the deletion of the C terminus in Vif protein severely loses the Vif-Vif binding activity. Further studies indicated that deletion at amino acid 151–164 would significantly decrease this binding ability (Fig. 2A). This result was further confirmed by native multimer formation assay. In the presence of 0.1% SDS, Vif mutants Δ151–192 and Δ151–164 were unable to form multimers, whereas other mutants were able to do so (Fig. 2B). It is notable that there are several positively charged amino acids in the 151–164 fragment. The mutants that substitute these positively charged amino acids, generated by Goncalves et al. (23), have been examined for this Vif-Vif binding. However, all these mutants still contain Vif-Vif binding ability (data not shown). It is also notable that there are several prolines (Pro156, Pro161, Pro162, and Pro164) in this fragment. Among these prolines, Pro161 is highly conserved in various strains of HIV-1 or simian immunodeficiency virus. Whether these prolines are important for Vif-Vif binding remains to be further verified.
Fig. 2. The effect of Vif mutants on Vif-Vif interactions.
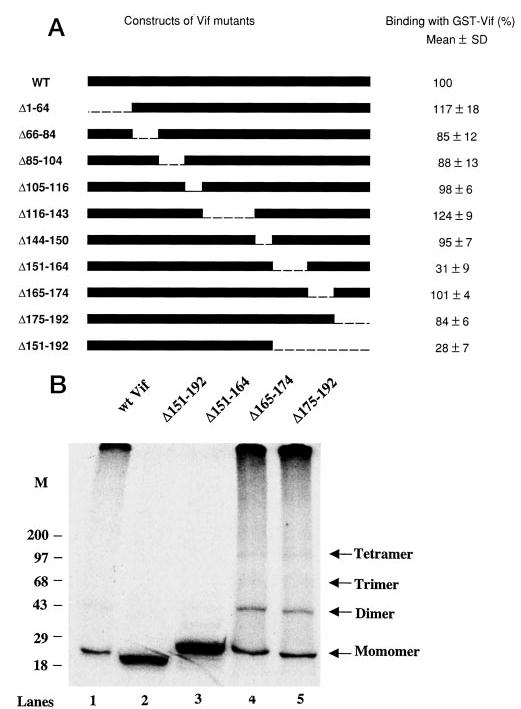
A, a series of deletions along the Vif 192 amino acids were generated via PCR-based mutagenesis and in vitro translation. The in vitro-translated, 35S-labeled HIV-1NL4–3 Vif protein and its mutants were allowed to bind to GST-Vif conjugated on agarose beads. The bead-associated, 35S-labeled Vif protein and its mutants were subjected to SDS-PAGE and visualized by direct autoradiography. The values were obtained by quantitation with densitometry of the autoradiographs. The ratio of bound Vif versus the input was then calculated. The ratio of GST-Vif-bound 35S-labeled wild-type Vif and 35S-labeled wild-type Vif input was further set as 100% (with the standard deviations). The relative binding ability of Vif mutants was thus determined. In most cases, the data reflect at least five independent experiments. WT, wild-type. B, in vitro-translated 35S-labeled HIV-1NL4–3 Vif protein and its mutants (50,000 cpm count for each) were loaded directly onto a 4–20% Tris-HCl gel (SDS-free), with loading buffer (62.5 mm Tris-HCl (pH 6.8), 20% glycerol) plus 0.1% SDS. Electrophoresis was performed with a Tris/glycine running buffer containing 0.05% SDS, followed by autoradiography.
Vif-Vif Interactions within a Cell
To elucidate the possibility that Vif self-association also occurs intracellularly, we utilized a coimmunoprecipitation method. The Vif protein was tagged with c-Myc or FLAG epitope at its C terminus, respectively, and expressed in the COS-1 cells. Fig. 3 indicated that the expression of c-Myc-tagged Vif and FLAG-tagged Vif could be detected via Western blotting, with mouse anti-c-Myc epitope antibody or goat anti-FLAG epitope antibody, respectively (top two panels). To study Vif-Vif interaction, the cell lysates were immunoprecipitated with anti-Myc antibody and then subjected to SDS-PAGE, followed by Western blotting. The goat anti-FLAG antibody was used to detect FLAG-tagged Vif. Fig. 3 demonstrated that the FLAG-tagged Vif was coprecipitated with Myc-tagged Vif when mouse anti-Myc antibody was utilized for the immunoprecipitation, suggesting a Vif-Vif interaction within a cell (Fig. 3, bottom panel).
Fig. 3. Coimmunoprecipitation method to study Vif-Vif interactions within cells.
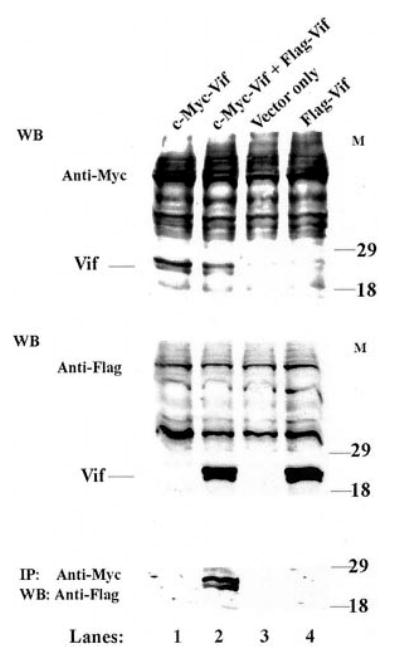
COS-1 cells were transfected with vectors harboring FLAG or c-Myc tagged Vif. After 54 h of incubation at 5% CO2, 37 °C, 20 μg of total cell lysates were resolved by 15% Tris-HCl gel. The Vif proteins were detected by Western blotting (WB) using an M2 anti-FLAG monoclonal antibody and A14 anti-c-Myc polyclonal antibody, respectively. For coimmunoprecipitation, the whole-cell lysates from the same batch were subjected to immunoprecipitation (IP) with A14 anti-c-Myc polyclonal antibody. Immunoprecipitates were resolved at 15% Tris-HCl gel, transferred onto a membrane, and then detected using an M2 anti-FLAG antibody.
Alternatively, the in vivo Vif-Vif interaction was examined by the mammalian two-hybrid system. A fusion protein composed of VP16 and Gal4 is able to activate the Gal4 response element-contained E1b promoter. Gal4 would function as a DNA binding domain, whereas VP16 will function as a DNA activation domain. HIV-1 Vif protein was allowed to replace VP16 or Gal4 domain, respectively (Fig. 4A). If the interaction between Vif proteins take place, the VP16 and Gal4 domain would be brought together, and the Gal4 binding sequence-contained E1b promoter would be activated. Fig. 4B indicated that, like Rev-Rev interactions, Vif in Vif-VP16 fusion protein could bind to Vif in the Gal4-Vif fusion protein and activate the expression of CAT (lane 6). As controls, pGal-Vif or pVif-VP alone were unable to activate CAT expression (lanes 3 and 4). Fig. 4B also shows that Vif mutant Δ151–164, which did not have the ability to interact with Vif protein in other systems, also could not interact with Vif in this system (lane 7).
Fig. 4. Mammalian two-hybrid system to study Vif-Vif interaction.
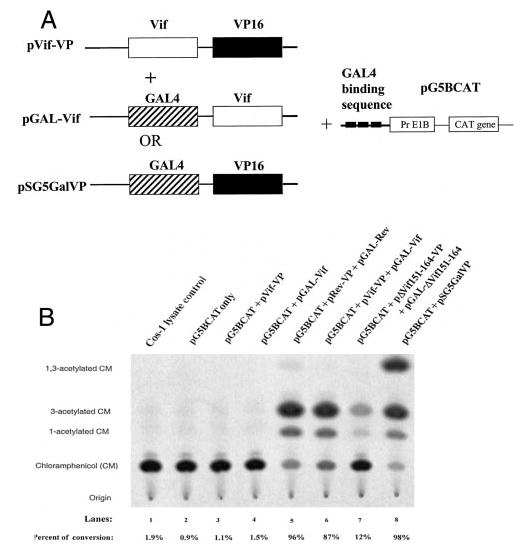
A, a schematic map showing the plasmids utilized in the experiments. B, COS-1 cells were transfected with plasmids combined with various vectors. After 48 h, cell lysates were harvested and subjected to CAT analyses.
Deletion of the Vif-Vif Binding Domain Severely Decreases the Vif Function in the Viral Life Cycle
As mentioned previously, Vif functions in the late events of HIV-1 life cycle and is required by nonpermissive cells, such as peripheral blood mononuclear cells, macrophages, and H9 T-cells (2–4). To investigate the physiological significance of Vif multimerization, we examined whether Vif mutant (Δ151–164), which is unable to form multimers in the cell-free system and within cells, is able to complement Vif function in the viral life cycle. To this end, a single-round viral infectivity assay was adapted. Wild-type Vif or its mutants were expressed in the nonpermissive H9 T-cells. At the same time, pseudotyped (with VSV envelope) HIV-1 viruses, without vif and env in their genome, were generated from these cells. After ultracentrifugation for enrichment, the recombinant viruses were allowed to infect the target cells (HelaCD4-CAT), which harbor an expression cassette containing the HIV-1 long terminal repeat promoter-driven CAT gene. The viral infectivity was measured by the level of CAT gene expression in the target cells, which is driven by the HIV-1 Tat protein expressed by the newly synthesized proviruses. Fig. 5 demonstrates that, when the wild-type vif gene was expressed in the vif-defective HIV-1 virus-producing nonpermissive H9 T-cells, the viral infectivity could reach a high level (lane 2). However, when VifΔ151–164 was expressed in the vif-defective HIV-1 virus-producing nonpermissive H9 T-cells, the viral infectivity was almost unaltered (lane 3), compared with the vif-defective HIV-1 viruses (lane 4). These data indicated that the 151–164 deletion severely decreased the function of Vif protein and made it unable to rescue the infectivity of the vif-defective HIV-1 viruses generated from nonpermissive T-cells. It is notable that another group also demonstrated that this fragment is essential for Vif function (24). This experiment demonstrated that multimerization of Vif proteins is required for Vif function.
Fig. 5. Viral infectivity affected by Vif or Vif mutants.
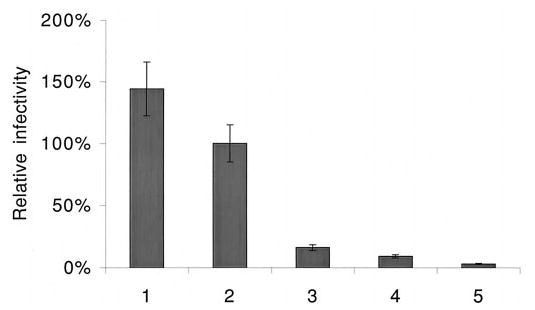
The pCI-Neo constructs, containing wild-type vif gene or its mutants, pNL4–3ΔenvΔvif plasmid and pMD.G (containing VSV envelope (env)), were cotransfected into H9 cells to generate the pseudotyped viral particles. After concentration via ultracentrifugation, the viral particles were normalized by HIV-1 p24 antigen. In the presence of polybrene (8 μg/ml), the viruses were used to infect HelaCD4-CAT cells. After 48 h, the cell lysates were collected and subjected to CAT analyses. Lane 1, pNL4–3; lane 2, pNL4–3ΔenvΔvif, VSV env plus wild-type vif; lane 3, pNL4–3ΔenvΔvif, VSV env plus vifΔ 151–164; lane 4, pNL4–3ΔenvΔvif, VSV env plus vifΔ 144–150; lane 5, pNL4–3ΔenvΔvif, VSV env plus pCI-Neo vector only. The value of wild-type vif complementation was set as 100%. The relative values of the other samples were calculated accordingly. This figure is representative of three independent experiments. Values are means ± standard deviations.
DISCUSSION
Many HIV-1 proteins, including Gag, protease, reverse transcriptase, integrase, glycoprotein 41 (gp41), Tat, Rev, Vpr, and Nef, have been shown to form dimers or multimers in vitro and in vivo. The formation of dimers or multimers has been demonstrated to be important for their functions in the lentiviral life cycle (25–28). In addition, multimerization is critical to the biological activity of many prokaryotic and eukaryotic proteins and is a common mechanism for the functional activation/inactivation of proteins. In this study, we analyzed the multimerization potential of HIV-1 Vif proteins via various complementary methods. The in vitro-translated, 35S-lableled Vif proteins were able to form multimers in the native environment. Conversely, GST-Vif fusion proteins, rather than GST proteins, which were generated from the bacterial expression system, were able to bind to the in vitro-translated, 35S-lableled Vif proteins. Further, coimmunoprecipitation and a mammalian two hybrid system also demonstrated a Vif-Vif interaction intracellularly. These in vitro and in vivo data strongly support the notion that Vif proteins are able to form multimers. As the deletion of the domain that is essential for the Vif-Vif binding severely decreases the function of Vif in the nonpermissive cells, multimerization of Vif could be important for its function in the HIV-1 life cycle. However, as the function of Vif protein in the life cycle remains largely unknown, the precise role of Vif multimerization and the active form(s) (i.e. monomer, dimer, or tetramer) of Vif protein in the virus-producing cells remains to be determined.
The domain for Vif multimerization has been located in a positively charged amino acid- and proline-enriched fragment (amino acid 151–164) (Fig. 2). As the positively charged amino acids in this region are not responsible for the Vif-Vif interaction, whether the prolines are important remains to be clarified. It is notable that a highly conserved motif, SLQYLAL (amino acid 144–150 for HIV-1NL4–3), is close to this domain. It has also been shown that Ser165 is phosphorylated by the mitogen-activated protein kinase (p44/42) of Vif, and this phosphorylation is important for Vif function (30). As these residues are close to the domain for multimerization, it is possible that the multimerization of Vif proteins is regulated by phosphorylation in the virus-producing cells. Interestingly, the positively charged amino acids (replaced in B4 and B7 mutants) in the C terminus of Vif are responsible for Vif-NCp7 binding in vitro (14). Recently, we demonstrated that HIV-1 Vif is an RNA binding protein and an integral component of a messenger ribonucleoprotein complex of viral RNA in the cytoplasm and could be involved in the viral RNA packaging process (31). In contrast to interactions with NCp7 via its C terminus, Vif binds to RNA via its N terminus. Although more RNA would bind to Vif than to Gag at the same conditions when RNA is mixed with Vif or Gag separately, RNA will only bind to Gag but not Vif when Vif protein is mixed together with RNA and NCp7 (31). This “displacement” could be because of various mechanisms and is under investigation. However, as the domains for Vif multimerization and for Vif-NCp7 binding are quite close in location or possibly overlap, it is possible that the interaction between Vif and Gag, as well as the interactions among Vif, RNA, and Gag, is regulated by Vif multimerization.
Thus, the finding of Vif multimerization may be helpful in understanding the structure-function relationship of Vif protein, identifying the molecular mechanism(s) of HIV-1 Vif in the viral life cycle. In addition, these data provide a promising intervention target for anti-HIV-1 agent development.
Acknowledgments
We thank Drs. Roger J. Pomerantz, Geethanjali Dornadula, Charvi A. Patel, and Jianhua Fang for critical review of the manuscript and valuable discussions. We also thank Dr. Dana Gabuzda for providing plasmids of Vif C-terminal mutants (B1-B7).
Footnotes
This work was supported by Thomas Jefferson University funds and the Margaret Q. Landenberger Research Foundation (to H. Z.).
The abbreviations used are: HIV-1, human immunodeficiency virus type I; GST, glutathione S-transferase; CAT, chloramphenicol acetyl-transferase; PCR, polymerase chain reaction; PAGE, polyacrylamide gel electrophoresis; VSV, vesicular stomatitis virus.
References
- 1.Emerman M, Malim MH. Science. 1998;280:1880–1884. doi: 10.1126/science.280.5371.1880. [DOI] [PubMed] [Google Scholar]
- 2.Gabuzda DH, Lawrence K, Langhoff E, Terwilliger E, Dorfman T, Haseltine WA, Sodroski J. J Virol. 1992;66:6489–6495. doi: 10.1128/jvi.66.11.6489-6495.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blanc D, Patience C, Schulz TF, Weiss R, Spire B. Virology. 1993;193:186–192. doi: 10.1006/viro.1993.1114. [DOI] [PubMed] [Google Scholar]
- 4.von Schwedler U, Song J, Aiken C, Trono D. J Virol. 1993;67:4945–4955. doi: 10.1128/jvi.67.8.4945-4955.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Courcoul M, Patience C, Rey F, Blanc D, Harmache A, Sire J, Vigne R, Spire B. J Virol. 1995;69:2068–2074. doi: 10.1128/jvi.69.4.2068-2074.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sova P, Volsky DJ. J Virol. 1993;67:6322–6326. doi: 10.1128/jvi.67.10.6322-6326.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dornadula G, Yang S, Pomerantz RJ, Zhang H. J Virol. 2000;74:2594–2602. doi: 10.1128/jvi.74.6.2594-2602.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gabuzda DH, Li H, Lawrence K, Vasir BS, Crawford K, Langhoff E. J Acquir Immune Defic Syndr. 1994;7:908–915. [PubMed] [Google Scholar]
- 9.Trono D. Cell. 1995;82:189–192. doi: 10.1016/0092-8674(95)90306-2. [DOI] [PubMed] [Google Scholar]
- 10.D’Aloja P, Olivetta E, Bona R, Nappi F, Pedacchia D, Pugliese K, Ferrari G, Verani P, Federico M. J Virol. 1998;72:4308–4319. doi: 10.1128/jvi.72.5.4308-4319.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Simon JH, Gaddis NC, Fouchier RA, Malim MH. Nat Med. 1998;4:1397–1400. doi: 10.1038/3987. [DOI] [PubMed] [Google Scholar]
- 12.Madani N, Kabat D. J Virol. 1998;72:10251–10255. doi: 10.1128/jvi.72.12.10251-10255.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Simon JH, Carpenter EA, Fouchier RA, Malim MH. J Virol. 1999;73:2667–2674. doi: 10.1128/jvi.73.4.2667-2674.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bouyac M, Courcoul M, Bertoia G, Baudat Y, Gabuzda D, Blanc D, Chazal N, Boulanger P, Sire J, Vigne R, Spire B. J Virol. 1997;71:9358–9365. doi: 10.1128/jvi.71.12.9358-9365.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Simon JH, Fouchier RA, Southerling TE, Guerra CB, Grant CK, Malim MH. J Virol. 1997;71:5259–5267. doi: 10.1128/jvi.71.7.5259-5267.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huvent I, Hong SS, Fournier C, Gay B, Tournier J, Carrière C, Courcoul M, Vigne R, Spire B, Boulanger P. J General Virol. 1998;79:1069–1081. doi: 10.1099/0022-1317-79-5-1069. [DOI] [PubMed] [Google Scholar]
- 17.Zhang H, Dornadula G, Alur P, Laughlin MA, Pomerantz RJ. Proc Natl Acad Sci U S A. 1996;93:12519–12524. doi: 10.1073/pnas.93.22.12519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shimano R, Iida S, Fukumori T, Yamamoto Y, Kawamura M, Furuta RA, Adachi A. Biochem Biophys Res Comm. 1998;242:313–316. doi: 10.1006/bbrc.1997.7963. [DOI] [PubMed] [Google Scholar]
- 19.Zhang H, Duan LX, Dornadula G, Pomerantz RJ. J Virol. 1995;69:3929–3932. doi: 10.1128/jvi.69.6.3929-3932.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bogerd H, Greene WC. J Virol. 1993;67:2496–2502. doi: 10.1128/jvi.67.5.2496-2502.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Naldini L, Blomer U, Gage FH, Trono D, Verma IM. Proc Natl Acad Sci U S A. 1996;93:11382–11388. doi: 10.1073/pnas.93.21.11382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ciminale V, Felber BK, Campbell M, Pavlakis GN. AIDS Res Hum Retroviruses. 1990;6:1281–1287. doi: 10.1089/aid.1990.6.1281. [DOI] [PubMed] [Google Scholar]
- 23.Goncalves J, Shi B, Yang X, Gabuzda D. J Virol. 1995;69:7196–7204. doi: 10.1128/jvi.69.11.7196-7204.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Simon JH, Sheehy AM, Carpenter EA, Fouchier RA, Malim MH. J Virol. 1999;73:2675–2681. doi: 10.1128/jvi.73.4.2675-2681.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Frankel AD, Young JA. Annu Rev Biochem. 1998;67:1–25. doi: 10.1146/annurev.biochem.67.1.1. [DOI] [PubMed] [Google Scholar]
- 26.Vaishnav YN, Wong-Staal F. Annu Rev Biochem. 1991;60:577–630. doi: 10.1146/annurev.bi.60.070191.003045. [DOI] [PubMed] [Google Scholar]
- 27.Zhao LJ, Wang L, Mukherjee S, Narayan O. J Biol Chem. 1994;269:32131–32137. [PubMed] [Google Scholar]
- 28.Liu L, Heveker N, Fackler OT, Arold S, Gall SL, Janvier K, Peterlin BM, Dumas C, Schwartz O, Benichou S, Benarous R. J Virol. 2000;74:5310–5319. doi: 10.1128/jvi.74.11.5310-5319.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Deleted in proof
- 30.Yang X, Gabuzda D. J Biol Chem. 1998;273:29879–29887. doi: 10.1074/jbc.273.45.29879. [DOI] [PubMed] [Google Scholar]
- 31.Zhang H, Pomerantz RJ, Dornadula G, Sun Y. J Virol. 2000;74:8252–8261. doi: 10.1128/jvi.74.18.8252-8261.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]