

Abstract
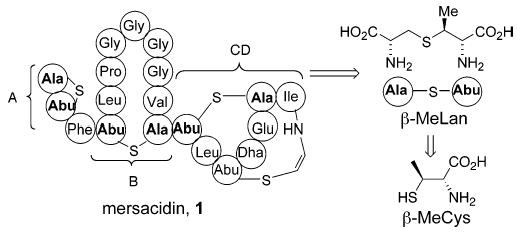
Lantibiotics are a class of lanthionine (and/or β-methyllanthionine)-containing peptides with antibioitic activity against Gram-positive bacteria. As part of our research effort directed toward the synthesis and mechanistic study of the lantibiotic peptide mersacidin (1), we report stereoselective syntheses of orthogonally protected β-methylcysteine (β-MeCys) and β-methyllanthionine (β-MeLan), two key nonnatural amino acid components of the mersacidin architecture.
The onset of vancomycin resistance, particularly among enterococcal and multidrug-resistant staphylococcal pathogens, has underscored the urgent need for an upgrade of the antibiotic armamentarium for the treatment of infections arising from Gram-positive pathogens. In this connection, we have initiated a broad research program that entails chemical synthesis and mechanistic studies of several peptide antibiotics that inhibit bacterial peptidoglycan (PG) biosynthesis via mechanisms distinct from that utilized by vancomycin. Specifically, we are interested in lantibiotic mersacidin (1), which has shown great promise in this area. Mersacidin is active against several bacterial strains including streptococci, bacilli, and methicillin-resistant Staphylococcus aureus with mimimum inhibitory concentration (MIC) values comparable to that of vancomycin.1 Even more interesting is the observation that mersacidin is active against vancomycin-resistant Enterococcus faecium, a strain that synthesizes PG precursors terminating in d-Ala-d-Lac.2
Mersacidin is believed to exert its antibiotic activity by inhibition of transglycosylation reaction in PG biosynthesis via complexation with lipid II at the disaccharide–pyrophosphate domain, a site that is not believed to be targeted by other known antibiotics.2,3 Its unique binding site as well as inhibition of the transglycosylation event make it an attractive target for development of novel therapeutics because (i) transglycosylation occurs on the cell surface so cellular penetration of the antibiotic is not a requirement for activity and (ii) the transglycosylation reaction utilized in PG biosynthesis is unique to bacterial cells. With the ultimate goal of delineating the exact mode of action of mersacidin and its variants with lipid substrates, as well as developing novel analogues with increased efficacy, we have embarked on the total synthesis of 1.
The presence of the sensitive (Z)-amidovinyl sulfide linkage in the CD ring system, as well as the unnatural amino acid (2S,3S)-β-methylcysteine (β-MeCys) and (2S,3S,6R)-β-methyllanthionine (β-MeLan) fragments in the A, B, and CD ring systems render mersacidin a challenging synthetic target. We initially focused our attention on development of an efficient and enantioselective synthesis of these two nonnatural amino acids since both are embedded in all four rings of mersacidin. Moreover, an orthogonal protective group scheme would be necessary for the regioselective manipulation of these core residues. To the best of our knowledge, despite the apparent simplicity in their structures, efficient and stereoselective methods for preparation of orthogonally protected versions of these two amino acids (2, 5, and 6; Scheme 1) are not currently available.4,5 Herein, we report versatile routes to β-MeCys and β-MeLan that fulfill all the aforementioned requirements.
Scheme 1.
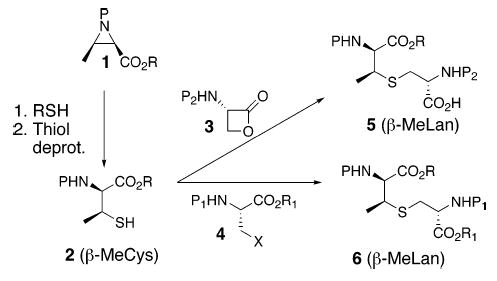
Our synthesis of protected β-MeLan (5 and 6) relies on the stereoselective alkylation of β-MeCys derivative 2 with serine-derived electrophiles of general representation 3 and 4 (Scheme 1). β-MeCys 2 would in turn be obtained by the regio- and stereoselective ring opening of d-threonine derived aziridine such as 1 by a thiol nucleophile.
Initially, we explored the use of benzyloxycarbonyl aziridine 10 (Scheme 2) as a precursor for synthesis of protected β-MeCys derivatives.6 Wakamiya has reported the acid-promoted ring opening of 10 by thiobenzoic acid. However, competitive ring-opening by oxygen led to formation of a thionoester byproduct along with the desired thioester.6 Hydrogen sulfide gas has also been used as the nucleophile, but its high toxicity makes its use inconvenient, especially on preparative scale.7 Thus, it became clear to us that the success of this strategy will depend on the regio-and stereoselective opening of the aziridine at C3 with an appropriate S-nucleophile as well as the ability to cleave the thiol protective group under mild conditions so as to avoid β-elimination and C2 epimerization.
Scheme 2.
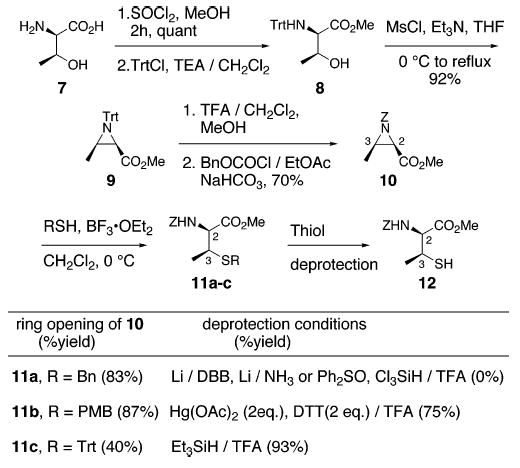
Aziridine 10 was prepared from d-threonine (7) by modification of reported procedures as outlined in Scheme 2.6,8 In each example, BF3·OEt2-mediated ring-opening reactions of 10 with various thiol nucleophiles smoothly afforded the corresponding β-MeCys derivatives (11a–c) as a single diastereomer.9 Subsequent thiol deprotection, however, proved to be more difficult. In case of 11a, both reductive as well as oxidative cleavage of the S-Bn group was incompatible with Cbz and methyl ester functionalities.10 Deblocking of S-PMB ether utilizing reported protocols resulted either in incomplete cleavage or decomposition of the starting material.11,12 After some experimentation, we found that by using 2 equiv each of Hg(OAc)2 and DTT in TFA, the PMB group could be removed cleanly to afford thiol 12 (75% yield).
The mild conditions for cleavage of S-trityl ethers prompted us to explore the use of triphenylmethyl thiol as a nucleophile in the aziridine ring-opening reactions. We were pleased to find that under optimized conditions, S-trityl protected β-MeCys (11c) was obtained in 40% yield along with an almost equal amount of free thiol 12. In addition, detritylation of 11c could be carried out separately with TFA/Et3SiH in excellent yields.13 Taken together, thiol 12 was obtained in 73% yield starting from aziridine 10 via S-Trt derivative 11c.
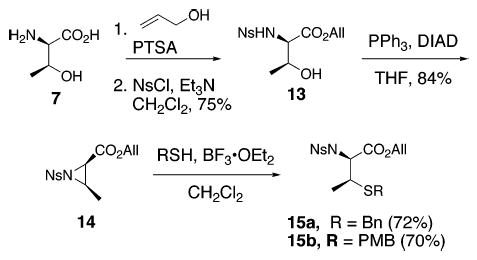
Although nucleophilic opening of N-alkoxycarbonyl aziri-dines allows efficient access to β-MeCys derivatives, it is restricted to the use of Cbz-protected aziridines rendering it unsuitable for Fmoc- or Boc-based peptide synthesis in solution or on the solid phase. To get around this limitation, we have utilized p-nitrophenylsulfonyl (Ns) aziridine (14, Scheme 3) as the electrophile, which has provided a more concise and versatile route to orthogonally protected β-MeCys derivatives. As shown in Scheme 3, 14 was prepared in only three steps starting from d-threonine (compared to five steps for Cbz aziridine 10). d-Threonine allyl ester was transformed to the p-nitrophenylsulfonamide 13 by treatment with NsCl/Et3N in 75% yield. Ring closure of 14 under Mitsunobu conditions afforded the desired aziridine in 84% yield as a single diastereomer.14 The ring-opening reaction of 14 by thiol nucleophiles under our optimized conditions provided the desired N-nitrosulfonyl β-MeCys derivatives (15a and 15b) with high regio- and stereoselectivity.
Scheme 3.
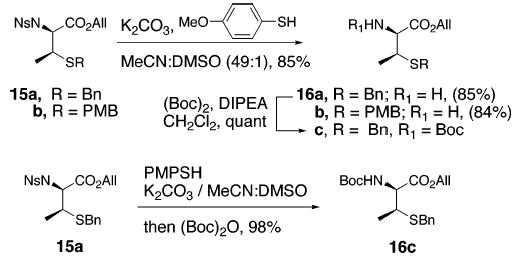
For the Ns-aziridine route to construct β-MeCys derivatives to be synthetically useful, it was critical to establish conditions for cleavage of the Ns group while avoiding any undesired side reactions. Maligres and co-workers found that deprotection of primary nitrophenylsulfonamides was incomplete under Fukuyama conditions and prolonged heating in the presence of PhSH and K2CO3 was needed.15 This was of particular concern to us since nosyl-protected β-MeCys derivatives (15a,b) could be prone to epimerization under the Maligres protocol. Fortunately, we were able to accomplish Ns cleavage of 15a and 15b in 3 h at room temperature simply by using p-methoxyphenyl thiol instead of PhSH, to afford the primary amines in 80–85% yields (16a,b, Scheme 4). In addition, primary amine 16a could be quantitatively reprotected as its Boc derivative (16c). Furthermore, a highly efficient, one-pot, Ns to Boc switch could also be carried out by simply adding (Boc)2O to the reaction mixture after Ns cleavage (15a to 16c, Scheme 4). In a similar manner, Fmoc or any other carbamate functionality can easily be incorporated in protected β-MeCys derivatives providing flexibility in choice of the amine protecting group. Thus, the use of N-nitrosulfonyl aziridine 14 has enabled a much more direct access to orthogonally protected β-MeCys derivatives such as 16c (5 steps, 45% overall yield starting from d-threonine). In contrast, preparation of β-methylcysteine derivatives via ring-opening of Cbz-aziridine intermediates requires additional amino protecting group manipulations for target structures in which Cbz protection is not desirable.16
Scheme 4.
Having accessed β-MeCys with the desired protective group scheme, we next explored the synthesis of differently protected β-MeLan. Van der Donk has employed a biomimetic route involving intramolecular Michael addition of MeCys to dehydrobutyrine (Dhb).16 This strategy is suitable only for cyclic β-methyllanthionines where the stereoselectivity of Michael addition originates from the intrinsic preference of the acyclic peptide precursor to produce a particular diastereomer upon cyclization. Another approach involving coupling of alanyl β-cation equivalents with β-MeCys has remained problematic due to the formation of norlanthionines.17,18 Finally, BF3·OEt2-mediated ring opening of aziridines such as 10 by cysteine-derived nucleophiles provided the corresponding β-MeLan derivative in only 12% yield.19
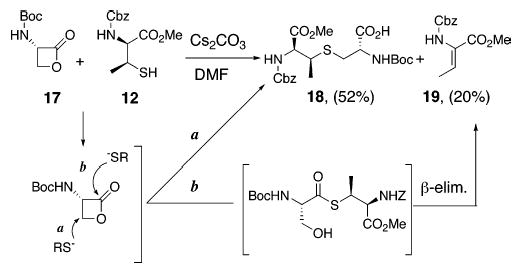
In light of these issues, we set out to investigate the methods reported by Goodman and Schmidt for synthesis of our target β-methyllanthionines (Schemes 5 and 6). Goodman utilized the ring-opening reactions of serine β-lactone derivatives such as 17 (Scheme 5) by cesium thiolates of penicillamine (β,β-dimethylcysteine) to access the corresponding β,β-dimethyllanthionine derivatives in high yields (78–92%).20 When the same protocol was applied to coupling of β-MeCys 12 with lactone 17, 52% of orthogonally protected β-MeLan 18 was obtained as a single diastereomer (as determined by 1H and 13C NMR analysis) along with 20% of dehydrobutyrine 19. Out of the two reaction pathways (a and b, Scheme 5) available for nucleophilic ring opening of 17, path a leads to formation of the desired product 18. On the other hand, path b generates the thioester intermediate, which likely undergoes β-elimination under the reaction conditions to produce 19. This strategy, though reasonably efficient, suffers from the limitation that the coupling conditions are not compatible with the Fmoc protecting group.
Scheme 5.
Scheme 6.
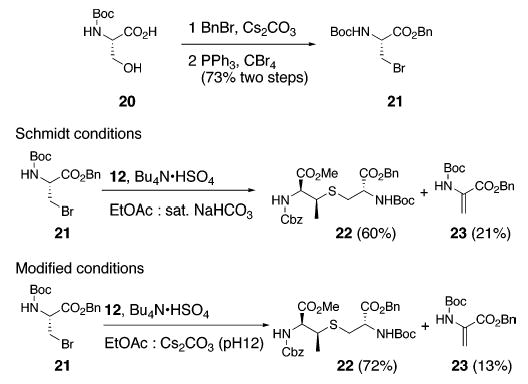
We therefore explored conditions reported by Schmidt and co-workers for the synthesis of unsubstituted, differently protected lanthionines.21 This method involved S-alkylation of protected cysteine derivatives by protected β-bromoala-nines under phase transfer conditions (Bu4NβHSO4, EtOAc, aq NaHCO3). Under Schmidt conditions, displacement of bromide 2121 by β-MeCys (12) afforded fully protected β-MeLan 22 in 60% yield. Although no dehydroalanine byproducts were reported in reactions utilizing unsubstituted cysteine nucleophiles, we found the β-elimination pathway to be competitive, resulting in the isolation of dehydroalanine 23 in 21% yield. This is likely a consequence of the diminished reactivity of β-MeCys derivative 12 as compared to its unsubstituted cysteine counterpart. Use of aqueous Cs2CO3 (pH 12) as base, in place of saturated NaHCO3, improved the yield of 22 to 72% while minimizing the formation of 23. The isomeric purity of 22 was established by 1H, 13C, and LCMS analysis (see the Supporting Information).
In conclusion, we have prepared orthogonally protected β-methylcysteines enantioselectively in an efficient manner via d-threonine-derived aziridine derivatives. Use of as yet unexplored Ns-aziridine derivative has allowed a more concise and versatile access to β-MeCys analogues. Synthesis of linear, orthogonally protected β-methyllanthionines has been accomplished for the first time by extension of previously reported methods for the synthesis of lanthionines. Modified Schmidt conditions (use of aqueous Cs2CO3 instead of NaHCO3) were found to provide the fully protected β-methyllanthionine derivative in the highest yield. With the two unnatural amino acids available in multigram quantities, we have initiated an effort toward the total synthesis of mersacidin. Our progress toward this end will be reported in due course.
Acknowledgments
This paper is dedicated to the memory of Professor Murray Goodman. Financial support provided by the National Institutes of Health (AI 059327), The Hellman Foundation, and the Regents of the University of California is gratefully acknowledged. M.S.V. also thanks Eli Lilly and Company for support in the form of an Eli Lilly and Company New Faculty Award.
Footnotes
Supporting Information Available: Experimental details and tabulated spectroscopic data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Chatterjee S, Chatterjee DK, Jani RH, Blumbach J, Ganguli BN, Klesel N, Limbert M, Siebert G. J Antibiot. 1992;45:839. doi: 10.7164/antibiotics.45.839. [DOI] [PubMed] [Google Scholar]
- 2.Brötz H, Bierbaum G, Leopold K, Reynolds PE, Sahl HG. Antimicrob Agents Chemother. 1998;42:154. doi: 10.1128/aac.42.1.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brötz H, Bierbaum G, Markus A, Molitor E, Sahl HG. Antimicrob Agents Chemother. 1995;39:714. doi: 10.1128/AAC.39.3.714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.For recent efforts directed toward synthesis of orthogonally protected lanthionines, see: (a) Bregant S, Tabor AB.J Org Chem 2005702430. [DOI] [PubMed] [Google Scholar]; b Mustapa MFM, Harris R, Bulic-Subanovic N, Chubb NAL, Gaffney PRJ, Driscoll PC, Tabor AB. J Org Chem. 2003;68:8185. doi: 10.1021/jo0346398. [DOI] [PubMed] [Google Scholar]; c Matteucci M, Bhalay G, Bradley M. Tetrahedron Lett. 2004;45:1399. [Google Scholar]; d Swali V, Matteucci M, Elliot R, Bradley M. Tetrahedron. 2002;58:9101. [Google Scholar]
- 5.For a recent review on chemical and enzymatic synthesis of lanthionines, see: Paul M, van der Donk WA.Mini-Rev Org Chem 2005223 [Google Scholar]
- 6.Wakamiya T, Shimbo K, Shiba T, Nakajima K, Neya M, Okawa K. Bull Chem Soc Jpn. 1982;55:3878. [Google Scholar]
- 7.Nakajima K, Okawa K. Bull Chem Soc Jpn. 1983;56:1565. [Google Scholar]
- 8.McKeever B, Pattenden G. Tetrahedron. 2003;59:2713. [Google Scholar]
- 9.Xiong CY, Wang W, Hruby VJ. J Org Chem. 2002;67:3514. doi: 10.1021/jo011172x. [DOI] [PubMed] [Google Scholar]
- 10.a Koide T, Otaka A, Suzuki H, Fujii N. Synlett. 1991:345. [Google Scholar]; b Akaji K, Tatsumi T, Yoshida M, Kimura T, Fujiwara Y, Kiso Y. J Chem Soc, Chem Commun. 1991:167. [Google Scholar]
- 11.Nishimura O, Kitada C, Fujino M. Chem Pharm Bull. 1978;26:1576. doi: 10.1248/cpb.26.539. [DOI] [PubMed] [Google Scholar]
- 12.Macmillan D, Anderson DW. Org Lett. 2004;6:4659. doi: 10.1021/ol048145o. [DOI] [PubMed] [Google Scholar]
- 13.Moreau X, Campagne JM. J Org Chem. 2003;68:5346. doi: 10.1021/jo034195f. [DOI] [PubMed] [Google Scholar]
- 14.Ibuka T, Mimura N, Aoyama H, Akaji M, Ohno H, Miwa Y, Taga T, Nakai K, Tamamura H, Fujii N, Yamamoto Y. J Org Chem. 1997;62:999. doi: 10.1021/jo962094u. [DOI] [PubMed] [Google Scholar]
- 15.Maligres PE, See MM, Askin D, Reider PJ. Tetrahedron Lett. 1997;38:5253. [Google Scholar]
- 16.Zhou H, van der Donk WA. Org Lett. 2002;4:1335. doi: 10.1021/ol025629g. [DOI] [PubMed] [Google Scholar]
- 17.Dugave C, Ménez A. Tetrahedron: Asymmetry. 1997;8:1453. [Google Scholar]
- 18.Mustapa MFM, Harris R, Mould J, Chubb NAL, Schultz D, Driscoll PC, Tabor AB. Tetrahedron Lett. 2002;43:8359. [Google Scholar]
- 19.Nakajima K, Oda H, Okawa K. Bull Chem Soc Jpn. 1983;56:520. [Google Scholar]
- 20.Shao H, Wang SHH, Lee CW, Osapay G, Goodman M. J Org Chem. 1995;60:2956. [Google Scholar]
- 21.Zhu XM, Schmidt RR. Eur J Org Chem. 2003:4069. [Google Scholar]