

Summary
Recent studies show that the melanocyte transcription factor MITF not only activates differentiation genes but also genes involved in the regulation of the cell cycle, suggesting that it provides a link between cell proliferation and differentiation. MITF, however, comes in a variety of splice isoforms with potentially distinct biological activities. In particular, there are two isoforms, (-) and (+) MITF, that differ in six residues located upstream of the DNA binding basic domain and show slight differences in the efficiency with which they bind to target DNA. Using in vitro BrdU incorporation assays and FACS analysis in transiently transfected cells, we show that (+) MITF has a strong inhibitory effect on DNA synthesis while (-) MITF has none or only a mild one. The strong inhibitory activity of (+) MITF is not influenced by a number of mutations that modulate MITF’s transcriptional activities and is independent of the protein’s carboxyl terminus but dependent on its aminoterminus. A further dissection of the molecule points to the importance of an aminoterminal serine, serine-73, which in both isoforms is phosphorylated to comparable degrees. The results suggest that one or several aminoterminal domains cooperate with the alternatively spliced hexapeptide to render MITF anti-proliferative in a way that does not depend on direct E box binding.
Keywords: melanocyte, transcription regulation, post-translational regulation, cell cycle
Introduction
For many cell types, terminal differentiation goes hand in hand with exit from the cell cycle. Some cells such as melanocytes and melanoma cells, however, can express cell differentiation genes and yet continue to divide. A link between continued cell proliferation and differentiation of melanocytes may possibly be provided by MITF, a basic helix-loop-helix-leucine zipper transcription factor that serves as a key regulator of both cell cycle genes and differentiation genes. For instance, Tyrosinase, whose product is critical in melanin biosynthesis, and Cdk2, whose product promotes G1 → S progression, are both stimulated by Mitf, and so is Tbx2, whose product suppresses a negative regulator of G1 → S progression, p21Cip1 (Du et al., 2004; Prince et al., 2004; Vance et al., 2005, reviewed in Arnheiter et al., in press; Steingrimsson et al., 2004; Steingrimsson et al., 2005; Vance and Goding, 2004). Nevertheless, p21 is also directly stimulated by Mitf (Carreira et al., 2005), as are other negative regulators of the cell cycle, such as INK4A which promotes cell cycle exit (Loercher et al., 2005). Moreover, MITF protein interacts with hypophosphorylated retinoblastoma protein-1 (Rb1) to cooperatively stimulate p21 and Tyrosinase (Carreira et al., 2005). Given these complexities, it is not surprising that MITF has been found, in vitro, to inhibit cell proliferation under some conditions and to promote it under others, and that in vivo genetic evidence has not provided a consistent answer either. For instance, for retinal pigment epithelium cells, Mitf seems to slow down proliferation, although not to abolish it (Nakayama et al., 1998; Packer, 1967), but for neural crest-derived melanocytes, a similar action is not easily demonstrable chiefly because Mitf mutations first and foremost affect melanoblast survival (Hornyak et al., 2001; Opdecamp et al., 1997). It is conceivable, therefore, that the regulation of Mitf itself might dictate to what extent positive and negative cell cycle regulators accumulate at different time points in melanocyte biology and hence control the balance between melanocyte proliferation and differentiation.
Mitf is regulated both at the transcriptional and post-translational levels. First, the gene is transcribed from multiple promoters giving rise to a variety of splice isoforms with potentially distinct biological activities (Arnheiter et al., in press; Steingrimsson et al., 2004). The promoter prominently and seemingly specifically used in the neural crest-derived melanocyte lineage is called the M-promoter which responds to melanogenic extracellular signals activating tyrosine kinase and G-coupled receptors. Second, M-Mitf mRNA comes in two major isoforms resulting from a differential splice acceptor use in exon 6. The (+) isoform, which includes 18 bases corresponding to exon 6a, is translated into a 419 residue protein that includes the sequence ACIFPT upstream of the DNA-binding basic domain. The (-) isoform lacks this sequence, encodes a 413 residue protein, and in mouse melanocytes and melanoma cells has been found to accumulate to similar levels as the (+) isoform (Hodgkinson et al., 1993; Steingrímsson et al., 1994). The levels to which the corresponding (+) and (-) MITF proteins accumulate, however, have not yet been determined because no easy separation techniques or isoform-specific immunoreagents are available. The two proteins may have distinct functions as (+) MITF binds target E boxes in promoter DNA with a slightly higher affinity than (-) MITF in vitro (Hemesath et al., 1994). This observation is consistent with the fact that the mouse allele Mitfmi-sp (microphthalmia-spotted), from which only (-) MITF can be expressed because of a supernumary base inserted into exon 6a (Steingrímsson et al., 1994), is associated with a mild pigmentation phenotype (Wolfe and Coleman, 1964). Besides these two isoforms of Mitf, a number of other isoforms have been described where individual exons are missing (Hallsson et al., 2000). The biological significance of many of these isoforms is yet to be determined and in no case is it clear that their expression is differentially regulated in a developmental or cell cycle-specific way.
MITF is also modified post-translationally. For example, extracellular signaling-mediated phosphorylations at serines 73, 298, and 307 have generally been found to enhance activity, but single phosphorylation at serine-73 or double serine-73/serine-409 phosphorylations have also been implicated in enhanced proteasome-mediated degradation, perhaps representing a negative feed-back loop (reviewed in Arnheiter et al., in press; Steingrimsson et al., 2004). Furthermore, lysine 182 and lysine 316 are sites of sumoylation and their mutation increases MITF activity. This increase in activity is more prominent on promoters containing multiple E box binding sites compared with those containing single binding sites, suggesting target gene-specific regulation (Miller et al., 2005; Murakami and Arnheiter, 2005). Moreover, acetylation can occur in lysines in the basic domain and in the leucine zipper domain (Steingrimsson et al., 2004, and unpublished observations), although the biological significance of these modifications is yet to be determined.
Here we sought to test the parameters by which different domains of MITF protein might regulate cell proliferation. To this end, we analyzed DNA synthesis and cell cycle characteristics in cells transfected with expression plasmids encoding wild type Mitf or various Mitf isoforms and mutants. These experiments were mainly done in HEK293 cells, a human embryonic kidney cell line with neuron-like gene expression profiles (Shaw et al., 2002). We chose this non-melanocytic cell line because it does not discernibly express endogenous MITF, allowing us to eliminate potential complications that might arise from interference by wild type MITF. Consistent with previous results obtained in melanoma and fibroblastic cell lines (Carreira et al., 2005; Loercher et al., 2005), we found that in HEK293 cells, (+) MITF leads to a reduction in DNA synthesis consistent with its role as an inhibitor of cell proliferation. Surprisingly, however, (-) MITF, although accumulating to comparable levels as (+) MITF in transfected cells, had no such inhibitory effect. Moreover, the anti-proliferative activity of (+) MITF did not depend on an intact DNA binding domain or on the carboxyl terminal domain, but depended on the aminoterminal domain. The results suggest that the regulation of cell proliferation by MITF may not exclusively rely on its direct regulation of cell cycle target genes and may depend on yet uncharacterized intra- or intermolecular interactions that involve the aminoterminus of MITF and its exon 6a-encoded hexapeptide.
Results
(+) MITF but not (-) MITF inhibits BrdU incorporation in transfected HEK293 cells
To assess the role of the (-) and (+) isoforms of MITF in cell proliferation, we transfected corresponding cDNAs into HEK293 cells and performed BrdU incorporation assays. HEK293 cells are adherent, can consistently be transfected with high efficiency (>60% transfectants), and show a short doubling time: in dishes transfected with either of the two isoforms of MITF, non-transfectants double every 16.2 h (data not shown). Immunofluorescent staining (Figure 1A) and western blot analysis (Figure 1B) did not reveal differences in the levels of expression or nuclear accumulation between (-) and (+) MITF. Notably, the intensity of the upper band in western blots (Figure 1B), representing MITF phosphorylated at serine-73 (Hemesath et al., 1998), is similar for both (+) and (-) isoforms, suggesting that the steady state level of phosphorylation at this position is similar for the two isoforms.
Figure 1.

(+) MITF but not (-) MITF inhibits BrdU incorporation in transfected cells. (A) MITF immunofluorescence of HEK293 cells transfected with (-) or (+) Mitf cDNA. (B) Western blot analysis of HEK293 cell extracts prepared 20 h after transfection with either (-) or (+) Mitf cDNA, using C5 anti-MITF antibody. Loading control corresponds to an unspecific band. (C) BrdU incorporation in HEK293 cells 20 h after transfection with (-) or (+) Mitf cDNA or pEGFP-C1 plasmid. Cells were incubated for 30 min with 10 μM BrdU and double-labeled with anti-BrdU and C5 anti-MITF antibodies. Double-positive cells were counted as a percentage of the total number of MITF-positive cells. (D) Similar assay as in C but done in BHK cells.
To assess BrdU incorporation, transfected cells were exposed to BrdU for 30 min, washed, fixed, and doubly stained for BrdU and MITF, using corresponding first and fluorescently labeled second antibodies. Each transfection was done in triplicate and the results combined from three independent experiments. As shown in Figure 1C, 64 ± 3% of control non-transfected cells were BrdU-positive, reflecting their rapid rate of proliferation. The percentage was similar in cells expressing (-) MITF where 67 ± 2% of MITF-positive cells were BrdU-positive. In contrast, among cells expressing (+) MITF, this percentage was only 44 ± 2%. Hence, in HEK293 cells, only (+) MITF induced a visible reduction in BrdU incorporation while (-) MITF did not. One might conclude, therefore, that (-) MITF has no effect on DNA synthesis. It is conceivable, however, that unrelated control proteins, expressed at high levels, may be inhibitory, and that if this were the case, (-) MITF would have to be considered pro-proliferative. We reasoned that green fluorescent protein (EGFP) may represent a suitable control as it is widely used for marking cells and convenient for visualization in microscopy. As shown in Figure 1C, however, cells expressing EGFP incorporated BrdU at lower levels compared with untransfected cells or cells expressing (-) MITF (57 ± 5% versus 64 ± 2%, respectively, P < 0.01) although the inhibition by (+) MITF was still stronger (44 ± 2%). Hence, compared with this control protein, (-) MITF appears to have a pro-proliferative activity in HEK293 cells.
Since HEK293 cells express the adenoviral E1A protein which binds p300, a transcriptional co-activator with histone acetyltransferase activity, at a site through which MITF also interacts with p300 (Price et al., 1998; Sato et al., 1997), it was conceivable that the above results reflect a difference in the efficacy with which E1A competes with (-) or (+) MITF for binding to p300. To exclude this possibility, we also tested the effects of (-) and (+) MITF on BrdU incorporation in BHK cells which do not express E1A and which, like HEK293 cells, also lack endogenous MITF (not shown). As shown in Figure 1D, in contrast to HEK293 cells, BHK cells divide more slowly, and BrdU incorporation in control non-transfected cells was only 34 ± 5%. BrdU incorporation was inhibited by both (-) and (+) MITF. Nevertheless, like in HEK293 cells, the inhibition by (+) MITF was greater (14 ± 3% BrdU/MITF double-positive cells per total number of MITF-positive cells) compared with that achieved by (-) MITF (24 ± 6% BrdU/MITF double-positive cells per total number of MITF-positive cells) (Figure 1D). These results suggest that the difference between (+) and (-) MITF in inhibiting BrdU incoporation in HEK293 cells is not simply because of the presence of E1A, and that the extent to which MITF, (-) or (+), inhibits DNA synthesis is cell type-dependent.
The difference between (+) and (-) MITF on BrdU incorporation is independent of direct DNA binding or modulation of transcriptional activity
To determine if the inhibitory effect on BrdU incorporation of the (+) MITF isoform was because of the activation of target genes important in cell cycle regulation, we tested the effect of MITF mutated in the DNA binding basic domain. In all bHLH and bHLH-Zip proteins, an invariant glutamic acid contacts the core bases of the respective E-box motifs in DNA, and mutations of this residue are known to abrogate specific binding (Fisher et al., 1993). Hence, we generated two mutated plasmids, encoding (-) MITF E213A and (+) MITF E213A (Figure 2A), and expressed them in HEK293 cells. Immunofluorescence (Figure 2B) and western blots (Figure 2C) indicated that the proteins were nuclear and expressed at comparable levels. As observed for the corresponding wild type MITFs, (-) MITF E213A did not inhibit BrdU incorporation (66 ± 1% BrdU/MITF double-positive cells per total number of MITF-positive cells) but (+) MITF E213A did inhibit it (46 ± 2% BrdU/MITF double-positive cells per total number of MITF-positive cells) (Figure 2D). The results suggest that the inhibitory effect of (+) MITF is independent of its capacity to directly bind DNA. They do not exclude the possibility, however, that MITF, by binding other proteins, might indirectly regulate the transcription of cell cycle genes.
Figure 2.
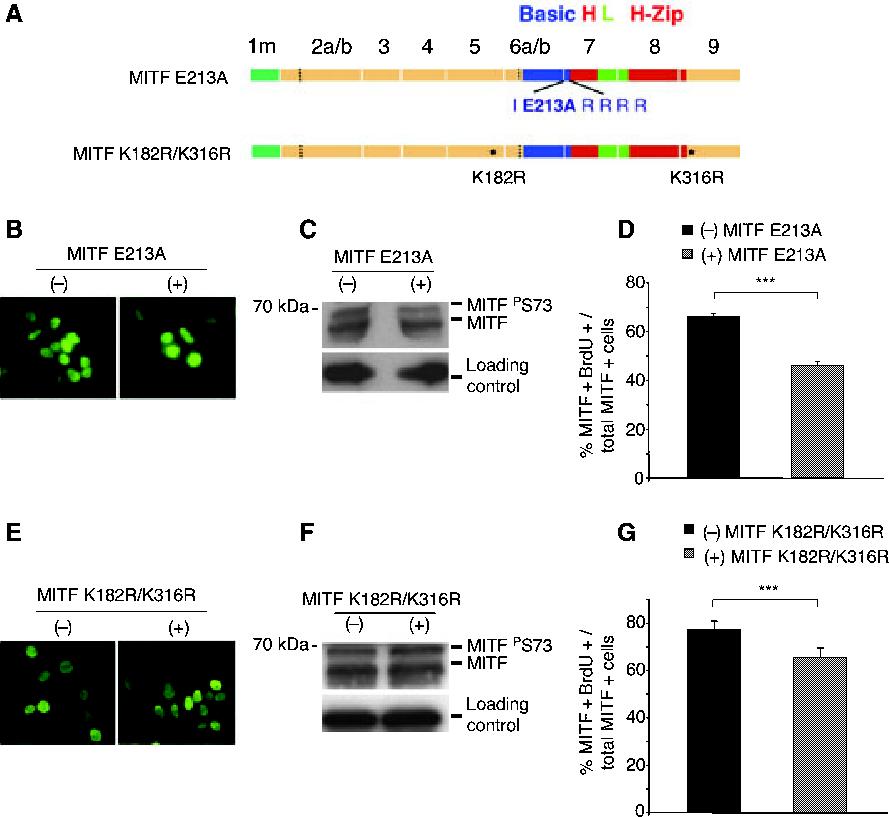
MITF mutations that affect DNA binding or transcriptional activity do not interfere with the enhanced inhibition of BrdU incorporation by (+) MITF. (A) schematic representation of MITF E213A and MITF K182R/316R. (B and E) Immunofluorescent labeling of cells transfected with (-) or (+) MITF E213A (B) or (-) or (+) MITF K182R/K316R (E). (C and F) Western blot analysis of cells transfected with (-) or (+) MITF E213A (C) or (-) or (+) MITF K182R/K316R (F). Loading controls correspond to beta-tubulin. (D and G) BrdU incorporation in cells transfected with (-) or (+) MITF E213A (D) or (-) or (+) MITF K182R/K316R (G). Cells were incubated with 10 μM BrdU and stained with anti-MITF and anti-BrdU. Double MITF/BrdU-positive cells were counted as in Figure 1.
It has recently been demonstrated that (+) MITF with mutations in the two sumoylation sites at lysines 182 and 316 has, compared with wild type, an increased transcriptional activity on promoters containing multiple E box motifs (Miller et al., 2005; Murakami and Arnheiter, 2005). This increased activity is not because of changes in DNA binding and may reflect decreased recruitment of transcriptional co-repressors, notwithstanding the fact that sumoylation, a dynamic process balanced by de-sumoylation, cannot easily be demonstrated at the steady state level. To assess the effect of increased transcriptional activity on cell proliferation, we tested (-) and (+) MITF with double lysine182/316-to-arginine mutations [(-) MITF K182R/K316R and (+) MITF K182R/K316R, Figure 2A]. Both mutants were nuclear (Figure 2E) and expressed at similar levels (Figure 2F). As shown in Figure 2G, 77 ± 3.6% of cells expressing (-) MITF K182R/K316R and 65 ± 3.9% of cells expressing (+) MITF K182R/K316R showed BrdU incorporation. Interestingly, these numbers were higher (77 ± 3.6% and 65 ± 3.9% for (-) and (+) MITF K182R/K316R, respectively) than those obtained with the corresponding wild type proteins (see Figure 1C), whereby BrdU incorporation in cells not expressing MITF (65.8 ± 4%, not shown) was comparable with that obtained in Figure 1C and subsequent experiments. Despite increased BrdU incorporation, however, the difference between (-) and (+) MITF was retained, suggesting that the increase in transcriptional activity associated with these mutants, or the proximity of the K182R mutation to the exon 6a-encoded residues, do not eliminate the influence of exon 6a on BrdU incorporation. We conclude, therefore, that neither a decrease in transcriptional activity resulting from a DNA-binding mutation, nor an increase in activity on promoters with multiple E boxes as seen with the sumoylation mutants, interfere with the role of the alternatively spliced six residues in regulating cell proliferation.
Inhibition of BrdU incorporation is reduced in mutant (+) MITF with impaired nuclear translocation
We then investigated if nuclear localization of MITF was critical for inhibition of BrdU incorporation. For this purpose, we reproduced the mutation present in Mitfmi/mi which is characterized by a deletion of one arginine in a run of four in the DNA-binding basic domain (Figure 3A) and is associated with one of the most severe pigmentation phenotypes in both mouse and man. This mutant protein has been reported to accumulate less efficiently in the nucleus than wild type protein, both in vitro (Takebayashi et al., 1996) and in vivo (Nakayama et al., 1998). Upon transfection of HEK293 cells with plasmids encoding (-) or (+) MITF DelR216, both proteins were more readily localized in the cytoplasm even though both were also present in the nucleus (Figure 3B). Despite expression to levels comparable with each other (Figure 3C) and to those seen with wild type protein (compare, for instance, with Figure 1B), (-) MITF DelR216 resulted in 57 ± 2% BrdU/MITF double-positive cells per total number of MITF positive cells, and (+) MITF DelR216 in 48 ± 5% BrdU/MITF double-positive cells per total number of MITF-positive cells. Hence, (-) MITF DelR216 was inhibitory compared with non-transfected cells, of which 64 ± 2% were BrdU-positive (not shown), perhaps because its increased cytoplasmic accumulation is deleterious to cells. Nevertheless, (+) MITF DelR216 was still more inhibitory, indicating that the presence of the six residues influences the anti-proliferative activity of the mutant protein independent of its increased presence in the cytoplasm.
Figure 3.

A mutation leading to decreased nuclear translocation reduces the inhibitory effect of (+) MITF. (A) schematic representation of MITF DelR216. (B) Cytoplasmic and nuclear localization of (-) and (+) MITF DelR216. (C) Western blot analysis of protein extracts of cells transfected with (-) and (+) MITF DelR216. Loading control corresponds to beta-tubulin. (D) BrdU incorporation assay in cells transfected with (-) or (+) MITF DelR216. Double MITF/BrdU-positive cells were counted as in Figure 1.
Deletion of the aminoterminal but not the carboxyl terminal domain abolishes inhibition of BrdU incorporation by (+) MITF
The above results indicated that direct binding of (+) MITF to E box motifs via its basic domain was not required for inhibition of BrdU incorporation. In order to test the role of other domains, we then prepared proteins with aminoterminal or carboxyl terminal deletions (schematic diagram in Figure 4A). Each of the mutant proteins accumulated in the nuclei of transfected cells and was expressed at similar levels (Figure 4B,C,E,F). The carboxyl terminal deletion started at residue 263, thus deleting the dimer-stabilizing leucine zipper. Such mutant protein is incapable of forming stable DNA binding dimers and in vivo leads to the severe MITF eye and coat pigmentation phenotype associated with the Mitfmi-ce allele. Upon transfection, (-) MITF R263Stop led to 67 ± 8% BrdU/MITF double-positive per total number of MITF-positive cells (Figure 4D), not significantly different from BrdU incorporation in untransfected cells (63 ± 4%, not shown). In contrast, expression of (+) MITF R263Stop reduced this percentage to 43 ± 3% (Figure 4D). Hence, neither the dimerization domain nor the remainder of the (sumoylatable and phosphorylatable) carboxyl terminal domain were necessary for the inhibitory effect of (+) MITF.
Figure 4.

Effects of truncated or serine-73-to-alanine mutated (-) and (+) MITF on BrdU incorporation. (A) Schematic representation of the different mutants. (B, E and H) MITF immunofluorescence of HEK293 cells transfected with the indicated (-) or (+) MITF mutants. (C, F and I) Western blot analyses of protein extracts from cells transfected with the indicated (-) or (+) MITF mutants. Loading control corresponds to beta-tubulin (C and F) or an unspecific band (I). (D, G and J) BrdU incoporation in cells transfected with the indicated (-) or (+) MITF mutants. BrdU incorporation and counting done as in Figure 1.
In order to test the role of the aminoterminal domain, mutant proteins with truncations of the first 178 residues were prepared. In these proteins, the truncation occurred within a region corresponding to exon 5 of the Mitf gene, thereby excluding the phosphorylatable domain encoded by exon 2b and the activation domain/p300 binding domain encoded by exon 4 but including the domain encoded by exon 6a and hence still allowing for a differential test of (+) and (-) MITF. As observed for the carboxyl terminal truncation, both (+) MITF Del1-178 and (-) MITF Del1-178 proteins were localized in the nucleus (Figure 4E) and were expressed at similar levels (Figure 4F). In contrast to the carboxyl terminal deletions, however, there was no significant difference in inhibition of BrdU incorporation between the aminoterminally deleted (-) and (+) MITF (the respective percentages being 71 ± 4% and 67 ± 8%, Figure 4G). This suggests that the six residue sequence ACIFPT in (+) MITF can exert its inhibitory effect on BrdU incorporation only in conjunction with a domain present within the first 178 residues.
Since transfection of (-) or (+) MITF in HEK293 cells leads to spontaneous phosphorylation of Ser73 (Figure 1B), and since, as shown above, the exon in which this residue resides was necessary for the inhibitory effect of (+) MITF, we also tested whether modification at this site alone might play a role in the inhibition of BrdU incorporation. We, therefore, mutated the corresponding codon to encode an alanine, a neutral and non-phosphorylatable residue. The mutant proteins [(+) MITF S73A and (-) MITF S73A, Figure 4A] were nuclear and expressed at comparable levels whereby the upper, phosphorylated band seen in wild type is absent in the mutants (Figure 4H,I). Interestingly, the difference between (+) and (-) MITF in affecting BrdU incorporation, although still significant at P < 0.01, is much smaller in these mutants [61 ± 3% for (-) and 54 ± 4% for (+) MITF S73A (Figure 4J), compared with 67 ± 2% for (-) and 44 ± 2% for (+) wild type MITF, see Figure 1C]. The smaller difference was due both to an increase in inhibitory activity of (-) MITF S73A [compared with wild type (-) MITF] and a decrease in inhibitory activity of (+) MITF S73A [compared with wild type (+) MITF]. This suggests that S73 phosphorylation is important in regulating DNA synthesis and that in conjunction with the exon 6a-encoded hexapeptide, might have distinct effects on biologically relevant intra- or intermolecular interactions.
(+) MITF interferes with S-phase
In order to confirm our immunofluorescent counting results and to determine at which phase of the cell cycle MITF expressing cells were blocked, FACS analyses of HEK293 cells transfected with either (-) or (+) MITF were performed. Twenty-four hours after transfection, cells were pulsed with BrdU for 30 min and labeled with anti-BrdU antibodies to measure DNA synthesis and propidium iodide to measure total DNA content. They were then subjected to two-color cell cycle analysis. Figure 5A shows a typical series of FACS plots for non-transfected control cells and cells transfected with (-) MITF or (+) MITF, as well as the gates for G0G1, early and late S phase, and G2M as indicated in the figure and corresponding legend. The same gates were used to quantitate the results from three independent experiments with comparable transfection efficiencies between experiments and between the (-) and (+) MITF group (Figure 5B-D). The analysis showed that 63.41 ± 7.12% of non-transfected control cells were in S phase. Upon transfection of cultures with (-) MITF, 59.81 ± 4.85% of cells were in S phase, and with (+) MITF, 76.41 ± 10.45% (Figure 5B). This increase in the percentage of cells in S-phase in (+) MITF transfected cultures occurred at the expense of the percentages of cells in G0G1 [11.94 ± 5.92% with (+) MITF WT, and 24.67 ± 5.01% with (-) MITF WT, P = 0.01] but not at the expense of the percentage of cells in G2M [3.68 ± 2.68% with (+) MITF WT and 5 ± 1.31% with (-) MITF WT, P = 0.24]. These results may seem surprising given that the immunofluorescent microscopic analysis has shown fewer (+) MITF/BrdU double-positive cells compared with (-) MITF/BrdU double positive cells. The analysis of the FACS results revealed, however, that more (+) MITF expressing cells were in early S-phase, and hence expressed very low levels of BrdU, compared with (-) MITF expressing cells. In fact, a significantly greater percentage of cells with low BrdU content (gate R9, Figure 5C) was seen after transfection with (+) MITF (58.53 ± 4.93%) versus (-) MITF (21.2 ± 0.62%) or in non-transfected cells (25.66 ± 7.29%). Taking into account DNA content, the results revealed that 40 ± 6.47% of (+) MITF expressing cells were in early S-phase (gate R7, Figure 5D), compared with 22.89 ± 0.42% of (-) MITF expressing cells and 26.35 ± 0.75% of non-transfected cells. The corresponding numbers for late S-phase (gate R8, Figure 5D) were 8.7 ± 0.63% in (+) MITF expressing cultures, 9.53 ± 2.14% in (-) MITF expressing cultures and 10.97 ± 2.85% in non-transfected cultures. We conclude from these results that in contrast to (-) MITF, (+) MITF efficiently impairs, although does not block entry into, S-phase, and that this effect results in an apparent reduction in BrdU fluorescence above the threshold required for visual microscopic inspection.
Figure 5.

Cell cycle FACS analysis on HEK293 cells transfected with wild type (-) and (+) MITF WT. Twenty hours after transfection, cells were incubated with BrdU for 30 min and processed for BrdU and propidium iodide labeling before FACS analysis. (A) FACS plots of representative assays. Control represents non-transfected cells, and (-) MITF and (+) MITF correspond to transfections with the respective plasmids. The gates (R1-R10) were determined empirically. Note that HEK293 cells are aneuploid (hypotriploid) but undergo normal DNA doubling with each round of replication. R1, apoptotic, (hypo-)hypotriploid cells with no BrdU incorporated; and R2, apoptotic, (hypo)-hypotriploid cells that have incorporated BrdU. R6, apoptotic cells corresponding to (hypo-)hypohexaploidy. R3, G0G1; R5, G2M; R7, early S-phase; R8, late S-phase; R9, S-phase with low BrdU content; R10, S-phase with high BrdU content. Note that total S-phase corresponds to R9 + R10. (B-D) FACS analysis was repeated in three independent experiments, using the same gating as shown in (A). The results were quantified using the Cellquest software. (B) Quantitative analysis of the percentage of cells in G0G1, S, and G2M phases of the cell cycle. Note a significant difference between (-) and (+) MITF on S-phase (P < 0.02). (C) Separation of S-phase into low and high BrdU content. Note a significant difference between (-) and (+) MITF in low BrdU content (P < 0.01). (D) Separation of cells into early and late S-phase. Note a significant difference between (-) and (+) MITF on early S-phase (P < 0.01).
Discussion
Here we used a simple transfection paradigm to assay effects of the melanocyte transcription factor MITF on cell proliferation. The experiments were prompted by previous observations that indicated that wild type MITF inhibits cell proliferation under some conditions (Carreira et al., 2000, 2005; Hornyak et al., 2001; Horsford et al., 2005; Müller, 1950; Nakayama et al., 1998; Packer, 1967) and promotes it under others (Du et al., 2004; Widlund et al., 2002). MITF, however, comes in two distinct, genetically relevant splice isoforms, (-) and (+) Mitf, and many of the in vitro studies have only used (+) MITF or have failed to indicate which isoform was used. Hence, we sought to systematically compare the effects of the two isoforms and found, surprisingly, that when expressed in heterologous HEK293 cells for the duration of a single cell cycle and assayed by immunofluorescence microscopy, (-) MITF did not discernibly inhibit BrdU incorporation compared with non-transfected cells while (+) MITF did. Also, using FACS analysis, we did not find any significant differences between cultures transfected with (-) MITF and non-transfected cultures. Hence, (-) MITF, differing from (+) MITF by only six residues and expressed in cells at levels comparable with those of (+) MITF, can be considered an excellent control for the anti-proliferative effects of (+) MITF. Nevertheless, transfection of an unrelated control plasmid, pEGFP, was slightly inhibitory, so that compared with EGFP, (-) MITF would seem to have a positive effect on DNA synthesis. In contrast, in the more slowly proliferating BHK cells, (-) MITF had a mild inhibitory effect, suggesting cell-type dependency of these subtle effects. Importantly, however, even subtle effects that can be observed during a single replication cycle may be compounded during subsequent cell cycles.
In contrast to the FACS results obtained with (-) MITF, FACS analyses of (+) MITF transfected cultures were seemingly at variance with the microscopic analyses, showing higher, and not lower, BrdU incorporation rates. A more detailed analysis of the corresponding FACS plots indicated, however, that (+) MITF transfected cultures contained significantly more cells that have incorporated low amounts of BrdU, and fewer that have incorporated high amounts. Low amounts of BrdU are registered by FACS but are expected to be below the threshold levels required for visual inspection of microscopic slides. Hence, the results are internally consistent and suggest that (+) MITF interferes with S-phase, not by inhibiting entry into S-phase, but by slowing its progression.
In this context, it is important to consider the phenotypes of mice containing the microphthalmia-spotted allele, Mitfmi-sp. This allele is only capable of expressing (-) Mitf, and compound heterozygotes in which an Mitfmi-sp allele is combined with an Mitf null allele have white spots in contrast to mice harboring one wild type and one null allele. White spots usually point to a local lack of pigment cells resulting from a developmental paucity of precursor cells (melanoblasts). If our in vitro results were at all applicable to the in vivo situation, one might assume that the lack of a major anti-proliferative factor, (+) MITF, should increase and not decrease the numbers of melanoblasts. Further analyses will show whether the phenotype associated with the Mitfmi-sp allele is because of the lower DNA binding and transcriptional activities of (-) MITF that are not sufficiently compensated in vivo by a reduction in the anti-proliferative effect normally exerted by (+) MITF.
The further analyses of mutant MITF proteins on BrdU incorporation revealed additional insights into MITF’s role in cell proliferation. First, we found that (+) MITF with an E213A mutation in the conserved DNA binding basic domain, which cannot interact in a sequence-specific way with E box motifs, still interferes with BrdU incorporation. This suggests that direct DNA binding is not a prerequisite for this effect. Interestingly, a distinct mutation in the DNA-binding domain, isoleucine212-to-asparagine (I212N), was found to impair the inhibitory activity of MITF because of a lack of stimulation of the Ink4A promoter (Loercher et al., 2005). The effects of this particular mutation, however, are complex since the corresponding mutant in its (+) isoform [although not in its (-) isoform] can form DNA-binding heterodimers with the related proteins TFEB, TFE3 and TFEC (Hemesath et al., 1994), and Loercher et al. indeed used (+) MITF. Also, in vivo, the corresponding mutant allele, Microphthalmia-white (MitfMi-wh), has a still unexplained strong heterozygous but a comparatively mild homozygous phenotype and is unique in its capacity to complement null alleles (Steingrimsson et al., 2003). In other words, the I212N mutation, affecting a residue that does not contact DNA, is not comparable with the E213A mutation, which based on the analysis of related bHLH-Zip proteins affects the core residue that contacts the central two bases of an E box (Ferre-D’Amare et al., 1993).
The notion that direct E-box-dependent transcription may not be the critical parameter controlling the distinction between the (-) and (+) isoforms in inhibiting BrdU incorporation gained further support from the use of sumoylation mutants that previously had been shown to be associated with increased transcriptional activities on target genes containing multiple E box motifs (Miller et al., 2005; Murakami and Arnheiter, 2005). If transcription were an all-important parameter controlling inhibition of DNA synthesis, then one might expect at least the corresponding (+) isoform to display enhanced inhibitory activities. This, however, was not the case and although the two isoforms still retained the difference observed with wild type MITF, they each led to a generally higher rate of BrdU incorporation. In the case of (-) MITF, this rate was higher still than that in non-transfected cells, suggesting that the sumoylation mutants actually had pro-proliferative activities that were only partially counteracted by the presence of the alternatively spliced six residues of the (+) isoform.
Interestingly, truncations of the carboxyl terminal domain that remove the dimer-stabilizing leucine zipper also did not lead to a reduced inhibitory activity of (+) MITF. This is in line with the above observations made with the E213A mutation since both the E213A and the truncated mutant proteins retain anti-proliferative activity and yet cannot bind DNA efficiently. Moreover, the truncated carboxyl terminus contains an activation domain, three phosphorylation sites, and one of the two sumoylation sites whose modifications have been shown to influence MITF’s transcriptional activity, and yet all of these are apparently dispensable for the anti-proliferative activity of (+) MITF. In contrast, an aminoterminally truncated (+) MITF protein that retained the sequences distinguishing it from the (-) isoform lost its inhibitory activity. This suggested that the aminoterminus contained a domain, or a single residue, that is required to function in conjunction with the alternatively spliced hexapeptide to produce an inhibitory MITF molecule, at least in HEK293 cells. The truncation included the melanocyte-specific aminoterminal 11 residues, a phosphorylatable serine (serine 73) encoded by exon 2b, and an activation domain/P300 binding domain encoded by exon 4. Since the above results suggested that E-box-dependent transcriptional activity may not be critical to (+) MITF’s anti-proliferative activity, we focused on the phosphorylatable serine-73 because of its involvement in regulating protein stability (Wu et al., 2000; Xu et al., 2000). Although we have no evidence that a serine-73-to-alanine mutation affects the proteins’ steady state levels in transfected HEK293 cells (data not shown), it considerably reduced the difference between (-) and (+) MITF on BrdU incorporation, with (-) MITF being more inhibitory, and (+) MITF being less so. Although a molecular explanation of this phenomenon needs further exploration, it is clear that serine-73, and one or several domains within the molecules’ aminoterminus, play key roles in conjunction with the six residues that allow MITF to control DNA synthesis.
Taken together, our findings suggest that even though MITF has been found to regulate promoters of cell cycle genes, it may not exert its control on cell proliferation exclusively through direct transcriptional activities. Interestingly, there is precedence for a transcription-independent regulation of cell proliferation by transcription factors. For instance, the homeodomain protein Six3 can promote cell proliferation, not by transcription regulation, but by sequestering a negative regulator of cell cycle progression, geminin (Del Bene et al., 2004). Also, the bHLH protein MyoD can induce cell growth arrest even when mutated in the DNA-binding basic domain (Crescenzi et al., 1990; Sorrentino et al., 1990). In fact, MyoD inhibits cell growth by sequestering CDK4 whose inhibition leads to the accumulation of hypo-phosphorylated Rb protein and thereby to the inhibition of cell cycle progression (for review, see Wei and Paterson, 2001). Hence, it is conceivable that MITF, belonging to the same superfamily of bHLH proteins, follows similar pathways and that the transcriptional regulation of cell cycle genes is not the only way by which MITF links control over cell proliferation with control over differentiation.
Methods
Cell cultures and transfection assays
HEK293 cells and BHK cells were grown in DMEM supplemented with 10% fetal bovine serum, non-essential amino acids, and penicillin-streptomycin (Invitrogen, Carlsbad, CA, USA). Cells were plated in two-well chamber slides (Nalge Nunc, Rochester, NY, USA) for proliferation assays or in 3 cm dishes for western and FACS analyses. Transfections were performed when the cells were at 80% confluence using lipofectamine reagent (Invitrogen) according to the manufacturer’s instructions, using 1 μg of DNA/chamber or dish. Twenty hours post-transfection, the cells were harvested and processed for immunofluorescence, Western blot, or FACS analysis.
Plasmid constructs
The plasmids pCMV-HA-MITF(+) and pCMV-HA-MITF(-), encoding HA-tagged wild type MITF with or without the alternatively spliced six amino acids, were obtained from C. Goding. The plasmids encoding (-) and (+) MITF K182R/K316R were a kind gift of H. Murakami. The plasmids encoding, MITF E213A, MITF DelR216, MITF R263Stop and MITF S73A were engineered using the quickchange site directed mutagenesis kit (Stratagene, La Jolla, CA, USA) and pCMV-HA-MITF (-) or (+) as templates. The plasmids encoding (-) and (+) MITF with amino terminal deletions, MITF Del1-178 were made by cloning PCR fragments into the SalI and BglII sites of pCMV-HA (Clontech, Mountain View, CA, USA). Hence, in all Mitf plasmids, the translational start sites are encoded by the ATG of the HA-tag. The plasmid pEGFP-C1 was purchased from Clontech.
Western blot analysis
Cells were washed with PBS and proteins were extracted using RIPA buffer containing a cocktail of protease inhibitors (EMD Bioscienes, San Diego, CA, USA). The protein amount of each extract was assessed by BCA assay (Pierce, Rockford, IL, USA). Twenty μg of total protein was loaded on a 7.5% or 12% gel (BioRad, Hercules, CA, USA). After blotting, the membranes were exposed to mouse monoclonal anti-MITF C5 (Lab Vision, Freemont, CA, USA) or mouse monoclonal anti-HA (Roche, Nutley, NJ, USA) or mouse monoclonal anti-tubulin (Sigma, St Louis, MO, USA), washed, and exposed to anti-mouse-HRP (Amersham, Piscataway, NJ, USA). Signals were revealed by super signal west pico chemiluminescent substrate (Pierce).
BrdU incorporation and staining
Twenty hours after transfection, cells were incubated with 10 μM BrdU (Sigma) for 30 min at 37°C, washed in PBS, fixed in 4% paraformaldehyde, permeabilized with 0.1% triton in PBS, and stained for MITF using C5 mouse monoclonal anti-MITF (Lab Vision) and rat-absorbed anti-mouse antibody coupled to FITC (Southern Biotech, Birmingham, AL, USA). After post-fixation with 2% paraformaldehyde, the cells were processed for BrdU staining which included incubation in 2N HCl for 15 min at room temperature, neutralization with 0.1 M sodium borate, and staining with a monoclonal rat anti-BrdU antibody (Accurate, Westbury, NY, USA) overnight at 4°C. The cells were then exposed to mouse-absorbed anti-rat antibody coupled with TRITC (Southern Biotech). Cell counting was done manually using a Polyvar microscope equipped for epifluorescence, and results were expressed as the ratio of the number of MITF/BrdU double-positive cells to the total number of MITF-positive cells. Each experiment was done in triplicate and repeated three times independently. The statistical significance of differences between (-) MITF and (+) MITF was analyzed using Student’s t-test. A value of P < 0.05 was considered statistically significant (*P < 0.05; **P < 0.02; ***P < 0.01; Ø non-significant).
Fluorescence-activated cell sorting
After a pulse of 30 min with 10 lμM BrdU, the cells were washed in PBS and fixed in 70% ice cold ethanol for 30 min on ice, washed in PBS containing 0.1% BSA, incubated in 2N HCl/Triton X-100 for 30 min at room temperature, and washed twice with 0.1 M sodium borate pH 8.5. Ten million cells were aliquoted and incubated with the monoclonal mouse anti-BrdU antibody (0.5 μg/106 cells) (Becton Dickinson, Mountain View, CA, USA) for 30 min at room temperature. The cells were then washed by centrifugation, incubated with anti-mouse-FITC (Sigma) in PBS/BSA/0.5% Tween 20, washed, and finally resuspended in 1 ml PBS containing 5 lg/ml propidium iodide (Sigma). Cell cycle analysis was performed by flow cytometer FACSVantage SE (Becton-Dickinson) and the results analyzed using Cellquest software. The gating was determined empirically in one experiment and then applied in exactly the same way to all subsequent experiments.
Acknowledgements
We thank Drs C. Goding and H. Murakami for plasmids, S. Skuntz for encouragement and technical advice and the members of the laboratory and Dr M. Dubois-Dalcq for critical comments on the manuscript.
References
- Arnheiter H, Hou L, Nguyen M-TT, Bismuth K, Csermely T, Murakami H, Skuntz S, Liu W, Bharti K.Mitf - A Matter of Life and Death for the Developing Melanocytes Humana Press; Totowa, NJ: in press [Google Scholar]
- Carreira S, Liu B, Goding CR. The gene encoding the T-box factor Tbx2 is a target for the microphthalmia-associated transcription factor in melanocytes. J. Biol. Chem. 2000;275:21920–21927. doi: 10.1074/jbc.M000035200. [DOI] [PubMed] [Google Scholar]
- Carreira S, Goodall J, Aksan I, La Rocca SA, Galibert MD, Denat L, Larue L, Goding CR. Mitf cooperates with Rb1 and activates p21Cip1 expression to regulate cell cycle progression. Nature. 2005;433:764–769. doi: 10.1038/nature03269. [DOI] [PubMed] [Google Scholar]
- Crescenzi M, Fleming TP, Lassar AB, Weintraub H, Aaronson SA. MyoD induces growth arrest independent of differentiation in normal and transformed cells. Proc. Natl. Acad. Sci. U. S. A. 1990;87:8442–8446. doi: 10.1073/pnas.87.21.8442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Bene F, Tessmar-Raible K, Wittbrodt J. Direct interaction of geminin and Six3 in eye development. Nature. 2004;427:745–749. doi: 10.1038/nature02292. [DOI] [PubMed] [Google Scholar]
- Du J, Widlund HR, Horstmann MA, Ramaswamy S, Ross K, Huber WE, Nishimura EK, Golub TR, Fisher DE. Critical role of CDK2 for melanoma growth linked to its melanocyte-specific transcriptional regulation by MITF. Cancer Cell. 2004;6:565–576. doi: 10.1016/j.ccr.2004.10.014. [DOI] [PubMed] [Google Scholar]
- Ferre-D’Amare AR, Prendergast GC, Ziff EB, Burley SK. Recognition by Max of its cognate DNA through a dimeric b/HLH/Z domain. Nature. 1993;363:38–45. doi: 10.1038/363038a0. [DOI] [PubMed] [Google Scholar]
- Fisher DE, Parent LA, Sharp PA. High affinity DNA-binding Myc analogs: recognition by an alpha helix. Cell. 1993;72:467–476. doi: 10.1016/0092-8674(93)90122-7. [DOI] [PubMed] [Google Scholar]
- Hallsson JH, Favor J, Hodgkinson C, et al. Genomic, transcriptional and mutational analysis of the mouse microphthalmia locus. Genetics. 2000;155:291–300. doi: 10.1093/genetics/155.1.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemesath TJ, Steingrimsson E, Mcgill G, Hansen MJ, Vaught J, Hodgkinson CA, Arnheiter H, Copeland NG, Jenkins NA, Fisher DE. Microphthalmia, a critical factor in melanocyte development, defines a discrete transcription factor family. Genes Dev. 1994;8:2770–2780. doi: 10.1101/gad.8.22.2770. [DOI] [PubMed] [Google Scholar]
- Hemesath TJ, Price ER, Takemoto C, Badelian T, Fisher DE. MAP kinase links the transcription factor Microphthalmia to c-Kit signalling in melanocytes. Nature. 1998;391:298–301. doi: 10.1038/34681. [DOI] [PubMed] [Google Scholar]
- Hodgkinson CA, Moore KJ, Nakayama A, Steingrímsson E, Copeland NG, Jenkins NA, Arnheiter H. Mutations at the mouse microphthalmia locus are associated with defects in a gene encoding a novel basic-helix-loop-helix-zipper protein. Cell. 1993;74:395–404. doi: 10.1016/0092-8674(93)90429-t. [DOI] [PubMed] [Google Scholar]
- Hornyak TJ, Hayes DJ, Chiu LY, Ziff EB. Transcription factors in melanocyte development: distinct roles for Pax-3 and Mitf. Mech. Dev. 2001;101:47–59. doi: 10.1016/s0925-4773(00)00569-4. [DOI] [PubMed] [Google Scholar]
- Horsford DJ, Nguyen MT, Sellar GC, Kothary R, Arnheiter H, Mcinnes RR. Chx10 repression of Mitf is required for the maintenance of mammalian neuroretinal identity. Development. 2005;132:177–187. doi: 10.1242/dev.01571. [DOI] [PubMed] [Google Scholar]
- Loercher AE, Tank EM, Delston RB, Harbour JW. MITF links differentiation with cell cycle arrest in melanocytes by transcriptional activation of INK4A. J. Cell Biol. 2005;168:35–40. doi: 10.1083/jcb.200410115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller AJ, Levy C, Davis IJ, Razin E, Fisher DE. Sumoylation of MITF and its related family members TFE3 and TFEB. J. Biol. Chem. 2005;280:146–155. doi: 10.1074/jbc.M411757200. [DOI] [PubMed] [Google Scholar]
- Müller G. Eine entwicklungsgeschichtliche Untersuchung über das erbliche Kolobom mit Mikrophthalmus bei der Hausmaus. Z. Mikrosk. Anat. Forsch. 1950;56:520–558. [Google Scholar]
- Murakami H, Arnheiter H. Sumoylation modulates transcriptional activity of MITF in a promoter-specific manner. Pigment Cell Res. 2005;18:265–277. doi: 10.1111/j.1600-0749.2005.00234.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama A, Nguyen MT, Chen CC, Opdecamp K, Hodgkinson CA, Arnheiter H. Mutations in microphthalmia, the mouse homolog of the human deafness gene MITF, affect neuroepithelial and neural crest-derived melanocytes differently. Mech. Dev. 1998;70:155–166. doi: 10.1016/s0925-4773(97)00188-3. [DOI] [PubMed] [Google Scholar]
- Opdecamp K, Nakayama A, Nguyen MT, Hodgkinson CA, Pavan WJ, Arnheiter H. Melanocyte development in vivo and in neural crest cell cultures: crucial dependence on the Mitf basic-helix-loop-helix-zipper transcription factor. Development. 1997;124:2377–2386. doi: 10.1242/dev.124.12.2377. [DOI] [PubMed] [Google Scholar]
- Packer SO. The eye and skeletal effects of two mutant alleles at the microphthalmia locus of Mus musculus. J. Exp. Zool. 1967;165:21–45. doi: 10.1002/jez.1401650103. [DOI] [PubMed] [Google Scholar]
- Price ER, Ding HF, Badalian T, Bhattacharya S, Takemoto C, Yao TP, Hemesath TJ, Fisher DE. J. Biol. Chem. 1998;273:17983–17986. doi: 10.1074/jbc.273.29.17983. [DOI] [PubMed] [Google Scholar]
- Prince S, Carreira S, Vance KW, Abrahams A, Goding CR. Tbx2 directly represses the expression of the p21(WAF1) cyclin-dependent kinase inhibitor. Cancer Res. 2004;64:1669–1674. doi: 10.1158/0008-5472.can-03-3286. [DOI] [PubMed] [Google Scholar]
- Sato S, Roberts K, Gambino G, Cook A, Kouzarides T, Goding CR. CBP/p300 as a co-factor for the Microphthalmia transcription factor. Oncogene. 1997;14:3083–3092. doi: 10.1038/sj.onc.1201298. [DOI] [PubMed] [Google Scholar]
- Shaw G, Morse S, Ararat M, Graham FL. Preferential transformation of human neuronal cells by human adenoviruses and the origin of HEK 293 cells. Faseb. J. 2002;16:869–871. doi: 10.1096/fj.01-0995fje. [DOI] [PubMed] [Google Scholar]
- Sorrentino V, Pepperkok R, Davis RL, Ansorge W, Philipson L. Cell proliferation inhibited by MyoD1 independently of myogenic differentiation. Nature. 1990;345:813–815. doi: 10.1038/345813a0. [DOI] [PubMed] [Google Scholar]
- Steingrímsson E, Moore KJ, Lamoreux ML, et al. Molecular basis of mouse microphthalmia (mi) mutations helps explain their developmental and phenotypic consequences [see comments] Nat. Genet. 1994;8:256–263. doi: 10.1038/ng1194-256. [DOI] [PubMed] [Google Scholar]
- Steingrimsson E, Arnheiter H, Hallsson JH, Lamoreux ML, Copeland NG, Jenkins NA. Interallelic complementation at the mouse Mitf locus. Genetics. 2003;163:267–276. doi: 10.1093/genetics/163.1.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steingrimsson E, Copeland NG, Jenkins NA. Melanocytes and the microphthalmia transcription factor network. Annu. Rev. Genet. 2004;38:365–411. doi: 10.1146/annurev.genet.38.072902.092717. [DOI] [PubMed] [Google Scholar]
- Steingrimsson E, Copeland NG, Jenkins NA. Melanocyte stem cell maintenance and hair graying. Cell. 2005;121:9–12. doi: 10.1016/j.cell.2005.03.021. [DOI] [PubMed] [Google Scholar]
- Takebayashi K, Chida K, Tsukamoto I, Morii E, Munakata H, Arnheiter H, Kuroki T, Kitamura Y, Nomura S. The recessive phenotype displayed by a dominant negative microphthalmia-associated transcription factor mutant is a result of impaired nucleation potential. Mol. Cell Biol. 1996;16:1203–1211. doi: 10.1128/mcb.16.3.1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance KW, Goding CR. The transcription network regulating melanocyte development and melanoma. Pigment Cell Res. 2004;17:318–325. doi: 10.1111/j.1600-0749.2004.00164.x. [DOI] [PubMed] [Google Scholar]
- Vance KW, Carreira S, Brosch G, Goding CR. Tbx2 is overexpressed and plays an important role in maintaining proliferation and suppression of senescence in melanomas. Cancer Res. 2005;65:2260–2268. doi: 10.1158/0008-5472.CAN-04-3045. [DOI] [PubMed] [Google Scholar]
- Wei Q, Paterson BM. Regulation of MyoD function in the dividing myoblast. FEBS Lett. 2001;490:171–178. doi: 10.1016/s0014-5793(01)02120-2. [DOI] [PubMed] [Google Scholar]
- Widlund HR, Horstmann MA, Price ER, Cui J, Lessnick SL, Wu M, He X, Fisher DE. Beta-catenin-induced melanoma growth requires the downstream target Microphthalmia-associated transcription factor. J. Cell Biol. 2002;158:1079–1087. doi: 10.1083/jcb.200202049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe HG, Coleman DL. Mi-spotted:a mutation in the mouse. Genet. Res. Camb. 1964;5:432–440. [Google Scholar]
- Wu M, Hemesath TJ, Takemoto CM, Horstmann MA, Wells AG, Price ER, Fisher DZ, Fisher DE. c-Kit triggers dual phosphorylations, which couple activation and degradation of the essential melanocyte factor Mi. Genes Dev. 2000;14:301–312. [PMC free article] [PubMed] [Google Scholar]
- Xu W, Gong L, Haddad MM, Bischof O, Campisi J, Yeh ET, Medrano EE. Regulation of microphthalmia-associated transcription factor MITF protein levels by association with the ubiquitin-conjugating enzyme hUBC9. Exp. Cell Res. 2000;255:135–143. doi: 10.1006/excr.2000.4803. [DOI] [PubMed] [Google Scholar]