

Abstract
The role of steroid hormone receptors in very early embryonic development remains unknown. Clearly, expression during organogenesis is important for tissue-specific development. However, progesterone receptor (PR) and estrogen receptors (ERα, ERβ), are expressed during early development through the blastocyst stage in mice and other species, and yet are not essential for embryonic viability. We have utilized the mouse embryonic stem (mES) cell model to investigate the regulated expression of these receptors during differentiation. Surprisingly, one of the earliest changes in gene expression in response to a differentiation signal observed is PR gene induction. It parallels the time course of expression for the patterning genes Hoxb1 and Hoxa5. Unexpectedly, PR gene expression is not regulated in an estrogen dependent manner by endogenous ERs or by transiently overexpressed ERα. Our results suggest a potentially novel mechanism of PR gene regulation within mES cells compared to adult tissues and the possibility of unique targets of PR action during early mES cell differentiation
Keywords: estrogen receptor, steroid receptors, development, embryonic stem cells, nuclear receptors, differentiation
INTRODUCTION
Embryonic stem (ES) cells are defined by the properties of pluripotency and self-renewal, and are capable of differentiating into cell lineages from all three germ layers as well as into germ cells [1-5]. They are derived from the inner cell mass of blastocysts and provide a powerful model system to study the molecular mechanisms regulating early and late lineage specific differentiation. Differentiation of mouse ES (mES) cells can be induced by withdrawal of leukemia inhibitory factor (LIF) from plated cells, the formation of embryoid bodies (EBs) derived by growth of non-adherent mES cells in the absence of LIF, or by treatment of either plated cells or EBs with retinoic acid (RA) [6-13]. In addition, thyroid hormone treatment can modulate the differentiation process in these cells [14, 15]. The early effects of RA are mediated predominantly by retinoic acid receptor α (RARα) and those of thyroid hormone by thyroid hormone receptor α (TRα), both of which are members of the nuclear receptor family.
Other members of the nuclear receptor family, estrogen receptor alpha (ERα), estrogen receptor beta (ERβ), and progesterone receptor (PR) are present in the early developing embryo of mice and other species [16-23]. In particular, ERα is present in the oocyte, shows variable expression through the morula stage, followed by increased expression in the inner cell mass of the blastocyst. In the mouse there is little expression of PR until the blastocyst stage [17]. Separate genes encode ERα and ERβ; both bind estrogen with high affinity, and show tissue specific expression patterns and activity in adult tissues. Two isoforms of PR, PRA and PRB, are encoded from a single gene with two separate promoters [24-26]. PRA shares identical sequence with PRB, except PRB is larger. Both PRA and PRB bind progesterone with high affinity and show tissue specific expression patterns and activity in adult tissues [26 for review]. PRA and PRB gene expression is highly regulated by estrogens in most adult tissues. Knockout mice have been created for ERα, ERβ, PRA, and PRB. While none of these sex steroid hormone receptors appears to be essential for embryonic survival, a range of defects are observed in mature tissues of knockout mice, primarily in the female reproductive tract [27, 28, 29 for reviews]. Excess estrogen during mouse development, given either exogenously or by knockout of the 5α-reductase gene, leads to fetal death at midgestation in utero [30]. The mechanism remains unknown, but indicates the presence of functional estrogen signaling pathways. The functional relevance of ERα, ERβ, and PR expression by the cells of the inner cell mass is also unknown, although physiologically appropriate levels of ligands necessary to activate these receptors are present at this stage of gestation.
We studied the changes in sex steroid hormone receptor expression induced by differentiation of mES cells. We want to identify the pathways regulated by the sex steroid hormone receptor family during this early period of differentiation and needed to establish the temporal expression profile and the effect of differentiation. In addition to possible insight into early differentiation, mES cells in culture provide a model system to determine whether hormonal manipulation (estrogens or progestins) can modulate the pathways of lineage specific differentiation attained during induction of differentiation by LIF withdrawal or treatment with RA.
Unexpectedly, PR expression was rapidly induced by either LIF withdrawal or RA treatment as a differentiation signal. PR induction was one of the earliest changes observed following a differentiation signal tracking with the induced expression of well characterized differentiation markers such as the Hoxb1 and Hoxa5 patterning genes. PR expression preceded expression of ERα. This was consistent with the second surprising result that, unlike in most mature tissues, the regulation of PR gene expression in mES cells occurs independently of estrogen receptor signaling. We did not observe hormonal regulation of a limited number of selected genes known to be progesterone targets in mature tissues. Rather those genes were strongly regulated by “differentiation” itself in these cells. We hypothesize that the set of target genes for steroid receptors may be quite different during early differentiation of mES cells than in highly differentiated adult tissues. We believe this mES cell culture model will provide insight into the mechanism of regulation and the functional relevance of sex steroid hormone receptor expression early in differentiation.
MATERIALS AND METHODS
Cell culture
CCE mES cells were used for all studies except where indicated and were obtained from Dr. John Gearhart (Johns Hopkins University) with permission from Dr. Gordon Keller [31, 32]. Plastic tissue culture dishes were pretreated with 0.1% porcine gelatin Type A (Sigma) in water for 30 min to 2 hrs at 37ºC. Undifferentiated mES cells were grown at 5% CO2 in DMEM (Gibco) containing high glucose, phenol red and glutamine and supplemented with 15% fetal bovine serum (FBS, mES qualified from Gibco), 3.7 gm/L sodium bicarbonate (pH to 7.4), 1 mM sodium pyruvate, 0.1 mM non-essential amino acids, 0.15 mM monothioglycerol (ICN Biomedicals Inc.), and 1800 units/mL LIF (Chemicon). Cells were passaged every three days at 1:100 and maintained at low cell density. Approximately 5-10% of cells undergo spontaneous differentiation even under these conditions. Cells were induced to differentiate by removal of LIF from the media (all other components were unchanged) or by the addition of 1 μM all trans RA (Sigma) either in the presence or absence of LIF. EBs were prepared from undifferentiated mES cells after trypsinization as follows. The cells were diluted into media without LIF containing FBS to inactivate trypsin, centrifuged, then resuspended in media without LIF at 40,000 to 50,000 cells/mL. Approximately 20 μL drops were placed on the lid of a Falcon petri dish (60-80 drops per lid) and DMEM or Ca++-Mg++-free Hank’s balanced salt solution (CMF) added to dish to maintain humidity. The lid containing the hanging drops was placed back on the dish and the EBs allowed to form for 2 days. On day 2 the EBs were harvested and placed into 10 mL of media without LIF in a petri dish (Bibby Sterilin, LTD, Staffordshire, UK; the EBs will not plate in these dishes) for 3 more days. On day 5 the floating EBs were transferred to a gelatin treated tissue culture plate in media without LIF for 1 - 4 additional days during which the EBs plated down. Alternatively, freshly harvested mES cells were placed directly into petri dishes (Sterilin) in media without LIF at 20,000 to 50,000 cells per mL for 5 days with frequent media changes and then plated in gelatinized dishes. The EBs obtained by this method had more variability in size. D3 mES cells were obtained from ATCC and adapted to grow under feeder free conditions in the same media as described for CCE cells, except the FBS concentration which was 10%. GH3 rat pituitary tumor cells (ATCC) were grown in DMEM without phenol red, with 10% FBS, 4 mM glutamine, and 0.6 μg/mL insulin in 5% CO2 at 37ºC. GH3 cells were transferred to estrogen-free media (5% dextran-coated charcoal stripped fetal bovine serum, 4 mM glutamine, phenol red-free DMEM) for 1-4 days prior to hormone treatments.
Transfection
Cells were transiently transfected with the expression vector for HA-tagged human ERα, pUHD10-3-ER-HA, generously provided by Elaine T. Alarid, Department of Physiology, University of Wisconsin-Madison [33]. Cells were plated at 20,000 cells per well in a six-well plate in standard media containing LIF on day 0. Cells were fed on day one and transfected on day 2 with 4 μg DNA in complex with Lipofectamine 2000 (Invitrogen) prepared as per manufacturer’s instructions for 2 hrs. Cells were then treated with ligand and harvested for RNA or protein 22 hrs later. Control transfections were performed using a vector without ERα sequences. Neither the transfection conditions nor the vector used induced differentiation of the mES cells.
Preparation of total RNA
RNA samples were isolated from cell culture (usually one 10 cm plate/sample) using Trizol® reagent (Invitrogen Technologies) per the manufacturer’s recommended protocol. RNA samples were further purified with RNase-free DNase (Ambion). Samples were treated with 4 U of DNase per sample for 20 min at 37ºC and the reaction was stopped with termination buffer containing 0.1 M EDTA and 1 mg/mL glycogen. RNA was purified with two phenol: chloroform: isoamyl alcohol (25:24:1) washes, one chloroform: isoamyl alcohol (24:1) wash, and precipitated using ammonium acetate and ethanol. The RNA was redisolved in water and stored at −80ºC.
Conventional and quantitative RT-PCR
The cDNA was synthesized from 5 μg total RNA with AMV-RT enzyme and random hexamers (Promega). Conventional PCR was performed using 5% of the RT reaction product with Taq DNA Polymerase (Promega), individually optimized concentrations of MgCl2, and forward and reverse primers listed in Table I. Amplification conditions for annealing temperature and cycle number are also listed in Table I. PCR products were separated by 1% agarose gel electrophoresis and visualized using ethidium bromide staining under UV light. GAPDH was amplified as a control. Parallel reactions using product from cDNA reactions without RT enzyme confirmed that all DNA was removed during DNase treatment. Cycle numbers for all primers were titrated so that the sample with the highest signal was still within the linear range. The relative mRNA levels for PR were analyzed by real-time quantitative RT-PCR using a Bio-Rad iCycler system (Bio-Rad, Hercules CA). Total RNA was reverse transcribed as described above. Real time PCR was performed using a SYBR supermix kit (Bio-Rad) and running 45 cycles of 95ºC for 20 sec and 61ºC for 1 min. Each cDNA sample was run in triplicate for two separate experiments. The PR mRNA level of each sample was normalized to that of GAPDH mRNA. The fold increase in PR was calculated with the formula 2[Ct(GAPDH) − Ct(PR)]. Real time primer sequences for PR were: forward 5′CAGATTCAGAAGCCAGCCAGAG and reverse 5′CCACAGGTAAGCACGCCATAG (product size 114 bp).
TABLE 1.
PRIMERS FOR RT-PCR
| cDNA Target | Forward Sequence Reverse Sequence | Product Size (bp) | °C | Cycle # | [Mg++] mM |
|---|---|---|---|---|---|
| αFP | CCT GTG AAC TCT GGT ATC AG GCT CAC ACC AAA GCG TCA AC | 410 | 55 | 35 | 1 |
| AR | TGT GTG GAA ATA GAT GGG TAC ATG TGG TCA AGT GGG | 615 | 55 | 35 | 1 |
| Areg | AGT GCT GTT GCT GCT GGT CTT AG GAT AAC GAT GCC GAT GCC AAT A | 613 | 60 | 35 | 1 |
| β-globin | CTC AAG GAG ACC TTT GCT CA AGT CCC CAT GGA GTC AAA GA | 265 | 55 | 35 | 1 |
| Brachy | TGC TGC CTG TGA GTC ATA AC AAG GGA GGA CAT TAG AGG TG | 726 | 60 | 32 | 2 |
| BRG1 | CAC CCA GGG GCC TGG AGG TCC TGT GGC GGA CAC TGA | 516 | 60 | 32 | 1 |
| CBP | CCA ACA AGA AGA AGC CC GAG CGG CGT AAG GAA GAG | 243 | 60 | 32 | 2 |
| ERα | TTC TGA CAA TCG ACG CCA GAA T CAT CAT GCC CAC TTC GTA ACA C | 294 | 65 | 35 | 4 |
| ERβ | CAG TAA CAA GGG CAT GGA AC GTA CAT GTC CCA CTT CTG AC | 242 | 65 | 35 | 2 |
| ERRα | GAG CCT CTC TAC ATC AAG GAC CAC TAT CTC TCG ATC | 686 | 60 | 35 | 2.5 |
| ERRβ | GGG ATG CTG AAG GAA GGT G CAT CGT ATG GGA GCG AGC G | 390 | 60 | 32 | 1 |
| ERRγ | GTG CTT AGT GTG TGG CGA CAT C GTA GGG TCA GGC ATG GCA TAG | 386 | 60 | 32 | 1 |
| GAPDH | ACG ACC CCT TCA TTG ACC TCA ACT ATA TTT CTC GTG GTT CAC ACC CAT | 320 | 65 | 20 | 1 |
| GR | TTC TGC GTC TTC ACC CTC A TCC CCA TCA CTT TTG TTT CG | 200 | 56 | 30 | 1 |
| Hoxb1 | AGC GCC TAC AGC GCC CCA ACC TCT TTT CTT GAC CTT CAT CCA GTC GAA GGT CCG | 550 | 60 | 35 | 2 |
| Hoxa5 | GTG CCA ATG TTG TGT GTT CAT ACA TCA CTG AAC TGC GC | 298 | 64 | 35 | 4 |
| Nestin | AGG ACA GGA CCA AGA GGA AC TCT TGG TGC TGC TCC CTC TC | 533 | 60 | 27 | 3 |
| Oct-3/4 | GCC GAC AAC AAT GAG AAC CTT CAG ATA GCC TGG GGT GCC AAA GTG GGG | 343 | 65 | 25 | 1 |
| PR | CCC ACA GGA GTT TGT CAA ACT C TAA CTT CAG ACA TCA TTT CCG G | 327 | 65 | 35 | 2 |
| RARα | GGC TTC ACC ACC CTC ACC AT GCT GAT GCT CCG AAG GTC TG | 422 | 65 | 32 | 5 |
| RARβ2 | GCA GGA ATG CAC AGA GAG CTA TGA GAT GAC GGT GAC TGA CTG ACT CCA CTG TTC TCC ACT | 842 | 65 | 32 | 5 |
| Rex1 | AAC CAA GGA GGA AAT AGA GC CGT ATG ATG CAC TCT | 820 | 55 | 25 | 1 |
| SRC1 | TTT CAA GAA GTG ATG ACT CGT GG GGG ATT GCT GCT CTG GGA AC | 500 | 60 | 32 | 2.5 |
| SRC2 | GAT GGG TTC TTC TTC GTT G CTT TGA TGG ACT TGG GCT G | 348 | 60 | 32 | 1 |
| SRC3 | AGT GTC CTC CTC AAC ATC AGG CTT CTT AGG ACT CAG CTG CTC C | 215 | 60 | 32 | 1 |
| TRα | CCG GAC GGA GAC AAG GTA GAC GTC CAA ACG CCA GCA GGT AG | 439 | 65 | 35 | 4 |
| TRβ | GAC ATT GGA CAA GCA CCC ATC AAT CAT CCG CAG GTC TGT CAC | 549 | 62 | 35 | 1 |
| VDR | ACG ATG GAT CTG AAT GAA G GGA ACG GTA CTG TTT GGA G | 600 | 62 | 35 | 2.5 |
| Wnt4 | GAG TGC CAA TAC CAG TTC CTG CCA GCC TCG TTG TTG | 355 | 60 | 32 | 1 |
| Wnt5b | CAC CGT GGA CAA CAC ATC GAG CCA GCA GGT CTT GAG | 414 | 60 | 32 | 1 |
Immunoprecipitation and immunoblotting
Cells were rinsed twice with cold CMF containing 1 mM PMSF. The cells were scraped, pelleted, and frozen at −80ºC. Two volumes of ice-cold immunoprecipitation buffer (IP: 10 mM Tris-Cl pH 7.5, 150 mM NaCl, 0.4% IGEPAL, 1 mM EDTA, 0.5 mM AEBSF, and 10 μM leupeptin) were added to the cell pellets and the cells ruptured using 20 strokes of a syringe with an 18-gauge needle followed by 20 strokes with a 23-gauge needle. The cell extract was centrifuged for 5 min at top speed in a microfuge at 4ºC. Immunoprecipitation for PR used equal amounts of total protein from each sample diluted 1:5 into IP buffer, 2 μg of antibody against PR (DAKO A0098), and protein-A agarose beads. This was mixed for 3 hrs at 4ºC. The beads were washed twice with IP buffer and sample eluted with 1× SDS-PAGE sample buffer at 85ºC for 10 min. Specificity of the immunoprecipitation was confirmed using an identical method for extraction of PR from GH3 pretreated with 17β-estradiol (E2). This resulted in recovery of both PRA and PRB protein isoforms (data not shown). Whole cell extracts were used directly for SRC3, CBP, BRG1, and ERα immunoblotting. All samples were separated by SDS-PAGE, transferred to nitrocellulose, and immunostained using standard methods. Primary antibodies were against PR (Stressgen SRA-1100), ERα (HC-20, Santa Cruz), SRC3/AIB1 (Transduction Labs 611105), CBP (C-20, Santa Cruz), and BRG1 (H-88, Santa Cruz). Appropriate conjugated secondary antibodies were used and binding detected by either enhanced chemiluminescence or chemiflourescence (Amersham).
Immunohistochemistry
Cells were grown in 4 well Lab-Tek chamber slides (Nalge Nunc International) pretreated with gelatin. The cells were treated with 20-100 nM progesterone for 30 min prior to fixation with paraformaldehyde. Paraformaldehyde in H2O at 8% (w/v) was prepared fresh daily, diluted with 2X PBS to 4%, and pH adjusted to 7.5. Cells were washed with PBS, 4% paraformaldehyde added for 10 min at room temperature (RT), and cells washed twice with PBS. Cells were permeabilized and nonspecific peroxidase activity blocked by adding 80% MeOH with 0.6% H2O2 for 20 min at RT, then rinsed with PBS 4 times over 5-10 min. Nonspecific antigen sites were blocked with 5% FBS in PBS with 0.2% Tween-20 for 60 min in a humidity chamber. Primary antibody (anti-PR from DAKO or normal rabbit IgG from Santa Cruz) was added directly to the block at 1:200 dilution for 60 min at RT in a humidity chamber, then cells washed with blocking agent and 0.2% Tween-20 3 times over 5 min. Secondary antibody and substrate development were per Vectastain ABC kit (Vector Laboratories) instructions. The slides were counter-stained with hematoxylin and mounted with a cover slip.
PR Ligand Binding Assay
PR content was measured by a whole cell uptake assay using the synthetic progestin 3H-R5020 (NEN Life Sciences). CCE cells were cultured in the absence of LIF and the presence of 5 nM E2 for 5 days. Cells were lifted with 6 mM EDTA in PBS, counted, and distributed to borosilicate glass tubes. Triplicate 2 mL assays contained 8 × 106 cells in PBS with concentrations of 3H-R5020 from 0.4 to 20 nM. A 200-fold excess of unlabeled progesterone was added to duplicate tubes to estimate non-specific binding. Cells were incubated at 37ºC for 20 minutes, transferred to ice and washed once with PBS - 0.1% methylcellulose, twice with PBS - 0.1% methylcellulose - 0.1% BSA, and twice with PBS - 0.1% methylcellulose. Each cell pellet was extracted with ethanol at RT and the extract subjected to liquid scintillation counting. Specific binding was calculated by subtraction of counts in assays with unlabeled progesterone from total counts in the 3H-R5020 only samples. A similar whole cell uptake assay was performed on GH3 cells following E2 treatment as a positive control for the assay protocol (data not shown). Non-linear regression analysis using a one site model was performed with the GraphPad Prism® 3.0 software package [34].
RESULTS
Progesterone receptor gene is expressed early during differentiation of mES cells.
We examined the importance of differentiation on the expression of a number of the nuclear hormone receptors in mES cells by measuring changes in mRNA accumulation by RT-PCR over a time course of differentiation as shown in Figure 1A. Expression levels in cells maintained in the undifferentiated state in the presence of LIF are shown in the first lane. Differentiation was induced by LIF withdrawal from the culture media for 1 to 5 days (plated cells) or cells were grown as EBs in the absence of LIF for 1 to 8 days or 16 days. PR mRNA accumulation can be detected as early as one day following LIF withdrawal in both plated cells and EBs. This accumulation increases up to about day 5 and then is maintained at a lower level. Real time quantitative RT-PCR data indicated a 16-fold increase in PR mRNA following 4 days of LIF withdrawal as compared to undifferentiated mES cells (Figure 1B).
Figure 1.
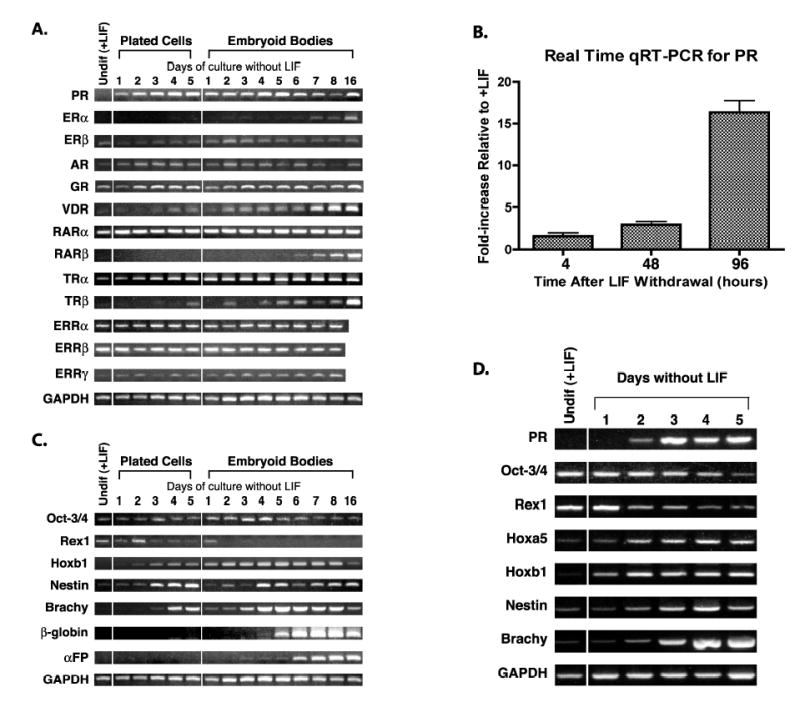
Expression of steroid/nuclear receptors in mES cells. RNA was prepared from cells cultured as described in Materials and Methods and RT-PCR performed using primers and conditions listed in Table I. Samples from mES cells maintained in the undifferentiated state by addition of LIF to the culture media are labeled Undif (+LIF). Differentiation was induced by LIF withdrawal for the time indicated in either plated cells or EBs. A. Detection of mRNA for a variety of steroid/nuclear receptors in CCE mES cells. B. Real time quantitative PCR analysis for PR expression during LIF withdrawal in CCE mES cells. Fold increases are 1.6 +/− 0.4 at 4h, 3.0 +/− 0.4 at 48h, and 16.3 +/− 1.4 at 96h. Fold increases are 1.6 +/− 0.4 at 4h, 3.0 +/− 0.4 at 48h, and 16.3 +/− 1.4 at 96h. C. Detection of mRNA for a series of mES cell differentiation markers in CCE cells. D. PR and cell differentiation marker expression patterns during differentiation of D3 mES cells. GAPDH is used as a normalization control.
The patterns of mRNA expression for the various nuclear receptors after a differentiation signal can generally be divided into three categories: those receptors showing 1) no change, 2) late (> 5 days) increased expression, and 3) early (< 3 days) increased expression. None of the nuclear receptors tested showed decreased expression levels. Those receptors constitutively expressed with little change over the time course shown here include ERβ, RARα, TRα, glucocorticoid receptor (GR), and estrogen receptor related proteins alpha, beta, and gamma (ERRα, ERRβ, and ERRγ). TRα did appear to increase with time of differentiation over the level seen in undifferentiated mES cells. ERα and retinoic acid receptor beta (RARβ2) showed no mRNA expression in the undifferentiated mES cells and only a small amount of induction late (greater than 5 days) following LIF withdrawal. Vitamin D receptor (VDR) and thyroid hormone receptor β (TRβ) also were not expressed in undifferentiated cells and showed late induction after LIF withdrawal in plated cells, but expression was more rapid in EBs. Those receptors showing little or no expression in the undifferentiated state, followed by rapid (1-2 days) mRNA accumulation upon LIF withdrawal in both plated cells and EBs, were PR and androgen receptor (AR).
LIF withdrawal results in nonspecific lineage differentiation down endoderm, mesoderm and ectoderm pathways. Characterization of the differentiation status of the mES cells at each time point, as shown in Figure 1C, is based on the expression of well-characterized markers for differentiation in this system. Rex1 and Oct-3/4 are known markers for undifferentiated mES cells. Down regulation of Rex1 occurred quickly in response to LIF withdrawal, whereas Oct-3/4 decreased more slowly as has been previously reported in cultured cells [35-37]. In developing embryos Oct-3/4 shows a much more rapid down regulation during differentiation. We demonstrate that mRNA for a known patterning gene, Hoxb1, accumulated rapidly (within 1-2 days). We also chose known markers for all three germ layers to demonstrate the pluripotent nature of this mES cell line. We observed mRNA within 2-3 days for nestin (an early neural ectoderm marker) and brachyury (Brachy, an early mesoderm marker), whereas β-H1 globin (β-globin, a mesoderm marker) and α-fetoprotein (α-FP, an endoderm marker) mRNAs accumulated later (days 5-6). These patterns of expression are as previously reported for multilineage differentiation following LIF withdrawal [8, 9, 32, 38-41].
To ensure that our observation of PR expression during early differentiation of cultured mES cells was not unique to this particular mES cell line (CCE), we tested another mES cell line, D3, for PR expression. The D3 line was adapted to grow under feeder free conditions for 11 passages. We then induced differentiation by LIF withdrawal. As can be seen in Figure 1D, PR mRNA accumulation occurred within 1 to 2 days in these cells as assayed by RT-PCR. Hoxa5 and Hoxb1 showed mRNA accumulation within 1 day and nestin and Brachy mRNA accumulated between days 2 and 3 following LIF withdrawal. Oct-3/4 and Rex1 levels decreased over 5 days. These patterns are similar to those observed in CCE mES cells. Therefore, PR gene expression was rapidly induced by differentiation signals in more than one mES cell line.
Comparison of figures 1A and 1C showed that PR expression was an early, strong marker of mES cell differentiation by LIF withdrawal. We therefore focused our studies on the regulation and functional significance of PR expression during mES cell differentiation in culture. PR and differentiation marker expression patterns were examined using other differentiation signals. High concentrations of RA, either in the presence of absence of LIF, also signal differentiation in mES cells. Figure 2 shows that PR mRNA accumulated within 1 day of addition of RA to the culture either in the presence or absence of LIF. RA is known to induce more lineage restricted differentiation than LIF withdrawal alone; in particular towards neural ectoderm and extraembryonic endoderm. The expression patterns of the differentiation markers confirmed this with nestin mRNA accumulation noted within 1 day and α-fetoprotein mRNA accumulation within 4-5 days. The two markers of mesoderm differentiation (Brachy in Fig 2, and β-globin, data not shown) were not induced by RA treatment. Hoxb1 mRNA showed rapid and strong accumulation following RA treatment as expected. Hoxb1 has previously been shown to be directly up-regulated by RA via a retinoic acid response element in the 3′ regulatory region of the gene [42]. Hoxa5, another patterning gene, was also rapidly induced by RA treatment. Oct-3/4 was more rapidly down-regulated with RA treatment than seen with LIF withdrawal. Rex1 down-regulation upon LIF withdrawal in the presence or absence of RA was similar. The presence of LIF with RA however, maintained Rex1 mRNA levels. In general, this was the expected pattern of marker expression for RA induced differentiation
Figure 2.
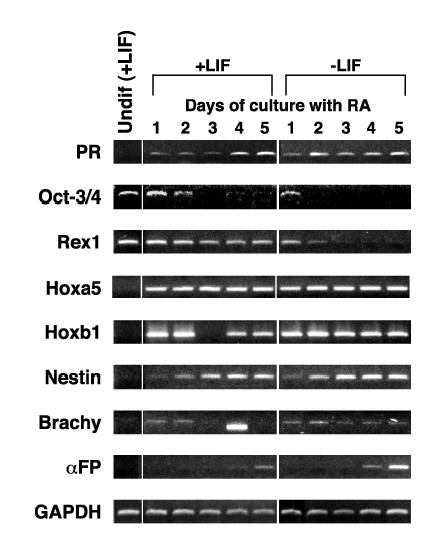
RA induction of PR expression in CCE mES cells. RNA was prepared from cells cultured as described in Materials and Methods and RT-PCR performed using primers and conditions listed in Table I. Samples from mES cells maintained in the undifferentiated state by addition of LIF to the culture media are labeled Undif (+LIF). Differentiation was induced by RA for the time indicated in plated cells grown in the presence or absence of LIF. The expression of a series of mES cell differentiation markers is also shown. Note that the band on day 4 of +LIF in the Brachy panel is smaller than the correct product and represents a primer dimer. GAPDH is used as a normalization control.
We used a short timecourse to determine how rapidly PR mRNA accumulated compared to our set of differentiation markers during the first 24 hrs following a differentiation signal (Figure 3). Within 8 to 12 hours following any of the three differentiation signals there was induction of PR mRNA expression by conventional RT-PCR. Quantitative RT-PCR (Figure 1B) shows a 1.6 fold increase as early as 4h after LIF withdrawal. Hoxa5 and Hoxb1 showed increased mRNA accumulation in the same time frame (within 8-12 hours following LIF withdrawal or within 4 hours after RA treatment). Oct 3/4 expression did not change and nestin showed a low expression at 24 hours following RA with LIF withdrawal. None of the other markers showed changes in mRNA accumulation over this short time course. PR was one of the most rapidly expressed genes following a differentiation signal in mES cells, closely paralleling the time course of accumulation of the patterning genes Hoxb1 and Hoxa5.
Figure 3.
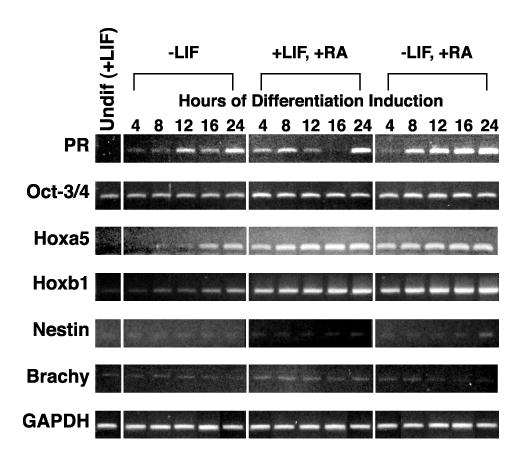
Short timecourse of PR expression in mES cells upon differentiation induction. RNA was prepared from cells cultured as described in Materials and Methods and RT-PCR performed using primers and conditions listed in Table I. Samples from mES cells maintained in the undifferentiated state by addition of LIF to the culture media are labeled Undif (+LIF). Differentiation was induced by either LIF withdrawal, addition of RA, or both for the times indicated in plated cells. The expression of a series of mES cell differentiation markers is also shown. GAPDH is used as a normalization control.
PR protein expression during mES cell differentiation.
While in general, the accumulation of mRNA for a particular gene will result in increased protein expression, we wanted to determine 1) whether PR protein is made in differentiating mES cells, 2) whether all of the differentiating cells make PR, 3) how much PR protein is produced, and 4) whether both PR protein isoforms (A and B) are generated. We performed immunohistochemistry (IHC) for PR expression in plated cells following LIF withdrawal for 5 days, EBs generated by LIF withdrawal, and EBs generated by LIF withdrawal with RA added (Figure 5A). We optimized the use of anti-PR antibodies for immunohistochemistry using the GH3 rat pituitary cell line, which are estrogen responsive for PR expression (data not shown). The panels 1 and 2 of Figure 4A are plated mES cells differentiated by LIF withdrawal for 5 days. Panel 1 shows the control staining pattern using normal rabbit IgG as the primary antibody. This compares directly with panel 2 in which most cells show nuclear staining with anti-PR antibody. Panels 3 and 4 of Figure 4A show EBs formed by LIF withdrawal with or without RA. We observed discrete clusters of cells within the EBs with variable positive nuclear staining for PR protein. In EBs created by simple LIF withdrawal (panel 3) many of these positively staining cells were on the periphery of the EBs and may represent cells of extraembryonic endoderm lineage. When RA was used during the generation of the EBs (panel 4), smaller clusters of elongated cells scattered throughout the EBs showed nuclear staining. By 5 days following LIF withdrawal most of the differentiated mES cells expressed PR by immunohistochemistry. Later, during EB formation, we observed specific sub-populations of cells expressing higher levels of PR. Undifferentiated mES cells could not be used as negative controls as their pattern of growth as tight balls of cells resulted in nonspecific antibody trapping with strong diffuse staining even in the absence of primary antibody (using secondary antibody only - data not shown).
Figure 5.
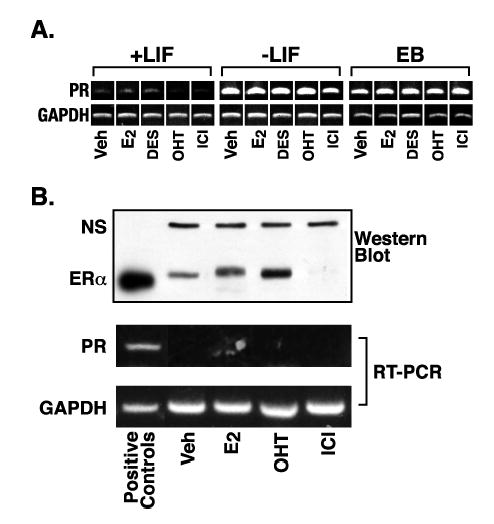
Progesterone receptor expression in mES cells is regulated by differentiation, rather than estrogen receptor signaling. A. RNA was prepared from cells cultured as described in Materials and Methods and RT-PCR performed using primers and conditions listed in Table I. Samples from mES cells were maintained in the undifferentiated state by addition of LIF to the culture media (+LIF). Differentiation was induced by LIF withdrawal for the 4 days in plated cells (−LIF) or 8 days in EBs (EB). Hormone treatments were vehicle control (veh), 5 nM E2, 3.7 μM DES, 100 nM OHT, or 100 nM ICI for 4 days in plated cells or the final 2 days in EBs. B. Undifferentiated mES cells were transfected with an hERα expression vector for 24 hrs as described in Materials and Methods. Cells were treated with hormone as in (A) for 22 hrs and replicate cultures harvested for protein or RNA. Western blot analysis for ERα was performed as described in Materials and Methods. The band labeled NS (non-specific) acts as a loading control for the mES cell extract lanes. The positive control is 10 fmoles purified hERα from PanVera. The positive control for PR expression in the RT-PCR panel is RNA from mES cells withdrawn from LIF for 4 days.
Figure 4.
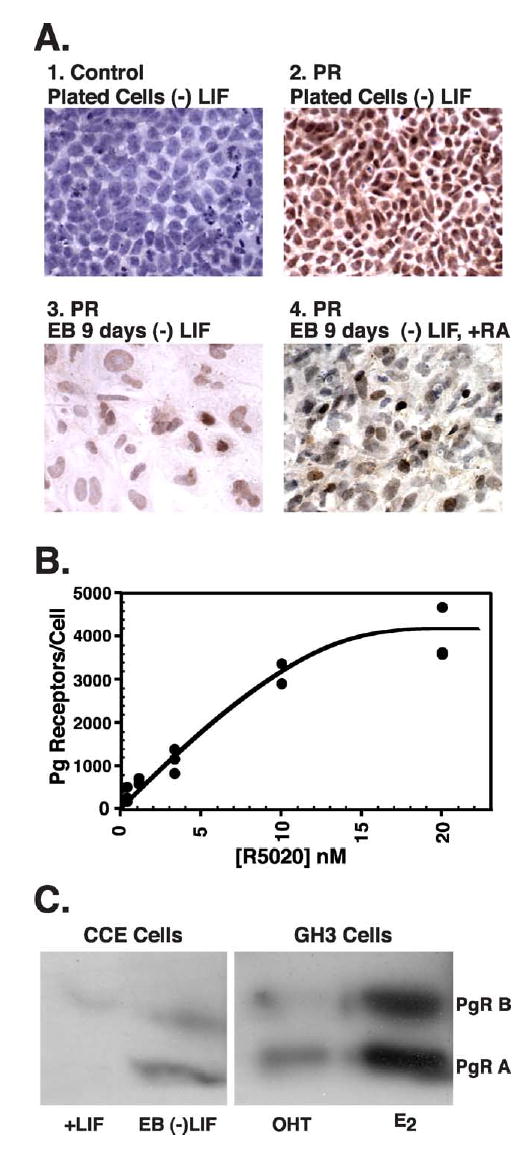
Expression of PR protein in mES cells. A. Immunohistochemistry for PR was performed as described in Materials and Methods. Plated cells induced to differentiate by LIF withdrawal for 5 days were subjected to staining with normal rabbit immunoglobulin (1. control) or anti-PR antibody (2. PR). Cells grown as EBs were induced to differentiate by LIF withdrawal (3. PR) or LIF withdrawal and addition of RA (4. PR), and both were stained for the presence of PR. The magnification is 400x for panels 1, 3, 4 and is at 200X for panel 2. B. Quantification of PR in mES cells by binding of a radiolabelled progestin (R5020). Cells were induced to differentiate by LIF withdrawal for 5 days. Whole cells were incubated with increasing concentrations of 3H-R5020, specifically bound ligand determined as described in Materials and Methods, and converted to receptor number per cell. Graphical presentation of the data as a saturation curve is shown. C. Equal amounts of whole cell extract protein from CCE mES or GH3 cells were subjected to immunoprecipitation for PR with one antibody (DAKO) and recovered protein subjected to western blot for PR with a different antibody (StressGen). CCE mES cells were undifferentiated (+LIF) or grown as EBs and induced to differentiate by LIF withdrawal for 8 days (EB (−)LIF). Control GH3 cells were treated with 1 nM E2 or 100 nM OHT for 3 days.
We used equilibrium binding analysis with a radiolabelled progestin (3H-R5020) to estimate the amount of PR protein in mES cells differentiated by LIF withdrawal for 5 days (Figure 4B). Non-linear regression analysis of the data shown in Figure 4B estimated 5700 +/− 900 PR molecules per cell averaged over the whole population with a binding affinity (Kd) of 13 +/− 4 nM. Non-specific binding was a large fraction of total binding due to the low concentration of PR, which made estimation of the binding affinity less accurate. The binding affinity was lower than expected for purified PR, but comparable to published values for PR binding R5020 in crude protein mixtures [43, 44]. Control binding experiments were performed with GH3 cells that are known to express PR and show increased expression with E2 treatment. In GH3 cells treated with ethanol vehicle we measured 13,000 receptors per cell and that increased to 104,000 receptors per cell with E2 treatment (data not shown).
We utilized western blotting to determine whether both isoforms of PR (A and B) were produced in differentiated mES cells (Figure 4C). We immunoprecipitated with one anti-PR antibody and then used a different anti-PR antibody for western blot detection. This reduced background and allowed us to detect the relatively low levels of PR protein in the mES cells. We used extracts from rat GH3 cells subjected to the same protocol as positive controls. Based on our radioligand binding studies, we expected the levels of PR in vehicle treated GH3 cells to be about twice the level in differentiated mES cells and to increase 8-10 fold with E2 treatment. We treated the GH3 cells with the anti-estrogen, 4-hydroxytamoxifen (OHT), to decrease the PR levels further. As can be seen in the right panel of Figure 4C, both PRA (MW 86,000) and PRB (MW 110,000) were present in the OHT treated GH3 cells and increased substantially with E2 treatment. The expected sizes for PR in mouse (MW 82,000 for PRA and MW 99,000 for PRB) are smaller than rat. As can be seen in the left panel of Figure 4C, a small amount of PRB was present in undifferentiated mES cells, which likely represents PR protein expressed in the 5-10% of spontaneously differentiating cells. EBs at day 8 showed a large induction of both PR isoforms (Figure 4C).
Mechanism of PR gene induction during differentiation induced by LIF withdrawal.
In most target organs (mammary gland, uterus, and pituitary) the expression of PR is up-regulated by estrogens whereas antiestrogens show tissue specific antagonistic or partial agonistic activity. In undifferentiated mES cells, ERβ was expressed constitutively at relatively low levels. We estimated approximately 400 ERβ molecules per cell by ligand binding assay using 3H-E2 (data not shown). ERα was not expressed in undifferentiated mES cells or in the immediate 5 days following LIF withdrawal, but was expressed by day 7-8 in EBs (Figure 1; immunohistochemistry data not shown). To determine whether estrogens and anti-estrogens could regulate PR gene expression in mES cells we used RT-PCR to detect changes in PR mRNA accumulation following various hormone treatments in either undifferentiated mES cells, plated cells or EBs differentiated by LIF withdrawal (Figure 5A). Treatments were ethanol vehicle control, E2, diethylstilbestrol (DES, a synthetic estrogen), OHT (partial agonist/antagonist), or ICI 182,780 (ICI, a pure antagonist). The first panel of Figure 5A shows RT-PCR results for PR expression when undifferentiated mES cells (+LIF) were treated with hormones (the number of PCR cycles was increased by 3 compared to Figure 1 in order to look for subtle changes in mRNA accumulation even in the presence of LIF). In the second and third panels the cells were treated with the same hormones but without LIF and PCR performed with the usual number of cycles. The data in Figure 5 are representative of at least three independent experiments and no consistent effect of any of the hormones was observed. Treatments were performed in regular growth media containing phenol red and 15% FBS that was not charcoal-stripped to remove low levels of endogenous steroids. This could have masked our ability to see PR induction by hormone, however we also did not observe significant down regulation of PR expression with high concentrations of the ER antagonists, OHT and ICI. It was difficult to maintain these mES cells using charcoal-stripped FBS for long periods of time without the occurrence of variable amounts of differentiation and cell death. We also performed the same study with cells in phenol red-free media with charcoal-stripped FBS and still failed to observe any consistent effect of ER agonists or antagonists on PR gene expression (data not shown).
We asked if high levels of ERα could induce PR expression in mES cells. We transiently transfected ERα into undifferentiated mES cells, then subjected the cells to various hormone treatments. We observed robust, transient expression of ERα in the mES cells under vehicle, E2, or OHT treatment (Figure 5B - western blot panel). We estimated mES cells averaged in excess of 100,000 receptors per cell in the vehicle treatment by comparison with the positive control lane, which contained 10 fmoles of purified ERα. The expression of ERα was higher in the E2 and OHT treated cells, and these ligands have been reported to stabilize ERα protein in other systems [45]. We saw a dramatic loss of ERα protein upon ICI treatment, and this ligand has been reported to promote rapid degradation of the receptor in other systems [46, 47]. A non-specific protein band (NS) was used as a loading control for mES cell samples. The response of ERα protein levels to these hormone treatments was evidence that the transfected ERα was functional. However, as can be seen in the RT-PCR panels of Figure 5B, even very high expression of ERα was unable to induce PR expression under any hormone treatment. Thus, the major regulator of PR expression in mES cells was downstream of the differentiation signal (LIF withdrawal or addition of RA) and estrogens/antiestrogens did not alter PR expression in this cell model of differentiation.
Functional relevance of PR expression during mES cell differentiation
We wanted to determine whether the PR protein, which was expressed so early in mES cell differentiation, was functional. Steroid hormone receptors activate transcription of target genes by interacting at DNA response elements with known coactivators following hormone treatment. Known coactivators of PR action include members of the p160 family of Steroid Receptor Coactivators (SRC1, SRC2, and SRC3) [48]. Other proteins are also important in activation of target gene transcription following progesterone binding to PR including CBP and ATP-dependent chromatin remodeling complexes containing BRG. We investigated the expression levels of these five coactivators in undifferentiated mES cells and during differentiation induced by LIF withdrawal using RT-PCR. All five coactivators were expressed under all conditions at fairly steady levels (data not shown). Western blot analysis showed SRC3, CBP, and BRG1 protein levels did not change during differentiation (data not shown). These data indicate that a variety of coregulators that PR might utilize were available in these cells.
In order to explore the functional role of PR protein in differentiating mES cells, we studied the effects of progesterone treatment on the expression of candidate target genes. We examined PR expression itself, since it has been reported to be down regulated by progesterone in some tissues [49]. We also examined the levels of expression of three genes that have been shown to be regulated by progesterone in mature tissues: Wnt4, Wnt5b, and amphiregulin (Areg) [50-56]. As can be seen in Figure 6, all four of these genes showed virtually no expression in undifferentiated mES cells (+LIF) and there was no effect of progesterone nor RU486 (a potent anti-progestin). LIF withdrawal in either plated cells for 4 days or during 8 days of EB formation resulted in strong mRNA accumulation for all four of these genes. However, treatment with progesterone or RU486 did not regulate the amount of accumulated mRNA for any of these candidate targets. We hypothesize that the set of target genes for progesterone in a differentiating mES cell model system may be considerably different than seen in a highly differentiated tissue such as breast. We also examined whether progesterone and/or RU486 could alter the selection of lineage specific differentiation pathways these cells underwent during LIF withdrawal at early time points of 4 days in plated cells and 8 days in EBs. We analyzed the effects of progesterone and RU486 on the overall RT-PCR signal for nestin (ectoderm marker) and brachyury (mesoderm marker) and saw no effect of hormone treatment on the signal intensity for these early differentiation markers, indicating a lack of major alteration in early lineage selection by PR signaling (Figure 6).
Figure 6.
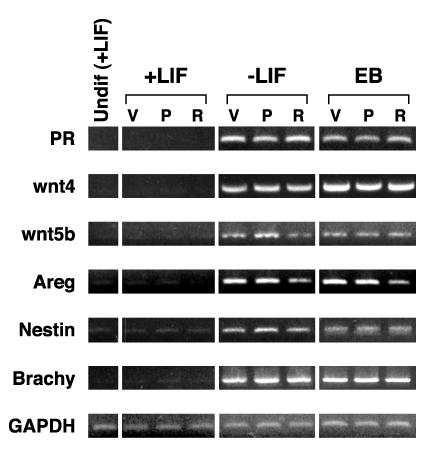
In mES cells, differentiation, not PR ligand, regulates the expression of three genes that are progesterone-responsive in other systems. RNA was prepared from cells cultured as described in Materials and Methods and RT-PCR performed using primers and conditions listed in Table I. Samples from mES cells maintained in the undifferentiated state by addition of LIF to the culture media are labeled Undif (+LIF). Differentiation was induced by LIF withdrawal for 4 days in plated cells (−LIF) or 8 days in EBs. Cells were treated with vehicle control (V), 200 nM progesterone (P), or 1 μM RU486 (R).
DISCUSSION
We have utilized the mES cell model to study the role of sex steroid receptors during early embryonic differentiation events. In the developing mouse embryo, ERα, ERβ, and PR are expressed at the blastocyst stage and thereafter [16-23]. The expression of these receptors in mES cells may not directly reflect the same timing as in development, but their expression in mES cells allows us to use it as a model system of in vitro differentiation. We also examined the three ERR proteins that do not have known endogenous ligands, however, ERRβ and ERRγ have been shown to bind DES and OHT at very high concentrations, with resulting alterations in activity. The mRNA for all three ERRs (α,β,γ) are constitutively expressed in undifferentiated mES cells and their levels do not significantly change during differentiation in culture. Unlike developing mouse embryos, which express ER α at the blastocyst stage, undifferentiated mES cells in culture do not express ERα mRNA and show little accumulation of ERα mRNA during differentiation induced by LIF withdrawal. ERβ, however, is constitutively expressed in undifferentiated mES cells and following LIF withdrawal.
The most surprising result of our studies was the rapid induction of PR (16 fold by real time RT-PCR) expression following LIF withdrawal. In this study we further characterized PR expression in differentiating mES cells in culture. We observe rapid PR expression in differentiating mES cells following any of three signals initiating differentiation: LIF withdrawal, RA treatment, or the combination. Besides mRNA accumulation, PRA and PRB proteins are clearly produced by day 8 of EB formation. The western blots and IHC are not sensitive enough to determine when accumulation of PR protein begins, but the ligand binding assay indicates that by four days following LIF withdrawal there are between 5000 to 6000 PR molecules per cell. While this number is lower than that seen in some differentiated tissues including breast cancer cells, it was in the range we observe for unstimulated GH3 rat pituitary cells (13,000 receptors per cell), and could be sufficient to elicit transcriptional activation of some target genes. The IHC results indicate that almost all of the plated differentiating cells at day four following LIF withdrawal express some PR protein. However, later during EB formation, when cells are more highly differentiated, PR is predominantly located within the cells on the outer rim of the EB (most likely representing extraembryonic endoderm [8, 9]) and in focal collections of cells on the interior. A recent report demonstrates a different pattern of expression for ERα, ERβ, and PR in human ES (hES) cells [57]. In hES cells PR is not expressed during the first 6 days of EB formation, whereas both ERα and ERβ are expressed in the undifferentiated state and levels increase with differentiation. The significance of increased AR expression in mES cells remains to be examined.
What is the functional significance of PR expression early in mES cell differentiation? PR expression is clearly important in normal female reproductive tract development, but is not essential for embryonic viability [24 for review]. Knockout of both PR isoforms results in multiple abnormalities of the reproductive tract in female mice. One model is that the primary role of PR during very early differentiation is to modulate expression levels of target gene transcription rather than act as an all or none regulator. Known target genes of PR in mature tissues that we examined in our study include Wnt4, Wnt5b and Areg, all of which are important developmental genes [50-56]. None of these known PR target genes are regulated by treatment with progesterone or the antiprogestin RU486 in mES cells. Clearly all of the transcription machinery necessary for mediating transcriptional activation via nuclear hormone receptors such as retinoic acid receptor alpha and thyroid hormone receptor alpha is present within ES cells. However, PR signaling is not detected at the limited number of genes examined here. This is not surprising as many nuclear hormone receptors show tissue specific effects in regulating transcription from target genes. Thus, RARα and TRα may signal normally in mES cells when ligand is added, but PR has a different set of target genes. This could be regulated at many levels including transcriptional repression of expected targets via DNA and/or histone tail methylation, via direct target gene repression by repressors such as NCoR or SMRT, by altered ratios of the available coactivators necessary for regulating the target genes we have thus far examined, or the presence of other regulators (that may override PR regulation) of the examined target genes not usually present in adult tissues. The later seems to be at least part of the explanation for the small number of PR targets we have examined, as they are transcriptionally activated during differentiation (withdrawal of LIF) but are not regulated by PR suggesting the presence of a differentiation-induced transcriptional regulator of these genes. Thus, PR at relatively low intracellular concentrations can no longer act as the primary transcriptional regulator of these genes. The identity of these regulators that are activated during differentiation remains unknown. It is also likely that the direct targets of PR function may be different in mES cells than reported for more highly differentiated tissues (uterus, ovary, mammary gland, pituitary). Therefore, cDNA microarray analysis will be necessary to determine whether novel gene targets exist in mES cells compared to mature tissues. Regulation by progesterone at very early times in embryonic differentiation may be important to the development of the female reproductive tract that is clearly disrupted in the PR knockout mouse. We did not observe major alterations in differentiation during the first few days following LIF withdrawal with high doses of progesterone or RU486 (data not shown). There also is no difference in the overall expression of early neural ectoderm (nestin) and early mesoderm (brachyury) differentiation markers (see Figure 6). Given that the defects observed in knock-out animals are in specific organ development, our future studies are to assess longer term progesterone treatment on lineage specific differentiation in the mES cell model. We are currently developing immunofluorescence detection for early markers of each cell lineage (endoderm, mesoderm, ectoderm) in order to determine whether progesterone can cause small but detectable shifts in the number of individual cells expressing lineage specific markers. Once again our inability to detect early effects on lineage selection is not surprising and later tissue specific effects may be more easily detected.
We examined possible mechanisms regulating PR gene transcription in this mES cell differentiation model. The first was based upon clear evidence that estrogen regulates PR gene expression in highly differentiated tissues such as breast and uterus. The PRA promoter has a partial estrogen response element (ERE) that is important in mediating estrogen action in human breast cancer cells [58]. We and others have recently mapped the site of ER interaction within the PRB promoter in human breast cancer cells, as well as some of the complexities of regulation of these promoters [59-62]. The mouse and human PR promoters share some similarity in promoter sequences (especially in the PRA promoter), but are not identical. Others have shown that stable overexpression of ERα in some cell lines followed by estrogen treatment is sufficient to activate endogenous PR expression [63, 64]. These data implicate estrogen and ER as important regulators of PR gene expression. ERβ is constitutively expressed both before and after differentiation signals in mES cells, albeit at relatively low levels (approximately 400 ERβ receptors per cell). ERα is expressed at 7-8 days of EB differentiation with LIF withdrawal. However, we see no effect of estrogen or antiestrogen on PR gene expression, even when we overexpress ER α by transfection. We also show that all three ERR proteins (α, β, γ) are expressed in undifferentiated and differentiating mES cells. As these three proteins can modulate known estrogen responsive genes in some model systems, it is possible that the ERRs can activate PR gene expression independent of estrogen. Although, why these constitutively expressed proteins would only activate PR expression following a differentiation signal is not clear. High concentrations of DES and OHT can bind to ERRβ and ERRγ and inhibit the ligand independent transcriptional activation functions of these proteins. When we treated mES cells (+/− LIF) with high doses of DES or OHT we saw no effect on PR expression levels (data not shown). We conclude that neither the estrogen nor estrogen related receptors play a major role in regulating PR gene activation in this mES cell model of early differentiation.
What is the transcriptional regulator of PR in mES cells, if not ERα or ERβ? One possibility would be repression of PR by the stat pathway mediated through LIF signalling. However, retinoic acid treatment even in the presence of LIF induces PR (Figure 3). In addition, adding LIF back to ES cells following 48 or more hours of LIF withdrawal does not repress PR gene expression (data not shown). PR gene expression has been shown to be directly regulated by overexpression of Hoxa5 in human breast cancer cells [65]. There are two consensus Hoxa5 DNA binding sites in the mouse PRB promoter region. The Hoxa5 consensus binding sequence of AAATAA is found at 297 bp and 984 bp 5′-upstream of the major transcriptional start site for the PRB gene (GenBank™ accession number U12644; [66]). In addition, Hoxa5 has been shown to activate transcription through TAAT sites in the human PR gene, more than one of which are conserved in the mouse PRB promoter [65]. We examined the mRNA accumulation of Hoxa5 in mES cells following a differentiation signal of either LIF withdrawal or addition of RA as shown in Figures 2 and 3. Rapid accumulation of Hoxa5 mRNA is observed following either method of differentiation, though it is more rapid (less than 4 hours) with RA than LIF withdrawal (8 to 12 hours). A similar pattern of Hoxa5 expression upon LIF withdrawal is also seen in the D3 mES cell line (Figure 1D). The pattern of Hoxa5 accumulation following LIF withdrawal is similar to the pattern seen for PR accumulation, leaving open the possibility that Hoxa5 may directly regulate PR gene expression early during mES cell differentiation. Unfortunately, due to limitations in the specificity of commercially available antibodies against Hoxa5, we could not confirm early Hoxa5 protein synthesis or utilize these antibodies for chromatin immunoprecipitation analysis. Therefore, Hoxa5 remains a potential, but unproven regulator of early PR gene expression during mES cell differentiation. If PR is downstream from Hoxa5, then possible functions for PR may be hypothesized from Hoxa5 function. In tumors, Hoxa5 expression can induce apoptosis, perhaps through regulation of p53 expression [67]. Hoxa5 knockout mice are viable but show alterations in mesenchymal-epithelial signaling [68]. Therefore, PR expression may play a role in modulating apoptosis during early differentiation and/or mesenchymal-epithelial signaling during later differentiation. However, progesterone and RU486 treated mES cells during LIF withdrawal have the same overall growth rate in culture (data not shown), suggesting a minimal effect on apoptosis or proliferation.
In conclusion, we have demonstrated that PR gene expression is induced very early during mES cell differentiation and the mechanism of induction is independent of estrogen signaling. PR is expressed within 24 hours of several differentiation signals in at least two mES cell lines. Important developmental regulators of PR expression remain to be identified. However, perhaps the most potent regulators in highly differentiated cell models, ERα/ERβ, do not play a role here. Finally, we are continuing to explore the pathways regulated by this very early PR gene expression during mES cell differentiation that may contribute to the reproductive tract abnormalities observed in the PR knockout mouse.
Acknowledgments
The authors thank Timothy J. Kruser for technical assistance. The authors also thank the members of the University of Wisconsin nuclear receptor club for helpful discussions. This work was supported in part by grants awarded to MKF: The American Cancer Society Mary Jo Boler and Alaska Run for Women Research Grant (#RSG-02-071-01) and NIH R01 DK064243-01.
References
- 1.Evans MJ, Kaufman MH. Establishment in culture of pluripotential cells from mouse embryos. Nature. 1981;292:154–156. doi: 10.1038/292154a0. [DOI] [PubMed] [Google Scholar]
- 2.Brook FA, Gardner RL. The origin and efficient derivation of embryonic stem cells in the mouse. Proc Natl Acad Sci U S A. 1997;94:5709–5712. doi: 10.1073/pnas.94.11.5709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chambers I, Smith A. Self-renewal of teratocarcinoma and embryonic stem cells. Oncogene. 2004;23:7150–7160. doi: 10.1038/sj.onc.1207930. [DOI] [PubMed] [Google Scholar]
- 4.West JA, Daley GQ. In vitro gametogenesis from embryonic stem cells. Curr Opin Cell Biol. 2004;16:688–692. doi: 10.1016/j.ceb.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 5.Hubner K, Fuhrmann G, Christenson LK, Kehler J, Reinbold R, De La Fuente R, Wood J, Strauss JF, 3rd, Boiani M, Scholer HR. Derivation of oocytes from mouse embryonic stem cells. Science. 2003;300:1251–1256. doi: 10.1126/science.1083452. [DOI] [PubMed] [Google Scholar]
- 6.Smith AG. Mouse embryo stem cells: Their identification, propagation and manipulation. Semin Cell Biol. 1992;3:385–399. doi: 10.1016/1043-4682(92)90010-s. [DOI] [PubMed] [Google Scholar]
- 7.Rathjen J, Rathjen PD. Mouse ES cells: Experimental exploitation of pluripotent differentiation potential. Curr Opin Genet Dev. 2001;11:587–594. doi: 10.1016/s0959-437x(00)00237-9. [DOI] [PubMed] [Google Scholar]
- 8.Doetschman TC, Eistetter H, Katz M, Schmidt W, Kemler R. The in vitro development of blastocyst-derived embryonic stem cell lines: Formation of visceral yolk sac, blood islands and myocardium. J Embryol Exp Morphol. 1985;87:27–45. [PubMed] [Google Scholar]
- 9.Lake J, Rathjen J, Remiszewski J, Rathjen PD. Reversible programming of pluripotent cell differentiation. J Cell Sci. 2000;113(Pt 3):555–566. doi: 10.1242/jcs.113.3.555. [DOI] [PubMed] [Google Scholar]
- 10.Strubing C, Ahnert-Hilger G, Shan J, Wiedenmann B, Hescheler J, Wobus AM. Differentiation of pluripotent embryonic stem cells into the neuronal lineage in vitro gives rise to mature inhibitory and excitatory neurons. Mech Dev. 1995;53:275–287. doi: 10.1016/0925-4773(95)00446-8. [DOI] [PubMed] [Google Scholar]
- 11.Weiss MJ, Orkin SH. In vitro differentiation of murine embryonic stem cells. New approaches to old problems. J Clin Invest. 1996;97:591–595. doi: 10.1172/JCI118454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen AC, Gudas LJ. An analysis of retinoic acid-induced gene expression and metabolism in AB1 embryonic stem cells. J Biol Chem. 1996;271:14971–14980. doi: 10.1074/jbc.271.25.14971. [DOI] [PubMed] [Google Scholar]
- 13.Bain G, Kitchens D, Yao M, Huettner JE, Gottlieb DI. Embryonic stem cells express neuronal properties in vitro. Dev Biol. 1995;168:342–357. doi: 10.1006/dbio.1995.1085. [DOI] [PubMed] [Google Scholar]
- 14.Lee LR, Mortensen RM, Larson CA, Brent GA. Thyroid hormone receptor-alpha inhibits retinoic acid-responsive gene expression and modulates retinoic acid-stimulated neural differentiation in mouse embryonic stem cells. Mol Endocrinol. 1994;8:746–756. doi: 10.1210/mend.8.6.7935490. [DOI] [PubMed] [Google Scholar]
- 15.Liu YY, Tachiki KH, Brent GA. A targeted thyroid hormone receptor alpha gene dominant-negative mutation (P398H) selectively impairs gene expression in differentiated embryonic stem cells. Endocrinology. 2002;143:2664–2672. doi: 10.1210/endo.143.7.8906. [DOI] [PubMed] [Google Scholar]
- 16.Wu TC, Wang L, Wan YJ. Expression of estrogen receptor gene in mouse oocyte and during embryogenesis. Mol Reprod Dev. 1992;33:407–412. doi: 10.1002/mrd.1080330406. [DOI] [PubMed] [Google Scholar]
- 17.Hou Q, Gorski J. Estrogen receptor and progesterone receptor genes are expressed differentially in mouse embryos during preimplantation development. Proc Natl Acad Sci U S A. 1993;90:9460–9464. doi: 10.1073/pnas.90.20.9460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hou Q, Paria BC, Mui C, Dey SK, Gorski J. Immunolocalization of estrogen receptor protein in the mouse blastocyst during normal and delayed implantation. Proc Natl Acad Sci U S A. 1996;93:2376–2381. doi: 10.1073/pnas.93.6.2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hiroi H, Momoeda M, Inoue S, Tsuchiya F, Matsumi H, Tsutsumi O, Muramatsu M, Taketani Y. Stage-specific expression of estrogen receptor subtypes and estrogen responsive finger protein in preimplantational mouse embryos. Endocr J. 1999;46:153–158. doi: 10.1507/endocrj.46.153. [DOI] [PubMed] [Google Scholar]
- 20.Ying C, Yang YC, Hong WF, Cheng WT, Hsu WL. Progesterone receptor gene expression in preimplantation pig embryos. Eur J Endocrinol. 2000;143:697–703. doi: 10.1530/eje.0.1430697. [DOI] [PubMed] [Google Scholar]
- 21.Ying C, Hsu WL, Hong WF, Cheng WT, Yang Y. Estrogen receptor is expressed in pig embryos during preimplantation development. Mol Reprod Dev. 2000;55:83–88. doi: 10.1002/(SICI)1098-2795(200001)55:1<83::AID-MRD11>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 22.Kowalski AA, Graddy LG, Vale-Cruz DS, Choi I, Katzenellenbogen BS, Simmen FA, Simmen RC. Molecular cloning of porcine estrogen receptor-beta complementary DNAs and developmental expression in periimplantation embryos. Biol Reprod. 2002;66:760–769. doi: 10.1095/biolreprod66.3.760. [DOI] [PubMed] [Google Scholar]
- 23.Lassiter CS, Kelley B, Linney E. Genomic structure and embryonic expression of estrogen receptor beta a (ERbetaa) in zebrafish (Danio rerio) Gene. 2002;299:141–151. doi: 10.1016/s0378-1119(02)01050-8. [DOI] [PubMed] [Google Scholar]
- 24.Conneely OM, Kettelberger DM, Tsai MJ, Schrader WT, O’Malley BW. The chicken progesterone receptor A and B isoforms are products of an alternate translation initiation event. J Biol Chem. 1989;264:14062–14064. [PubMed] [Google Scholar]
- 25.Kastner P, Krust A, Turcotte B, Stropp U, Tora L, Gronemeyer H, Chambon P. Two distinct estrogen-regulated promoters generate transcripts encoding the two functionally different human progesterone receptor forms A and B. EMBO J. 1990;9:1603–1614. doi: 10.1002/j.1460-2075.1990.tb08280.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Conneely OM, Mulac-Jericevic B, DeMayo F, Lydon JP, O’Malley BW. Reproductive functions of progesterone receptors. Recent Prog Horm Res. 2002;57:339–355. doi: 10.1210/rp.57.1.339. [DOI] [PubMed] [Google Scholar]
- 27.Couse JF, Korach KS. Estrogen receptor null mice: What have we learned and where will they lead us? Endocr Rev. 1999;20:358–417. doi: 10.1210/edrv.20.3.0370. [DOI] [PubMed] [Google Scholar]
- 28.Pettersson K, Gustafsson JA. Role of estrogen receptor beta in estrogen action. Annu Rev Physiol. 2001;63:165–192. doi: 10.1146/annurev.physiol.63.1.165. [DOI] [PubMed] [Google Scholar]
- 29.Conneely OM, Mulac-Jericevic B, Lydon JP, De Mayo FJ. Reproductive functions of the progesterone receptor isoforms: Lessons from knock-out mice. Mol Cell Endocrinol. 2001;179:97–103. doi: 10.1016/s0303-7207(01)00465-8. [DOI] [PubMed] [Google Scholar]
- 30.Mahendroo MS, Cala KM, Landrum DP, Russell DW. Fetal death in mice lacking 5alpha-reductase type 1 caused by estrogen excess. Mol Endocrinol. 1997;11:917–927. doi: 10.1210/mend.11.7.9933. [DOI] [PubMed] [Google Scholar]
- 31.Robertson E, Bradley A, Kuehn M, Evans M. Germ-line transmission of genes introduced into cultured pluripotential cells by retroviral vector. Nature. 1986;323:445–448. doi: 10.1038/323445a0. [DOI] [PubMed] [Google Scholar]
- 32.Keller G, Kennedy M, Papayannopoulou T, Wiles MV. Hematopoietic commitment during embryonic stem cell differentiation in culture. Mol Cell Biol. 1993;13:473–486. doi: 10.1128/mcb.13.1.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fowler AM, Solodin N, Preisler-Mashek MT, Zhang P, Lee AV, Alarid ET. Increases in estrogen receptor-alpha concentration in breast cancer cells promote serine 118/104/106-independent af-1 transactivation and growth in the absence of estrogen. FASEB J. 2004;18:81–93. doi: 10.1096/fj.03-0038com. [DOI] [PubMed] [Google Scholar]
- 34.H. J. Motulsky (1999). Analyzing data with Graphpad Prism. GraphPad Software, Inc., San Diego CA.
- 35.Minucci S, Botquin V, Yeom YI, Dey A, Sylvester I, Zand DJ, Ohbo K, Ozato K, Scholer HR. Retinoic acid-mediated down-regulation of Oct3/4 coincides with the loss of promoter occupancy in vivo. EMBO J. 1996;15:888–899. [PMC free article] [PubMed] [Google Scholar]
- 36.Pesce M, Scholer HR. Oct-4: Gatekeeper in the beginnings of mammalian development. Stem Cells. 2001;19:271–278. doi: 10.1634/stemcells.19-4-271. [DOI] [PubMed] [Google Scholar]
- 37.Leahy A, Xiong JW, Kuhnert F, Stuhlmann H. Use of developmental marker genes to define temporal and spatial patterns of differentiation during embryoid body formation. J Exp Zool. 1999;284:67–81. doi: 10.1002/(sici)1097-010x(19990615)284:1<67::aid-jez10>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 38.Kabrun N, Buhring HJ, Choi K, Ullrich A, Risau W, Keller G. Flk-1 expression defines a population of early embryonic hematopoietic precursors. Development. 1997;124:2039–2048. doi: 10.1242/dev.124.10.2039. [DOI] [PubMed] [Google Scholar]
- 39.Choi K, Kennedy M, Kazarov A, Papadimitriou JC, Keller G. A common precursor for hematopoietic and endothelial cells. Development. 1998;125:725–732. doi: 10.1242/dev.125.4.725. [DOI] [PubMed] [Google Scholar]
- 40.Fehling HJ, Lacaud G, Kubo A, Kennedy M, Robertson S, Keller G, Kouskoff V. Tracking mesoderm induction and its specification to the hemangioblast during embryonic stem cell differentiation. Development. 2003;130:4217–4227. doi: 10.1242/dev.00589. [DOI] [PubMed] [Google Scholar]
- 41.Kubo A, Shinozaki K, Shannon JM, Kouskoff V, Kennedy M, Woo S, Fehling HJ, Keller G. Development of definitive endoderm from embryonic stem cells in culture. Development. 2004;131:1651–1662. doi: 10.1242/dev.01044. [DOI] [PubMed] [Google Scholar]
- 42.Huang D, Chen SW, Langston AW, Gudas LJ. A conserved retinoic acid responsive element in the murine Hoxb-1 gene is required for expression in the developing gut. Development. 1998;125:3235–3246. doi: 10.1242/dev.125.16.3235. [DOI] [PubMed] [Google Scholar]
- 43.Hurd C, Moudgil VK. Characterization of R5020 and RU486 binding to progesterone receptor from calf uterus. Biochemistry. 1988;27:3618–3623. doi: 10.1021/bi00410a014. [DOI] [PubMed] [Google Scholar]
- 44.Mockus MB, Lessey BA, Bower MA, Horwitz KB. Estrogen-insensitive progesterone receptors in a human breast cancer cell line: Characterization of receptors and of a ligand exchange assay. Endocrinology. 1982;110:1564–1571. doi: 10.1210/endo-110-5-1564. [DOI] [PubMed] [Google Scholar]
- 45.Pink JJ, Jordan VC. Models of estrogen receptor regulation by estrogens and antiestrogens in breast cancer cell lines. Cancer Res. 1996;56:2321–2330. [PubMed] [Google Scholar]
- 46.Preisler-Mashek MT, Solodin N, Stark BL, Tyriver MK, Alarid ET. Ligand-specific regulation of proteasome-mediated proteolysis of estrogen receptor-alpha. Am J Physiol Endocrinol Metab. 2002;282:E891–898. doi: 10.1152/ajpendo.00353.2001. [DOI] [PubMed] [Google Scholar]
- 47.Parker MG. Action of "pure" antiestrogens in inhibiting estrogen receptor action. Breast Cancer Res Treat. 1993;26:131–137. doi: 10.1007/BF00689686. [DOI] [PubMed] [Google Scholar]
- 48.Smith CL, O’Malley BW. Coregulator function: A key to understanding tissue specificity of selective receptor modulators. Endocr Rev. 2004;25:45–71. doi: 10.1210/er.2003-0023. [DOI] [PubMed] [Google Scholar]
- 49.Alexander IE, Clarke CL, Shine J, Sutherland RL. Progestin inhibition of progesterone receptor gene expression in human breast cancer cells. Mol Endocrinol. 1989;3:1377–1386. doi: 10.1210/mend-3-9-1377. [DOI] [PubMed] [Google Scholar]
- 50.Das SK, Chakraborty I, Paria BC, Wang XN, Plowman G, Dey SK. Amphiregulin is an implantation-specific and progesterone-regulated gene in the mouse uterus. Mol Endocrinol. 1995;9:691–705. doi: 10.1210/mend.9.6.8592515. [DOI] [PubMed] [Google Scholar]
- 51.Bui TD, Zhang L, Rees MC, Bicknell R, Harris AL. Expression and hormone regulation of wnt2, 3, 4, 5a, 7a, 7b and 10b in normal human endometrium and endometrial carcinoma. Br J Cancer. 1997;75:1131–1136. doi: 10.1038/bjc.1997.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brisken C, Heineman A, Chavarria T, Elenbaas B, Tan J, Dey SK, McMahon JA, McMahon AP, Weinberg RA. Essential function of wnt-4 in mammary gland development downstream of progesterone signaling. Genes Dev. 2000;14:650–654. [PMC free article] [PubMed] [Google Scholar]
- 53.Robinson GW, Hennighausen L, Johnson PF. Side-branching in the mammary gland: The progesterone-wnt connection. Genes Dev. 2000;14:889–894. [PubMed] [Google Scholar]
- 54.Humphreys RC, Lydon J, O’Malley BW, Rosen JM. Mammary gland development is mediated by both stromal and epithelial progesterone receptors. Mol Endocrinol. 1997;11:801–811. doi: 10.1210/mend.11.6.9891. [DOI] [PubMed] [Google Scholar]
- 55.Rudolph MC, McManaman JL, Hunter L, Phang T, Neville MC. Functional development of the mammary gland: Use of expression profiling and trajectory clustering to reveal changes in gene expression during pregnancy, lactation, and involution. J Mammary Gland Biol Neoplasia. 2003;8:287–307. doi: 10.1023/b:jomg.0000010030.73983.57. [DOI] [PubMed] [Google Scholar]
- 56.Martinez-Lacaci I, Saceda M, Plowman GD, Johnson GR, Normanno N, Salomon DS, Dickson RB. Estrogen and phorbol esters regulate amphiregulin expression by two separate mechanisms in human breast cancer cell lines. Endocrinology. 1995;136:3983–3992. doi: 10.1210/endo.136.9.7649107. [DOI] [PubMed] [Google Scholar]
- 57.Hong SH, Nah HY, Lee YJ, Lee JW, Park JH, Kim SJ, Lee JB, Yoon HS, Kim CH. Expression of estrogen receptor-alpha and -beta, glucocorticoid receptor, and progesterone receptor genes in human embryonic stem cells and embryoid bodies. Mol Cells. 2004;18:320–325. [PubMed] [Google Scholar]
- 58.Petz LN, Nardulli AM. Sp1 binding sites and an estrogen response element half-site are involved in regulation of the human progesterone receptor A promoter. Mol Endocrinol. 2000;14:972–985. doi: 10.1210/mend.14.7.0493. [DOI] [PubMed] [Google Scholar]
- 59.Petz LN, Ziegler YS, Loven MA, Nardulli AM. Estrogen receptor alpha and activating protein-1 mediate estrogen responsiveness of the progesterone receptor gene in MCF-7 breast cancer cells. Endocrinology. 2002;143:4583–4591. doi: 10.1210/en.2002-220369. [DOI] [PubMed] [Google Scholar]
- 60.Xu X, Murdoch FE, Curran EM, Welshons WV, Fritsch MK. Transcription factor accessibility and histone acetylation of the progesterone receptor gene differs between parental MCF-7 cells and a subline that has lost progesterone receptor expression. Gene. 2004;328:143–151. doi: 10.1016/j.gene.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 61.Schultz JR, Petz LN, Nardulli AM. Cell- and ligand-specific regulation of promoters containing activator protein-1 and Sp1 sites by estrogen receptors {alpha} and {beta} J Biol Chem. 2005;280:347–354. doi: 10.1074/jbc.M407879200. [DOI] [PubMed] [Google Scholar]
- 62.Petz LN, Ziegler YS, Schultz JR, Kim H, Kemper JK, Nardulli AM. Differential regulation of the human progesterone receptor gene through an estrogen response element half site and Sp1 sites. J Steroid Biochem Mol Biol. 2004;88:113–122. doi: 10.1016/j.jsbmb.2003.11.008. [DOI] [PubMed] [Google Scholar]
- 63.Kaneko KJ, Gelinas C, Gorski J. Activation of the silent progesterone receptor gene by ectopic expression of estrogen receptors in a rat fibroblast cell line. Biochemistry. 1993;32:8348–8359. doi: 10.1021/bi00083a039. [DOI] [PubMed] [Google Scholar]
- 64.Yang X, Phillips DL, Ferguson AT, Nelson WG, Herman JG, Davidson NE. Synergistic activation of functional estrogen receptor (ER)-alpha by DNA methyltransferase and histone deacetylase inhibition in human ER-alpha-negative breast cancer cells. Cancer Res. 2001;61:7025–7029. [PubMed] [Google Scholar]
- 65.Raman V, Tamori A, Vali M, Zeller K, Korz D, Sukumar S. Hoxa5 regulates expression of the progesterone receptor. J Biol Chem. 2000;275:26551–26555. doi: 10.1074/jbc.C000324200. [DOI] [PubMed] [Google Scholar]
- 66.Hagihara K, Wu-Peng XS, Funabashi T, Kato J, Pfaff DW. Nucleic acid sequence and DNase hypersensitive sites of the 5′ region of the mouse progesterone receptor gene. Biochem Biophys Res Commun. 1994;205:1093–1101. doi: 10.1006/bbrc.1994.2778. [DOI] [PubMed] [Google Scholar]
- 67.Raman V, Martensen SA, Reisman D, Evron E, Odenwald WF, Jaffee E, Marks J, Sukumar S. Compromised Hoxa5 function can limit p53 expression in human breast tumours. Nature. 2000;405:974–978. doi: 10.1038/35016125. [DOI] [PubMed] [Google Scholar]
- 68.Aubin J, Dery U, Lemieux M, Chailler P, Jeannotte L. Stomach regional specification requires Hoxa5-driven mesenchymal-epithelial signaling. Development. 2002;129:4075–4087. doi: 10.1242/dev.129.17.4075. [DOI] [PubMed] [Google Scholar]