

Abstract
Previous studies indicate that Krüppel-like factor 5 (KLF5), also known as intestinal-enriched Krüppel-like factor (IKLF), is a positive regulator of cell proliferation and gives rise to a transformed phenotype when over-expressed. Here we demonstrate that levels of KLF5 transcript and protein are significantly elevated in oncogenic H-Ras-transformed NIH3T3 cells. These cells display an accelerated rate of proliferation in both serum-containing and serum-deprived media and form anchorage-independent colonies in soft agar assays. H-Ras-transformed cells also contain elevated mitogen-activated protein kinase (MAPK) activity. When treated with inhibitors of MEK (MAPK kinase), H-Ras-transformed cells lose their growth advantage and no longer form colonies. Significantly, levels of KLF5 transcript and protein are substantially reduced in H-Ras-transformed cells treated with MEK inhibitors. Moreover, inhibition of KLF5 expression in H-Ras-transformed cells with KLF5-specific small interfering RNA (siRNA) leads to a decreased rate of proliferation and a significant reduction in colony formation. H-Ras-transformed cells also contain elevated levels of Egr1 that are diminished by MEK inhibitors. Inhibition of Egr1 by siRNA results in a reduced level of KLF5, indicating that Egr1 mediates the inductive action of MAPK on KLF5. Lastly, KLF5 activates expression of cyclin D1. These findings indicate that the increased expression of KLF5 in H-Ras-transformed cells is secondary to increased MAPK activity from H-Ras overexpression and that the elevated level of KLF5 is in part responsible for the proproliferative and transforming activities of oncogenic H-Ras.
Keywords: IKLF, transformation, mitogen-activated protein kinase, Egr1, cyclin D1, proliferation, small interfering RNA, anchorage-independent growth
Abbreviations: DMEM, Dulbecco’s modified Eagle’s medium; ERK, extra-cellular signal-regulated kinase; FBS, fetal bovine serum; GAPDH, glyceraldehydes-3-phosphate dehydrogenase; GKLF, gut-enriched Krüppel-like factor; IKLF, intestinal Krüppel-like factor; KLF, Krüppel-like factor; MAPK, mitogen-activated protein kinase; MEK, MAPK/ERK kinase; PBS, phosphate-buffered saline; PI, propidium iodide; PMA, phorbol-12-myristate-13-acetate; RT—PCR, reverse transcription—polymerase chain reaction; siRNA, small interfering RNA
Introduction
Krüppel-like factors (KLFs) are Sp1-like transcription factors that have regulatory functions in diverse physiologic processes including cell proliferation and differentiation (Dang et al., 2000; Black et al., 2001; Simmen and Simmen, 2002; Kaczynski et al., 2003). KLFs contain multiple conserved C2H2 zinc-finger motifs and bind to GC-rich DNA elements to regulate transcription (Crossley et al., 1996; Philipsen and Suske, 1999; Shi et al., 1999; Turner and Crossley, 1999; Dang et al., 2002). Two members of the KLF family have been extensively characterized in the epithelium of the gastrointestinal tract. KLF4 or GKLF (gut-enriched Krüppel-like factor) is primarily present in the post-mitotic, terminally differentiated epithelial cells of the gut (Shields et al., 1996). In contrast, KLF5 or IKLF (intestinal-enriched Krüppel-like factor) is primarily found in the proliferating population of crypt epithelial cells (Conkright et al., 1999; Ohnishi et al., 2000). Similar to this complimentary pattern of expression in the intestinal epithelium, the two KLFs exhibit opposite biological behaviors in cultured cells. While expression of KLF4 is primarily associated with a growth-arrest state (Garrett-Sinha et al., 1996; Shields et al., 1996; Shie et al., 2000), that of KLF5 is associated with proliferation (Sun et al., 2001). These findings suggest that KLF4 and KLF5 may function to coordinate proliferation and differentiation of intestinal epithelial cells.
The mechanisms that regulate expression of KLF5 were investigated by several studies. For example, vascular endothelial growth factor (VEGF) and basic fibroblast growth factor (bFGF) were shown to be potent activators of KLF5 in vitro (Kawai-Kowase et al., 1999; Hoshino et al., 2000; Ogata et al., 2000). Activation of KLF5 expression has also been linked to the Wnt/Wg signaling pathway. Wnt-1, a member of the Wnt family, stimulates expression of KLF5 in a pathway that is independent of β-catenin and dependent on protein kinase C (PKC) (Ziemer et al., 2001). Another example is the stimulation of KLF5 expression by treatment with phorbol-12-myristate-13-acetate (PMA) (Kawai-Kowase et al., 1999), a potent activator of PKC and known to activate the mitogen-activated protein kinase (MAPK) (Dekker and Parker, 1994; Troppmair et al., 1994). Inhibition of MAPK inhibits expression of KLF5, indicating the importance of the MAPK pathway in regulating KLF5 expression (Kawai-Kowase et al., 1999). Consistent with these observations, we recently showed that KLF5 is a delayed early response gene in response to treatment of cells with PMA or serum (Sun et al., 2001). Importantly, we showed that overexpression of KLF5 in transfected cells resulted in an increased rate of proliferation and eventually led to a transformed phenotype as evidenced by the loss of contact inhibition and anchorage dependence (Sun et al., 2001).
The Ras family of proteins was initially identified as important regulators of cell growth (Barbacid, 1987). Ras, represented by one of three protooncogene products, H-Ras, K-Ras and N-Ras, is constitutively activated in many tumors due to point mutation (Downward, 2003). Physiologically activated Ras proteins play a critical role in numerous cellular processes including proliferation, survival and apoptosis (Dobrowolski et al., 1994; Muszynski et al., 1995; Khosravi-Far et al., 1998; Downward, 2003). The expression of several target genes important for cell cycle regulation, including cyclin D1, p21cip1, p27kip1, cdc25 and c-Myc, are regulated by Ras (Kerkhoff and Rapp, 1998). Tumors that possess constitutively activated Ras exhibit an increase in metastatic invasiveness and angiogenesis with a corresponding loss of anchorage-dependent growth (Shields et al., 2000). Similarly, forced expression of activated Ras in cultured NIH3T3 fibroblasts results in anchorage-independent proliferation (Kuo et al., 1993; Chen et al., 1995; Yang et al., 1998; Sun et al., 2001).
The canonical Ras pathway involves several downstream mediators. A major substrate of Ras is Raf-1, a serine/threonine protein kinase (Leevers et al., 1994; Marais et al., 1995; Shields et al., 2000). Activation of Raf-1 results in a downstream cascade that involves phosphorylation of several proteins including mitogen-activated protein kinase kinases (MAPKK or MEK), which consecutively activate extracellular signal-regulated kinases (ERK), also known as p44/42 MAPK (Daum et al., 1994; Kerkhoff and Rapp, 1998; Downward, 2003). Phosphorylated ERK is translocated into the nucleus where it activates several transcription factors. There have been various reports on the PMA-induced activation of specific components of the Ras pathway. For example, Ras is directly involved in PMA-induced MAPK phosphorylation (Nori et al., 1992; Thomas et al., 1992). However, conflicting reports on a Ras-independent mechanism of activation have led to the conclusion that PMA could trigger both Ras-dependent and Ras-independent pathways depending on the cell type (de Vries-Smits et al., 1992; Howe et al., 1992; Ming et al., 1994). In the mouse embryonic NIH3T3 fibroblasts, PMA stimulation was shown to result in the downstream activation of Raf-1 and MEK (Troppmair et al., 1994) although the entry point for PMA in this process is still debated. PKC is known to directly stimulate Raf-1 by phosphorylation (Kolch et al., 1993). However, this phosphorylation apparently does not lead to the direct activation of ERK (MacDonald et al., 1993; Wartmann et al., 1997). Instead, PMA-mediated phosphorylation of Shc and the consequent activation of Sos lead to the stimulation of Ras/ERK pathway (El-Shemerly et al., 1997).
Our previous observations that PMA stimulates expression of KLF5 and that KLF5 overexpression causes transformation (Sun et al., 2001) led us to investigate whether KLF5 is a target of the Ras signaling pathway. In the current study, we present evidence that in vitro transformation of mouse fibro-blasts with oncogenic H-Ras results in the increased expression of KLF5. We also show that the elevated expression of KLF5 in H-Ras-transformed cells is significantly reduced by inhibition of MEK in the transformed cells. Importantly, inhibition of KLF5 expression by either MEK inhibitors or small interfering RNA (siRNA) results in a similar reduction in the rate of cell proliferation and anchorage-independent growth. These findings indicate the KLF5 is an important mediator of the proproliferative and transforming activities of oncogenic H-Ras.
Results
Oncogenic H-Ras-transformed NIH3T3 cells exhibit an increased rate of proliferation
Two independent clones of NIH3T3 cells transformed by oncogenic H-Ras, designated as Ras 2 and Ras 7, were examined for their rate of proliferation. When maintained in the presence of 10% fetal bovine serum (FBS), both Ras 2 and Ras 7 exhibited an increased rate of proliferation when compared to the untransformed parent NIH3T3 cells (Figure 1a). In addition, Ras 2 and Ras 7 cells exhibited serum-independent proliferation as compared to NIH3T3 cells when maintained in serum-deprived conditions (Figure 1b). When examined by FACS analysis, both Ras 2 and Ras 7 cells contained a statistically significant higher proportion of S-phase cells than NIH3T3 cells beginning at day 1 after serum deprivation (Figure 1c). Finally, Ras 2 and Ras 7 cells but not NIH3T3 cells were capable of anchorage-independent proliferation as evidenced by formation of colonies in soft agar (Figure 1d).
Figure 1.
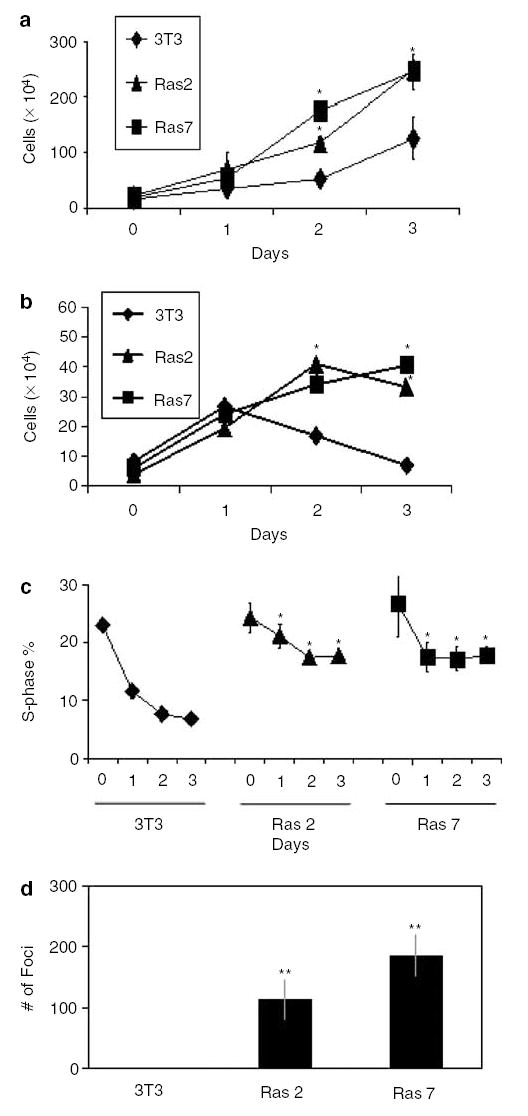
Effects of oncogenic H-Ras on cell proliferation in mouse fibroblasts. Panels a–d show results of experiments performed on control, untransformed NIH3T3 cells and two independently derived oncogenic H-Ras-transformed clones, designated as Ras 2 and Ras 7. In panels a and b, cells were seeded at 104 cells/well on day 0 and total cell numbers were counted every 24 h after seeding. Cells in panel a were supplemented with 10% FBS, while those in panel b were supplemented with 0.5% FBS. In panel c, cells were stained with PI and DNA content analysed by flow cytometry. The percentages of cells in the S-phase population were plotted over time. Panel d is the result of colony formation assay in soft agar in 10-cm dishes. N = 3 in all experiments. *P < 0.05; **P < 0.01 when compared with control NIH3T3 cells
KLF5 is overexpressed in oncogenic H-Ras-transformed NIH3T3 cells
To determine whether elevated expression of KLF5 is associated with transformation, we compared the levels of KLF5 mRNA and protein in oncogenic H-Ras-transformed clones and untransformed NIH3T3 cells. As seen in Figure 2a, the two stable cell lines, Ras 2 and Ras 7, contained elevated levels of H-Ras mRNA when compared to untransformed cells, with Ras 7 containing a higher amount of H-Ras transcript than Ras 2. Importantly, the levels of KLF5 mRNA were also elevated in the two transformed cell lines when compared to untransformed NIH3T3 cells (Figure 2a). The quantitative difference in expression levels of KLF5 in the different cell lines was further investigated by semiquantitative reverse transcription–polymerase chain reaction (RT–PCR) (Figure 2b) and real-time PCR (Figure 2c). The latter assay showed that the relative abundance of KLF5 mRNA in Ras 2 and Ras 7 cells was approximately four- and eightfold, respectively, of that in NIH3T3 cells (Figure 2c). Lastly, Western blot analysis of proteins extracted from the three cell lines also revealed a quantitative difference in the levels of KLF5 protein, which correlated with the abundance of H-Ras protein (Figure 2d).
Figure 2.
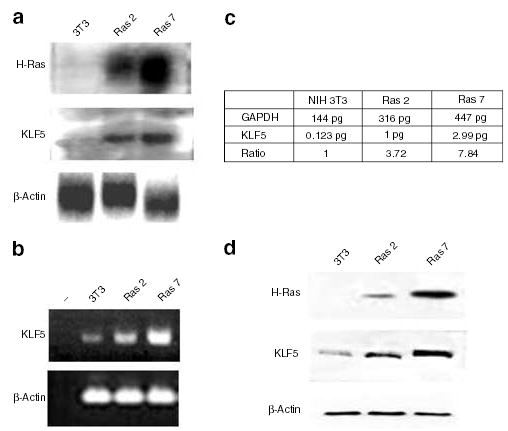
Activation of KLF5 expression in NIH3T3 cells transformed by oncogenic H-Ras. Expression levels of H-Ras, KLF5 and β-actin were analysed in control NIH3T3 cells and the two H-Ras-transformed cells. Panel a shows the result of Northern blot analysis and panel b that of semiquantitative RT–PCR. Panel c is the result of quantitative (realTime) PCR on reverse-transcribed KLF5 and GAPDH cDNAs. The ratio denotes the relative level of expression of KLF5 after normalization with the GAPDH concentration. Panel d shows the results of Western blot analysis using antibodies specific to H-Ras, KLF5 and β-actin
The elevated expression of KLF5 in oncogenic H-Ras-transformed cells is due to increased MAPK activities
ERK1/2, or p44/42 MAPK, activated by numerous growth factors and mitogens, is an important downstream component of the oncogenic H-Ras signaling cascade (Daum et al., 1994; Kerkhoff and Rapp, 1998; Downward, 2003). Upon stimulation, ERK1/2 becomes phosphorylated by MAPK kinase or MEK and is translocated into the nucleus to activate transcription of downstream effector genes. The result in Figure 3a demonstrates that both Ras 2 and Ras 7 cells contained significantly elevated pERK1/2 levels when compared to the control NIH3T3 cells. Since there were minimal pERK1/2 levels in the untransformed cells, which possessed an equal amount of total ERK1/2 as compared to the transformed cells, the elevated MAPK activity in Ras 2 and Ras 7 was a result of over-expression of oncogenic H-Ras. Importantly, treatment of Ras 2 and Ras 7 cells with two MEK inhibitors, PD98059 and U0126, resulted in a significant reduction in the levels of KLF5 mRNA (Figures 3b and c, respectively) and protein (Figures 3d and e, respectively). These results indicate that the elevated expression of KLF5 in H-Ras-transformed cells is due to the increased MAPK activities in these cells.
Figure 3.
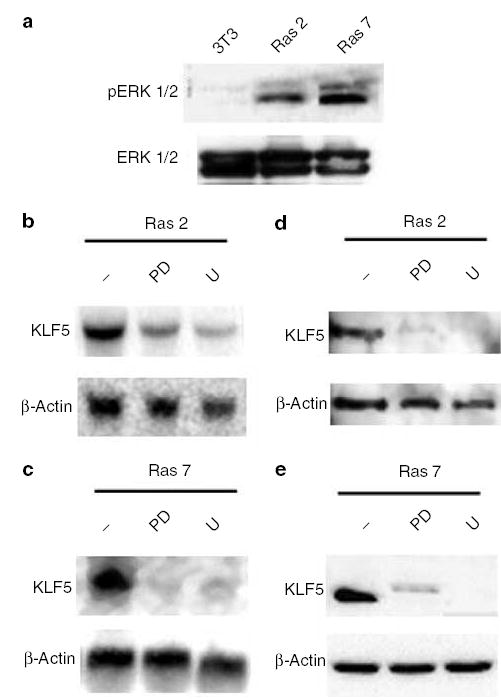
Effect of MEK inhibition on KLF5 expression in H-Ras-transformed cells. Panel a shows the MAPK activity in NIH3T3, Ras 2 and Ras 7 cells as determined by the level of pERK1/2 (top row). The total amounts of ERK1/2 were determined by Western blot analysis using specific antibodies (bottom row). Panels b and c show the results of Northern blot analysis of RNA extracted from Ras 2 and Ras 7 cells, respectively, after treatment with two MEK inhibitors, PD98059 (PD) and U0126 (U). − indicates untreated cells. Panels d and e show the results of Western blot analysis of proteins extracted from Ras 2 and Ras 7 cells, respectively, after treatment with two MEK inhibitors, PD98059 (PD) and U0126 (U). − indicates untreated cells
MEK inhibition results in a reduced rate of proliferation of oncogenic H-Ras-transformed cells
To examine the phenotypic changes effected by MEK inhibitors in H-Ras-mediated transformation, we measured cell proliferation and performed cell cycle analyses in Ras 7 cells in the presence or absence of inhibitors. As shown in Figure 4a, the rate of proliferation of Ras 7 cells was significantly reduced when they were maintained in the presence of PD98059 and U0126 relative to that without any inhibitors. Similarly, the proportion of cells in the S-phase was significantly reduced in cells treated with either inhibitor when compared to the untreated cells (Figure 4b). Cells treated with the two inhibitors also displayed a substantial reduction in anchorage-independent growth when compared to untreated cells (Figure 4c). Similar effects of MEK inhibitors on proliferation and foci formation were also observed in Ras 2 cells (results not shown). These findings indicate that the proproliferative effect of oncogenic H-Ras is mediated by the MAPK pathway of signal transduction.
Figure 4.
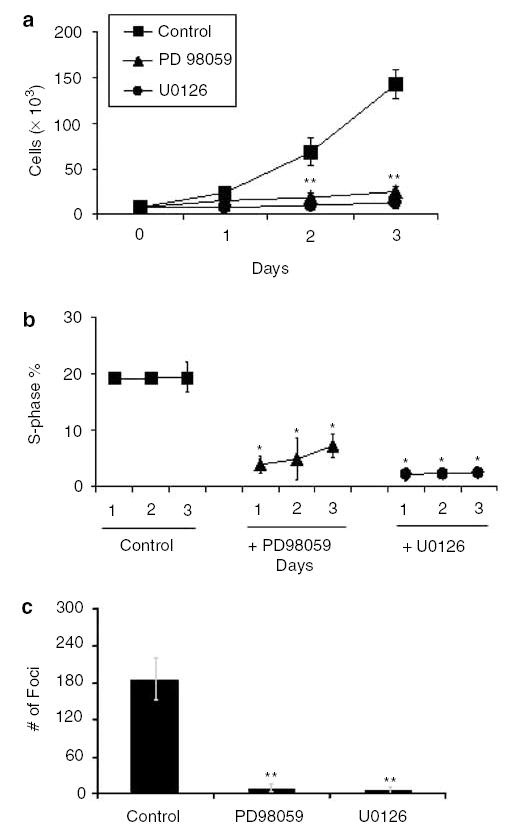
Effect of MEK inhibition on the oncogenic H-Ras transformed phenotype. Panel a shows the total cell count in Ras 7 cells treated with MEK inhibitors, PD98059 and U0126. Cells were initially seeded at 5 × 103 cells/well and supplemented with 10% FBS. Panel b shows the percentage of S-phase subpopulations of the cell cycle in cells treated with MEK inhibitors over a 3-day period. Panel c shows the result of soft agar assays, expressed as mean numbers of colonies per 10-cm dishes. Control represents untreated cells. N = 3 in all experiments. *P < 0.05; **P < 0.01 when compared with control Ras 7 cells
Inhibition of KLF5 expression in oncogenic H-Ras-transformed cells results in a decreased rate of proliferation and anchorage-independent growth
KLF5 has previously been shown to have a proproliferative effect in stably transfected fibroblasts (Sun et al., 2001). The results in Figure 3 indicate that KLF5 is a target of oncogenic H-Ras signaling through the MAPK pathway. To determine whether KLF5 may mediate the proproliferative effect of oncogenic H-Ras, we inhibited KLF5 expression in Ras 7 cells using siRNA and monitored the effect of such treatments. As seen in Figures 5a and b, KLF5-specific siRNA significantly reduced the levels of KLF5 mRNA and protein, respectively. In contrast, a scrambled or nonspecific (NS) siRNA had no effect on the KLF5 protein level in Ras 7 cells (Figure 5b, NS siRNA). Importantly, the rate of proliferation of Ras 7 cells treated with KLF5-specific siRNA was significantly reduced as compared to those treated with the NS siRNA or untreated cells (Figure 5c). Cells treated with KLF5-specific siRNA also exhibited a significant reduction in the proportion of cells in S phase of cell cycle when compared to cells treated with NS siRNA or those left untreated (Figure 5d). Lastly, there was a marked reduction in anchorage-independent growth among the cells treated with the KLF5-specific siRNA in comparison with the control treatments (Figure 5e). The colonies that did form in cells treated with KLF5-specific siRNA were considerably smaller in size when compared to the control treatments (results not shown). These findings strongly indicate that KLF5 plays an essential role in mediating the proproliferative and transforming activities of oncogenic H-Ras.
Figure 5.
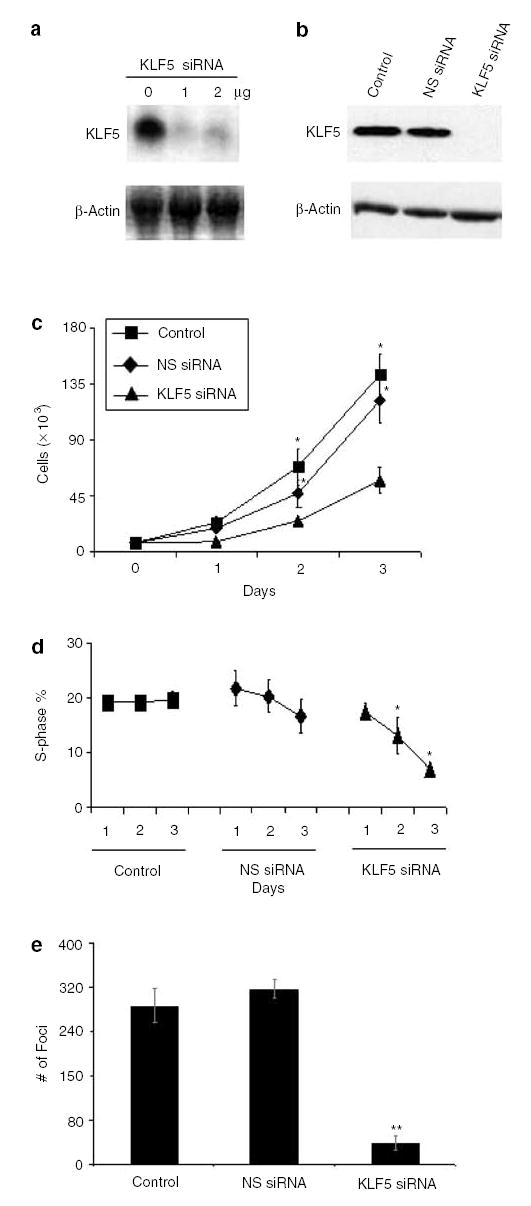
siRNA inhibition of KLF5 in H-Ras-transformed cells results in reduced proliferation. siRNA was used to inhibit KLF5 expression in Ras 7 cells. Panel a shows the result of Northern blot analysis of RNA isolated from Ras 7 cells treated with 0, 1, and 2 μg/10-cm dish of KLF5-specific siRNA. Panel b is the result of Western blot analysis of proteins from untreated control Ras 7 cells, and Ras 7 cells treated with 1 μg NS siRNA or KLF5-specific siRNA. Panel c shows the total cell count of untreated Ras 7 cells and Ras 7 cells treated with 1 μg of NS siRNA or KLF5-specific siRNA. Panel d shows the S-phase subpopulations of Ras 7 cells with different treatment conditions. Panel e is the result of colony formation in soft agar by Ras 7 cells with different treatment conditions. N = 3 in all experiments. *P < 0.05; **P < 0.01 when compared with control Ras 7 cells
The inductive effect of oncogenic H-Ras on KLF5 is mediated by the immediate early gene Egr1
In an attempt to further define the causal-effective relationship between MAPK and KLF5, we treated Ras 7 cells with either NS or KLF5-specific siRNA and measured pERK1/2 levels. As seen in Figure 6a, the levels of both pERK1/2 and total ERK1/2 remained unaltered in Ras 7 cells treated with either siRNA. These results suggest that KLF5 is a downstream target of MAPK and does not itself alter either MAPK activity or stability of signaling intermediates. To further identify the upstream mediator of KLF5 induction by MAPK, we measured the levels of the immediately early gene product, Egr1, in Ras 7 cells under various conditions. Egr1 was selected as a candidate regulator of KLF5 as it was previously reported that the KLF5 promoter contains an Egr1-binding site (Kawai-Kowase et al., 1999). Figure 6b shows that Ras 7 contained abundant levels of Egr1 as compared to untransformed NIH3T3 cells (data not shown). The levels of Egr1 were significantly reduced in Ras 7 cells treated with either MEK inhibitor, PD98059 or U0126 (Figure 6b), indicating that Egr1 is controlled by MAPK. In contrast, levels of Egr1 were unaltered in Ras 7 cells treated with either NS or KLF5-specific siRNA (Figure 6b), indicating the KLF5 does not influence Egr1 expression. Importantly, inhibition of Egr1 expression in Ras 7 cells resulted in a significant reduction in the levels of KLF5 as compared to untreated cells or cells treated with NS siRNA (Figure 6c). These results indicate that Egr1 is an upstream regulator of KLF5 expression.
Figure 6.
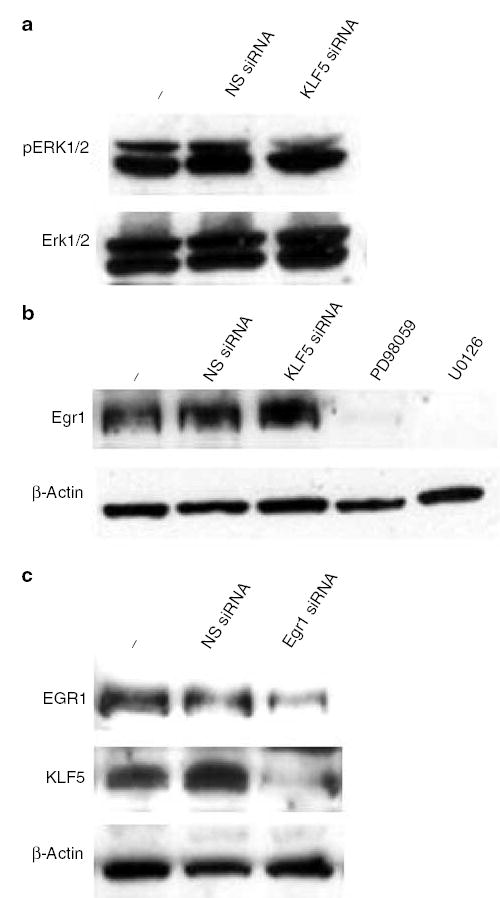
The relationship among MAPK, Egr1 and KLF5 in the signaling pathway of oncogenic H-Ras. Panel a depicts the MAPK activity in Ras 7 cells treated with NS or KLF5-specific siRNA. pERK1/2 (top row) and ERK1/2 (bottom row) levels are shown. − indicates untreated cells. Panel b is the result of Western blot analysis for Egr1 in proteins extracted from Ras 7 cells that were untreated (−), or treated with NS siRNA, KLF5-specific siRNA, PD98059 or U0126. Panel c demonstrates the result of Western blot analysis of Egr1 and KLF5 in Ras 7 cells treated with NS or Egr1-specific siRNA. − indicated untreated cells
KLF5 activates cyclin D1 gene expression
To investigate the mechanism by which KLF5 increases cell proliferation, we determined the levels of cyclin D1 in Ras 7 cells under various conditions. As seen in Figure 7a, levels of cyclin D1 were significantly elevated in Ras 7 cells as compared to untransformed 3T3 cells and could be reduced by treating cells with MEK inhibitors. Importantly, the levels of cyclin D1 were also reduced in Ras 7 cells treated with KLF5-specific siRNA but not NS siRNA (Figure 7a). These findings suggest that the elevated cyclin D1 level is a consequence of increased KLF5 expression. Indeed, cotransfection studies in 3T3 cells involving a luciferase reporter gene linked to the cyclin D1 promoter showed that full-length but not truncated KLF5 was able to stimulate cyclin D1 promoter activity (Figure 7b). These results demonstrate that KLF5 is a transcriptional activator of the cyclin D1 gene.
Figure 7.
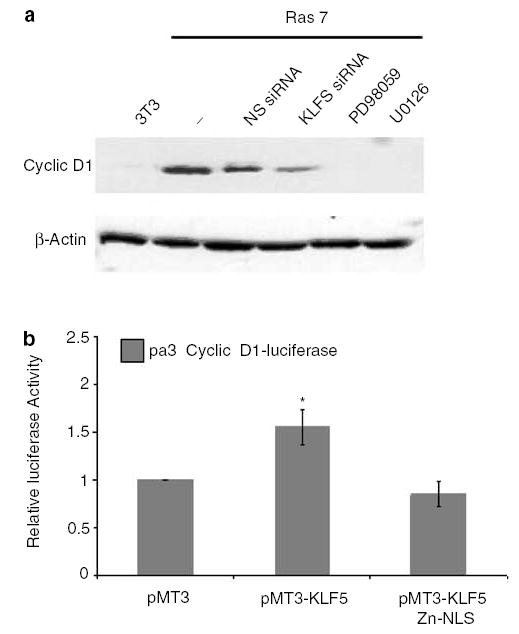
KLF5 regulates cyclin D1 expression. Panel a is a Western blot analysis of cyclin D1 in untransformed NIH3T3 cells or Ras 7 cells treated with the various conditions indicated in the figure. Panel b is the result of luciferase assays in NIH3T3 cells cotransfected with the cyclin D1 promoter-luciferase reporter and vector alone (PMT3), a construct containing full-length KLF5 (PMT3-KLF5) or truncated KLF5 (PMT3-KLF5 Zn-NLS). Relative luciferase activities after normalizing to an internal renilla luciferase are shown. N = 3. *P < 0.05 when compared to PMT3-transfected cells
Discussion
Increase in cellular proliferation is a hallmark of the in vitro transformation process mediated by oncogenic Ras (Kerkhoff and Rapp, 1998; Shields et al., 2000; Downward, 2003). H-Ras, a member of the oncogenic group of Ras proteins, has previously been described to have a potent enhancing effect on cell proliferation, anchorage-independent growth and G1–S cell cycle transition (Kerkhoff and Rapp, 1998; Malumbres and Pellicer, 1998). In addition, the ability of H-Ras-transformed NIH3T3 fibroblasts to maintain proliferation in the absence of serum has been well documented (Filmus et al., 1994; Liu et al., 1995). Results of our study demonstrate that stable transfection of NIH3T3 cells with oncogenic H-Ras resulted in increased levels of H-Ras mRNA and protein, as expected. The transformed phenotype of the two independently derived clones, Ras 2 and Ras 7, was evident as they displayed an increased rate of cell proliferation, serum-independent growth and the ability to form anchorage-independent colonies. Importantly, the relative abundance of the KLF5 mRNA and protein was correlated to that of H-Ras in the transformed cells. These findings indicate that KLF5 is a direct target of oncogenic H-Ras.
Ras activation is accompanied by the stimulation of several downstream cascades, which result in subsequent transcriptional regulation within the cell (Shields et al., 2000). Ras regulates expression of several important factors that contribute to its transforming effect, including c-Myc and cyclin D1 (Cleveland et al., 1994; Liu et al., 1995; Winston et al., 1996). Previous studies have extensively described the activation of MEK and ERK when Ras expression is triggered in vitro (Gallego et al., 1992; Robbins et al., 1992; MacDonald et al., 1993; Downward, 2003). Consistent with these studies, our study also showed that MAPK activity, as measured by the level of pERK, was much higher in both Ras 2 and Ras 7 cells than untransformed NIH3T3 cells. Significantly, inhibition of ERK activity in the two H-Ras-transformed cells by two MEK inhibitors, PD98059 and U0126 (Dudley et al., 1995; DeSilva et al., 1998; Favata et al., 1998), resulted in a considerable reduction in the levels of KLF5 mRNA and protein. These findings provide strong evidence that KLF5 is regulated by an activated MAPK pathway elicited by oncogenic H-Ras. The importance of the MAPK pathway in mediating the proliferative effect of H-Ras is further demonstrated by the loss of growth advantage of Ras 7 cells treated with MEK inhibitors.
The present study identified Egr1 as a mediator of KLF5 induction in H-Ras-transformed cells. Thus, the ability of MEK inhibitors to inhibit Egr1 expression (Figure 6b) and the ability of Egr1-specific siRNA to inhibit KLF5 expression (Figure 6c) in Ras 7 cells place Egr1 in the intermediate step between MAPK and KLF5. This is consistent with previous studies that demonstrate the presence of an Egr1-response element in the KLF5 promoter that mediates MAPK induction of KLF5 expression (Kawai-Kowase et al., 1999). In addition, our study also shows that KLF5 activation does not influence either MAPK activity (Figure 6a) or Egr1 level (Figure 6b), thus positioning KLF5 at the end of the signaling pathway beginning with oncogenic H-Ras followed by MAPK and Egr1.
Since KLF5 is a proproliferative transcription factor (Sun et al., 2001), we surmise that the transformed phenotype caused by oncogenic H-Ras could at least in part be due to KLF5 overexpression. Indeed, inhibition of KLF5 expression using KLF5-specific siRNA in Ras 7 cells impeded proliferation and transformation in a manner reminiscent of the same cells treated with MEK inhibitors. Specifically, KLF5 inhibition reduced the number of cells in the S phase of the cell cycle. This suggests that KLF5 may facilitate the entry of cells into the S phase and raises the possibility that KLF5 may regulate some of the same downstream mediators of Ras function in cell proliferation. The findings that KLF5-specific siRNA reduced cyclin D1 level (Figure 7a) and that KLF5 transactivated cyclin D1 promoter (Figure 7b) indicate that cyclin D1 is a target of KLF5 and mediates KLF5’s effect on cell proliferation. Combined with results of previous studies showing that cyclin D1 level is elevated secondary to oncogenic H-Ras overexpression and that this increased cyclin D1 expression is necessary for H-Ras-mediated cell transformation (Liu et al., 1995), a model of signal transduction initiated by oncogenic H-Ras that leads to increased cell proliferation of transformation can be proposed as illustrated in Figure 8. As different oncogenic Ras proteins converge on the MAPK pathway to exert their cellular effects, the ability of MEK inhibitors to inhibit KLF5 expression suggests that KLF5 induction is independent of the type of activating Ras mutation. Other activators, like PMA, are also known to stimulate MAPK activity (El-Shemerly et al., 1997) and in the specific case of KLF5, PMA is known to be an effector (Sun et al., 2001). Our results could also explain the mechanism of activation of KLF5 by PMA.
Figure 8.
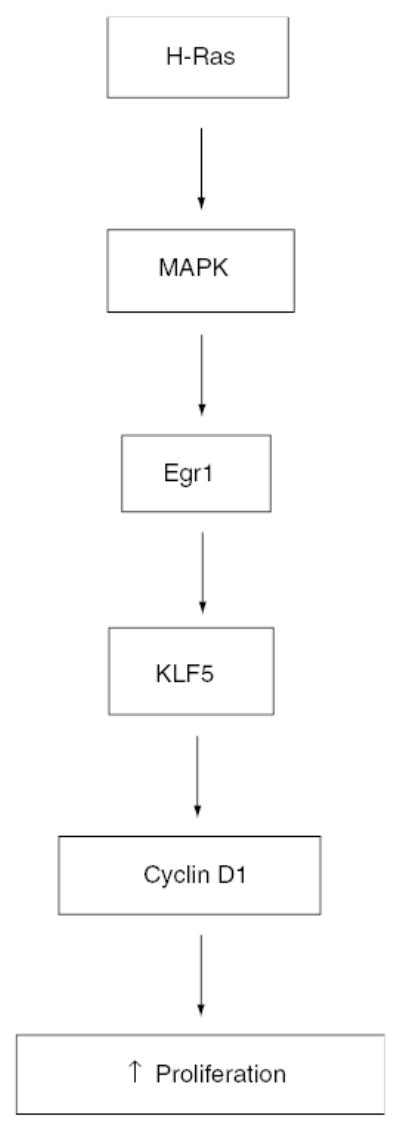
A model of signaling pathway from oncogenic H-Ras to increased cell proliferation and transformation
Since our findings indicate that KLF5 expression is controlled by oncogenic H-Ras, it would be very interesting to examine expression of KLF5 in various primary tumors and cancer cell lines that possess mutated Ras. For example, between 30 and 50% of colorectal cancers exhibit oncogenic Ras mutations, a majority of which are K-Ras (Bos et al., 1987; Vogelstein et al., 1988). It would therefore be interesting to determine whether KLF5 expression is elevated in relation to K-Ras mutation. In contrast, two recent studies have referred to KLF5 as a putative tumor suppressor in prostate and breast cancers, as the chromosomal locus on which KLF5 resides is frequently deleted in these tumors (Chen et al., 2002, 2003). Whether KLF5 functions in a different capacity in the context of different tissues to regulate proliferation and differentiation needs to be investigated.
Materials and methods
Reagents
Culture media and FBS were purchased from Mediatech, Inc. (Herndon, VA, USA). Radioisotopes were obtained from Perkin-Elmer Life Sciences (Boston, MA, USA). MAPK/ERK kinase (MEK) inhibitors, PD98059 and U0126, were purchased from Promega (Madison, WI, USA). The expression construct containing the oncogenic H-Ras was generously provided by Dr Raul Urrutia (Gebelein et al., 1998). Complementary DNA (cDNA) clone encoding KLF5 was provided by Dr Jerry Lingrel (Conkright et al., 1999). The pA3 cyclin D1-luciferase reporter construct that contained 1745 bp of the cyclin D1 promoter sequence was a generous gift of Dr Richard Pestell (Watanabe et al., 1998). The monoclonal antibody against H-Ras was purchased from Oncogene Research (San Diego, CA, USA). Polyclonal antibodies against pERK1/2, ERK1/2, Egr1 and cyclin D1 were purchase from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA).
Cell lines
The mouse fibroblast cell line NIH3T3 was maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% FBS, 1% penicillin–streptomycin at 37°C in a 5% CO2 atmosphere. Stable clones of NIH3T3 transformed by oncogenic H-Ras were selected from foci formed in soft agar as a result of anchorage-independent growth as described previously (Sun et al., 2001). The two clones selected for further studies were designated as Ras 2 and Ras 7, respectively. For experiments involving serum starvation, cells were seeded in media containing 10% FBS and then substituted with media containing 0.5% FBS to impose serum starvation. Treatments with PD98059 and U0126 were carried out in DMEM containing 10% FBS. Cells, in 10 cm or six-well dishes, were rinsed with phosphate-buffered saline (PBS) and then treated with media containing 50 μm PD98059 or U0126. The medium was replaced every 24 h.
Production of KLF5 in bacteria and generation of anti-KLF5 serum
The carboxyl terminal portion of KLF5, between amino acids 242 and 446 was cloned into pET101/D (Dang et al., 2002), and the resulting plasmid construct introduced into the BL21 (DE3) strain of Escherichia coli cells. The solubilized protein from the transformed cells was purified by nickel affinity chromatography and separated using SDS–polyacrylamide gel electrophoresis. The band representing overexpressed KLF5 was excised from the gel and used to raise a rabbit polyclonal antiserum by Covance Research Products (Denver, PA, USA).
Northern and Western blot analyses
RNA was isolated using the Trizol method (Invitrogen), resolved using denaturing agarose gel electrophoresis and transferred to nylon membrane (Hybond-N; Amersham). [α-32P]dATP labeled cDNA probes against H-Ras, KLF5 and β-actin, were labeled using the Prime It II random prime labeling kit (Stratagene). Hybridization and washing were performed under high-stringency conditions as per the QuikHyb hybridization solution protocol (Stratagene). Western blot analysis was performed as previously described (Yoon et al., 2003). The blots were analysed for H-Ras, KLF5, β-actin, pERK1/2 or ERK1/2 using the respective primary and secondary antibodies.
Semiquantitative and real-time RT–PCR
In all, 5 μg total RNA were treated with DNase I per protocol from Ambion, Inc. (Austin, TX, USA). In all, 1 μg of the treated RNA was used for first strand cDNA synthesis using the SuperScript First-Strand Synthesis System for RT–PCR from Invitrogen, Inc. (Carlsbad, CA, USA) as per protocol. The resulting first-strand cDNA was subsequently treated with RNase H (Invitrogen). Primers were designed against mouse KLF5 (Genbank accession number NM_009769), β-actin (Genbank accession number M12481) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH, Genbank accession number NM_008084), the latter two used as internal controls and synthesized by Integrated DNA Technologies, Inc. (Coralville, IA, USA). For mouse KLF5, the forward primer is 5′-ACCATTTTCAGCCACCAGAG-3′, the reverse primer is 5′-GTCTGGTGGGAGCTGAAGA-3′ and the expected PCR product is 100 bp. For β-actin, the forward primer is 5′-GTGGGCCGCTCTAGGCACCAA-3′, the reverse primer is 5′-CTCTTTGATGTCACGCACGATTTC-3′ and the expected PCR product is 540 bp. For GAPDH, the forward primer is 5′-ACCCAGAAGACTGTGGATGG-3′, the reverse primer is 5′-GGATGCAGGGATGATGTTCT-3′ and the amplified PCR product is 81 bp. The primers targeting KLF5 and GAPDH, both used for quantitative PCR were analysed against their secondary RNA structures using software from the European Mfold Server (http://bibiserv.techfak.uni-biele-feld.de/mfold/), to confirm optimal amplification at selected annealing temperatures. The number of cycles was determined from the exponential phase of the amplification reaction for all the primer pairs.
Semiquantitative PCR reaction was carried out with cDNA from 500 ng of total initial RNA. The PCR reaction was carried out with the Advantage cDNA PCR kit as per protocol (BD Biosciences Clontech) using 100 pmol of each primer. The reaction was started with an initial denaturation at 95°C for 5 min followed by cycles of amplification and finished with a final extension step at 72°C for 10 min. The amplification cycle included denaturation at 95°C for 45 s, primer annealing at 56°C for 45 s (KLF5) or 62°C for 45 s (β-actin), and primer extension at 72°C for 1 min and amplification was carried on for 24 cycles (KLF5) or 22 cycles (β-actin). The PCR products were visualized on a 1.5% agarose gel stained with ethidium bromide.
For quantitative or ‘real-time’ PCR, amplifications were detected using SYBR Green 1 in reactions performed with the iCycler iQ real-time PCR Detection System (Bio-Rad). Each 25 μl PCR reaction contained 1 × iQ SYBR Green Supermix (Bio-Rad), 0.4 μm of each primer pair and 500 ng of each cDNA. PCR reactions were started with an initial denaturation at 95°C for 5 min followed by 45 cycles (KLF5) or 40 cycles (GAPDH) consisting of denaturation at 95°C for 30 s, annealing at 67°C (KLF5) or 57°C (GAPDH) for 45 s and extension at 72°C for 1 min, followed by a final extension at 72°C for 5 min. A melting curve analysis was performed after amplification was completed. A standard curve with known quantities of plasmid cDNA templates were used to quantify the amounts of KLF5 and GAPDH from each sample. KLF5 quantities were then normalized to the respective control GAPDH amounts and presented as a ratio among the different cell lines.
Synthesis of KLF5 and Egr1 siRNA and transfection
A sequence corresponding to nucleotides between aa 875 and 895 in the KLF5 coding region, 5′-AACCAGACGGCAGUAAUGGAC-3′ (GenBank accession number NM_009769), was selected to synthesize siRNA. Sequences specific for Egr1 is a mixture of two oligonucleotides, 5′-AACAUCGCUCUGAAUAAUGAG-3′ and 5′-AAACGCAAGAGGCAUACCAAA-3′, that correspond to aa 312–339 and 1224–1242, respectively, in the Egr1 coding region (GenBank accession number NM_007913). Double-stranded siRNA oligonucleotides and an NS, ‘scramble’ duplex (Kullmann et al., 2002; Rajendran et al., 2003), used as control, were synthesized at Dharmacon, Inc. (Lafayette, CO, USA). The cells were grown to 40% confluence in six-well plates before transfection with siRNA using DMRIE-C reagent (Invitrogen) for 4 h in Opti-MEM I Reduced Serum Medium (Invitrogen). Following transfection, the cells were supplemented with DMEM, containing 10% FBS and 1% penicillin–streptomycin. For cell cycle and cell proliferation experiments, DMEM and FBS were replaced daily.
Cell proliferation and anchorage-independent focus formation assays
Cell proliferation was analysed as previously described (Sun et al., 2001). Cells were seeded onto six-well plates and supplemented with 10% or 0.5% FBS (serum-starved), at a density of 105 cells per well, and the medium was replaced every day. The cells were trypsinized each day and counted using a Bright-Line Hemacytometer (Sigma). Anchorage-independent focus formation assays were also performed as previously reported (Sun et al., 2001). Cells with a density of 104 per 10-cm plate were seeded in a 0.3% top agar suspension overlaid on a 0.5% bottom agar layer. The cells were fed fresh medium (DMEM with 10% FBS) every 3 days and incubated at 37°C in 5% CO2 atmosphere. For experiments involving treatment of cells, the medium was supplemented with the appropriate inhibitors or siRNA. Colonies were counted at the end of the 3-week incubation period.
Cell cycle analysis
Cells were analysed for different phases of the cell cycle as previously described (Yoon et al., 2003). In short, cells were rinsed, trypsinized, pelleted and then resuspended in 70% ethanol for fixing overnight at −20°C. Before analysis, cells were pelleted again and resuspended in propidium iodide (PI) solution consisting of 50 μg/ml PI, 50 μg/ml RNase A, 0.1% Triton X-100 and 0.1 mm EDTA for 30 min. Cells were then analysed using a FACSCalibur flow cytometer (Becton Dickinson).
Reporter assays
NIH3T3 cells were cotransfected with an equal amount of pA3 cyclin D1-luciferase reporter (Watanabe et al., 1998) and the empty expression construct PMT3, or PMT3 containing full-length KLF5 (PMT3-KLF5) or truncated KLF5 that contains only the zinc fingers and nuclear localization signal portion of the protein (PMT3-KLF5 Zn-NLS). Included in the transfection was an internal standard, CMV-renilla luciferase reporter. Luciferase activities were determined 48 h following transfection and normalized to the renilla luciferase activities.
Acknowledgments
We thank Drs Raul Urrutia, Jerry Lingrel and Richard Pestell for kindly providing the various plasmid constructs in the study. This work was in part supported by grants from the National Institutes of Health (DK52230, DK64399 and CA84197). VWY is a recipient of a Georgia Cancer Coalition Distinguished Cancer Clinician Scientist Award.
References
- Barbacid M. Annu Rev Biochem. 1987;56:779–827. doi: 10.1146/annurev.bi.56.070187.004023. [DOI] [PubMed] [Google Scholar]
- Black AR, Black JD, Azizkhan-Clifford J. J Cell Physiol. 2001;188:143–160. doi: 10.1002/jcp.1111. [DOI] [PubMed] [Google Scholar]
- Bos JL, Fearon ER, Hamilton SR, Verlaan-de Vries M, van Boom JH, van der Eb AJ, Vogelstein B. Nature. 1987;327:293–297. doi: 10.1038/327293a0. [DOI] [PubMed] [Google Scholar]
- Chen C, Bhalala HV, Qiao H, Dong JT. Oncogene. 2002;21:6567–6572. doi: 10.1038/sj.onc.1205817. [DOI] [PubMed] [Google Scholar]
- Chen C, Bhalala HV, Vessella RL, Dong JT. Prostate. 2003;55:81–88. doi: 10.1002/pros.10205. [DOI] [PubMed] [Google Scholar]
- Chen JY, Penco S, Ostrowski J, Balaguer P, Pons M, Starrett JE, Reczek P, Chambon P, Gronemeyer H. EMBO J. 1995;14:1187–1197. doi: 10.1002/j.1460-2075.1995.tb07102.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleveland JL, Troppmair J, Packham G, Askew DS, Lloyd P, Gonzalez-Garcia M, Nunez G, Ihle JN, Rapp UR. Oncogene. 1994;9:2217–2226. [PubMed] [Google Scholar]
- Conkright MD, Wani MA, Anderson KP, Lingrel JB. Nucleic Acids Res. 1999;27:1263–1270. doi: 10.1093/nar/27.5.1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crossley M, Whitelaw E, Perkins A, Williams G, Fujiwara Y, Orkin SH. Mol Cell Biol. 1996;16:1695–1705. doi: 10.1128/mcb.16.4.1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang DT, Pevsner J, Yang VW. Int J Biochem Cell Biol. 2000;32:1103–1121. doi: 10.1016/s1357-2725(00)00059-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang DT, Zhao W, Mahatan CS, Geiman DE, Yang VW. Nucleic Acids Res. 2002;30:2736–2741. doi: 10.1093/nar/gkf400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daum G, Eisenmann-Tappe I, Fries HW, Troppmair J, Rapp UR. Trends Biochem Sci. 1994;19:474–480. doi: 10.1016/0968-0004(94)90133-3. [DOI] [PubMed] [Google Scholar]
- de Vries-Smits AM, Burgering BM, Leevers SJ, Marshall CJ, Bos JL. Nature. 1992;357:602–604. doi: 10.1038/357602a0. [DOI] [PubMed] [Google Scholar]
- Dekker LV, Parker PJ. Trends Biochem Sci. 1994;19:73–77. doi: 10.1016/0968-0004(94)90038-8. [DOI] [PubMed] [Google Scholar]
- DeSilva DR, Jones EA, Favata MF, Jaffee BD, Magolda RL, Trzaskos JM, Scherle PA. J Immunol. 1998;160:4175–4181. [PubMed] [Google Scholar]
- Dobrowolski S, Harter M, Stacey DW. Mol Cell Biol. 1994;14:5441–5449. doi: 10.1128/mcb.14.8.5441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downward J. Nat Rev Cancer. 2003;3:11–22. doi: 10.1038/nrc969. [DOI] [PubMed] [Google Scholar]
- Dudley DT, Pang L, Decker SJ, Bridges AJ, Saltiel AR. Proc Natl Acad Sci USA. 1995;92:7686–7689. doi: 10.1073/pnas.92.17.7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Shemerly MY, Besser D, Nagasawa M, Nagamine Y. J Biol Chem. 1997;272:30599–30602. doi: 10.1074/jbc.272.49.30599. [DOI] [PubMed] [Google Scholar]
- Favata MF, Horiuchi KY, Manos EJ, Daulerio AJ, Stradley DA, Feeser WS, Van Dyk DE, Pitts WJ, Earl RA, Hobbs F, Copeland RA, Magolda RL, Scherle PA, Trzaskos JM. J Biol Chem. 1998;273:18623–18632. doi: 10.1074/jbc.273.29.18623. [DOI] [PubMed] [Google Scholar]
- Filmus J, Robles AI, Shi W, Wong MJ, Colombo LL, Conti CJ. Oncogene. 1994;9:3627–3633. [PubMed] [Google Scholar]
- Gallego C, Gupta SK, Heasley LE, Qian NX, Johnson GL. Proc Natl Acad Sci USA. 1992;89:7355–7359. doi: 10.1073/pnas.89.16.7355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett-Sinha LA, Eberspaecher H, Seldin MF, de Crombrugghe B. J Biol Chem. 1996;271:31384–31390. doi: 10.1074/jbc.271.49.31384. [DOI] [PubMed] [Google Scholar]
- Gebelein B, Fernandez-Zapico M, Imoto M, Urrutia R. J Clin Invest. 1998;102:1911–1919. doi: 10.1172/JCI1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshino Y, Kurabayashi M, Kanda T, Hasegawa A, Sakamoto H, Okamoto E, Kowase K, Watanabe N, Manabe I, Suzuki T, Nakano A, Takase S, Wilcox JN, Nagai R. Circulation. 2000;102:2528–2534. doi: 10.1161/01.cir.102.20.2528. [DOI] [PubMed] [Google Scholar]
- Howe LR, Leevers SJ, Gomez N, Nakielny S, Cohen P, Marshall CJ. Cell. 1992;71:335–342. doi: 10.1016/0092-8674(92)90361-f. [DOI] [PubMed] [Google Scholar]
- Kaczynski J, Cook T, Urrutia R. Genome Biol. 2003;4:206. doi: 10.1186/gb-2003-4-2-206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai-Kowase K, Kurabayashi M, Hoshino Y, Ohyama Y, Nagai R. Circ Res. 1999;85:787–795. doi: 10.1161/01.res.85.9.787. [DOI] [PubMed] [Google Scholar]
- Kerkhoff E, Rapp UR. Oncogene. 1998;17:1457–1462. doi: 10.1038/sj.onc.1202185. [DOI] [PubMed] [Google Scholar]
- Khosravi-Far R, Campbell S, Rossman KL, Der CJ. Adv Cancer Res. 1998;72:57–107. doi: 10.1016/s0065-230x(08)60700-9. [DOI] [PubMed] [Google Scholar]
- Kolch W, Heidecker G, Kochs G, Hummel R, Vahidi H, Mischak H, Finkenzeller G, Marme D, Rapp UR. Nature. 1993;364:249–252. doi: 10.1038/364249a0. [DOI] [PubMed] [Google Scholar]
- Kullmann M, Gopfert U, Siewe B, Hengst L. Genes Dev. 2002;16:3087–3099. doi: 10.1101/gad.248902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo ML, Kang JJ, Yang NC. Cancer Lett. 1993;74:197–202. doi: 10.1016/0304-3835(93)90243-3. [DOI] [PubMed] [Google Scholar]
- Leevers SJ, Paterson HF, Marshall CJ. Nature. 1994;369:411–414. doi: 10.1038/369411a0. [DOI] [PubMed] [Google Scholar]
- Liu JJ, Chao JR, Jiang MC, Ng SY, Yen JJ, Yang-Yen HF. Mol Cell Biol. 1995;15:3654–3663. doi: 10.1128/mcb.15.7.3654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdonald SG, Crews CM, Wu L, Driller J, Clark R, Erikson RL, McCormick F. Mol Cell Biol. 1993;13:6615–6620. doi: 10.1128/mcb.13.11.6615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malumbres M, Pellicer A. Front Biosci. 1998;3:d887–d912. doi: 10.2741/a331. [DOI] [PubMed] [Google Scholar]
- Marais R, Light Y, Paterson HF, Marshall CJ. EMBO J. 1995;14:3136–3145. doi: 10.1002/j.1460-2075.1995.tb07316.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ming XF, Burgering BM, Wennstrom S, Claesson-Welsh L, Heldin CH, Bos JL, Kozma SC, Thomas G. Nature. 1994;371:426–429. doi: 10.1038/371426a0. [DOI] [PubMed] [Google Scholar]
- Muszynski KW, Ruscetti FW, Heidecker G, Rapp U, Troppmair J, Gooya JM, Keller JR. J Exp Med. 1995;181:2189–2199. doi: 10.1084/jem.181.6.2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nori M, L’Allemain G, Weber MJ. Mol Cell Biol. 1992;12:936–945. doi: 10.1128/mcb.12.3.936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogata T, Kurabayashi M, Hoshino Y, Sekiguchi K, Ishikawa S, Morishita Y, Nagai R. J Thorac Cardiovasc Surg. 2000;119:983–989. doi: 10.1016/S0022-5223(00)70093-6. [DOI] [PubMed] [Google Scholar]
- Ohnishi S, Laub F, Matsumoto N, Asaka M, Ramirez F, Yoshida T, Terada M. Dev Dyn. 2000;217:421–429. doi: 10.1002/(SICI)1097-0177(200004)217:4<421::AID-DVDY9>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Philipsen S, Suske G. Nucleic Acids Res. 1999;27:2991–3000. doi: 10.1093/nar/27.15.2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajendran RR, Nye AC, Frasor J, Balsara RD, Martini PG, Katzenellenbogen BS. J Biol Chem. 2003;278:4628–4638. doi: 10.1074/jbc.M210066200. [DOI] [PubMed] [Google Scholar]
- Robbins DJ, Cheng M, Zhen E, Vanderbilt CA, Feig LA, Cobb MH. Proc Natl Acad Sci USA. 1992;89:6924–6928. doi: 10.1073/pnas.89.15.6924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H, Zhang Z, Wang X, Liu S, Teng CT. Nucleic Acids Res. 1999;27:4807–4815. doi: 10.1093/nar/27.24.4807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shie JL, Chen ZY, O’Brien MJ, Pestell RG, Lee ME, Tseng CC. Am J Physiol Gastrointest Liver Physiol. 2000;279:G806–G814. doi: 10.1152/ajpgi.2000.279.4.G806. [DOI] [PubMed] [Google Scholar]
- Shields JM, Christy RJ, Yang VW. J Biol Chem. 1996;271:20009–20017. doi: 10.1074/jbc.271.33.20009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shields JM, Pruitt K, McFall A, Shaub A, Der CJ. Trends Cell Biol. 2000;10:147–154. doi: 10.1016/s0962-8924(00)01740-2. [DOI] [PubMed] [Google Scholar]
- Simmen RC, Simmen FA. Front Biosci. 2002;7:d1556–d1565. doi: 10.2741/simmen. [DOI] [PubMed] [Google Scholar]
- Sun R, Chen X, Yang VW. J Biol Chem. 2001;276:6897–6900. doi: 10.1074/jbc.C000870200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas SM, DeMarco M, D’Arcangelo G, Halegoua S, Brugge JS. Cell. 1992;68:1031–1040. doi: 10.1016/0092-8674(92)90075-n. [DOI] [PubMed] [Google Scholar]
- Troppmair J, Bruder JT, Munoz H, Lloyd PA, Kyriakis J, Banerjee P, Avruch J, Rapp UR. J Biol Chem. 1994;269:7030–7035. [PubMed] [Google Scholar]
- Turner J, Crossley M. Trends Biochem Sci. 1999;24:236–240. doi: 10.1016/s0968-0004(99)01406-1. [DOI] [PubMed] [Google Scholar]
- Vogelstein B, Fearon ER, Hamilton SR, Kern SE, Preisinger AC, Leppert M, Nakamura Y, White R, Smits AM, Bos JL. N Engl J Med. 1988;319:525–532. doi: 10.1056/NEJM198809013190901. [DOI] [PubMed] [Google Scholar]
- Wartmann M, Hofer P, Turowski P, Saltiel AR, Hynes NE. J Biol Chem. 1997;272:3915–3923. doi: 10.1074/jbc.272.7.3915. [DOI] [PubMed] [Google Scholar]
- Watanabe G, Albanese C, Lee RJ, Reutens A, Vairo G, Henglein B, Pestell RG. Mol Cell Biol. 1998;18:3212–3222. doi: 10.1128/mcb.18.6.3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winston JT, Coats SR, Wang YZ, Pledger WJ. Oncogene. 1996;12:127–134. [PubMed] [Google Scholar]
- Yang JJ, Kang JS, Krauss RS. Mol Cell Biol. 1998;18:2586–2595. doi: 10.1128/mcb.18.5.2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon HS, Chen X, Yang VW. J Biol Chem. 2003;278:2101–2105. doi: 10.1074/jbc.M211027200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziemer LT, Pennica D, Levine AJ. Mol Cell Biol. 2001;21:562–574. doi: 10.1128/MCB.21.2.562-574.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]