

Abstract
T-cell tolerance is a critical element of tumor escape. However, the mechanism of tumor-associated T-cell tolerance remains unresolved. Using an experimental system employing the adoptive transfer of transgenic T cells into naïve recipients, we found that the population of Gr-1+ immature myeloid cells (ImC) from tumor-bearing mice was able to induce CD8+ T-cell tolerance. These ImC accumulate in large numbers in spleens, lymph nodes, and tumor tissues of tumor-bearing mice and are comprised of precursors of myeloid cells. Neither ImC from control mice nor progeny of tumor-derived ImC including tumor-derived CD11c+ DCs were able to render T cells non-responsive. ImC are able to take-up soluble protein in vivo, process it, and present antigenic epitopes on their surface and induce antigen-specific T-cell anergy. Thus, this is a first demonstration that in tumor-bearing mice CD8+ T-cell tolerance is induced primarily by ImC that may have direct implications for cancer immunotherapy.
Introduction
T-cell tolerance is one of the major mechanisms of tumor escape and one of the major factors limiting the success of cancer vaccines. Although there are some debates regarding whether this tolerance is important at very early stages of tumor development, there is clear evidence now that it plays a major role in advanced stages of cancer (1). Previous studies have established that bone marrow derived antigen presenting cells (APC) are primarily responsible for the induction of tumor-induced T-cell tolerance (2, 3). However, the exact nature of these cells as well as the mechanism of this phenomenon remains unclear. In physiological conditions APC consist of primarily dendritic cells (DC) and macrophages. In tumor-bearing mice, another group of bone marrow derived cells may be a part of APC population. These cells identified as Gr-1+CD11b+ cells are comprised of precursors of macrophages, granulocytes, DC, and myeloid cells at earlier stages of differentiation (rev. (4–6)). These cells are present in bone marrow and in small numbers in spleens of healthy mice, and under normal conditions differentiate into mature myeloid cells (7). However, they accumulate in large numbers in spleens, lymph nodes, and tumor tissues of tumor-bearing mice due to the effect of various tumor-derived factors(8–14). These cells can suppress T cells in vitro. Because of the nature of these cells they are called immature myeloid cells (ImC) or myeloid suppressive cells.
These data suggested a possible role of ImC in tumor-associated immune suppression. However, whether ImC are able to take up antigens in vivo, process them, and induce lasting antigen-specific T-cell tolerance is unknown. The investigation of T-cell tolerance in tumor-bearing host is limited by the nature of the experimental models since the persistent effect of tumor-derived factors makes it very difficult to investigate the role of defined cell populations. Therefore, in this study we used a model with adoptive transfer of different cell populations isolated from tumor-bearing mice into tumor-free recipients. This study, for the first time, has demonstrated that tumor-bearing host derived ImC, but not their progeny or Gr-1+ cells from control mice, are not only able to migrate into the tumor site and pick-up soluble proteins, but also process them and induce antigen-specific tolerance of CD8+ T-cells.
Materials and Methods
Mice and reagents.
Female C57BL/6 mice (6–8 weeks of age) were obtained from the National Cancer Institute (Frederick, MD). OT-1 TCR-transgenic mice (C57Bl/6–Tg(TCRαTCRβ)1100mjb), were purchased from Jackson Laboratories (Bar Harbor, ME), F1 C57Bl/6xBALB/c mice from Charles Rivers Laboratories (Wilmington, MA). 2C TCR transgenic mice were a gift from Dr. Schneck (John Hopkins University, Baltimore, MD). OVA-derived peptide (H-2Kb-restricted, aa 257–264, SIINFEKL), 2C-derived peptide (H-2Kb restricted SIYRYYGL (SIY)), control H-2Kb restricted RAHYNIVTF, and H-2Ld restricted QLSPFPFDL (QL9) peptides were obtained from SynPep Corporation, Dublin, CA. Complete and incomplete Freund’s adjuvants (CFA and IFA, respectively) were purchased from Sigma Chemical Co, St. Louis, MO. Ovalbumin-FITC and CFSE were obtained from Molecular Probes (Eugene, OR). All antibodies used for flow cytometry were obtained from BD Pharmingen with the exception of anti-F4/80 (Serotec, Raleigh, NC). Murine hybridoma 25-D1.16 secreting an IgG1 mAb specific for the pOV8·H-2Kb complex (15) was kindly provided by Drs. Germain and Porgador.
Cell isolation and generation.
Gr-1+ or CD11c+ cells were isolated from spleens of tumor-bearing mice using magnetic beads and MiniMACS columns ((Miltenyi Biotec GmbH, Auburn, CA) and biotinylated anti-Gr-1 or anti-CD11c mAbs. The purity of isolation was more than 90% as evaluated by flow cytometry. Consistent with previous observations >98% of Gr-1+ cells were also CD11b+. T lymphocytes were isolated from spleens using T cell enrichment columns (MTCC-525; R&D Systems, Minneapolis, MN). DCs were generated from murine bone marrow using 20 ng/ml murine recombinant GM-CSF and 10 ng/ml IL-4 (both from RDI, Flanders, NJ) as described before (16). DCs were activated on day 6 of culture by overnight incubation with LPS (1μg/ml).
Adoptive cell transfer and immunization.
3–5 x106 of purified T cells from OT-1 or 2C TCR transgenic mice were injected i.v. into naive C57BL/6 recipient mice. Two-three days later these mice were injected i.v. with 3–5 x 106 Gr-1+ cells. Within one hour after cell transfer mice were immunized s.c. with 100 μg of OVA-derived peptide SIINFEKL or 2C specific peptide SIYRYYGL emulsified in IFA. Ten days later cells from LN and spleens were isolated, re-stimulated in vitro with specific or control peptide and analyzed (Fig. 1A).
Figure 1. Induction of CD8 T cell non-responsiveness by adoptive transfer of Gr-1+ ImC derived from tumor-bearing host.
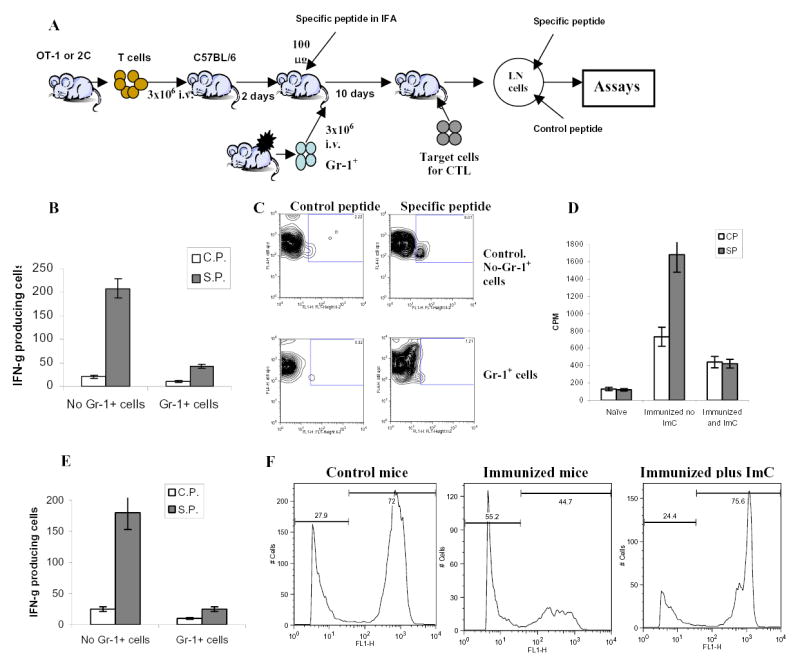
A. Adoptive transfer model.
B. LN cells isolated from mice after adoptive transfer of OT-1 T cells were incubated in triplicates in the presence of specific (SIINFEKL) (S.P.) or control (RAHYNIVTF) (C.P.) peptides for 24 h. Number of IFN-γ producing cells was scored in ELISPOT assay. Results presented as Mean±SD. Six experiments with the same results were performed.
C. Antigen specific IL-2 production. LN cells isolated from mice as described above were cultured in the presence of specific or control peptide for 24 hr. Cells were then labeled with anti-CD8-APC and anti-Valpha2- PE antibodies, and then, fixed, permeabilized and stained with anti-IL-2-FITC antibody. The percentage of IL-2 positive cells amongst the CD8+Vα2+ population is shown at the upper corner. Two experiments with the same results were performed.
D. Antigen-specific proliferation. LN cells isolated from mice as described above were cultured for 4 days in triplicates in U-bottom 96-well plates (105 cells per well) in the presence of 10 μg/ml of specific or control peptide. Eighteen hours prior harvesting 1 μCi 3H-thymidine was added in each well and radioactivity was measured on liquid scintillation counter. Results presented as Mean±SD.
E. LN cells isolated from mice after adoptive transfer of 2C T cells were incubated in triplicates in the presence of specific or control peptides for 24 h. Number of IFN-γ producing cells was evaluated in ELISPOT assay. Results presented as Mean±SD. Four experiments with the same results were performed.
F. Cytotoxic activity in vivo. Splenocytes from syngeneic C57BL/6 mice loaded with fluorescent dye CFSE were used as targets. One group of splenocytes was pulsed for 2 hr with 10 μg/ml of the specific peptide (SIINFEKL), washed and loaded with 10 μM CFSE. The other group was pulsed with the control peptide (RAHYNIVTF) and loaded with 1 μM CFSE. Cells were then mixed together at 3:1 ratio and injected i.v. into mice 10 days after immunization and ImC administration as described in Fig. 1A. Thirty-six hours later spleens were collected and CFSE positive cells were evaluated. Left peak on histogram reflects the population of splenocytes pulsed with the control peptide, right peak – cells pulsed with the specific peptide. Two experiments with the same results were performed.
ELISPOT.
The number of IFN-γ producing cells in response to stimulation to the specific or control peptides (10 μg/ml) was evaluated in ELISPOT assay performed as described earlier (12). Each well contained 2 x 105 LN cells. The number of spots was counted in triplicates and calculated using an automatic ELISPOT counter (Cellular Technology, Ltd, Cleveland, OH).
Flow cytometry.
Flow cytometry data were acquired using a FACSCalibur flow cytometer and analyzed with CellQuest software. For intracellular staining, cells were labeled with anti-CD8 and anti-Vα2 antibodies, fixed and permeabilized according to the manufacturer's protocol with a Golgi Stop kit (BD Pharmingen), and then stained for intracellular-IFN-γ 2 or isotype control (BD Pharmingen). The samples were analyzed by flow cytometry.
Results
Gr-1+ ImC from tumor-bearing mice induce T cell non-responsiveness in vivo
To investigate the nature of cells responsible for tumor-induced tolerance we selected the model that utilizes tumor-free mice after adoptive transfer of transgenic T cells and ImC. The goal was to avoid a potential effect of tumor-derived factors on APC and T cells and ascertain that ImC will not be present during the analysis of T-cell function. Our previous study has demonstrated that ImC differentiate into macrophages or DCs within 5 days after their transfer into congeneic tumor-free recipient and Gr-1+CD11b+ ImC became undetectable in LN ((7) and data not shown). Thus, antigen-specific immune response in LN could be assessed without the presence of ImC. H2Kb OT-1 TCR transgenic T cells specific to OVA-derived peptide (SIINFEKL) were adoptively transferred into naïve C57BL/6 mice. After 2–3 days these mice were immunized s.c. with this peptide in IFA. At the time of immunization mice were injected i.v. with PBS (control) or with non-manipulated Gr-1+ ImC isolated from spleens of EL-4 tumor-bearing mice. Ten days later draining lymph nodes were collected and cells were re-stimulated in vitro with cognate or irrelevant control peptides (Fig 1A).
LN cells from control mice demonstrated potent response (IFN-γ, IL-2 production, and proliferation) to the specific antigen, whereas the response of LN cells isolated from mice after adoptive transfer of ImC was either dramatically reduced or completely eliminated (Figs. 1 B, C, and D). To ascertain that these findings are not restricted to only a single experimental model, similar experiments were performed with adoptive transfer of T cells from transgenic 2C mice. Recipient mice were immunized with 2C specific peptide (SIY). As was in case of OT-1 T cells, Gr-1+ImC rendered T-cells from 2C mice non-responsive to the specific peptide (Fig. 1E). Gr-1+ ImC isolated from C3-fibrosarcoma showed the same effect on T cells as ImC from EL-4 tumor (data not shown).
To test the cytotoxic activity of CD8+ T cells in vivo, splenocytes from syngeneic C57BL/6 mice loaded with fluorescent dye CFSE were used as targets. Splenocytes pulsed with the specific peptide (SIINFEKL) were loaded with 10 μM CFSE and splenocytes pulsed with the control peptide were loaded with 1 μM CFSE. Cells were then mixed together at 3:1 ratio and injected i.v. into mice 10 days after immunization and ImC administration as described in Fig. 1A. Thirty-six hours later spleens were collected and the ratio of labeled target cells was calculated. Specific CTLs recognize and kill splenocytes pulsed with the specific but not with the control peptide, which should significantly decrease the ratio of these two target populations. In our experiments in control non-immunized mice the ratio of splenocytes pulsed with specific vs. control peptides was 2.6:1 (Fig. 1F). In mice immunized with the specific peptide the population of splenocytes pulsed with SIINFEKL was significantly reduced and the ratio was markedly decreased to 0.8:1 (Fig. 1F), whereas in immunized mice that received ImC that ratio remained at the original level (3.1:1) (Fig. 1F). Thus, these results showed the induction of antigen specific CTLs in immunized mice. Administration of ImC completely abrogated that CTL activity.
Antigen-specific nature of ImC mediated CD8+ T cell tolerance
Next, we verified that inhibition of T-cell response was due to the presentation of the specific peptide by ImC. Gr-1+ ImC were pulsed in vitro with control or specific peptides and then injected into recipient mice 8 days after the immunization. Mice were sacrificed in 24 hr and LN cells were re-stimulated with control or specific peptides. Administration of ImC loaded with the control peptide did not significantly affect the response of T-cells to immunization. In contrast, ImC loaded with the specific peptide significantly reduced T-cell response (Fig. 2A). In another set of experiments F1 (BALB/cxC57BL/6) mice were used as recipients of OT-1 T cells. Mice were immunized with specific peptide and received ImC at the time of immunization and response was evaluated 10 days later. Two sources of ImC were used in these experiments: EL-4 tumor-bearing C57BL/6 mice or MethA sarcoma-bearing BALB/c mice. If inhibition of T-cell response was MHC class I restricted then ImC from BALB/c mice (H2d) should not suppress the response of OT-1 T cells (H2b) to H2Kb matching peptide. This was indeed the case. No inhibition of T-cell response to the specific peptide was found in immunized mice after administration of Gr-1+ ImC isolated from tumor-bearing BALB/c mice. In contrast, administration of ImC from tumor-bearing C57BL/6 mice resulted in almost a complete block of antigen-specific T-cell response (Fig. 2B). Taken together these results demonstrate that Gr-1+ ImC induce MHC class I restricted antigen-specific tolerance of CD8+ T cells.
Figure 2. Antigen-specific nature of ImC mediated CD8+T-cell tolerance.
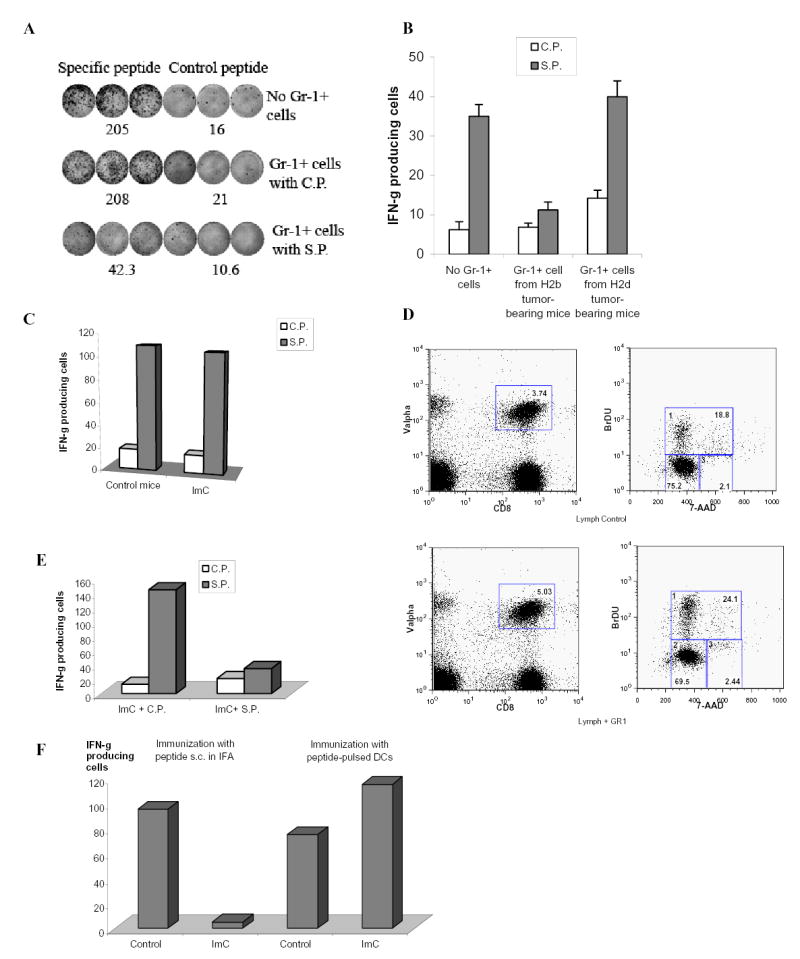
A. Naïve mice recipients of OT-1 T cells were immunized with SIINFEKL. Gr-1+ cells were isolated from spleens of EL-4 tumor-bearing mice, pulsed with specific or control peptides and injected i.v. (3x106 cells) 8 days after immunization. LNs were collected 24 hr later. Cells were re-stimulated with control or specific peptides and IFN-γ production was evaluated in ELISPOT assay. Actual results of one experiment are shown. Numbers represent average of three wells. Three experiments with the same results were performed.
B. OT-1 transgenic T cells (H2b) were transferred to F1 hybrid (C57BL/6(H2b) x BALB/c (H2d)) mice. Three days later these mice have received Gr-1+ cells from either EL-4 tumor-bearing C57BL/6 mice (H2b) or from MethA sarcoma-bearing BALB/c mice (H2d) and at the same time were immunized with SIINFEKL peptide. After 10 days LN cells from these mice were tested for production of IFN-γ in response to specific or control peptide using ELISPOT assay. Two independent experiments with the same results were performed.
C. OT-1 T cells (3x106) were adoptively transferred into naïve recipient mice. Two days later these mice were injected with Gr-1+ cells (3x106) from EL-4 tumor-bearing mice and immunized s.c. with SIINFEKL peptide. Three days later LN cells were collected and stimulated with control or specific peptide as described in Fig. 1B. The number of IFN-γ producing cells was evaluated in ELISPOT assay.
D. OT-1 T cells (3x106) were adoptively transferred into naïve recipient mice. Two days later these mice were injected with Gr-1+ cells (3x106) from EL-4 tumor-bearing mice and immunized s.c. with SIINFEKL peptide. Three days after immunization mice were injected i.p. with 1mg of BrdU. The next day regional LNs of immunized mice were isolated and cells were stained with anti-CD8-APC antibody, anti-Valpha2-PE antibody, anti-BrDU-FITC antibody and 7 AAD. CD8+Valpha+ cells were gated (left two panels) and BrDU positive proliferating cells were evaluated (gate # 1 in two right panels).
E. OT-1 T cells (3x106) were adoptively transferred into naïve recipient mice. Two days later these mice were immunized s.c. with SIINFEKL peptide. Gr-1+ ImC isolated from EL-4 tumor-bearing mice were loaded (2 hr incubation at 37°C) with control (C.P.) or specific (S.P.) peptides, washed and then injected i.v. (3x106 cells). Three days later LN cells were collected and the number of IFN-γ producing cells was evaluated in ELISPOT assay.
F. After adoptive transfer of OT-1 cells mice were immunized with either s.c. injection of specific peptide (SIINFEKL) with IFA or with 2x105 DCs pulsed with specific peptide. ImC (3x106 cells) were injected i.v. at the time of immunization. LN were collected 10 days after immunization and cells were stimulated with control or specific peptides as described above. The number of IFN-γ producing cells was measured in ELISPOT assay. To simplify the presentation of the data the values of IFN-γ producing cells after stimulation with control peptide were subtracted from the values of IFN-γ producing cells after stimulation with specific peptide.
The development of T-cell tolerance required time. If LN cells were collected 3 days after immunization and ImC administration no defect in antigen-specific CD8+T-cell response was found (Fig. 2C). In contrast to cells isolated 10 days after ImC administration - antigen-specific T cells demonstrated normal level of proliferation (Fig. 2D). We asked whether this effect was caused by time requirement for ImC to pick up process and antigen. To test this hypothesis Gr-1+ ImC were loaded with control or specific peptide in vitro and then administered 3 days after immunization of mice. ImC loaded with specific peptide completely abrogated antigen-specific CD8+ T-cell response (Fig. 2E). This data confirm critical role of antigen availability for ImC mediated T-cell non-responsiveness. To investigate this issue further, we used for immunization the peptide loaded on DCs. In contrast to immunization with free peptide in IFA, in this case ImC would not have access to a substantial amount of free peptide. Mice after adoptive transfer of OT-1 cells were immunized with either peptide-pulsed DCs or with s.c. injection of the peptide in IFA. ImC were administrated i.v. at the time of immunization. Ten days later the response of CD8+T cells to the specific peptide was measured in ELISPOT assay. ImC failed to induce tolerance in mice immunized with peptide-pulsed DCs (Fig. 2F).
CD8+ T cells tolerance is mediated by tumor-derived ImC but not their control counterparts or progenies
Gr-1+ myeloid cells are intrinsic part of normal myelopoiesis. Therefore, we compared the ability of ImC isolated from tumor-free and tumor-bearing hosts to induce T-cell tolerance. In contrast to ImC from tumor-bearing mice, Gr-1+ ImC from naïve mice were not able to inhibit T-cell response (Fig. 3A). DCs are professional bone marrow derived APC. Recent data demonstrated that immature DCs could induce T-cell tolerance in tumor-free host (17). We asked whether DCs directly isolated from tumor-bearing mice could induce T-cell tolerance. CD11c+ DCs isolated from spleens of EL-4 tumor-bearing mice failed to induce T-cell tolerance (Fig. 3A). It was plausible that in tumor-free environment DCs quickly differentiate and lose the ability to suppress T-cells. Therefore we modified the experimental conditions. CD11c+ DCs isolated from tumor-bearing mice were pulsed with SIINFEKL peptide and then injected into immunized mice on day 8 after the immunization. LN cells were collected 24 hr later and stimulated with control or specific peptide. Under these conditions ImC demonstrated dramatic inhibition of CD8+ T cell response (Fig. 2A). However, CD11c+DCs again failed to suppress T cells (Fig. 3B). Although these experiments cannot rule out the possible role of DC in induction of tumor-associated CD8+T-cell tolerance, they indicate that direct effect of DCs on induction of tolerance at least is not as strong as that of ImC.
Figure 3. The nature of cells responsible for ImC mediated CD8+ T cell tolerance.
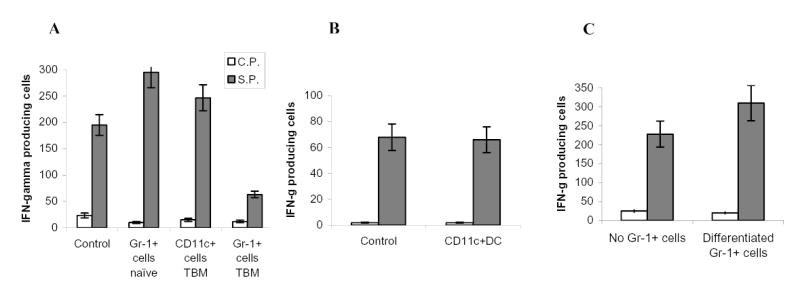
A. Experiments were performed as described in Fig. 1A. Gr-1+ cells were isolated from spleens of naïve tumor-free mice or EL-4 tumor-bearing mice. CD11c+ cells were isolated from spleens of EL-4 tumor-bearing mice. Ten days later LN cells were isolated, re-stimulated with specific (S.P.) or control (C.P.) peptide and the number of IFN-γ producing cells was analyzed by ELISPOT assay. Results presented as Mean±SD. Two experiments with the same results were performed.
B. OT-1 T cells were transferred i.v. into naïve C57BL/6 mice. Mice were immunized with SIINFEKL. CD11c+ cells were isolated from EL-4 tumor-bearing mice, pulsed with the specific peptide, and administered i.v. into mice 8 days after immunization. LN cells were collected 24 hr later and stimulated with either control or specific peptide. The number (Mean ± SD) of IFN-γ producing cells per 106 LN cells is shown.
C. Gr-1+ cells isolated from tumor-bearing mice were transferred first into naïve recipient mice. Three days later these mice received OT-1 T cells and 2 days after that, mice were immunized with SIINFEKL peptide. LN cells were collected 10 days after immunization and were re-stimulated with control or specific peptides and the number of IFN-γ producing cells was analyzed in ELISPOT assay. Results presented as Mean±SD.
Tumor-bearing mice derived ImC differentiate into mature myeloid cells within 5 days after adoptive transfer into congenic tumor-free recipient (7). To investigate the role of ImC progeny in the induction of T-cell tolerance Gr-1+ cells isolated from EL-4 tumor-bearing mice were transferred i.v. into naïve C57BL/6 recipient 5 days before the immunization. Under these experimental conditions ImC did not affect peptide-specific immune response (Fig.3C). Thus, it appears that the ability to induce T-cell non-responsiveness was restricted primarily to the population of ImC.
The mechanism of CD8+ T-cell tolerance induced by Gr-1+ myeloid cells
T-cell tolerance could be caused by the deletion of antigen-specific T cells or by the induction of T-cell anergy. We traced the presence of antigen-specific transgenic CD8+ cells expressing TCRVα2 in mice after adoptive transfer. Ten days after immunization and ImC administration, mice treated with Gr-1+ ImC had the same proportion of CD8+Vα2+ as control mice (Fig. 4A). Next, we asked whether T-cell non-responsiveness was limited only to the specific antigen. Cells collected from draining lymph nodes 10 days after immunization were re-stimulated with either specific peptide or anti-CD3 antibody. Production of IFN-γ was measured using intracellular staining inside the population of antigen-specific CD8+Vα2+ cells. Re-stimulation of LN cells with the specific peptide resulted in a significant increase in the proportion of IFN-γ positive antigen-specific cells, whereas no such increase was observed in the cells isolated from mice that received ImC (Fig. 4B). However, cells from both control and ImC treated mice responded equally well to the non-specific stimulation with anti-CD3 antibody (Fig. 4B).
Figure 4. The mechanisms of CD8+ T-cell tolerance induction by ImC.
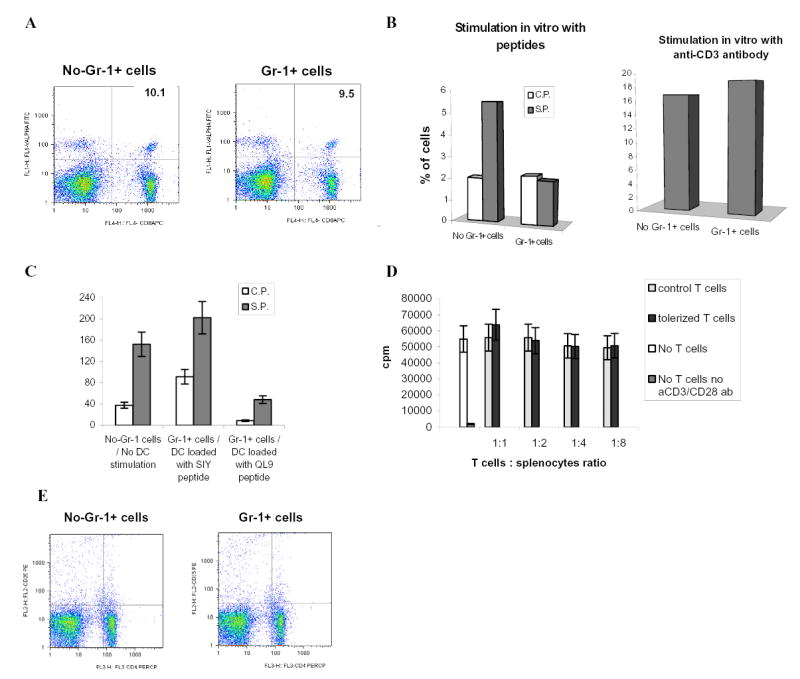
A. Adoptive transfer of OT-1 cells, Gr-1+ cells and immunization with peptide was done as shown above in Fig.1A. LN cells were isolated on day 10 after immunization and percentage of antigen-specific CD8+Valpha2+ T cells was determined by flow cytometry. A representative experiment of three performed is shown.
B. Adoptive transfer of OT-1 cells, Gr-1+ cells and immunization with peptide was done as shown in Fig.1A. Draining LNs were re-stimulated in vitro with specific (S.P.) or control (C.P.) peptides (10 μg/ml) or anti-CD3 antibody (1μg/ml) for 48 h. Production of IFN-γ was measured using intracellular staining within antigen-specific CD8+Vα2+ T cells. Typical results of one of two performed experiments are shown.
C. 2C T cells (3 x 106) were adoptively transferred i.v. into naive C57BL/6 recipients followed by 3 days later immunization with 2C specific peptide SIYRYYGL (100 μg) and injection of Gr-1+ cells (3 x 106) from EL-4 tumor-bearing mice. DCs generated in vitro from control C57BL/6 mice were activated with LPS, pulsed with either specific (SIY) or control (QL9) peptides, and injected s.c. (4x105) into mice 10 days after first immunization. In 7 days after that mice were sacrificed, LN cells isolated and re-stimulated with specific (S.P.) or control (C.P.) peptides. The number of IFN-γ producing cells was measured by ELISPOT. Results presented as Mean±SD.
D. T cells isolated from OT-1 mice were transferred i.v. into naïve C57BL/6 mice. Mice were immunized with SIINFEKL together with the transfer of Gr-1+ isolated from spleens of EL-4 tumor-bearing mice. Ten days later LN cells were collected and T cells were isolated and added in triplicates at indicated ratio to 105 splenocytes isolated from naïve mice and incubated for 4 days with 0.5 μg/ml anti-CD3 antibody and 0.1 μg/ml anti-CD28 antibody. Cell proliferation was evaluated using 3H-thymidine uptake. Results presented as Mean±SD. Control T cells – T cells isolated from immunized mice without transfer of Gr-1+ImC, tolerized T cells – T cells isolated from mice after adoptive transfer of Gr-1+ cells.
E. Adoptive transfer of OT-1 cells, Gr-1+ cells and immunization with peptide was done as shown in Fig.1A. LN cells were isolated on day 9 after immunization and percentage of CD4+CD25+ cells was determined by flow cytometry. Typical results of one of two performed experiments are shown.
We investigated the possibility of reversing this T-cell tolerance in vivo. 2C T cells were adoptively transferred into C57BL/6 mice. All mice were immunized s.c. with the specific peptide and ImC were administered at the time of immunization. Ten days later mice were immunized s.c. with LPS activated DCs generated from control C57BL/6 mice. DCs were loaded in vitro with either specific (SIY) or control (QL9) peptides. Seven days after the immunization with DCs, LN were isolated and cells were re-stimulated with specific or control peptides. Control mice (no ImC administration) immunized with specific SIY peptide retained strong response to this specific peptide 17 days after initial immunization. Mice that received Gr-1+ ImC and immunized with control DCs (QL9) had very low level of T-cell response to the specific peptide indicating the persistence of T-cell tolerance in these mice (Fig. 4C). However, immunization of mice with DCs loaded with the specific peptide completely restored antigen-specific T-cell response. This data indicate that at least in this model T-cell tolerance could be overcome with activated DCs.
Naturally arising CD4+CD25+ regulatory T cells (Treg) play an important role in induction and maintenance of T-cell tolerance. It was reported that their presence is increased in tumor-bearing hosts (rev. in (18). We investigated whether ImC could induce Treg. The main functional characteristic of Treg is their ability to suppress other T cells. T cells were isolated on day 9 after adoptive transfer of ImC and were mixed together with naïve syngeneic splenocytes at different ratios. Cells were then stimulated with anti-CD3/CD28 mAb. Even at 1:1 ratio T cells from ImC treated (tolerized mice) mice did not affect anti-CD3/CD28 antibodies-induced proliferation of syngeneic T cells (Fig. 4D). Similar results were obtained with cells isolated on day 4 after adoptive transfer (data not shown). Furthermore, no differences were found in the presence of CD4+CD25+ T cells in spleens of control or tolerant mice (Fig. 4E). Taken together these data argue against the possibility that ImC might induce tolerance via up-regulation of Treg.
Gr-1+ myeloid cells induce tolerance to tumor-associated antigens in tumor-bearing mice
The important question is whether the above described findings are directly relevant to the situation in tumor-bearing mice. First, we investigated whether ImC can migrate to a tumor site. Tumor-bearing mice derived ImC were labeled with cell tracker orange CMTMR and injected intravenously into syngeneic EL-4 tumor-bearing mice. Brightly fluorescent ImC were readily detectable in tumor tissues 16 hr after administration (Fig. 5A). Their number slightly decreased with time from 9.4 cells per field after 16 hr to 5.1 cells after 48 hr (Fig. 5A). Next, we tested the ability of ImC to pick up soluble protein. OVA protein, conjugated with FITC was injected i.p. to EL-4 tumor-bearing mice. Two hours later, a significant proportion (>70%) of Gr-1+CD11b+ ImC in spleens became FITC positive, indicating that they were able to take-up soluble OVA (Fig.5B).
Figure 5. ImC ability to induce CD8+ T-cell tolerance in tumor-bearing mice.
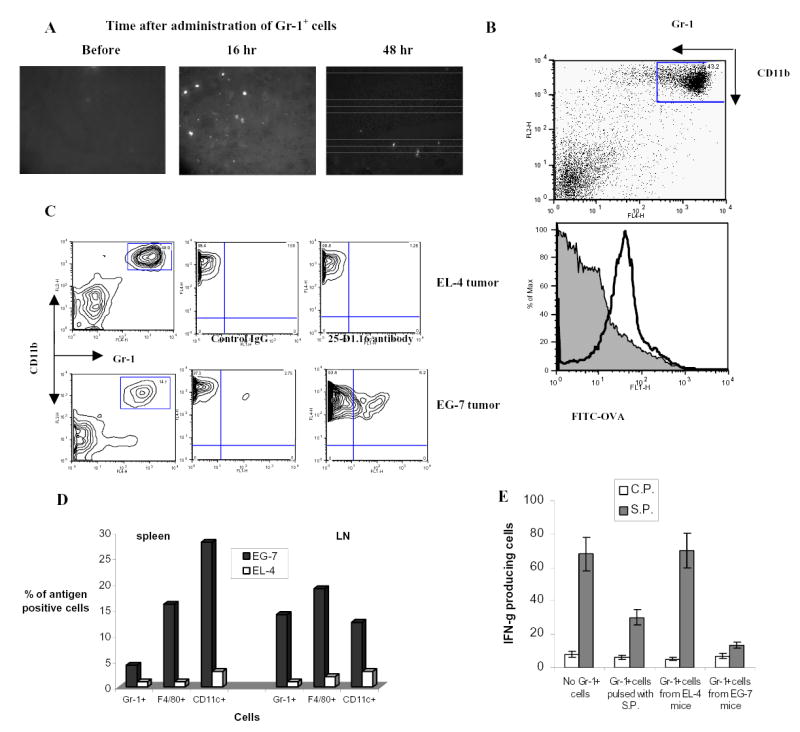
A. Cell detection in tumor tissues. Gr-1+ cells were labeled with 20 μM cell tracker orange 5-(and-6)-(((4-chloromethyl) benzoyl) amino) tetramethylrhodamine (CMTMR, Molecular Probes, Eugene, OR) and then injected (5x106/mouse) intravenously. Tumor tissues were taken out at different time points and snap-frozen in tissue freezing media (Triangle Biomedical Sciences, Durham, NC) at −80 °C. Frozen tissue sections were examined by fluorescence microscopy using a 540 nm filter. Labeled cells were counted in 10 fields at the total magnification x200.
B. ImC take up soluble protein in vivo. Tumor-bearing mice were injected i.p. with 0.5 mg of OVA-FITC. Two hours later spleens were isolated and percentage of OVA-positive cells was determined within Gr-1+CD11b+ population of ImC. Typical results of one of two performed experiments are shown.
C and D. C57BL/6 mice were inoculated s.c. with EG-7 or parent EL-4 tumor cells. Two weeks later when tumor reached 1.5 cm in diameter spleens and LNs were isolated and used in further experiments. B, C. Splenocytes and LN cells were stained with anti-Gr-1-APC and CD11b+-PE antibodies (Gr-1+), anti-CD11c-APC antibody (CD11c+), or anti-F4/80 antibody (F4/80+) in combination with anti-OVA 25-D1.16 antibody followed by staining with anti-mouse IgG-FITC. OVA-epitope positive cells were calculated after subtraction of the proportion of cells stained positive with isotype control IgG from cells stained positive with 25-D1.16 antibody. C. The result of typical experiment is shown (splenocytes). D. The proportion of OVA positive cells was calculated within each population of cells. Three experiments with similar results were performed.
E. Recipients of OT-1 cells were immunized with SIINFEKL as described above. Gr-1+ cells were isolated from spleens of EL-4 or EG-7 tumor-bearing mice and these non-manipulated cells were injected into mice 8 days after immunization. As positive control Gr-1+ cells isolated from EL-4 tumor-bearing mice and pulsed with specific peptide were used. LN cells were collected 24 hr later and re-stimulated in triplicates with control (C.P.) or specific (S.P.) peptide. The number of IFN-γ producing cells was calculated per 106 LN cells.
The ability of Gr-1+ myeloid cells to pick up and process TAA in vivo was addressed using tumors containing model antigen. EG-7 tumor cells derived from EL-4 lymphoma by stable transfection with OVA or parent EL-4 tumor cells were inoculated s.c. into C57BL/6 mice. Two weeks later when tumor size reached 1–1.5 cm in diameter APC in spleens and LN were analyzed using flow cytometry. The proportion of the cells expressing OVA-derived epitope was calculated within the populations of CD11c+ DCs, F4/80+ macrophages, or Gr-1+CD11b+ ImC using 25-D1.16 antibody that recognizes H2-Kb-OVA257–264 complex. In EG-7 tumor-bearing mice about 5% of ImC in spleen and about 14 % in LN were recognized by 25-D1.16 antibody (Fig. 5C and D). Considerably higher proportion of F4/80+ and CD11c+ cells in spleens of EG-7 tumor-bearing mice expressed OVA-derived epitope, whereas in LN their proportion was at the same level as of ImC (Fig. 5D). This data indicates that ImC can capture and process TAA. Gr-1+ cells isolated from spleens of EL-4 or EG-7 tumor-bearing mice were injected i.v. into immunized mice as described in Fig. 2A. The response of LN cells to the specific peptide was evaluated 24 hr later. ImC cells isolated from EL-4 tumor-bearing mice did not affect SIINFEKL-specific CD8+T-cell response, whereas ImC cells isolated from EG-7 mice almost completely abrogated that response (Fig. 5E). These data indicate that ImC in tumor-bearing mice can process TAA and suppress single epitope-specific T cells.
Effect of ImC on tumor growth
We asked whether spontaneous tumor rejection could be associated with accumulation of ImC. In these experiments we used methylcholantrene-induced (MethA) sarcoma, which if inoculated at cell dose less than 3x105 per mouse is characterized by a significant rate of spontaneous rejection (> 30% of mice usually reject tumor within 10 days after tumor inoculation). Our experiments demonstrated close association between tumor rejection and accumulation of ImC (Fig. 6A, B). On day 12 after tumor inoculation mice with progressing tumor had significantly higher level of ImC than mice with regressing tumors (Fig. 6B). As was shown in previous studies accumulation of ImC became significant on relatively late stages of tumor growth. What effect ImC may have during early stages of tumor growth? MethA sarcoma or CT-26 colon adenocarcinoma cells were inoculated s.c. into BALB/c mice. On day 5, when tumors became palpable mice were injected i.v. with 4x106 unmodified Gr-1+ImC. In tumor-bearing mice ImC are generated in bone marrow and spleens and migrate to tumor site via circulation. Therefore ImC were administered not to the site of tumor but intravenously. ImC isolated from CT-26 tumor-bearing mice were transferred into MethA sarcoma-bearing mice and vice versa. Tumor-bearing mice that have received PBS comprised control groups. Within a week after ImC administration differences in tumor size became statistically significant and 10 days after ImC administration the tumor size in mice that received ImC was more than 3-fold larger than in control mice (Fig. 6C,D). The differences became smaller one week later, apparently because of accumulation of endogenous ImC in control tumor-bearing mice. These data indicate that ImC from circulation can promote tumor growth in different tumor models.
Figure 6. Effect of ImC on tumor growth.
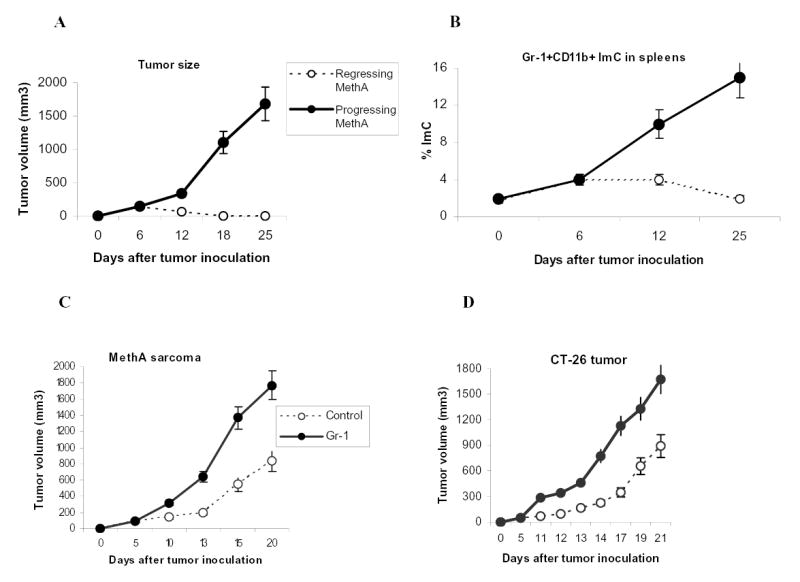
A. MethA sarcoma cells were inoculated s.c. (3x105 cells per mice) into 12 BALB/c mice. Tumor volume was monitored as indicated and calculated as follows: Tumor Volume= 0.5 (length) (breadth) (breadth). Five mice eventually rejected tumor (regressing MethA) and 7 had progressing tumor (progressing MethA). Tumor size for each group was traced back to day 6 after tumor inoculation.
B. Splenocytes were labeled with anti-Gr-1-APC and anti-CD11b-PE antibodies and the percentage of double-positive cells was evaluated by flow cytometry. Background level of Gr-1+CD11b+ ImC was established in naïve tumor-free mice (designates as day 0). On day 6 after MethA sarcoma inoculation the presence of ImC was evaluated in 6 mice. All mice showed the same slightly elevated level of ImC. On day 12, mice were split into two groups (with progressing and regressing tumors). Three mice per group were evaluated. The same groups were analyzed on day 25.
C and D. MethA sarcoma cells (C) or CT-26 tumor cells (D) (both 5x105 cells per mouse) were inoculated s.c. into BALB/c mice. Five days later mice were split into two groups (4 mice per group for each tumor model) with equal tumor volume. Control groups have received i.v. injection of 0.2 ml PBS, Gr-1 groups have received 4x106 Gr-1+ ImC derived either from CT-26 tumor-bearing mice (injected into MethA sarcoma-bearing mice) or from MethA tumor-bearing mice (injected into CT-26 tumor-bearing mice). Tumor volume was monitored as indicated. Mean ± SD are shown.
Discussion
Studies during recent years provide clear understanding that host immunity plays a significant role in control of tumor development and progression(1, 19). At the same time, tumor has developed elaborate mechanisms to avoid recognition and elimination by the immune system(20). Altered function of antigen-specific T cells is one of the major factors responsible for tumor escape. T cell ignorance apparently plays the most critical role at early stages of tumor development (21, 22), whereas acquired CD4+ and CD8+ T-cell tolerance to TAA is more prominent at later stages(2, 3, 23–27). Previous studies have established that induction of CD4+ T-cell tolerance in cancer requires tumor antigen capture and processing by bone marrow-derived APCs (3, 28). However, the nature of these cells remained unknown. Here, we focused on recently a defined population of Gr-1+CD11b+ myeloid cells. They exerted immunosuppressive activity in different in vitro tests and their removal from tumor-bearing hosts improved immune response (8, 10, 12–14, 16, 29–31). Suppressive activity of freshly isolated ImC was limited to CD8+ T cells and mediated via MHC class I primarily due to the fact that in a number of tumor models ImC don’t express MHC class II (12, 32). These facts form a basis for our hypothesis that tumor-induced CD8+T cell-tolerance might be mediated by Gr-1+ ImC.
Testing this hypothesis represented a technical challenge since presence of tumors stimulate generation of ImC and affect APC, which would make analysis of the role of the specific cell populations very difficult. Gr-1+ cells are part of a normal pathway of myeloid cell differentiation and elimination of these cells with anti-Gr-1 antibody would affect this process with the consequences that will be hard to interpret. Use of pharmacological agents like all-trans-retinoic acid resulted in only partial elimination of these cells(16). Therefore, we selected the model that utilizes tumor-free mice after adoptive transfer of transgenic T cells and ImC. Our previous studies have shown that LN from tumor-bearing mice contained substantial proportion of Gr-1+ ImC. In contrast, ImC differentiate into macrophages or DCs within 5 days after their transfer into congeneic tumor-free recipient (7) and Gr-1+CD11b+ ImC were undetectable in LN ((7) and data not shown). This provides us with the opportunity to assess antigen-specific immune response in LN without the presence of ImC.
Our experiments using adoptive transfer of OT-1 and 2C transgenic T-cells into tumor-free recipients have demonstrated that administration of tumor-derived ImC dramatically inhibit CD8+ T cell response to the specific antigen. This effect was observed 10 days after the administration of specific peptide and ImC, when ImC were no longer present during the in vitro re-stimulation of CD8+T cells. This non-responsiveness was antigen-specific and restricted to MHC class I. Taken together these data indicate that ImC induced CD8+ T-cell tolerance. CD8+ T cells retained the ability to respond to non-specific stimulus (anti-CD3 antibody) and to stimulation with functionally competent specific antigen-loaded DCs. These data indicate that ImC induce state of anergy in antigen-specific T cells.
Gr-1+ myeloid cells from tumor-free mice have failed to produce this effect. This was consistent with previous observations in vitro where T-cell suppressive activity was restricted only to ImC derived from tumor-bearing but not from control mice(32). One of the possible mechanisms of this effect could be that ImC from tumor-bearing mice have higher arginase I activity and produce substantially higher level of ROS than their control counterparts (32). Important role of ROS in T-cell defects has been demonstrated previously (33, 34). Several studies demonstrated an important role of arginase I in immune suppression by myeloid cells, which is mediated by depletion of L-arginine (35–37). Both ROS and L-arginin depletion can down-regulate CD3ζ expression, which is critically important for signaling in T-cells.(33,38). It also may affect a down-stream adaptor proteins mediating T-cell signaling like ZAP-70 or p56lck. ROS can also affect the affinity of antigen-specific TCR. This could explain the specificity of the tolerance induced by ImC. In addition to the effect of arginase I and ROS, ImC may affect CD8+ T cells via inhibitory molecules B7-H1, and B7-H4. In this study we could not address all questions regarding the molecular mechanisms of ImC mediated T-cell tolerance. All these potential mechanisms are subjects of ongoing investigation.
Our data with functional maturation of ImC indicate that their progeny did not render T-cells unresponsive. It is important to point out that differentiation of ImC in tumor-bearing host is different from that in naïve mice(7). Therefore it was possible that the progeny of ImC in tumor-bearing mice might have different functional features. We have specifically focused on CD11c+ DC population since DCs are most potent APC, the presence of immature DCs is increased in tumor-bearing hosts (6), and recent data have demonstrated that immature DCs in steady state loaded with antigen could induce T-cell tolerance in tumor-free host(17). We isolated CD11c+ DCs from tumor-bearing mice and immediately administered them to immunized mice instead of ImC. We hypothesized that if tumor-associated DCs were able to suppress T-cell response it would be detectable under these experimental conditions. However, CD11c+DCs derived from tumor-bearing mice failed to induce T-cell tolerance or suppress T-cell response in vivo. These experiments cannot completely rule out the possible role of DC in the induction of tumor-associated CD8+T-cell tolerance. However, they indicate that direct effect of DCs on induction of tolerance is not as strong as of ImC.
In this study activated DCs were used as a tool to overcome T-cell tolerance in the absence of Gr-1+ ImC. The fact that DCs can overcome T-cell tolerance is encouraging from the standpoint of cancer immunotherapy. However, it underscores the important point that vaccination effort in tumor-bearing hosts will not be fully successful until ImC will be eliminated or their presence substantially reduced.
It is unlikely that TAA are presented to ImC in an already processed form. ImC probably encounter those antigens in the form of soluble proteins or in association with dying tumor cells. Our experiments with OVA-expressing tumor-bearing mice demonstrated that ImC were able to migrate into tumor site, pick-up tumor associated antigens, process, and present epitopes on the cell surface and induce antigen-specific T-cell non-responsiveness. These facts were confirmed in two other tumor models (MethA sarcoma and CT-26 colon adenocarcinoma). Previous studies have demonstrated that accumulation of ImC in these tumor-bearing mice did not start until 10 days after tumor inoculation (7, 32). We proposed that if our hypothesis is correct then administration of ImC shortly after tumor inoculation should affect the rate of tumor growth. And indeed ImC injected i.v. 5 days after s.c. inoculation of tumor significantly increased tumor growth in mice. This difference starts disappearing after 2 weeks apparently when accumulation of endogenous ImC took place.
Thus, this study, for the first time, has provided evidence in favor of a role of ImC in induction of CD8+T cell tolerance in cancer. This not only helps to clarify the component responsible for this important phenomenon but also suggests direct approach to overcome this tolerance and improve the effect of cancer vaccines.
Acknowledgments
We thank Dr. M. Androlewitz with help in detection of OVA-derived epitope on cell surface.
Footnotes
This work has been supported in part by the Flow Cytometry Core at the H. Lee Moffitt Cancer Center. This work was supported by NIH grant CA84488 to DIG
This is an author-produced version of a manuscript accepted for publication in The Journal of Immunology (The JI). The American Association of Immunologists, Inc. (AAI), publisher of The JI, holds the copyright to this manuscript. This manuscript has not yet been copy edited or subjected to editorial proofreading by The JI; hence it may differ from the final version published in The JI (online and in print). AAI (The JI) is not liable for errors or omissions in this author-produced version of the manuscript or in any version derived from it by the United States National Institutes of Health or any other third party. The final, citable version of record can be found at www.jimmunol.org.”
References
- 1.Pardoll D. Does the immune system see tumors as foreign or self? Annu Rev Immunol. 2003;21:807. doi: 10.1146/annurev.immunol.21.120601.141135. [DOI] [PubMed] [Google Scholar]
- 2.Sotomayor EM, Borrello I, Tubb E, Rattis FM, Bien H, Lu Z, Fein S, Schoenberger S, Levitsky HI. Conversion of tumor-specific CD4+ T-cell tolerance to T-cell priming through in vivo ligation of CD40. Nat Med. 1999;5:780. doi: 10.1038/10503. [DOI] [PubMed] [Google Scholar]
- 3.Cuenca A, Cheng F, Wang H, Brayer J, Horna P, Gu L, Bien H, Borrello IM, Levitsky HI, Sotomayor EM. Extra-lymphatic solid tumor growth is not immunologically ignored and results in early induction of antigen-specific T-cell anergy: dominant role of cross-tolerance to tumor antigens. Cancer Res. 2003;63:9007. [PubMed] [Google Scholar]
- 4.Bronte V, Serafini P, Appoloni E, Zanovello P. Tumor-induced immune dysfunctions caused by myeloid suppressor cells. J Immunoth. 2001;24:431. doi: 10.1097/00002371-200111000-00001. [DOI] [PubMed] [Google Scholar]
- 5.Kusmartsev S, Gabrilovich DI. Immature myeloid cells and cancer-associated immune suppression. Cancer Immunol Immunoth. 2002;51:293. doi: 10.1007/s00262-002-0280-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gabrilovich D. The mechanisms and functional significance of tumour-induced dendritic-cell defects. Nat Rev Immunol. 2004;4:941. doi: 10.1038/nri1498. [DOI] [PubMed] [Google Scholar]
- 7.Kusmartsev S, Gabrilovich DI. Inhibition of myeloid cell differentiation in cancer: The role of reactive oxygen species. J Leukoc Biol. 2003;74:186. doi: 10.1189/jlb.0103010. [DOI] [PubMed] [Google Scholar]
- 8.Danna EA, Sinha P, Gilbert M, Clements VK, Pulaski BA, Ostrand-Rosenberg S. Surgical removal of primary tumor reverses tumor-induced immunosuppression despite the presence of metastatic disease. Cancer Res. 2004;64:2205. doi: 10.1158/0008-5472.can-03-2646. [DOI] [PubMed] [Google Scholar]
- 9.Nefedova Y, Huang M, Kusmartsev S, Bhattacharya R, Cheng P, Salup R, Jove R, Gabrilovich D. Hyperactivation of STAT3 is involved in abnormal differentiation of dendritic cells in cancer. J Immunol. 2004;172:464. doi: 10.4049/jimmunol.172.1.464. [DOI] [PubMed] [Google Scholar]
- 10.Terabe M, Matsui S, Park JM, Mamura M, Noben-Trauth N, Donaldson DD, Chen W, Wahl SM, Ledbetter S, Pratt B, Letterio JJ, Paul WE, Berzofsky JA. Transforming growth factor-beta production and myeloid cells are an effector mechanism through which CD1d-restricted T cells block cytotoxic T lymphocyte-mediated tumor immunosurveillance: abrogation prevents tumor recurrence. J Exp Med. 2003;198:1741. doi: 10.1084/jem.20022227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bronte V, Wang M, Overwijk W, Surman D, Pericle F, Rosenberg SA, Restifo NP. Apoptotic death of CD8+ T lymphocytes after immunization: induction of a suppressive population of Mac-1+/Gr-1+ cells. J Immunol. 1998;161:5313. [PMC free article] [PubMed] [Google Scholar]
- 12.Gabrilovich DI, Velders M, Sotomayor E, Kast WM. Mechanism of immune dysfunction in cancer mediated by immature Gr-1+ myeloid cells. J Immunol. 2001;166:5398. doi: 10.4049/jimmunol.166.9.5398. [DOI] [PubMed] [Google Scholar]
- 13.Melani C, Chiodoni C, Forni G, Colombo MP. Myeloid cell expansion elicited by the progression of spontaneous mammary carcinomas in c-erbB-2 transgenic BALB/c mice suppresses immune reactivity. Blood. 2003;102:2138. doi: 10.1182/blood-2003-01-0190. [DOI] [PubMed] [Google Scholar]
- 14.Li Q, Pan PY, Gu P, Xu D, Chen SH. Role of immature myeloid Gr-1+ cells in the development of antitumor immunity. Cancer Res. 2004;64:1130. doi: 10.1158/0008-5472.can-03-1715. [DOI] [PubMed] [Google Scholar]
- 15.Porgador A, Yewdell JW, Deng Y, Bennink JR, Germain RN. Localization, quantitation, and in situ detection of specific peptide-MHC class I complexes using a mAb. Immunity. Immunity. 1997;6:715. doi: 10.1016/s1074-7613(00)80447-1. [DOI] [PubMed] [Google Scholar]
- 16.Kusmartsev S, Cheng F, Yu B, Nefedova Y, Sotomayor E, Lush R, Gabrilovich DI. All-trans-retinoic acid eliminates immature myeloid cells from tumor-bearing mice and improves the effect of vaccination. Cancer Res. 2003;63:4441. [PubMed] [Google Scholar]
- 17.Bonifaz L, Bonnyay D, Mahnke K, Rivera M, Nussenzweig MC, Steinman RM. Efficient targeting of protein antigen to the dendritic cell receptor DEC-205 in the steady state leads to antigen presentation on major histocompatibility complex class I products and peripheral CD8+ T cell tolerance. J Exp Med. 2002;196:1627. doi: 10.1084/jem.20021598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zou W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nature Reviews Cancer. 2005;5:263. doi: 10.1038/nrc1586. [DOI] [PubMed] [Google Scholar]
- 19.Dunn GP, Old LJ, Schreiber RD. The three Es of cancer immunoediting. Annu Rev Immunol. 2004;22:329–360. doi: 10.1146/annurev.immunol.22.012703.104803. [DOI] [PubMed] [Google Scholar]
- 20.Gabrilovich D, Pisarev V. Tumor escape from immune response: mechanisms and targets of activity. Curr Drug Targets. 2003;4:525. doi: 10.2174/1389450033490849. [DOI] [PubMed] [Google Scholar]
- 21.Speiser DE, Miranda R, Zakarian A, Bachmann MF, McKall-Faienza K, Odermatt B, Hanahan D, Zinkernagel RM, Ohashi PS. Self antigens expressed by soild tumors do not efficiently stimulate naive or activated T cells: implications for immunotherapy. J Exp Med. 1997;186:645. doi: 10.1084/jem.186.5.645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Spiotto MT, Yu P, Rowley DA, Nishimura MI, Meredith SC, Gajewski TF, Fu YX, Schreiber H. Increasing tumor antigen expression overcomes "ignorance" to solid tumors via crosspresentation by bone marrow-derived stromal cells. Immunity. 2002;17:737. doi: 10.1016/s1074-7613(02)00480-6. [DOI] [PubMed] [Google Scholar]
- 23.Yang Y, Huang CT, Huang X, Pardoll DM. Persistent Toll-like receptor signals are required for reversal of regulatory T cell-mediated CD8 tolerance. Nat Immunol. 2004;5:508. doi: 10.1038/ni1059. [DOI] [PubMed] [Google Scholar]
- 24.Bogen B. Peripheral T cell tolerance as a tumor escape mechanism: deletion of CD4+ T cells specific for a monoclonal immunoglobulin idiotype secreted by a plasmacytoma. Eur J Immunol. 1996;26:2671. doi: 10.1002/eji.1830261119. [DOI] [PubMed] [Google Scholar]
- 25.Staveley-O'Corrol K, Sotomayor E, Mongomery J, Borrelo I, Hwang L, Fein S, Pardoll D, Levitsky H. Induction of antigen-specific T cell anergy: an early event in the course of tumor progression. Proc Natl Acad Sci USA. 1998;95:1178. doi: 10.1073/pnas.95.3.1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Colella TA, Bullock TN, Russell LB, Mullins DW, Overwijk WW, Luckey LC, Pierce RA, Restifo NP, Engelhard VH. Self-tolerance to the murine homologue of a tyrosinase-derived melanoma antigen: implications for tumor immunotherapy. J Exp Med. 2000;191:1221–1232. doi: 10.1084/jem.191.7.1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zippelius A, Batard P, Rubio-Godoy V, Bioley G, Lienard D, Lejeune F, Rimoldi D, Guillaume P, Meidenbauer N, Mackensen A, Rufer N, Lubenow N, Speiser D, Cerottini JC, Romero P, Pittet MJ. Effector function of human tumor-specific CD8 T cells in melanoma lesions: a state of local functional tolerance. Cancer Res. 2004;64:2865. doi: 10.1158/0008-5472.can-03-3066. [DOI] [PubMed] [Google Scholar]
- 28.Sotomayor EM, Borrello I, Rattis FM, Cuenca AG, Abrams J, Staveley-O'Carroll K, Levitsky HI. Cross-presentation of tumor antigens by bone marrow-derived antigen-presenting cells is the dominant mechanism in the induction of T-cell tolerance during B-cell lymphoma progression. Blood. 2001;98:1070. doi: 10.1182/blood.v98.4.1070. [DOI] [PubMed] [Google Scholar]
- 29.Kusmartsev S, Li Y, Chen SH. Gr-1+ myeloid cells derived from tumor-bearing mice inhibit primary T cell activation induced through CD3/CD28 costimulation. J Immunol. 2000;165:779. doi: 10.4049/jimmunol.165.2.779. [DOI] [PubMed] [Google Scholar]
- 30.Almand B, Clark JI, Nikitina E, English NR, Knight SC, Carbone DP, Gabrilovich DI. Increased production of immature myeloid cells in cancer patients. A mechanism of immunosuppression in cancer. J Immunol. 2001;166:678. doi: 10.4049/jimmunol.166.1.678. [DOI] [PubMed] [Google Scholar]
- 31.Almand B, Resser J, Lindman B, Nadaf S, Clark J, Kwon E, Carbone D, Gabrilovich D. Clinical significance of defective dendritic cell differentiation in cancer. Clin Cancer Res. 2000;6:1755. [PubMed] [Google Scholar]
- 32.Kusmartsev S, Nefedova Y, Yoder D, Gabrilovich DI. Antigen-specific inhibition of CD8+ T cell response by immature myeloid cells in cancer is mediated by reactive oxygen species. J Immunol. 2004;172:989. doi: 10.4049/jimmunol.172.2.989. [DOI] [PubMed] [Google Scholar]
- 33.Otsuji M, Kimura Y, Aoe T, Okamoto Y, Saito T. Oxidative stress by tumor-derived macrophages suppresses the expression of CD3 zeta chain of T-cell receptor complex and antigen-specific T-cell responses. Proc Natl Acad Sci USA. 1996;93:13119. doi: 10.1073/pnas.93.23.13119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cemerski S, van Meerwijk JP, Romagnoli P. Oxidative-stress-induced T lymphocyte hyporesponsiveness is caused by structural modification rather than proteasomal degradation of crucial TCR signaling molecules. Eur J Immunol. 2003;33:2178. doi: 10.1002/eji.200323898. [DOI] [PubMed] [Google Scholar]
- 35.Bronte V, Serafini P, De Santo C, Marigo I, Tosello V, Mazzoni A, Segal DM, Staib C, Lowel M, Sutter G, Colombo MP, Zanovello P. IL-4-induced arginase 1 suppresses alloreactive T cells in tumor-bearing mice. J Immunol. 2003;170:270. doi: 10.4049/jimmunol.170.1.270. [DOI] [PubMed] [Google Scholar]
- 36.Rodriguez PC, Quiceno DG, Zabaleta J, Ortiz B, Zea AH, Piazuelo MB, Delgado A, Correa P, Brayer J, Sotomayor EM, Antonia S, Ochoa JB, Ochoa AC. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res. 2004;64:5839. doi: 10.1158/0008-5472.CAN-04-0465. [DOI] [PubMed] [Google Scholar]
- 37.Rodriguez PC, Zea AH, DeSalvo J, Culotta KS, Zabaleta J, Quiceno DG, Ochoa JB, Ochoa AC. L-arginine consumption by macrophages modulates the expression of CD3 zeta chain in T lymphocytes. J Immunol. 2003;171:1232. doi: 10.4049/jimmunol.171.3.1232. [DOI] [PubMed] [Google Scholar]
- 38.Rodriguez PC, Zea AH, Culotta KS, Zabaleta J, Ochoa JB, Ochoa AC. Regulation of T cell receptor CD3zeta chain expression by L-arginine. J Biol Chem. 2002;277:21123. doi: 10.1074/jbc.M110675200. [DOI] [PubMed] [Google Scholar]