

Abstract
The molecular mechanisms regulating the amount of dietary cholesterol retained by the body, as well as the body’s ability to exclude other dietary sterols selectively, are poorly understood. An average Western diet will contain approximately 250–500 mg of dietary cholesterol and approximately 200–400 mg of non-cholesterol sterols, of which plant sterols are the major constituents. Approximately 50–60% of dietary cholesterol is absorbed and retained by the normal human body, but less than 1% of the non-cholesterol sterols are retained. There thus exists a subtle mechanism that allows the body to distinguish between cholesterol and non-cholesterol sterols. In sitosterolemia, a rare autosomal recessive disorder, affected individuals hyperabsorb and retain not only cholesterol but also all other sterols, including plant and shellfish sterols from the intestine. Consequently, patients with this disease have very high levels of plant sterols in the plasma, and develop tendon and tuberous xanthomas, accelerated atherosclerosis, and premature coronary artery disease. The STSL locus has been mapped to human chromosome 2p21. Mutations in two tandem ABC genes, ABCG5 and ABCG8, encoding sterolin-1 and -2, respectively, are now known to be mutant in sitosterolemia. The identification of these genes should now lead to a better understanding of the molecular mechanism(s) governing the highly selective absorption and retention of cholesterol by the body. Indeed, it is the very existence of this disease that has given credence to the hypothesis that there is a molecular pathway that regulates dietary cholesterol absorption and sterol excretion by the body.
Abbreviations: ABC adenosine triphosphate binding cassette, FH familial hypercholesterolemia
Introduction
Under normal circumstances, our diets contain almost equal amounts of cholesterol and plant sterols. However, normal individuals retain on average approximately 40– 50% of dietary cholesterol, but less than 5% of the dietary plant sterols [1–3]. As sitosterol constitutes the major species of plant sterol, this term will be utilized. The normal body is thus able to discriminate exquisitely between cholesterol and non-cholesterol sterols, although the mechanism by which this takes place is not known. This ‘discrimination’ applies to a variety of non-cholesterol sterols, such as shellfish sterols [4], and appears to suggest that the molecular mechanism may specifically retain cholesterol over non-cholesterol sterols in the body.
Sitosterolemia (also known as phytosterolemia, MIM 210250) is a rare autosomal recessively inherited lipid metabolic disorder, first described in two affected sisters by Bhattacharyya and Connor in 1974 [5]. Sitosterolemia is characterized by the presence of tendon xanthomas, premature coronary artery disease and atherosclerotic disease, hemolytic episodes, arthralgias and arthritis [6]. The hallmark of sitosterolemia is diagnostically elevated levels of plant sterols in the plasma [6–8]. The sitosterolemia locus, STSL, has been mapped to chromosome 2p21 [8], and further localized to a region bounded by markers D2S2294 and Afm210xe9 [9•]. The defective gene in sitosterolemia is hypothesized to play an important role in regulating dietary sterol absorption and biliary secretion [5,8,10]. Therefore, the elucidation of this gene disorder is important to identify selective exclusion mechanisms of non-cholesterol from the body. Mutations in two ‘half’ adenosine triphosphate binding cassette (ABC) transporters, ABCG5 and ABCG8, encoding proteins sterolin-1 and -2, respectively, are now known to cause this disease [11••,12••].
Clinical features of sitosterolemia
Sitosterolemia patients develop tendon and tuberous xanthomas, hemolytic episodes, arthralgias and arthritis, and premature coronary and aortic atherosclerosis leading to cardiac fatalities [5,7,13–16]. Sitosterolemia shares several clinical characteristics with the well-characterized homozygous familial hypercholesterolemia (FH), such as the development of tendon xanthomas in the first 10 years of life and the development premature atherosclerosis [6]. However, in contrast to FH patients, sitosterolemia patients usually have normal to moderately elevated total sterol levels and very high levels of plant sterols in their plasma (Fig. 1) [6,8,17]. More recently, we have noted that when children are diagnosed with sitosterolemia, their cholesterol levels can be very high [8]. Previously, such children were misdiagnosed as ‘pseudo-homozygous’ FH (as they did not have defects of the LDL receptor, yet had the clinical features of FH). We have been able to confirm that they have sitosterolemia, in some cases many years after their initial presentation (S.B. Patel and G. Salen, unpublished observations). It is unclear if all affected sitosterolemia individuals go through a period in childhood with very elevated plasma cholesterol levels, because none of the affected individuals, diagnosed as adults, shows such high plasma cholesterol levels (S.B. Patel and G. Salen, unpublished observations).
Figure 1. Plasma sitosterol and cholesterol levels in affected individuals, their parents, clinically unaffected siblings, and normal controls.
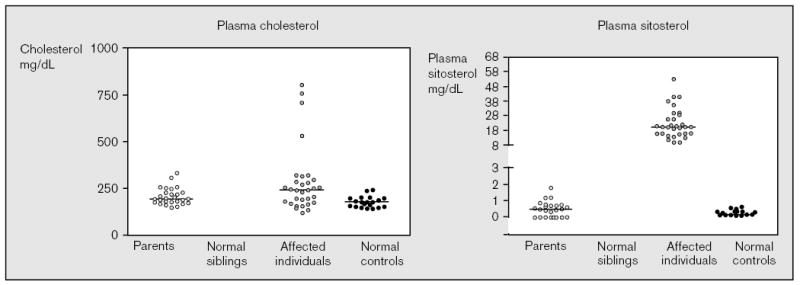
The plasma sitosterol levels were determined using capillary gas liquid or high performance liquid chromatography. In general, most unaffected individuals had plasma sitosterol levels of less than 1 mg/dl. Of the three parents and three siblings who had values higher than 1 mg/dl, none was higher than 2 mg/dl. All of the affected individuals had plasma sitosterol values greater than 8 mg/dl, and many of these values were obtained while the patients were on treatment. Note that the plasma cholesterol levels are not helpful. Three children all have mutations.
In sitosterolemia, affected individuals show very high levels of plasma plant sterols (sitosterol, campesterol, stigmasterol, avenosterol) and 5α-saturated stanols [7]. Plasma sitosterol levels are 10–25 times higher than in normal individuals (8–60 mg/dl). Affected individuals appear to hyperabsorb all sterols, including cholesterol, and have an inability to excrete sterols into the bile. At least two organ systems, the intestine and liver, are thus directly affected by this disorder. Parents and unaffected siblings show normal levels of cholesterol and sitosterol in their plasma (Fig. 1), suggesting that even with one functional copy of the gene, the body can adequately exclude non-cholesterol sterols.
Sitosterol is the major plant sterol species, hence the name sitosterolemia, although many other plant sterols and their metabolites (primarily 5α-stanols) are also significantly elevated in the plasma. Therefore, the definitive diagnosis of sitosterolemia is made by the detection of elevated phytosterols in the plasma using either capillary gas liquid or high performance liquid chromatography [18]. The requirement for specialized techniques to detect plasma phytosterols may lead to the under-detection of sitosterolemia cases.
Genetics studies of sitosterolemia
Only 45 cases have been reported in the literature worldwide, hence this condition appears to be relatively rare. The true prevalence of the disorder is not known. However, under-reporting may play a role, as the diagnosis requires both an initial suspicion on the part of the physician and a specialized test. On the basis of linkage analyses of 10 well-characterized pedigrees, sitosterolemia locus STSL was mapped to human chromosome 2p21, between microsatellite markers D2S1788 and D2S1352 [8]. We have assembled a unique collection of sitosterolemia pedigrees (Fig. 2). Genetic analyses of these pedigrees allowed us to refine the STSL locus to an interval less than 2 cM, between microsatellite markers D2S2294 and Afm210xe9 [9,19]. The pedigrees are from a number of different geographical and ethnic origins; seven are Japanese (700, 800, 2100, 2800, 3300, 3500 and 3700), two are Amish/Mennonite (2200 and 2700), two are Finnish (400 and 3900), one Norwegian (2900), two are Caucasian South African (600 and 3600), one Asian South African family (500), one Colombian family (3400, 2nd cousin marriage) and one Dutch family (2500) [9]. The remaining families are from the United States, and have Caucasian origins, except for family 3800, which is African American [9].
Figure 2. Sitosterolemia pedigrees.
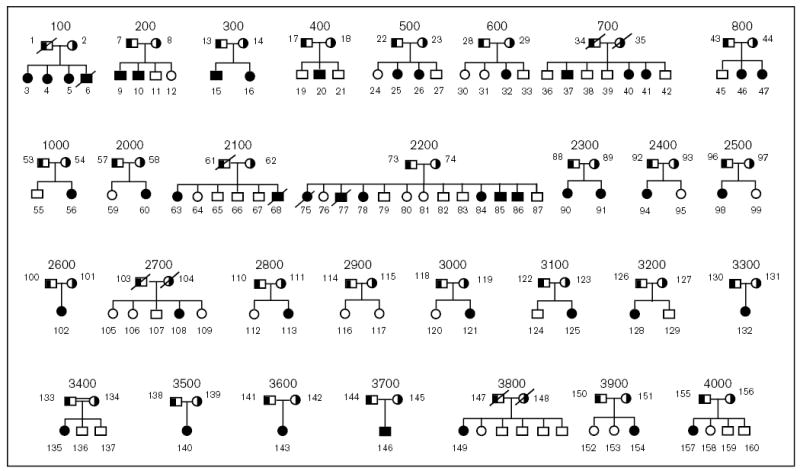
The pedigrees analyzed in this study are shown above. All parents are shown as obligate carriers. The one known consanguineous marriage, pedigree 3400, is as indicated. Affected individuals are indicated by the filled symbols, the obligate carriers (parents) by the half-filled symbols. All unaffected siblings are shown by unfilled symbols.
Homozygosity and founder effects
Many of the probands, despite coming from non-consanguineous marriages, showed homozygosity for a number of markers spanning the STSL locus [9]. Only one pedigree, 3400, is of known consanguineous marriage. All but one of the Japanese probands (pedigrees 700, 800, 2100, 2800, 3500 and 3700) showed homozygosity for a number of contiguous markers [9]. Only the proband from family 3300 was heterozygous for many of the markers in the critical region. The proband from one of the white South African families (pedigree 600) and one Asian South African family (pedigree 500) were also homozygous for a number of microsatellite markers. Three of the US Caucasian families (pedigrees 2300, 3100 and 4000) also showed homozygosity (Fig. 3). Such a level of homozygosity suggests either a high degree of consanguinity, or more likely, ‘founder’ effects within these groups.
Figure 3. Homozygosity detected in all of the pedigrees and haplotype sharing in some of the pedigrees.
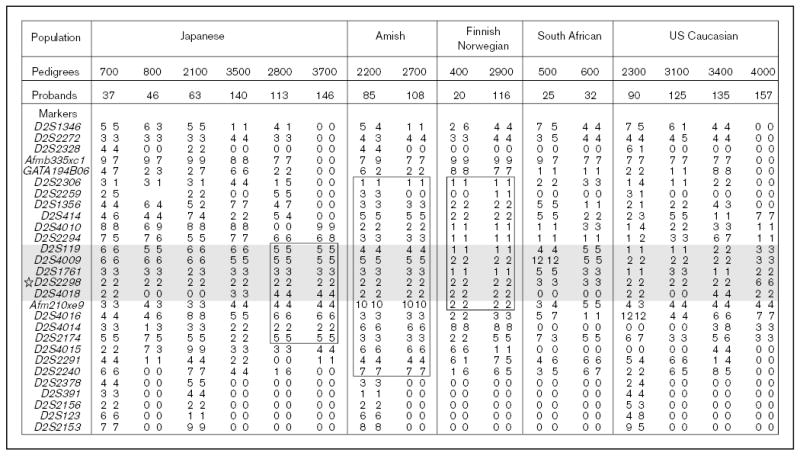
Genotypes for markers are shown for probands across the critical STSL area. The shaded regions indicate the minimal region of homozygosity present in all probands, bounded by markers D2S2294 and Afm210xe9. Note that only pedigree 3400 was known to be consanguineous. In addition to homozygosity, haplotype sharing, boxed areas, was detected in two Japanese probands, the Amish and Mennonite families and Norwegian and Finnish probands.
Interestingly, in addition to homozygosity, we also detected haplotype sharing. The two Amish/Mennonite families (pedigrees 2200 and 2700) showed extensive haplotype sharing across the STSL locus (Fig. 3). Initially, these two families were not known to be related to each other, but a concerted genealogical search allowed us to trace their lineages back to a potential of five founders (Fig. 4), dating back to around the early 1700s [9]. A historical background of the Amish and Mennonites, as well as their genealogy, has been well-documented [20] and has allowed us to trace back lineages of the affected individuals. The fewest number of generations linking any two of the obligate carriers is three, with a minimum of six generations linking all four together (Fig. 4), indicating that the ‘Amish’ mutation may have been introduced to the USA by the very first Mennonite settlers. Although many of the Amish/Mennonite founders originated from German-speaking Switzerland, many had lived in the Alsace and Palatinate regions before emigrating to the USA [20]. The closest geographical family is a Dutch family (pedigree 2500), which did not share any haplotypes with the affected Amish/Mennonite probands. We have three families/probands from Scandinavia (400, 2900 and 3900). Of these pedigrees, 400 and 3900 are Finnish, and pedigree 2900 is Norwegian [21]. Interestingly, pedigrees 2900 and 400 from Norway/Finland not only showed homozygosity but also exhibited haplotype sharing (Fig. 3) [9]. This would suggest that a single founder gave rise to the ‘Finnish/Norwegian’ mutation, and is responsible for the occurrence of the disease. The markers from the region of interest showed a significantly different allele frequency compared with randomly chosen individuals from Norway and Finland. However, the two ‘Finnish’ families do not share any of the haplotypes and appear to be unrelated to each; the proband from Finnish family 3900 is heterozygous for informative markers in this region. Founder effects have been identified for the Finnish population for other disease loci [22–24], but none, to date, links the Norwegian and Finnish populations. Finally, although several of the Japanese probands show homozygosity across the region of interest, only two of them (pedigrees 2800 and 3700) share the haplotypes (Fig. 3). These families also do not appear to be related by marriage, and a ‘founder’ effect is therefore postulated. Based upon haplotype sharing, as well as homozygosity, the prediction is that most probands will be homozygous for the mutations, and a number of these will be identical for them, to account for the genetic data.
Figure 4. Genealogical analyses of the Amish and Mennonite families in this study.
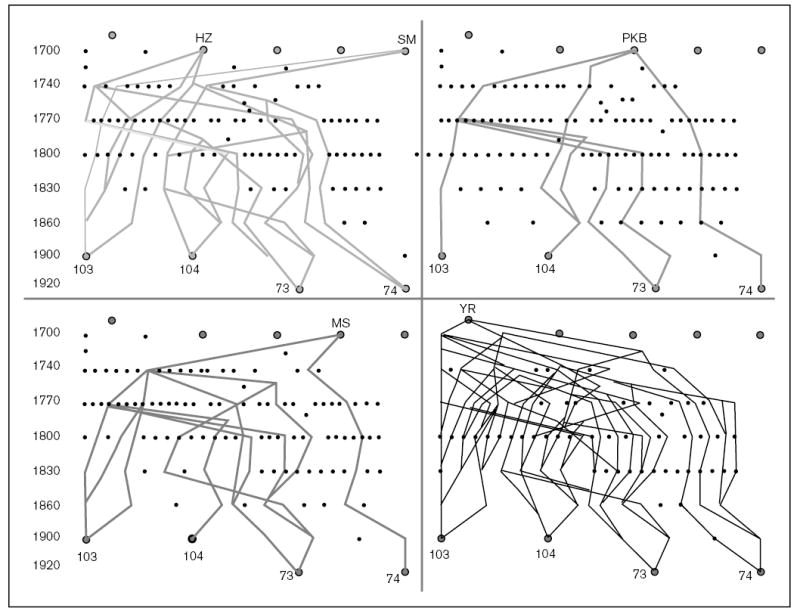
A genealogical analysis of the parents from pedigrees 2700 and 2200 was performed as described in the methods section. Five possible founders, who link all four parents, were identified. An approximate time index is as shown in the left margin for temporal orientation. As can be seen, individual 74 is the least related to all of the remaining three obligate carriers.
Identification of genes that cause sitosterolemia
To refine the minimal critical region and isolate sitosterolemia candidate gene(s), we constructed high-resolution YAC and BAC contigs, and built a transcript map [19]. A number of expressed sequence tags (ESTs) and genes that mapped into the region of interest were screened, on the basis of whether they were expressed in the liver or intestine, the organs important in dietary cholesterol retention. Two were selected for further study, because they were expressed in the liver and gut. Analyses of the full length complementary DNA showed these encoded a ‘half-ABC’ transporter each [25], and were assigned the names ABCG5 [26] and ABCG8 [11••]. We have named the protein products of these genes sterolin-1 and sterolin-2, respectively [12••]. The genes consist of 13 exons, and encode for six putative membrane-spanning domains that contain the characteristic ABC signature motifs at their N-terminal end (Fig. 5). Phylogenetic analyses indicated that these proteins are related to the ‘ABCG’ family [12••,26], members of which contain the white proteins in Drosophila, involved in retinal pigment metabolism, and ABC8, involved in cholesterol transfer in macrophages in the mouse [27].
Figure 5. Gene structure for ABCG5 and ABCG8.
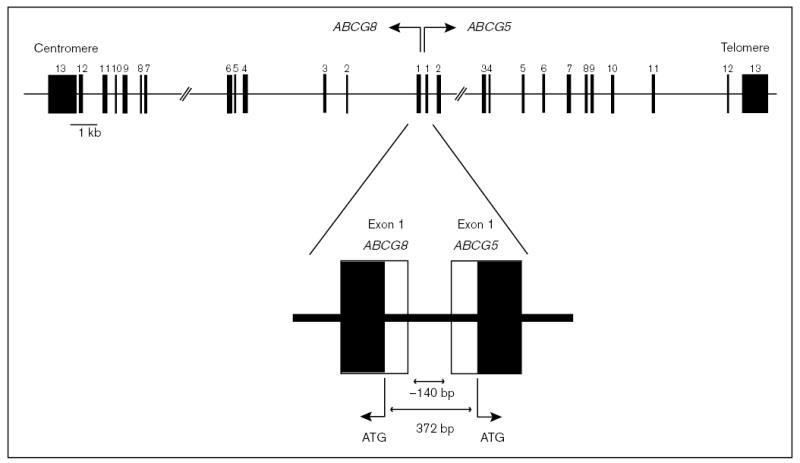
The gene structures for ABCG5 and ABCG8 are as depicted. Each gene consists of 13 exons and the genes are arranged in a head-to-head configuration, with no more than 150 bases separating the start-transcription sites, with only 372 bases separating the two respective ‘ATG’. No TATA box is present in the 140 bases separating the two genes.
Mutational analyses showed that both ABCG5 and ABCG8 are mutated in sitosterolemia [11••,12••] and Lee et al. (manuscript in preparation). A summary of these mutations is shown in Table 1. A number of polymorphisms have also been identified and are documented in Table 2. To date, all probands are either mutated for ABCG5 or ABCG8, and none shows mutations in both. It would thus appear that both sterolins are required for normal function. As both sterolins are half-ABC transporters, one speculation is that they function in concert, and may physically interact with each other. To date, no dominant negative mutations have been reported. Further investigations will undoubtedly shed light on these issues.
Table 1.
Mutations in ABCG5 and ABCG8 in sitosterolemia
| Patients | Nationality/ethnicity | ABCG5 | ABCG8 | ||
|---|---|---|---|---|---|
| 4 | US/Caucasian | Arg412X | Trp361X | ||
| 9 | US/Caucasian | Arg543Ser | Gln172X | ||
| 56* | US/Caucasian | Tyr658X | Trp361X | ||
| 60 | US/Caucasian | Trp361X | – | ||
| 90* | US/Caucasian | Trp361X | Trp361X | ||
| 94 | US/Caucasian | Trp361X | Arg184His | ||
| 120 | US/Caucasian | Leu501Pro | Trp361X | ||
| 125 | US/Caucasian | Trp361X | Trp361X | ||
| 128 | US/Caucasian | Leu596Arg | – | ||
| 32 | SA Caucasian | Arg121X | Arg121X | ||
| 98 | Dutch Caucasian | Gly574Glu | Trp361X | ||
| 102* | US/Caucasian | Trp361X | – | ||
| 135 | Columbian/Caucasian | Trp536X | Trp536X | ||
| 20 | Finnish | Trp361X | Trp361X | ||
| 116 | Norwegian | Trp36X | Trp361X | ||
| 84* | US/Amish/Mennonite | Gly574Arg | Gly574Arg | ||
| 108 | US/Amish/Mennonite | Gly574Arg | Gly574Arg | ||
| 25 | SA/Asian | Arg243X | Arg243X | ||
| 40 | Japanese | Arg419His | Arg419His | ||
| 46 | Japanese | Arg389His | Arg389His | ||
| 63 | Japanese | del Exon 3 | del Exon 3 | ||
| 113 | Japanese | Arg389His | Arg389His | ||
| 132 | Japanese | Arg419His | – | ||
| 140 | Japanese | Arg408X | Arg408X | ||
| 146 | Japanese | Arg389His | Arg389His | ||
| 157 | US/Caucasian | Arg419Pro | Arg419Pro | ||
| 15 | US/Caucasian | – | – | ||
| 143 | SA/Indian Asian | – | – | Arg121X | Arg164X |
| 149 | African American | Glu145Gln | – | ||
| 1* | German/Swiss | Trp361X | Trp361X | ||
| 2* | US/Amish | Gly574Arg | Gly574Arg | ||
| 3* | US/Caucasian | Trp361X | Tyr658X | ||
| 5* | US/Caucasian | Trp361X | Arg412X | ||
| 6* | US/Caucasian | Leu596Arg | – | ||
| 7 | US/Hispanic | Arg412X | del547C>191X | ||
| 8* | NZ/Caucasian | Trp361X | – | ||
| 4 | Chinese | Arg263Gln | Pro231Thr | ||
| 9 | Chinese | Arg408X | – | ||
This is a compilation of the mutations identified in ABCG5 and ABCG8. The top half shows mutations identified by Lee et al. [12••, and manuscript in preparation] and the bottom half by Berge et al. [11••]. The asterisks indicate probands that were available to both groups. In a few cases, only one mutation has been identified (indicated by a dash).
Table 2.
Polymorphisms in ABCG5 and ABCG8
| Gene | Amino acid changes | Nucleotide changes |
|---|---|---|
| ABCG5 | Pro 9 Pro | CCC>CCT |
| Gln 604 Glu | CAA>GAA | |
| ABCG8 | Glu 189 Glu | GAA>GAG |
| Gln 340 Glu | CAG>GAG | |
| Lys 400 Thr | AAG>ACG | |
| Ala 565 Ala | GCC>GCT | |
| Ala 632 Val | GCC>GTC | |
| Intron 9–19C>T | ||
| Intron10 2bp deletion at +35C | ||
| Intron10–50C>T | ||
| In12 +22C>G |
Polymorphic changes detected in ABCG5 and ABCG8 are as documented. These changes were judged to be ‘polymorphic’ on the basis that these were detected in normal unaffected individuals drawn from the general population.
Conclusion
The identification of the defective genes in sitosterolemia opens a new field for investigation of sterol regulation. It also gives credence to the hypothesis that specific molecular mechanisms regulate cholesterol entry into and out of the body, and that this mechanism allows for exquisite differentiation between sterol species that are very similar in their physicochemical properties. The surprise is that there are two genes in tandem, separated by less than 200 bases, transcribed in opposite directions and encoding ABC half-transporters (ABCG5 and ABCG8), which are involved in this process. We have named the proteins sterolins, to reflect their putative functions (‘sterol in’). Interestingly, to cause sitosterolemia, both copies of only one gene have to be defective: either ABCG5 or ABCG8. To date, no person with sitosterolemia has been identified who has a mutation in one allele of ABCG5 and one allele of ABCG8. In addition, carriers for mutations in either ABCG5 or ABCG8 do not appear to manifest any overt phenotype with respect to sitosterol exclusion from the body. The net effect of the complete loss of either sterolin function therefore results in increased cholesterol and sitosterol absorption, and decreased sitosterol and cholesterol excretion into the bile. If these observations hold true, we hypothesize that sterolin-1 and sterolin-2 function as heterodimers, regulating non-cholesterol sterols out of the cell.
A few models can be put forward to explain how sterolins may function, two of which are illustrated in Fig. 6. Model A shows that the function of the heterodimer is to pump sterols out. This putative sterolin complex would exhibit a greater affinity for pumping sitosterol out of the cell (enterocyte or hepatocyte) versus cholesterol, but it can pump cholesterol out in the absence of sitosterol. As this ‘pumping’ mechanism is likely to be rate-limited by substrate availability (non-cholesterol sterols are not normally elevated in the cells), heterozygous individuals would be predicted to be normal. A second model that can also be put forward is one in which the heterodimer complex (of sterolin-1 and sterolin-2) has a dual function; it has a greater affinity for pumping (or facilitating) cholesterol (versus non-cholesterol sterols) into the cells and a greater affinity for pumping non-cholesterol sterols out (Fig. 6B). Both of these models rely upon the apical expression of sterolins, and that both are expressed in hepatocytes and enterocytes. Other models are also possible; most ABC half-transporters are localized to membranes within the cell, and thus sterolins may regulate the transport of sterols in and out of specific compartments. In this respect, sterolins would be expected to function with other proteins. One such protein proposed by Repa et al. is ABC1 [28••], although the expression of ABC1 has not been shown to be enterocyte-specific [29]. These and more complex models are now amenable for testing.
Figure 6. Possible models of action of sterolins.
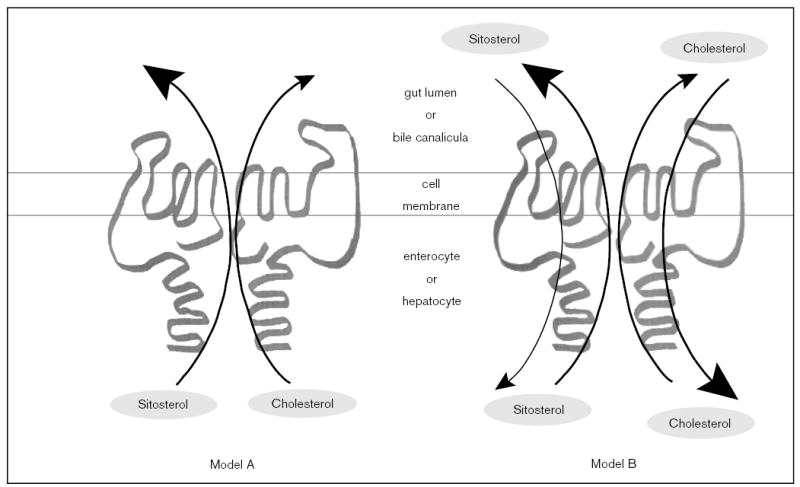
We propose two models that may explain how sterolins function to prevent non-cholesterol sterols (represented by sitosterol) from being retained by the body. In both models, both sterolin-1 and sterolin-2 function as a heterodimer. Model A predicts that the heterodimer is responsible for the exclusion of sterols by actively pumping them out of the cells (the enterocyte or the hepatocyte) at the apical border. However, this pump has a much higher affinity for non-cholesterol sterols compared with cholesterol. Note that this mode of action allows for the sterolins to pump cholesterol into bile, in the absence of non-cholesterol sterols in the liver. However, under model A, sterolins do not regulate the entry of sterols into the enterocyte or the hepatocyte. Model B predicts that the sterolin heterodimer acts in a bi-functional direction, again at the apical surfaces of the enterocyte or the hepatocyte. In this case, the heterodimer shows a higher affinity for the entry of cholesterol versus non-cholesterol sterols into the cells, and a higher affinity for pumping non-cholesterol sterols out of the cells.
Acknowledgments
This work was supported by the National Institutes of Health grant HL60613 and by the Medical University of South Carolina. The author is grateful to all the sitosterolemia patients, their families and their physicians for their continued support, participation and encouragement of his work.
References
Papers of particular interest, published within the annual period of review, have been highlighted as:
• of special interest
• • of outstanding interest
- 1.Gould RG, Jones RJ, LeRoy GV, et al. Absorbability of beta-sitosterol in humans. Metabolism. 1969;18:652–662. doi: 10.1016/0026-0495(69)90078-x. [DOI] [PubMed] [Google Scholar]
- 2.Salen G, Ahrens Jr E, Grundy SM. Metabolism of beta-sitosterol in man. J Clin Invest. 1970;49:952–967. doi: 10.1172/JCI106315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Salen G, Tint GS, Shefer S, et al. Increased sitosterol absorption is offset by rapid elimination to prevent accumulation in heterozygotes with sitosterolemia. Arterioscler Thromb. 1992;12:563–568. doi: 10.1161/01.atv.12.5.563. [DOI] [PubMed] [Google Scholar]
- 4.Gregg RE, Connor WE, Lin DS, Brewer Jr H. Abnormal metabolism of shellfish sterols in a patient with sitosterolemia and xanthomatosis. J Clin Invest. 1986;77:1864–1872. doi: 10.1172/JCI112513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bhattacharyya AK, Connor WE. Beta-sitosterolemia and xanthomatosis. A newly described lipid storage disease in two sisters. J Clin Invest. 1974;53:1033–1043. doi: 10.1172/JCI107640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bjorkhem I, Boberg KM. Inborn errors in bile acid biosynthesis and storage of sterols other than cholesterol. In: The metabolic basis of inherited disease, 7th ed., vol. 2. Scriver CR, Beaudet AL, Sly WS, Valle D (editors). New York: McGraw-Hill Inc.; 1995. pp. 2073–2102.
- 7.Salen G, Shefer S, Nguyen L, et al. Sitosterolemia [Review] J Lipid Res. 1992;33:945–955. [PubMed] [Google Scholar]
- 8.Patel SB, Salen G, Hidaka H, et al. Mapping a gene involved in regulating dietary cholesterol absorption. The sitosterolemia locus is found at chromosome 2p21. J Clin Invest. 1998;102:1041–1044. doi: 10.1172/JCI3963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9•.Lee M-H, Gordon D, Ott J, et al Fine mapping of a gene responsible for regulating dietary cholesterol absorption; founder effects underlie cases of phytosterolemia in multiple communities. Eur Hum Genet 2001; in press.A more complete description of the genetics of sitosterolemia and the founder effects identified in a number of different ethnic groups. [DOI] [PMC free article] [PubMed]
- 10.Patel SB, Honda A, Salen G. Sitosterolemia: exclusion of genes involved in reduced cholesterol biosynthesis. J Lipid Res. 1998;39:1055–1061. [PubMed] [Google Scholar]
- 11••.Berge KE, Tian H, Graf GA, et al. Accumulation of dietary cholesterol in sitosterolemia caused by mutations in adjacent ABC transporters. Science. 2000;290:1771–1775. doi: 10.1126/science.290.5497.1771. This paper demonstrates the power of deductive reasoning, the internet, the rich databases and the collaboration between biotechnology and academia in rapidly advancing science. [DOI] [PubMed] [Google Scholar]
- 12••.Lee M-H, Lu K, Hazard S, et al. Identification of a gene, ABCG5, important in the regulation of dietary cholesterol absorption. Nat Genet. 2001;27:79–83. doi: 10.1038/83799. Using a combination of positional cloning and genome databases, these authors identified mutations in ABCG5 in a sub-set of families. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miettinen TA. Phytosterolaemia, xanthomatosis and premature atherosclerotic arterial disease: a case with high plant sterol absorption, impaired sterol elimination and low cholesterol synthesis. Eur J Clin Invest. 1980;10:27–35. doi: 10.1111/j.1365-2362.1980.tb00006.x. [DOI] [PubMed] [Google Scholar]
- 14.Hidaka H, Nakamura T, Aoki T, et al. Increased plasma plant sterol levels in heterozygotes with sitosterolemia and xanthomatosis. J Lipid Res. 1990;31:881–888. [PubMed] [Google Scholar]
- 15.Hidaka H, Yasuda H, Kobayashi M, et al. Familial spinal xanthomatosis with sitosterolemia. Intern Med. 1992;31:1038–1042. doi: 10.2169/internalmedicine.31.1038. [DOI] [PubMed] [Google Scholar]
- 16.Shefer S, Salen G, Bullock J, et al. The effect of increased hepatic sitosterol on the regulation of 3-hydroxy-3-methylglutaryl-coenzyme A reductase and cholesterol 7 alpha-hydroxylase in the rat and sitosterolemic homozygotes. Hepatology. 1994;20:213–219. doi: 10.1016/0270-9139(94)90155-4. [DOI] [PubMed] [Google Scholar]
- 17.Brown MS, Goldstein JL. A receptor-mediated pathway for cholesterol homeostasis [Review] Science. 1986;232:34–47. doi: 10.1126/science.3513311. [DOI] [PubMed] [Google Scholar]
- 18.Kuksis A, Myher JJ, Marai L, et al. Usefulness of gas chromatographic profiles of plasma total lipids in diagnosis of phytosterolemia. J Chromatogr. 1986;381:1–12. doi: 10.1016/s0378-4347(00)83559-8. [DOI] [PubMed] [Google Scholar]
- 19.Lu K, Lee M-H, Carpten JD, et al. High-resolution physical and transcript map of human chromosome 2p21 containing the sitosterolemia locus. Eur Hum Genet 2001; in press. [DOI] [PMC free article] [PubMed]
- 20.Gingerich HF, Kreider RW. Amish and Amish Mennonite genealogies. Gordonville, PA: Pequea Publishers; 1986.
- 21.Lutjohann D, Bjorkhem I, Ose L. Phytosterolaemia in a Norwegian family: diagnosis and characterization of the first Scandinavian case. Scand J Clin Lab Invest. 1996;56:229–240. doi: 10.3109/00365519609088612. [DOI] [PubMed] [Google Scholar]
- 22.Hastbacka J, de la Chapelle A, Kaitila I, et al. Linkage disequilibrium mapping in isolated founder populations: diastrophic dysplasia in Finland [published erratum appears in Nat Genet 1992; 2:343] Nat Genet. 1992;2:204–211. doi: 10.1038/ng1192-204. [DOI] [PubMed] [Google Scholar]
- 23.Peltonen L. Molecular background of the Finnish disease heritage. Ann Med. 1997;29:553–556. doi: 10.3109/07853899709007481. [DOI] [PubMed] [Google Scholar]
- 24.de la Chapelle A, Wright FA. Linkage disequilibrium mapping in isolated populations: the example of Finland revisited. Proc Natl Acad Sci USA. 1998;95:12416–12423. doi: 10.1073/pnas.95.21.12416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Klein I, Sarkadi B, Varadi A. An inventory of the human ABC proteins. Biochim. Biophys Acta. 1999;1461:237–262. doi: 10.1016/s0005-2736(99)00161-3. [DOI] [PubMed] [Google Scholar]
- 26.Shulenin S, Schriml LM, Remaley A, et al A liver-specific ATP-binding cassette gene (ABCG5) from the ABCG (White) gene subfamily maps to human chromosome 2p21 in the region of the sitosterolemia locus. Cyto Cell Genet 2000; in press. [DOI] [PubMed]
- 27.Klucken J, Buchler C, Orso E, et al. ABCG1 (ABC8), the human homolog of the Drosophila white gene, is a regulator of macrophage cholesterol and phospholipid transport. Proc Natl Acad Sci USA. 2000;97:817–822. doi: 10.1073/pnas.97.2.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28••.Repa JJ, Turley SD, Lobaccaro JA, et al. Regulation of absorption and ABC1-mediated efflux of cholesterol by RXR heterodimers [see Comments] Science. 2000;289:1524–1529. doi: 10.1126/science.289.5484.1524. An intriguing study that suggests that dietary cholesterol absorption may be regulated by the transcriptional factors LXR and FXR, and that ABC1, the protein defective in Tangier disease, may be involved. However, ABC1 may not be expressed at high levels in the enterocytes, but is expressed in the lamina propria (see ref. [29]), thus making this a more complex story. [DOI] [PubMed] [Google Scholar]
- 29.Lawn RM, Wade DP, Couse TL, Wilcox JN. Localization of human ATP-binding cassette transporter (ABC1) in normal and atherosclerotic tissue. Arteriscler Thromb Vasc Biol. 2001;21:378–385. doi: 10.1161/01.atv.21.3.378. [DOI] [PubMed] [Google Scholar]