

Abstract
The molecular mechanisms regulating the amount of dietary cholesterol retained in the body, as well as the body’s ability to exclude selectively other dietary sterols, are poorly understood. An average western diet will contain about 250–500 mg of dietary cholesterol and about 200–400 mg of non-cholesterol sterols. About 50–60% of the dietary cholesterol is absorbed and retained by the normal human body, but less than 1% of the non-cholesterol sterols are retained. Thus, there exists a subtle mechanism that allows the body to distinguish between cholesterol and non-cholesterol sterols. In sitosterolemia, a rare autosomal recessive disorder, affected individuals hyperabsorb not only cholesterol but also all other sterols, including plant and shellfish sterols from the intestine1,2. The major plant sterol species is sitosterol; hence the name of the disorder. Consequently, patients with this disease have very high levels of plant sterols in the plasma and develop tendon and tuberous xanthomas, accelerated atherosclerosis, and premature coronary artery disease3. We previously mapped the STSL locus to human chromosome 2p21 (ref. 4) and further localized it to a region of less than 2 cM bounded by markers D2S2294 and D2S2291 (M.-H.L. et al., manuscript submitted). We now report that a new member of the ABC transporter family, ABCG5, is mutant in nine unrelated sitosterolemia patients.
A number of ESTs and genes were mapped into the region of interest and screened, based upon whether they were expressed in the liver and/or intestine, the organs important in dietary cholesterol retention (K.L. et al., manuscript submitted). Three ESTs were expressed in the liver and intestine only, of which one, T99836, was found to encode a ‘half-ABC’ transporter (for ATP binding cassette), and was studied further. A corresponding full-length cDNA was isolated and the gene structure characterized5. The gene (called ABCG5) consists of 13 exons, and encodes a 651-residue, 70 kD protein with 6 putative membrane-spanning domains that contains the characteristic ABC signature motifs at its amino-terminal end (in general, ABC proteins consist of 12 membrane spanning domains, hence the term ‘half-transporter’). We screened probands of nine families (Fig. 1) for mutations using a combination of SSCP and direct sequencing of PCR products. Based on their haplotype analyses, eight were expected to carry a homozygous mutation and one (family 3300, proband 132) was predicted to be a compound heterozygote (M.-H.L. et al., manuscript submitted). Analyses by single strand conformation polymorphism (SSCP) indicated alterations in exons 3, 6, 9 and 13 of the gene (data not shown). Of these, a similar variant in exon 13 was also detected in control samples, suggesting it to be a polymorphic change (Table 1).
Fig. 1.
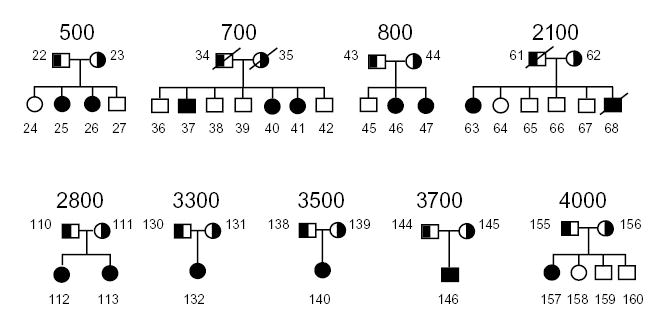
Pedigrees with sitosterolemia. The parents are depicted as obligate carriers, and the affected individuals are depicted as filled symbols. None of these pedigrees are consanguineous.
Table 1.
Frequency of nucleotide changes in unrelated Japanese and North Americans of European descent
| Nucleotide change | Predicted consequences | Carrier frequency | Restriction endonuclease changes |
|---|---|---|---|
| C167T | P9P | not screened | gain of BstNI site |
| Δ20 bp at 402 | frameshift & truncated protein | no carriers in 55 Japanese controls | - |
| C867T | R243X | not screened | gain of AlwNI site |
| G1306A | R389H | no carriers in 145 Japanese and 156 Caucasians | Loss of BstUI site |
| C1362T | R408X | not screened | loss of AvaI site |
| G1396A | R419H | no carriers in 145 Japanese and 156 Caucasians | Loss of BstUI site |
| G1396C | R419P | no carriers in 145 Japanese and 156 Caucasians | Loss of BstUI site |
| C1950G | Q604E | 36% carriers in Caucasians | loss of SmlI site |
Mutations (R243X, R408X, and R419H/P) or polymorphism Q604E were screened in unrelated Japanese and North Americans of European descent, using the restriction assays described in Fig. 2.
As potential sequence variants in exons 3, 6 and 9 were detected only in affected individuals and not in controls, they were directly sequenced. We identified five point mutations: R243X (exon 6, proband 25), R389H (exon 9, probands 46, 113 and 146), R408X (exon 9, proband 140), R419H (exon 9, probands 40 and 132) and R419P (exon 9, proband 157) (Fig. 2a). A complex deletion mutation (exon 3) was identified in one proband (Fig. 2b). To confirm that the missense nucleotide changes were mutations and not polymorphisms, we used the altered restriction endonuclease recognition sequences as an assay to perform segregation analyses, and to screen normal populations (Fig. 2 and Table 1). Mutations resulting in R243X, R408X, R389H and R419H/P altered cleavage sites of restriction enzymes. R243X mutation segregated with disease in pedigree 500; both parents are carriers and both affected children are homozygous (Fig. 2a) for the mutation. Similarly, correlations are observed with other mutations (Fig. 2), except R408X, in which the mutation was found in a single individual whose parents were not available for analysis.
Fig. 2.
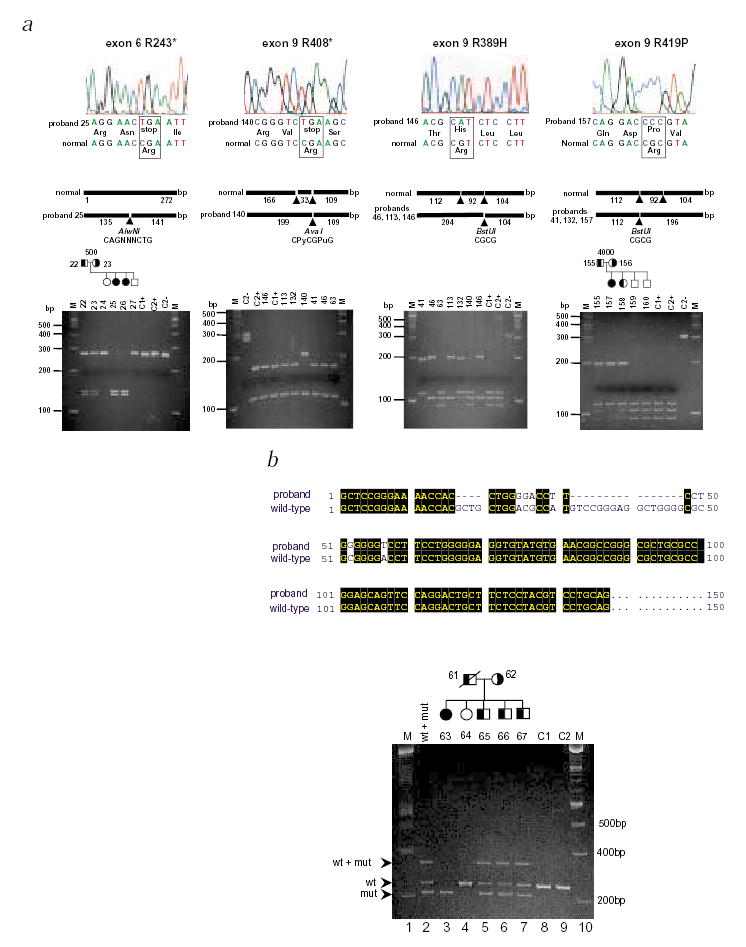
Mutational analyses in ABCG5 in sitosterolemia probands. a, The top panels show the sequence electropherograms; the middle panels, the sizes and restriction enzyme fragments; and the bottom panels, the gel electrophoresis of digested PCR fragments. Mutation R243X (left-hand panels) results in a gain of the AlwnI site. Using this as an assay, this mutation segregated within pedigree 500. Similarly, mutation R419P causes a loss of the second BstUI site and can be shown to segregate appropriately in pedigree 4000 (right-hand panels). BstUI can also be used to screen for R419H (data not shown). Additionally, the first BstUI site is altered by mutation R389H (middle right panels). Thus, digestion of the exon 9 PCR product with BstUI allows screening of all three mutations. Mutation R408X alters AvaI site (middle left panels). Control samples are indicated by the letters C1 and C2, with + or – indicating digested and undigested samples. Marker tracks (M) are as indicated. b, Following the detection of an abnormal SSCP for exon 3, direct sequencing of the PCR product revealed complex deletion and base substitutions, as depicted by the CLUSTALW alignment of the mutant product with the wild-type sequence. After re-designing the primer-pairs (exon3F and exon3R) to better detect the 20-bp size difference, the mutant alleles could be shown to segregate within pedigree 2100. Lane 2 contains a mixture of wild-type and mutant PCR products that were mixed, denatured and annealed before loading. The slower migrating band is therefore a hybrid between a wild-type strand and a mutant strand. Its presence is detected in heterozygotes only (lanes 5, 6 and 7). Based upon haplotype analyses, siblings 65, 66 and 67 are predicted to be carriers and sibling 64 is homozygous wild type, in good agreement with the segregation of the deletion. Maternal non-paternity was detected in this family and her sample was excluded from analyses (data not shown).
A homozygous complex deletion and base substitution mutation within exon 3 was identified for proband 63 (Fig. 2b). Based upon haplotype analyses (M.-H.L. et al., manuscript submitted), we correctly predicted siblings 65, 66 and 67 to be carriers of the mutation and sibling 64, free of it. The predicted effect of this mutation is a frameshift, resulting in an ORF that terminates in exon 5, and the translation of a truncated protein of approximately 20 kD devoid of the transmembrane domains (Fig. 3). It is more probable, however, that such nonsense mutations are likely to lead to rapid mRNA degradation via the nonsense-mediated mRNA decay pathway6. We screened 145 normal Japanese and 156 US Caucasians for these missense nucleotide changes mutations (Table 1); all tested negative.
Fig. 3.
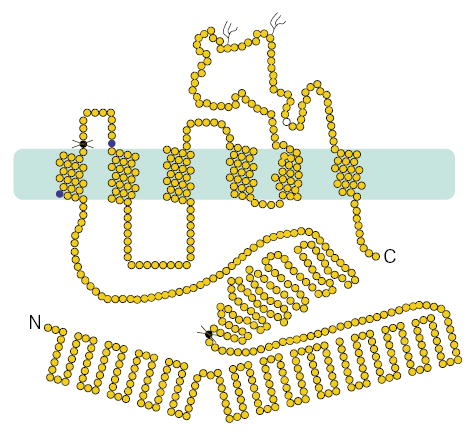
Predicted sterolin structure and position of the mutations. The polypeptide is predicted to have a very large N termini and both the N and C termini are predicted to be cytosolic. The transmembrane domains are contained within the second half of the polypeptide, encoded by exons 9–13. Two potential N-glycosylation sites are as indicated on a loop that faces the lumenal or cell surface. The polymorphic variant, Q604E (open circle) is also within this loop. The mutations (dark filled circles) are as indicated, with stop codon mutations indicated by the crosses.
The N-terminal region of ABCG5 is predicted to be cytosolic and contains characteristic ABC motifs; its sequence predicts six transmembrane domains and two potential N-glycosylation (Fig. 3). We propose the use of the name ‘sterolin’ for the protein product of ABCG5, as this reflects its putative function. Of the two missense mutations of ABCG5, both are located very close to transmembrane regions. These may lead to disruption of protein stability, as has been previously reported for mutations in DHCR7 leading to the Smith-Lemli-Opitz syndrome7.
Selectivity for sterol absorption is a feature of other mammals (such as mice, rats and dogs); cholesterol is absorbed and retained by the body whereas non-cholesterol sterols are not. One might therefore expect the gene whose mutation results in sitosterolemia to be highly conserved among these species. We isolated and sequenced the full-length mouse ABCG5 cDNA and carried out phylogenetic analyses with other known ABC proteins (Fig. 4). Mouse sterolin shows 80% conservation relative to human sterolin at the nucleotide level and 85% identity at the protein level. The nearest non-mammalian neighbor is a yeast putative ABC protein (YOLO75C, Genbank IDs Z74816, Z74817), to which no function has been assigned; its homology with the mouse and human sterolins is confined to its carboxy-terminal region only. And so sterolins would seem to represent a separate ‘family’ of ABC transporters, and are not closely related to either ABC1, mutated in Tangier disease8–10, or the multiple drug resistance proteins, MDRs (refs. 11,12), involved in the transport of phospholipid into bile. However, they are closely related to the ABC8/white family, the mammalian homologues of which have been shown to be involved in macrophage cholesterol and phospholipid accumulation13.
Fig. 4.
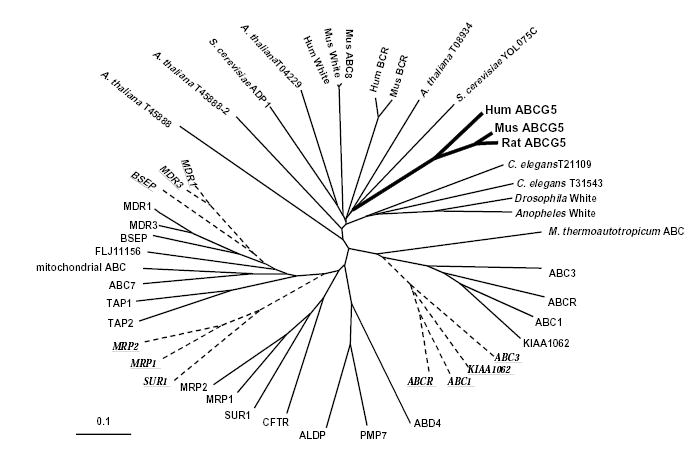
Phylogenetic analysis of human, mouse and yeast ABC proteins. Phylogenetic analyses of sterolin and ABC proteins were carried out as described in the text. Each ABC family member was included on the basis of identification by its ATP-binding domain and the inclusion of approximately 10 aa upstream and 120 aa downstream of this site. Some ABC proteins contain two ATP-binding sites. In these cases, each site was analyzed independently; the first site is depicted in by the continuous lines, and the second site by the broken lines. Sterolin forms a distinct family (bold solid lines), with its nearest neighbor being the White gene family, the mammalian homologues of which include ABC8 family members.
Based on the clinical defects in sitosterolemia, the gene is predicted to be expressed in the liver and/or the intestine. Northern-blot analysis showed ABCG5 expression was detected in the liver only5. However, by RT–PCR analysis, expression is detected in human intestine, and adult and fetal liver (Fig. 5a). Consistent with this finding is northern-blot analysis of RNA extracted from mouse tissues: we observed expression in both liver and intestine (Fig. 5b). Thus, sterolin is expressed in a tissue-specific manner, consistent with the disease profile.
Fig. 5.
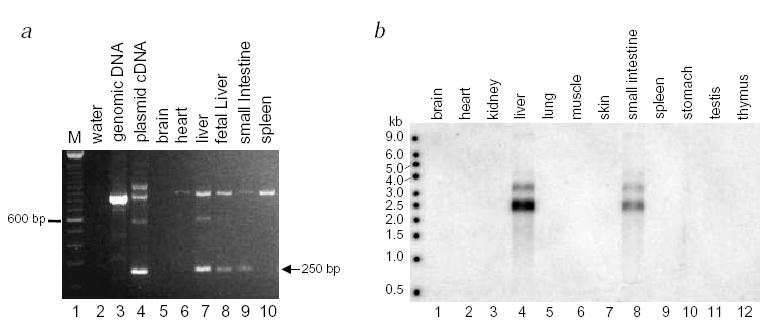
Analysis of Abcg5 expression. a, PCR products were obtained from cDNA synthesized from RNA extracted from different human tissues. A correctly spliced 250-bp fragment is expected for detection of expression (indicated by arrow), compared with a genomic fragment of 838 bp (compare cloned cDNA fragment, track 4, with genomic DNA fragment, track 3). A number of other PCR products were also consistently obtained, but did not contain ABCG5 sequence. b, Tissue expression of mouse Abcg5 was analyzed by northern-blot analysis. An expected 2.5-kb message was detected, with a fainter band of about 3.5 kb, in liver and small intestine only (tracks 4 and 8).
Although we have screened 30 families with sitosterolemia, we have been able to identify mutations in only 9 families. Preliminary analyses, by direct sequencing of all the exons, failed to identify mutations in the remaining probands. This suggests that either more subtle mutations, involving promoter sites or intronic regions may be affected, or that another closely-linked second locus may be involved. Sterolin is probably involved in the selective transport of dietary cholesterol in and out of enterocytes, and in selective sterol excretion by the liver into bile, as evidenced by the consequences when it is deficient. The identification of the specific mechanisms by which these processes come about will be greatly facilitated by the identification of ABCG5 as the gene which, when mutated, causes sitosterolemia.
Methods
Pedigrees
All pedigrees were recruited, based upon previously defined criteria4,14. Clinical features of some of the probands and their family members have been described4,14. Briefly, all probands had both clinical features compatible with a diagnosis of sitosterolemia and had diagnostically elevated plasma sitosterol levels. The pedigrees include six Japanese families (700, 800, 2100, 2800, 3300, 3500 and 3700), one South African family of Asian origin (500) and one United States family (4000). Informed consent was obtained from all participants, in accordance with local Institutional Review Board guidelines.
Exon amplification and DNA sequencing
Exons were amplified by PCR using oligonucleotide primers located in the flanking intronic area, as described5. Additional primers used were as follows: Wh3F1, 5′–CCCAACT GAAGCCACTCTGGGGA–3′ (in 5′ flanking region); Wh3R4, 5′–GCAA GATGTGATGTCCCACCA–3′ (in exon 2); exon3F, 5′–CTCTAGGGCCTT CTGTTG–3′; exon3R, 5′–GCGTCAGTGTAGCCT–3′; exon4F, 5′–CTTAG GCTACACTGACGC–3′; exon4R, 5′–GGGTGCAAAGGTACTCAG–3′; exon8F, 5′–CACATGGGTGACATCTTT–3′; exon8R, 5′–TCTCACATTTGT-GAGCCT–3′; exon9F, 5′–GAGGTCTTTAGCCATCCC–3′; exon9R, 5′–AGA AAGAGGTGCACCTCC–3′; exon12F, 5′–CTACTGAATTTCATTTTTGT TTTC–3′; exon12R, 5′–CATGCAAAAATAATATCCCCA–3′; exon13F, 5′–ACACCTTGACACTGTCAA–3′; exon13R, 5′–TTTCCCAGCCATGGCT TT–3′. SSCP variants were identified by incorporation PCR-SSCP (IPS) as described15. Direct sequencing of PCR products was performed using Amplicycle Sequencing kit (Perkin Elmer) and analyzed by ABIPRISM 377 Genetical Analyzer (Perkin Elmer). Both strands were sequenced to confirm the identified mutations. Sequence alignment was aided by the use of MacVector software (Oxford Molecular) running on an Apple iMac (Apple Corp). PCR products were digested with appropriate restriction endonucleases as described and analyzed by agarose or acrylamide gel electrophoresis16.
Isolation of mouse and rat sterolin cDNAs
Isolation of the human sterolin cDNA and its genomic organization has been described5. To identify the mouse cDNA, two primers, located in exons 4 and 10 respectively were used to amplify a fragment from cDNA synthesized from mouse liver. The resultant PCR product was directly sequenced, and a full-length cDNA obtained by 5′- and 3′-RACE–PCR. A partial rat cDNA clone was identified using the above primers and a rat enterocyte cDNA library as template.
Expression analyses
Northern-blot analysis was performed as described17. A multiple-tissue northern blot, containing 2 μg of poly(A)+ RNA (Origene) was hybridized with a full-length mouse cDNA for ABCG5. The hybridized filter was washed stringently with 0.1 × SSC/0.1 %SDS at 68 °C, exposed to a phosphorimager cassette, striped and re-probed with either mouse β-actin or GAPDH probed for comparison of RNA loading. For RT–PCR, human cDNAs (Origene) were used to amplify a fragment spanning exon 1 and 2 using oligonucleotides Wh3f1 and Wh3r4. A 250-bp product from cDNA is expected, compared with an 838-bp fragment from the genomic DNA.
Sequence alignments and phylogenetic analyses
Sterolin protein sequences were aligned with other ABC-transporter proteins using CLUSTALW (v 1.7; ref. 18). ABC protein sequences included in the alignment were based on a variety BLAST and TFASTX searches of databases. To ensure appropriate comparison of ABC proteins that included ‘half-transporters’, such as sterolin, to ABC proteins that contain two ATP-binding sites and ABC motifs, alignment was performed by identifying the ATP-binding site, selecting ~10 amino acids upstream through to 120 amino acids downstream of this site, and analyzing each such segment independently. Thus ABC1, which has two such segments is represented twice, the second site is indicated by the broken lines and the protein underlined (Fig. 4). Note that those ABC proteins that associate with the first site, are also closely grouped using the second site (Fig. 4). The alignment was assessed with 1000 bootstrap re-samplings using the integral options of CLUSTALW. In addition, the alignment was subjected to the heuristic bootstrap analysis option of the maximum parsimony program PAUP (v 4.0; ref. 19). A further bootstrap re-sampling was performed with the protein distance programs of the PHYLIP package (v3.5c; ref. 20). In all three analyses, the sterolin proteins associate on branches of the resulting phylogenetic trees that distinguish them from the WHITE proteins of various other species, but place within this family members as their closest relatives (Fig. 4). The trees produced by the programs were transferred to TreeView (v1.5; ref. 21) for manipulation and printing.
Accession numbers
Human ABCG5 cDNA, AF312715; mouse Abcg5, AF312713; rat partial cDNA, AF312714.
Acknowledgments
We thank the families for participation; Y. Zhou for technical assistance; the General Clinical Research Center, MUSC, for assistance with sequencing; and the members of the Patel lab for discussions. This work was supported by grants from the NIH and USPHS.
Footnotes
Note added in proof: Shortly before acceptance of our manuscript, Berge et al.22 reported identification of one mutation in ABCG5, and nine in ABCG8, in probands with sitosterolemia. ABCG5 and ABCG8 are genes in tandem, separated by less than 400 bases.
References
- 1.Bhattacharyya AK, Connor WE. β-sitosterolemia and xanthomatosis. A newly described lipid storage disease in two sisters. J Clin Invest. 1974;53:1033–1043. doi: 10.1172/JCI107640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gregg RE, Connor WE, Lin DS, Brewer H., Jr Abnormal metabolism of shellfish sterols in a patient with sitosterolemia and xanthomatosis. J Clin Invest. 1986;77:1864–1872. doi: 10.1172/JCI112513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bjorkhem, I. & Boberg, K.M. Inborn errors in bile acid biosynthesis and storage of sterols other than cholesterol. in The Metabolic Basis of Inherited Disease (eds. Scriver, C.R., Beaudet, A.L., Sly, W.S. & Valle, D.) 2073–2102 (McGraw-Hill, New York, 1995).
- 4.Patel SB, et al. Mapping a gene involved in regulating dietary cholesterol absorption. The sitosterolemia locus is found at chromosome 2p21. J Clin Invest. 1998;102:1041–1044. doi: 10.1172/JCI3963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shulenin, S. et al. A liver-specific ATP-binding cassette gene (ABCG5) from the ABCG (White) gene subfamily maps to human chromosome 2p21 in the region of the sitosterolemia locus. Cyto. Cell Genet. (in press). [DOI] [PubMed]
- 6.Frischmeyer PA, Dietz HC. Nonsense-mediated mRNA decay in health and disease. Hum Mol Genet. 1999;8:1893–1900. doi: 10.1093/hmg/8.10.1893. [DOI] [PubMed] [Google Scholar]
- 7.Witsch-Baumgartner M, et al. Mutational spectrum in the δ7-sterol reductase gene and genotype-phenotype correlation in 84 patients with Smith-Lemli-Opitz syndrome. Am J Hum Genet. 2000;66:402–412. doi: 10.1086/302760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brooks-Wilson A, et al. Mutations in ABC1 in Tangier disease and familial high-density lipoprotein deficiency. Nature Genet. 1999;22:336–345. doi: 10.1038/11905. [DOI] [PubMed] [Google Scholar]
- 9.Bodzioch M, et al. The gene encoding ATP-binding cassette transporter 1 is mutated in Tangier disease. Nature Genet. 1999;22:347–351. doi: 10.1038/11914. [DOI] [PubMed] [Google Scholar]
- 10.Rust S, et al. Tangier disease is caused by mutations in the gene encoding ATP-binding cassette transporter 1. Nature Genet. 1999;22:352–355. doi: 10.1038/11921. [DOI] [PubMed] [Google Scholar]
- 11.Erlinger S. Review article: new insights into the mechanisms of hepatic transport and bile secretion. J Gastroenterol Hepatol. 1996;11:575–579. doi: 10.1111/j.1440-1746.1996.tb01705.x. [DOI] [PubMed] [Google Scholar]
- 12.Klein I, Sarkadi B, Varadi A. An inventory of the human ABC proteins. Biochim Biophys Acta. 1999;1461:237–262. doi: 10.1016/s0005-2736(99)00161-3. [DOI] [PubMed] [Google Scholar]
- 13.Klucken J, et al. ABCG1 (ABC8), the human homolog of the Drosophila white gene, is a regulator of macrophage cholesterol and phospholipid transport. Proc Natl Acad Sci USA. 2000;97:817–822. doi: 10.1073/pnas.97.2.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Patel SB, Honda A, Salen G. Sitosterolemia: exclusion of genes involved in reduced cholesterol biosynthesis. J Lipid Res. 1998;39:1055–1061. [PubMed] [Google Scholar]
- 15.Sossey-Alaoui K, et al. Molecular cloning and characterization of TRPC5 (HTRP5), the human homologue of a mouse brain receptor-activated capacitative Ca2+ entry channel. Genomics. 1999;60:330–340. doi: 10.1006/geno.1999.5924. [DOI] [PubMed] [Google Scholar]
- 16.Yu H, Tint GS, Salen G, Patel SB. Detection of a common mutation in the RSH or Smith-Lemli-Opitz syndrome by a PCR-RFLP assay: IVS8-G—>C is found in over sixty percent of US propositi. Am J Med Genet. 2000;90:347–350. doi: 10.1002/(sici)1096-8628(20000214)90:4<347::aid-ajmg16>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 17.Wu J, Zhu YH, Patel SB. Cyclosporin-induced dyslipoproteinemia is associated with selective activation of SREBP-2. Am J Physiol. 1999;277:E1087–1094. doi: 10.1152/ajpendo.1999.277.6.E1087. [DOI] [PubMed] [Google Scholar]
- 18.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Swofford, D.L. & Olsen, G.J. Phylogeny reconstruction. in Molecular Systematics (eds. Hillis, D.M. & Moritz, C.) 411–501 (Sinauer Asociates, Sunderland, 1990).
- 20.Felsenstein J. Inferring phylogenies from protein sequences by parsimony, distance, and likelihood methods. Methods Enzymol. 1996;266:418–427. doi: 10.1016/s0076-6879(96)66026-1. [DOI] [PubMed] [Google Scholar]
- 21.Page RD. TreeView: an application to display phylogenetic trees on personal computers. Comput Appl Biosci. 1996;12:357–358. doi: 10.1093/bioinformatics/12.4.357. [DOI] [PubMed] [Google Scholar]
- 22.Berge KE, et al. Accumulation of dietary cholesterol in sitosterolemia caused by mutations in adjacent ABC transporters. Science. 2000;290:1771–1775. doi: 10.1126/science.290.5497.1771. [DOI] [PubMed] [Google Scholar]