

SUMMARY
The invasive ability of tumor cells plays a key role in prostate cancer metastasis and is a major cause of treatment failure. Urokinase plasminogen activator (uPA) and its receptor (uPAR)-mediated signaling have been implicated in tumor cell invasion, survival and metastasis in a variety of cancers. This study was undertaken to investigate the biological roles of uPA and uPAR in prostate cancer cell invasion, survival and the potential of uPA and uPAR as targets for prostate cancer therapy. uPA and uPAR expression correlates with the metastatic potential of prostate cancer cells. Thus, therapies designed to inhibit uPA and uPAR expression would be beneficial. LNCaP, DU145 and PC3 are prostate cancer cell lines with low, moderate and high metastatic potential, respectively, as demonstrated by their capacity to invade extracellular matrix (ECM). In this study we utilized small hairpin RNAs (shRNAs), also referred to as small interfering RNAs (siRNAs), to target human uPA and uPAR. These siRNA constructs significantly inhibited uPA and uPAR expression at both the mRNA and protein levels in the highly metastatic prostate cancer cell line PC3. Our data demonstrate that uPA-uPAR knockdown in PC3 cells resulted in a dramatic reduction of tumor cell invasion as indicated by a matrigel invasion assay. Furthermore, simultaneous silencing of the genes for uPA and uPAR using a single plasmid construct expressing shRNAs for both uPA and uPAR significantly reduced cell viability and ultimately resulted in the induction of apoptotic cell death. RNAi for uPA and uPAR also abrogated uPA-uPAR signaling to downstream target molecules such as extracellular-signal regulated kinases 1/2 (ERK1/2) and the signal transducer and activator of transcription 3 (Stat 3). In addition, our results demonstrate that intratumoral injection with the plasmid construct expressing shRNAs for uPA and uPAR almost completely inhibited established tumor growth and survival in an orthotopic mouse prostate cancer model. These findings uncovered evidence of a complex signaling network operating downstream of uPA-uPAR that actively advances tumor cell invasion, proliferation and survival of prostate cancer cells. Thus, RNAi-directed targeting of uPA and uPAR is a convenient and novel tool for studying the biological role of the uPA-uPAR system and raises the potential of its application for prostate cancer therapy.
Keywords: RNAi, prostate cancer, invasion, survival, uPA and uPAR
The abbreviations used are: siRNA (small interfering RNA), shRNA (small hairpin RNA), RNAi (RNA interference), uPA (urokinaseplasminogen activator), uPAR (uPA receptor), ANOVA (analysis of variance), RT (reverse transcription), TPBS (tween-20 (0.1%) phospate buffered saline), BSA (bovine serum albumin)
INTRODUCTION
Prostate cancer is the second most common malignancy in American men, with estimates of 230,110 new cases and approximately 30, 000 deaths in 2004 (1–4). As such, prostate cancer poses a major public health problem in the United States and worldwide (4,5). Currently, metastatic prostate cancer is incurable and ultimately claims the life of patients (6–10). An important factor in the relative seriousness of prostate cancer is the invasiveness of the constituent tumor cells causing metastasis (7,11,12). The invasive nature of tumor cells is crucial for cancer metastasis (13,14). Tumor cell invasion and metastasis are complex processes with three prominent stages: adhesion to the extracellular matrix, digestion of the matrix to release cells from the primary tumor mass, and migration of the tumor cells to secondary targets. A high level of expression of one or more proteases often correlates with the migration of cancer cells through the digested extracellular matrix (ECM) and contributes to tumor cell invasion and metastasis of virtually all malignancies including prostate cancer (5,15). One such key protease, urokinase plasminogen activator (uPA), and its receptor uPAR (CD87) play important roles in processes which lead to cancer cell invasion and metastasis (16–20).
Binding of uPA with its receptor uPAR activates downstream signaling molecules through a number of pathways, including the mitogen-activated protein kinases (MAPK) and signal transducer and activator of transcription (Stat) pathways (19,21–24). Ample evidence suggests that enhanced signal transduction via the binding of uPA to uPAR contributes to the malignant phenotype of many cancers and the particular nature of the response may be specific to cell-type (16,19,24–25). In multiple cells, binding of uPA to uPAR activates the extracellular-signal regulated kinase 1/2 (ERK1/2) pathway, which controls cancer cell invasion, proliferation and survival (26,27). Furthermore, many reports suggest that uPA-uPAR signaling activates the Jak/Stat signaling cascade (15,18, 23). Members of the Stat family regulate the expression of a variety of genes involved in proliferation, survival and apoptosis (28–30). Moreover, uPA-uPAR mediated signaling can upregulate the production of matrix metalloproteases (MMPs), which induce ECM degradation, and in turn, tumor invasion and metastasis (31,32).
Since uPA, uPAR and its downstream signaling pathways are implicated in many cancers and regulate essential cellular functions that contribute to the malignancy of tumor cells, targeting this system has become a matter of interest for cancer therapy. uPA or uPAR neutralizing antibodies (33), protease inhibitors (34) urokinase specific inhibitors (5,35, 36), uPA or uPAR antisense oligonucleotides (11,16,37,38), and small molecules that target uPA or uPAR (39, 40), are being investigated as possible strategies to block either uPA or uPAR activity, and thereby causing the suppression of tumor growth, invasion and metastasis.
Recent discovery of RNA interference (RNAi) has opened new avenues in cancer therapy (41–43). RNAi is a sequence-specific, post-transcriptional gene-silencing mechanism that is affected through double-stranded RNA molecules homologous to the sequence of the target gene (44). To study the key roles of uPA and uPAR in vitro and in vivo, we developed a bicistronic plasmid vector expressing small hairpin RNAs (shRNAs) directed against both uPA and uPAR under the control of a human CMV promoter. We show that RNAi driven by these shRNA-based plasmid constructs can effectively silence uPA and uPAR gene expressions at the mRNA level as well as protein level. Functional analyses revealed that the abrogation of both uPA and uPAR expression not only inhibits invasion and proliferation strongly but also induces apoptotic cell death and inhibits tumorigenicity. These results indicate that the simultaneous knockdown of uPA and uPAR using RNAi vectors is a promising tool for analysis of the function of downstream signaling pathways as well as the potential vectors for prostate cancer gene therapy.
EXPERIMENTAL PROCEDURES
Construction of small hairpin RNAs expressing plasmids
Small interfering oligonucleotides specific for uPA from 346 to 367 bases (agcttGagagccctgctggcgcgccatatataatggcgcgccagcagggctctca) and for uPAR from 77 to 98 bases (gatccTacagcagtggagagcgattatatataataatcgctctccactgctgtag) were synthesized and annealed. An uPA-uPAR RNAi plasmid vector that expresses shRNAs for both uPA and uPAR under the control of a human CMV promoter was constructed by inserting pairs of the annealed DNA oligonucleotides specific for uPA at the Hind III site and uPAR at BamHI site sequentially into the pcDNA3 vector (sh-uPAuPAR). Also, shRNA expression vectors for uPA (sh-uPA) and uPAR (sh-uPAR) singly were constructed. A pcDNA3-scrambled vector with an imperfect sequence, which does not form a perfect hairpin structure, was used to develop the scrambled vector for use as a control. The empty vector (EV) and scrambled vector (SV) controls have been tested in multiple cell lines and does not demonstrate any toxicity to cells as demonstrated by MTT assay after transfection as well as having no effect on the expression of housekeeping genes, GAPDH and β-actin.
Cell culture and transfection conditions
Human prostate cancer cell lines LNCaP, DU145 and PC3 were obtained from the American Type Culture Collection (Manassas, VA). LNCaP cells were grown in RPMI medium supplemented with 2mM L-glutamine, 1.5 g/L sodium bicarbonate, 4.5 g/L glucose, 10 mM HEPES, and 1.0 mM Sodium pyruvate (Invitrogen, Carlsbad, CA). PC3 and DU145 cells were grown in minimum essential medium (28). Both media contained 10% fetal bovine serum (GIBCO BRL, Lewisville, TX) and 5% penicillin/streptomycin and were maintained in a 37°C incubator in a 5% CO2 humidified atmosphere.
Transfections were performed using LipofectamineTM 2000 reagent (Life technologies, Rockville, MD) per the manufacturer’s instructions. After 72 h of transfection, cells were used for cell proliferation assays, immunoblot analysis, RT-PCR analysis, Matrigel invasion assay, DNA fragmentation assay, EMSA assay and caspase activity assay. For DAPI and double immunostaining, transfections were carried out in Lab-Tek II chamber slides (Nalge Nunc International, Naperville, IL).
Fibrin zymography
The enzymatic activity and molecular weight of electrophoretically separated forms of uPA were determined in conditioned medium of prostate cancer cell lines LNCaP, DU145 and PC3 by SDS-PAGE as described previously (16). Briefly, the SDS-PAGE gel contains acrylamide to which purified plasminogen and fibrinogen were substrates before polymerization. After polymerization, equal amounts of proteins in the samples were electrophoresed and the gel was washed and stained as described previously (16).
Reverse transcription-PCR analysis
Cellular RNA was isolated using the Qiagen RNeasy kit and 1 μg of RNA was DNase treated (10 units/μg of RNA, 1h) and used as a template for the reverse transcription reaction (RT, 20 μl). RT reaction mix (Invitrogen) contained 1 μl (10 pm) of primers. The resultant cDNA was then used in PCR reactions and analyzed by gel electrophoresis. The following primers were used: uPA-sense: 5’TGCGTCCTGGTCGTGAGCGA 3’; uPA-antisense: 5’CTACAGCGCTGACACGCTTG 3’; uPAR-sense: 5’CATGCAGTGTAAGACCCAACGGGGA 3’; uPAR-antisense: 5’AATAGGTGACAGCCCGGCCAGAGT 3’; GAPDH-sense: 5’CGGAGTCAACGGATTTGGTCGTAT 3’; and GAPDH-antisense: 5’AGCCTTCTCCATGGTGGTGAAGAC3’.
PCR conditions were as follows: 95°C for 5 minutes, followed by 35 cycles of 95°C for 1 min, 55°C for 1 minute, and 72°C for 1 minute. The final extension was at 72°C for 5 min.
Immunoblot analysis
After 72 h of transfection, cells were washed and lysed in 1X cell lysis buffer (Cell Signaling Technology Inc., Beverly, MA). Protein concentrations were determined on diluted samples using a bicinchoninic acid procedure (Pierce Biochemical Company, Rockford, IL). Equal amounts of protein were separated on SDS-PAGE and transferred to polyvinylidene difluoride membranes (BioRad). Membranes were blocked in phosphate-buffered saline (PBS) solution with 5% non-fat dry milk and incubated with primary antibodies in blocking solution overnight at 4°C, and washed three times with TPBS at 10-min intervals. Horseradish peroxidase conjugated secondary antibodies (Biomeda, Foster City, CA) were used for detection of immunoreactive proteins by chemiluminescence (Amersham, Biosciences, and Piscataway, NJ). The following antibodies were used: anti-uPA (Biomeda, Foster City, CA), anti-uPAR (American Diagnostics Inc., Greenwich, CT), anti-Bax (Santa Cruz Biotechnology, Santa Cruz, CA), anti-Bcl-XS/L (Santa Cruz Biotechnology, Santa Cruz, CA), anti-caspase 9 (Cell Signaling Technology Inc., Beverly, MA), and anti-GAPDH (Abcam, Cambridge, MA). Antibodies against total and phospho forms of ERK, JNK, p38 and Stat 3 were obtained from Santa Cruz Biotechnology (Santa Cruz, CA).
Immunofluorescence detection
PC3 cells transfected with various shRNA plasmids were fixed with 4% paraformaldehyde and incubated with anti-uPA (1: 500; Biomeda, Foster City, CA) and/or anti-uPAR (1: 500; American Diagnostics Inc., Greenwich, CT). After washing, fluorescent secondary antibodies (Santa Cruz Biotechnology, Santa Cruz, CA) were added at a 1:500 dilution. The cells were again washed three times with PBS, and counter-stained with DAPI. Fluorescent images were acquired using a charge-coupled device RT Slider Spot Camera (Diagnostic Instruments Inc, Burroughs Sterling Heights, MI) connected to a microscope (Olympus, Melville, NY) and managed by a computer equipped with the spot RT software v3.5 (Diagnostic instruments, Burroughs Sterling Heights, MI).
Matrigel invasion assay
After transfection, cells were detached and washed twice in PBS. 5×105 cells were seeded in the upper chamber of a Transwell insert (12 μM pores) coated with Matrigel (0.7 mg/ml) (Collaborative Research Inc., Boston, MA). The lower chamber was filled with 400 μl of RPMI medium. After a 24 h incubation period, the non-migrated cells in the upper chamber were gently scraped away and adherent cells present on the lower surface of the insert were stained with Hema-3 and photographed.
Cell proliferation assays
Viability of cells 72 h after transfection was evaluated using a MTT assay. MTT [3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide] (Sigma) was added to the culture medium in each well at a concentration of 500 μg/ml, and plates were incubated for 4 h at 37°C. Acid-isopropanol (0.04 N HCl/isopropanol) was immediately added to all wells and mixed vigorously so that the dark blue crystals dissolved effectively. Absorbance was measured at 570 nm (Benchmark, BIORAD, Hercules, CA).
In situ caspase activity assay
Caspase activation was detected using the polycaspase detection kit (Immunochemistry Technologies, Bloomington, IL) per manufacturer’s instructions. In this assay, the cell permeable, non-cytotoxic Fluorochrome Inhibitors of Caspases (FLICA) binds covalently to a reactive cysteine residue on the large subunit of the active caspase heterodimer, thereby inhibiting further enzymatic activity. This kit uses a carboxyfluorescein-labeled fluoromethyl ketone peptide inhibitor of many caspases (caspase 1, −3, −4, −5, −6, −7, −8 and –9; FAM-VAD-FMK), which is a generic probe for the detection of most caspases and emits green fluorescence. The green fluorescent signal is a direct measure of the amount of active caspase in the cell at the time the reagent was added. After 72 h of transfection, caspase activation was detected by staining the cells with the FAM-VAD-FMK dye (in situ marker). The bound marker was localized by fluorescence detection as observed with a confocal microscope. DAPI was used for nuclear staining.
DNA laddering assay
After transfection, cells were harvested and washed twice in PBS. Cell pellets were resuspended in lysis buffer (10mM Tris-HCl, 400mM NaCl, 1mM EDTA and 1% TritonX-100) containing 0.1 mg/ml Proteinase K (Invitrogen) and then incubated at 37°C for 2 h. DNA was cleared from the lysates by centrifugation and then extracted using an equal volume of phenol/chloroform and precipitated by adding absolute ethanol and 0.3 M sodium acetate (pH 5.2) at –80°C for 2 h. The DNA was resuspended in Tris-EDTA buffer (10mM Tris-HCl; pH 7.5, 1mM EDTA), treated with RNase A at 37 °C for 1 h, and then resolved on a 1.5% agarose gel stained with ethidium bromide (0.5 μg/ml).
Electrophoretic mobility shift assay (EMSA)
After transfection, nuclear proteins were extracted using a protein extraction kit (Ambion, Austin, TX) as per the manufacturer’s instructions. Concentrations of nuclear proteins were determined on diluted samples using a bicinchoninic acid procedure (Pierce Biochemical Company, Rockford, IL). Interaction between Stat 3 in the protein extract and DNA probe was investigated using an electrophoretic mobility shift assay (EMSA) kit from Panomics (Redwood City, CA) as per the manufacturer’s instructions.
DNA fragment end labeling assay
shRNA-treated or control prostate tumor tissue sections (5 μM thick) were deparaffinized and rehydrated. Next, the tissue sections were permeabilized by covering the entire specimen with Proteinase K solution (20 μg/ml Proteinase K in 10 mM Tris, pH 8) and incubated for 20 min at room temperature. The tissue sections were then washed in Tris-buffered saline (1x TBS, 20 mM Tris pH 7.6, 140 mM NaCl). Inactivation of endogenous peroxidases was accomplished by immersing the tissue sections in 3% hydrogen peroxide diluted in methanol for 5 min at room temperature. The glass slides were then placed in Klenow equilibration buffer (50 mM Tris pH 8, 50 mM NaCl, 10 mM MgCl2) for 30 min. The tissue sections were then incubated with 60 μl of a solution containing a mixture of labeled and unlabeled deoxynucleotides at a ratio optimum for DNA fragment end labeling with Klenow, according to the manufacture’s instructions (Klenow-FragEL DNA fragmentation detection kit, Oncogene Research Products, Cambridge, MA) at 37°C for 90 min in a humidified chamber. The enzymatic reaction was stopped by incubation with EDTA (0.5 M, pH 8) for 5 min at room temperature. The slides were then washed with TBS and immersed in blocking buffer for 10 min (4% BSA in PBS) followed by incubation with 100 μl of a solution containing peroxidase streptavidin for 30 min in a humidified chamber at room temperature. The tissue sections were then washed in TBS and covered with a solution containing 3, 3’ diaminobenzidine (DAB, 0.7 mg/ml), hydrogen peroxide and urea (0.6 mg/ml). Next, the slides were washed with distilled water and counterstained with methyl green (0.3%) for 30 sec and examined under an Olympus fluorescence microscope. The positive DNA fragment end labeled staining was scored from six randomly captured images/sample using spot RT software v3.5 (Diagnostic instruments, MI).
Orthotopic mouse prostate treatment model
Athymic male nude mice (nu/nu; 6–8 weeks of age) were obtained from Harlan Sprague-Dawley (Indianapolis, IN). Animal handling and experimental procedures were approved by the University of Illinois College of Medicine animal experiments committee. Orthotopic implantation was carried out as described previously (7). Briefly, after total body anesthesia with ketamine (50 mg/kg) and xylazine (10 mg/kg), a low midline incision was made in the lower abdomen. A suspension of PC3 cells (1×106) in 30 μl PBS was injected into a lateral lobe of the prostate and the wound was closed with surgical metal clips. This cell concentration was necessary to achieve consistent local tumor growth within 7 days of implantation. Mice were divided in to five treatment groups with six mice per treatment group. At days 7 and 14 post-implantation, a low midline incision was performed and the tumors were injected with plasmid constructs expressing sh-uPA, sh-uPAR, sh-uPA-uPAR or EV/SV controls (75 μg/150 μg each). In another set of experiments, the orthotopically-implanted mice were intratumorally coinjected with sh-uPA and sh-uPAR plasmids (150 μg each) on days 7 and 14. Mice were sacrificed 14–15 days after the final shRNA plasmid injection and the primary tumor growth and sites of metastasis were determined by visual inspection and photographed. We then excised, measured and weighed the primary tumors. Specimens were fixed in formalin and embedded in paraffin for H&E staining. Also, some of the tissue was snap frozen immediately for immunoblotting.
Statistical Analysis
Statistical comparisons were performed using ANOVA for analysis of significance between different values using GraphPad Prism software (San Diego, CA). Values are expressed as mean ± SD from at least three separate experiments and differences were considered significant at a P value of less than 0.05.
RESULTS
Endogenous uPA and uPAR protein expression is associated with in vitro invasiveness of human prostate cancer cells
Previous studies have shown that PC3 cells are highly metastatic, whereas DU145 and LNCaP cells are moderately and poorly metastatic, respectively (45–48). Because uPA and its receptor uPAR are involved in tumor invasion and metastasis, we first compared the levels of these proteins in the three human prostate cancer cell lines with different metastatic potentials. As shown in Fig. 1A, uPA and uPAR protein levels were significantly higher in PC3 and DU145 cells as compared with the poorly metastatic LNCaP cells, which expressed undetectable levels of these proteins. A similar trend was seen in uPA activity as assessed by fibrin zymography (Fig. 1B). Thus, uPA and uPAR protein levels as well as uPA activity were positively correlated with their known metastatic potential. To determine whether these protein expression findings correlate with the biological activity of the prostate cancer cell lines used, we examined the ability of these cells to invade matrigel, a gel layer composed of basement membrane proteins. This assay is a well-established in vitro model for assessing tumor invasiveness. We found that the highly metastatic prostate cancer cell line PC3 showed the greatest levels of invasiveness followed by the DU145 and LNCaP cell lines, an order consistent with their known metastatic potentials (Fig. 1C and 1D). PC3 cells were 14-fold more invasive and DU145 were 9-fold more invasive than LNCaP cells. Taken together, these results demonstrated a strong correlation between uPA and uPAR protein levels and the invasive ability of human prostate cancer cells with differing metastatic potentials. These initial findings prompted us to pursue the question of whether modulation of uPA and/or uPAR could affect metastatic prostate tumor progression.
Figure 1. uPA and uPAR protein expression levels and uPA activity correlate with invasive potential of human prostate cancer cell lines.

A. Endogenous uPA and uPAR protein expression was examined by immunoblot analysis of total cellular protein isolated from the following prostate cancer cell lines: LNCaP, DU145 and PC3. Equal amounts of isolated protein from cell extracts of all three cell lines were subjected to immunoblot with anti-uPA, anti-uPAR and anti-GAPDH antibodies. GAPDH was utilized as a loading control.
B. uPA activity in prostate cancer cell lines was assessed by fibrin zymography. Equal amounts of protein from prostate cancer cells in serum-free media were separated by SDS-PAGE on 10% gels containing fibrinogen and plasminogen under non-reducing conditions. After exchange of SDS with Triton X-100 washing, the gel was incubated in glycine buffer (0.1 M, pH 8.0). Fibrinolytic activity was detected as clear lysis bands after amido black staining and subsequent destaining with methanol-acetic acid.
C. Comparison of the in vitro invasive potentials of prostate cancer cell lines. Invasion assays were performed in 12-well transwell chambers containing polycarbonate filters with 12 μm pores coated with matrigel. Cells that had passed to the undersurface of the filters were stained and photographs were taken under microscope at a 200X magnification.
D. Cells invading through the matrigel were counted under a microscope in three random fields at a 200X magnification. Each bar represents the mean ± SD of three fields where significant differences from low or non-metastatic LNCaP cells, which exhibited undetectable uPA and uPAR protein expression, are represented by asterisks * (P <0.05).
(All results are representative of three separate experiments.)
Efficient knockdown of uPA and uPAR gene expression in human PC3 prostate cancer cells using RNAi
To study the biological role of uPA and uPAR in prostate tumor progression, we used small hairpin RNAs to knockdown endogenous uPA and uPAR gene expression in the human prostate cancer cell line PC3, which has been shown to have robust expression of uPA and uPAR as well as a high metastatic potential. First, we developed pcDNA3-CMV vectors containing small hairpin constructs capable of generating 19 or 21-nt duplex RNAi oligonucleotides corresponding to either uPA or uPAR. Also, a single bicistronic construct driven by cytomegalovirus (CMV) promoter to deliver dual small hairpins targeted against both uPA and uPAR was constructed to test the effectiveness of simultaneously inhibiting expression of two endogenous genes (Fig. 2A). Next, the vectors expressing shRNAs for uPA, uPAR and the uPA-uPAR combination were transfected into PC3 cells.
Figure 2. RNAi knockdown of uPA and uPAR expression in the prostate cancer cell line PC3.

A. Schematic representation of the sh-uPAuPAR plasmid construct. The construct consists of a human CMV promoter and homology sequences targeted against uPA and uPAR. Following expression, the strong CMV promoter would drive the formation of short hairpin molecules specific for uPA and uPAR. The bovine growth hormone (BGH) poly adenylation sequence serves as a RNA pol II-based CMV promoter termination signal. Dicer/Drosha processes the shRNA-specific for uPA and uPAR and the resulting siRNA molecules interact with the target genes uPA and uPAR. This interaction results in the simultaneous knockdown of uPA and uPAR gene expression.
B. Semi-quantitative reverse transcription-PCR of RNA extracted from shRNA-transfected PC3 cells as described in methods. The glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA was co-amplified as a control.
C. Immunoblotting of total protein lysates extracted from shRNA-transfected PC3 cells. Both uPA and uPAR bands are present in mock, EV and SV-transfected cells. Accordingly, each gene-specific shRNA lane shows a significant decrease of the appropriate band. GAPDH was included as a loading control.
D. uPA and uPAR protein expression levels were also detected using indirect immunofluorescence in PC3 cells. PC3 cells transfected with the EV, SV and mock cells stained positive for immunofluorescent detection of uPA (colored in green by FITC) and uPAR (colored in red by Texas Red). Gene-specific shRNA-transfected cells substantially changed the cell staining profiles of uPA and uPAR as compared to EV/SV-transfected and mock cells. Nuclear counterstaining (blue) was obtained with DAPI.
(Results are representative of at least three separate experiments.)
As shown in Fig. 2B, analysis of the shRNA-transfected cells for uPA and uPAR expression via semi-quantitative reverse transcription-PCR demonstrated a specific reduction in mRNA levels for each gene relative to the EV/SV-transfected cells or mock cells. However, the RNAi effect was slightly more with the shRNA vector simultaneously targeting uPA and uPAR (Fig. 2B). Immunoblot analysis of cell extracts was carried out to determine whether decreased mRNA expression, as observed, correlated with decreased translation of the gene product. A similar trend was observed by immunoblot assay as well (Fig. 2C). No effects of RNAi were observed on the expression of GAPDH, which was used as an internal control for specificity and loading at mRNA level as well as protein level. In addition, EV/SV-transfected cells also showed that RNAi-directed uPA and uPAR knockdown is specific (Fig. 2B & 2C).
In addition, we analyzed the effects of gene-specific shRNAs on uPA and uPAR protein expression in PC3 cells using double immunostaining with anti-uPA and anti-uPAR antibodies. As shown in Fig. 2D, uPA and uPAR staining was drastically reduced by gene-specific shRNAs in comparison to EV/SV-transfected cells. uPA and uPAR double immunostaining was totally diminished in cells transfected with sh-uPAuPAR (Fig. 2D). In contrast, the PC3 cells transfected with EV and SV exhibited a similar staining intensity and pattern as the mock cells. These immunofluorescence studies confirmed the RT-PCR and immunoblot analyses.
Knockdown of uPA and uPAR expression by RNAi inhibited matrigel invasion of PC3 cells
One of the key functions of uPA and uPAR is promotion of invasion, a process crucial for tumor metastasis. To evaluate the impact of uPA and uPAR knockdown on PC3 cellular invasion, we performed a matrigel invasion assay using the shRNA-transfected cells. When compared with mock cells or cells transfected with EV/SV, sh-uPAuPAR-transfected cells showed a substantial reduction in invasive capacity (Fig. 3A). Invasion of PC3 cells was reduced to 75% of that of the controls (i.e., mock or EV/SV-transfected cells) by sh-uPA and to 90% by sh-uPAuPAR (Fig. 3B). Although knockdown of uPAR alone did not show a significant decrease in invasion, knockdown of uPA as well as uPAR had a very significant effect (Fig. 3A & 3B), suggesting that PC3 cell invasion into matrigel is substantially regulated by coordinated function of uPA and uPAR. These results indicate that uPA and uPAR expression is required for prostate cancer invasion as well as metastasis. Notably, the sh-uPAuPAR effect was so dramatic that the cells could hardly invade through the matrigel membrane, suggesting that RNAi had significantly interfered with the uPA-uPAR system mediating proteolytic activity and cell viability.
Figure 3. RNAi knockdown of uPA and uPAR expression inhibits the invasive potential of PC3 cells.
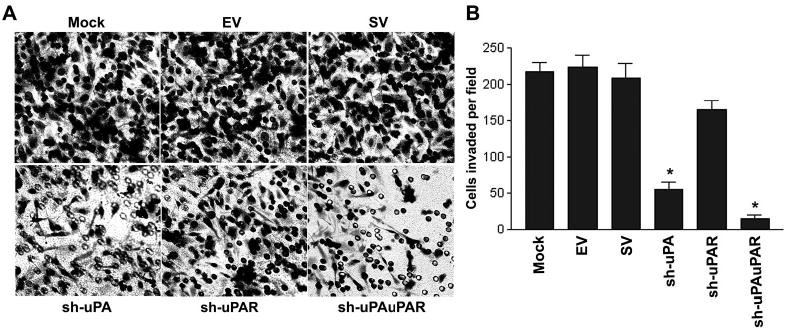
A. The invasive potential of mock and cells transfected with the indicated shRNA plasmids were examined by matrigel invasion assay (visual field representative of one experiment). Invasion assays performed as described above (see Fig. 1C).
B. Representative number of invading cells through the matrigel was counted under microscope in three random fields at 200X. Each bar represents the mean ± SD of three fields counted. Significant difference from controls (i.e., mock or scrambled vector-transfected cells) is indicated by asterisks * (P <0.05).
(Results are representative of three separate experiments.)
Knockdown of uPA and uPAR expression by RNAi inhibits cell proliferation and induces apoptosis
To access the potential effects of RNAi-mediated uPA and uPAR silencing on cell proliferation and survival, MTT analysis was performed 72 h after transfection with shRNA-specific to uPA and uPAR (Fig. 4A). RNAi-targeting against uPA had no effect on the proliferative ability of PC3 cells, whereas RNAi-specific to uPAR had a low inhibitory effect. In contrast, a dramatic reduction in proliferation of PC3 cells was observed with RNAi simultaneously targeting uPA and uPAR (Fig. 4A). The percentages of viable cells were reduced in the presence of uPAR and uPA-uPAR RNAi by approximately 30% and 60% on average, respectively, as compared to the control cells. These results suggest that increased uPA and/or uPAR levels in tumor cells might endow cells with enhanced growth and survival capacity. As such, reducing uPA and uPAR levels may induce apoptosis in cancer cells.
Figure 4. RNAi knockdown of uPA and uPAR expression inhibits cell proliferation and induces apoptosis in PC3 cells.
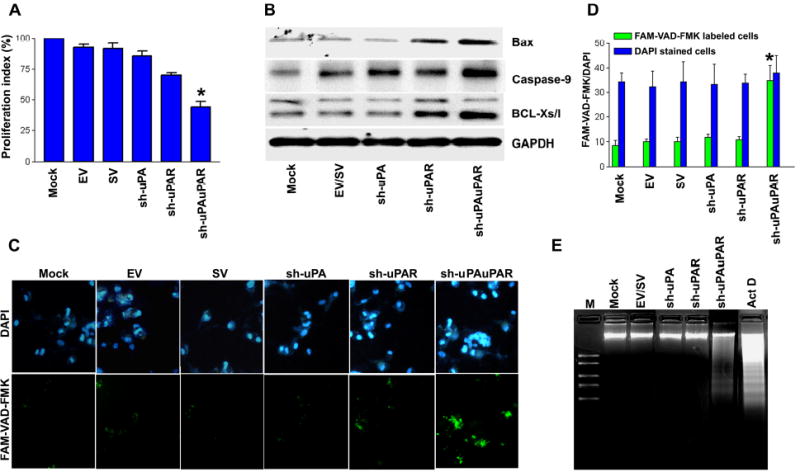
A. Viability of PC3 cells transfected with either gene-specific shRNA plasmids or controls (mock or EV/SV-transfected cells) was revealed by MTT assay. Each bar represents triplicate analyses of mean ± SD where significant difference from controls is represented by an asterisk * (P <0.05).
B. Representative immunoblots show changes in pro-apoptotic gene expression in uPA-uPAR knockdown PC3 cells. GAPDH was used as a loading control.
C. Caspase activation was detected in situ with fluorescence labeling (Green: lower panel) using FAM-VAD-FAK, a cell permeable caspase inhibitor that binds to activated caspases. Nuclear staining was performed with DAPI (Blue: upper panel). It is apparent that a significant number of cells transfected with sh-uPAuPAR displayed green fluorescence.
D. Bar diagram showing quantitative data of DAPI/FMK-VAD-FAK labeled cells ratio from three random fields under a confocal microscope. The ratio of DAPI to FMK-VAD-FAK was significantly increased in cells transfected with sh-uPAuPAR. Significant differences from mock or EV/SV-transfected cells are indicated by asterisks * (P <0.05).
E. DNA laddering was observed in cells transfected with uPA-uPAR shRNA and cells treated with actinomycin D (ActD, 0.2 μg/ml). An agarose gel was stained with ethidium bromide and photographed under UV light. DNA markers were electrophoresed as a kilobase pair reference with standard bands of 2.0, 1.5, 1.0, 0.75 and 0.5 kb (lane M).
(All results are representative of at least three separate experiments.)
To examine this possibility, we transfected PC3 cells with plasmids expressing sh-uPA, sh-uPAR or sh-uPAuPAR. Molecular analysis of PC3 cell protein extracts revealed that the sh-uPAuPAR transfection induced pro-apoptotic genes, including Bax, Bcl-XS/L, caspase 9 (Fig. 4B). Also, fluorescent dye staining of sh-uPAuPAR-transfected PC3 cells with FAM-VAD-FMK revealed enhanced caspase activity which was not detected in either the mock cells or EV/SV-transfected cells (Fig. 4C & 4D). DNA fragmentation analysis provided further evidence of apoptotic induction. As Fig. 4E indicates, sh-uPAuPAR-transfected PC3 cells exhibited DNA laddering, a typical hallmark of apoptosis, on agarose gel electrophoresis that was not detected in either mock or EV/SV-transfected cells. This DNA laddering was similar to that of apoptosis induced by actinomycin D treatment (Fig. 4E), thereby confirming that the observed cell death was a result of apoptosis. In addition, DNA laddering was not observed in cells transfected with either sh-uPA or sh-uPAR. These results are also in agreement with caspase 9 induction and enhanced caspase activity in general as determined by FAM-VAD-FMK.
Knockdown of uPA and uPAR expression by RNAi inhibits its downstream signaling and tumorigenesis in nude mice
The biological consequences occasioned by uPA and uPAR silencing may be a result of changes in uPA-uPAR-mediated signaling and subsequent downstream functions. Since increased expression of uPA and uPAR activates ERK1/2 signaling (49,50), we decided to examine the status of mitogen-activated protein kinases in uPA-uPAR knockdown cells. Immunoblot analysis shows that ERK 1/2 phosphorylation was completely abolished in the sh-uPAuPAR-transfected cells, but not in the control cells (Fig. 5A). In contrast, the ERK phosphorylation did not change in cells transfected with either sh-uPA or sh-uPAR. In addition, the total, including phosphorylation, activity of Stat 3 was substantially suppressed in cells transfected with sh-uPAuPAR when compared with control cells (Fig. 5B). Furthermore, electrophoretic mobility shift assay (EMSA) with nuclear extracts from cells transfected with sh-uPAuPAR demonstrated that knockdown of uPA and uPAR expression inhibited binding of the extracts to the labeled Stat 3 binding sites (Fig. 5C). Since Stat 3 activation contributes to the stimulation of the anti-apoptotic pathway, the reduced level of phospho-Stat 3 as well as DNA binding activity may explain the increased susceptibility of sh-uPAuPAR-transfected PC3 cells to apoptotic cell death. Alternatively, constitutive ERK activation may contribute to cell survival.
Figure 5. RNAi knockdown of uPA and uPAR expression inhibits downstream signaling in PC3 cells.
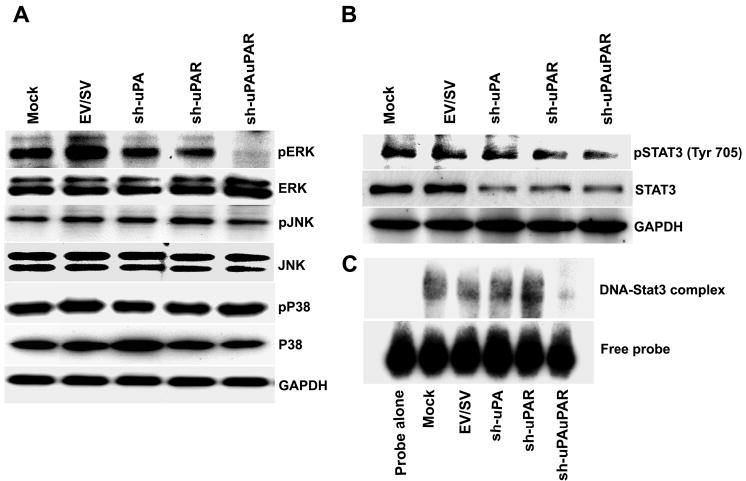
A. Immunoblot analysis of total and phosphorylated forms of ERK, p38 and JNK in mock and shRNA-transfected cells. PC3 cells transfected with mock, EV, SV, sh-uPA, sh-uPAR and sh-uPAuPAR were lysed 72 h later and subjected to SDS-PAGE followed by immunoblotting with total and phosphorylated forms of ERK, p38 and JNK antibodies. GAPDH antibodies were used to verify that similar amounts of protein were loaded in each lane.
B. Immunoblot analysis of Stat 3 protein in mock and shRNA-transfected cells. Equal amounts of protein were loaded and immunoblotting was carried out using phospho-specific Stat 3 antibodies against tyrosine 705 and antibodies against a non-phosphorylated form of Stat 3. GAPDH was included as a loading control.
C. The electrophoretic mobility shift assay of mock and shRNA-transfected cells. Protein-DNA complexes were separated on a 6% polyacrylamide gel, dried and autoradiographed. Shown above is specific DNA binding activity of nuclear extracts prepared from the indicated shRNA-transfected cells. Position of free probe is shown below.
(All results are representative of three separate experiments.)
Having established the significant effects of uPA-uPAR knockdown by RNAi in PC3 cells in vitro, we then asked whether uPA-uPAR RNAi would also suppress the tumorigenicity of pre-established PC3 orthotopic tumors in nude mice. PC3 cells were inoculated intraprostatically in the lateral lobe of prostate. On days 7 and 14 post-implantation, the tumors were injected with plasmid constructs expressing sh-uPA, sh-uPAR or sh-uPAuPAR. The mice were then sacrificed 14–15 days after the second dose of RNAi treatment, as was necessitated by the morbidity resulting from the tumors that had formed in control groups. The gross morphology of primary tumors and sites of metastasis were examined (Fig. 6A). No secondary tumors were observed visually in mice treated with the sh-uPA, sh-uPAR and sh-uPAuPAR plasmids, whereas mice treated with EV, SV and mock presented with secondary tumors in addition to the primary tumor within the prostate gland (Fig. 6A). Tumors were dissected and weighed (Fig. 6B). A 90–100% incidence of primary as well as secondary tumors was observed in mock, EV and SV-treated groups. Although sh-uPA and sh-uPAR treatments did not inhibit tumor growth completely, the weights of the tumor masses formed from these treatments groups were smaller than tumors from the control treatment groups (Fig. 6B). A drastic reduction in tumor weight was observed in mice treated with sh-uPAuPAR. Immunoblot analysis for protein levels in tumor samples confirmed that the tumors treated with sh-uPAuPAR had significantly decreased uPA and uPAR levels (Fig. 6C). Further 150 μg of sh-uPAuPAR completely regressed the pre-established prostate cancer (data not shown).
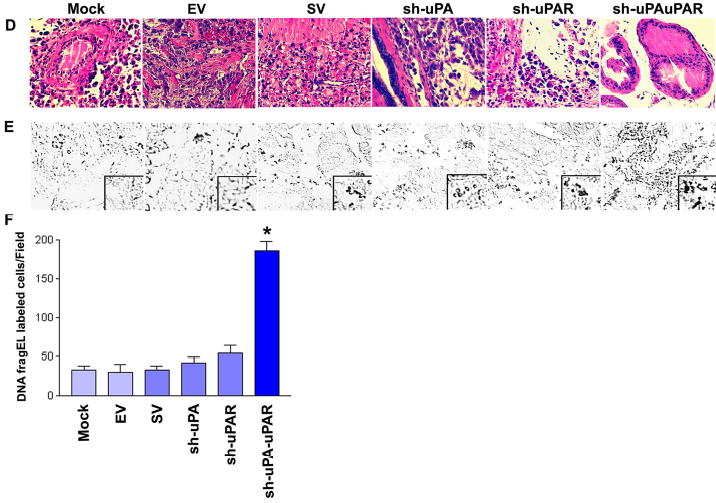
Figure 6. RNAi knockdown of uPA-uPAR expression abrogates tumor growth in an orthotopic mouse prostate tumor model.
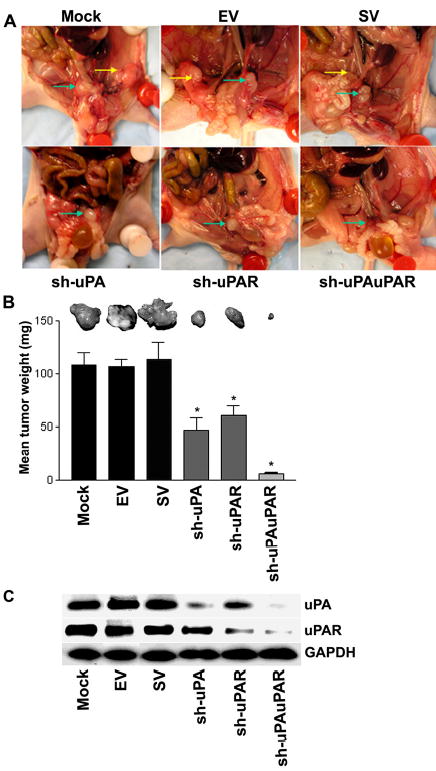
A. Representative in situ pictures from each treatment group of mice bearing orthotopic PC3 tumors. The primary prostate tumor is labeled with green arrows and yellow arrows indicate the position of metastases. PC3 cells were transplanted intraprostatically into nude mice and established PC3 prostate tumors were treated with shRNA-specific for uPA, uPAR and uPA-uPAR. After 4 weeks of the treatment of these constructs, the mice were sacrificed and evaluated for primary prostate tumor growth and metastases visually.
B. A comparison of dissected prostate tumors from each shRNA treatment group. Each bar represents the mean tumor weight ± SD of six animals per group. Significant differences from control groups (i.e., mock or EV/SV-treated) are represented by asterisks * (P <0.05).
C. Protein samples extracted from PC3 prostate tumors of six animals per group were analyzed using immunoblotting for uPA and uPAR expression levels. GAPDH was included as a loading control.
D. Representative hematoxylin and eosin sections of the orthotopic PC3 prostate mouse tumors. Primary prostate tumors were harvested from each treatment group at the conclusion of the experiment. Tumors were fixed in formalin and embedded in paraffin. Tissue sections (5 μm) were prepared and stained with H&E for histopathological analysis.
E. DNA fragEL staining of microdissected paraffin sections from established prostate tumors from the indicated treatment groups. DNA fragment end labeling assays were performed as described in Materials and Methods. Results are shown at a 40X magnification except for the box, which is at a 200X magnification.
F. Bar diagram showing quantitative data of DNA fragEL-labeled cells from six random fields per treatment group. Significant differences from control groups are indicated by asterisks * (P <0.05).
(Results are representative of three separate experiments.)
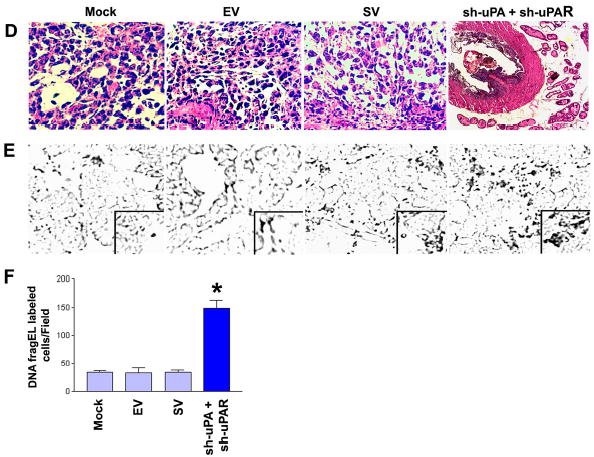
Immunohistochemical analysis was performed on the harvested paraffin-embedded tumor tissues to assess the effects of sh-uPAuPAR on the in vivo behavior of PC3 cells. sh-uPAuPAR-directed RNAi expression did not change the general architecture of the prostate gland and H&E staining showed largely normal histology, whereas staining revealed both tumor and host cells in the control groups (Fig. 6D). RNAi-targeted against either uPA or uPAR alone slightly reduced tumor cells relative to the control groups. Presumably, there was no in vivo rescue from the uPA-uPAR RNAi induced apoptosis. To test this, the paraffin-embedded tumor sections from all treatment groups were stained for apoptotic markers using Klenow-FragEL DNA fragmentation analysis. This end labeling for apoptotic cells demonstrates significant differences between treatment groups. Tumors of mock, EV and SV-treated groups showed generalized, low level staining for FragEL, therefore indicating that the tumor cells were healthy (Fig. 6E & 6F). In contrast, most of the areas of the sh-uPAuPAR-treated PC3 prostate tumors were positive for Klenow staining (Fig. 6E & 6F). Intratumoral coinjection of sh-uPA and sh-uPAR also resulted in almost complete regression of pre-established prostate tumor growth, whereas control groups of mock, EV and SV show reproducible and significant tumors (Fig. 7A & 7B). Immunoblot analysis demonstrated that the selective knockdown of uPA and uPAR protein levels in tumors cotreated with sh-uPA and sh-uPAR constructs (Fig. 7C). This cotreatment exhibited largely normal histology by H & E staining while tumors of mock, EV and SV treated groups displayed observable change in the general architecture of the prostate gland and H & E staining showed both tumor and host cells (Fig. 7D). Importantly, the knockdown of uPA and uPAR via cotreatment also resulted in a significant induction of apoptotic cell death, as revealed by positive Klenow staining (Fig. 7E & 7F). When combined with the data presented in Figs 6A–F and 7A–F, these results show that treatment with sh-uPAuPAR, however, has potent RNAi effect when compared to cotreatment with sh-uPA and sh-uPAR, which causes significant reduction in established tumor size.
Figure 7. RNAi knockdown of uPA and uPAR expression simultaneously abrogates tumor growth in an orthotopic mouse prostate tumor model.
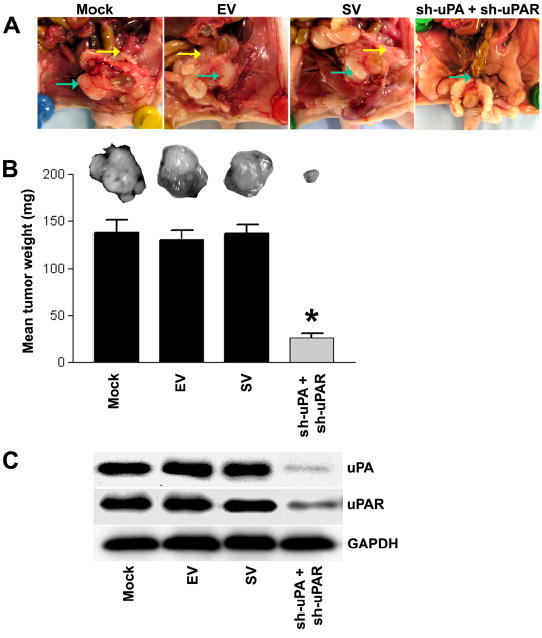
A. Representative in situ pictures from each treatment group of mice bearing orthotopic PC3 tumors. The primary prostate tumor is labeled with green arrows and yellow arrows indicate the position of metastases. PC3 cells were transplanted intraprostatically into nude mice and established PC3 prostate tumors were coinjected with both the sh-uPA and sh-uPAR vectors. After 4 weeks of the treatment of these constructs, the mice were sacrificed and evaluated for primary prostate tumor growth and metastases visually.
B. A comparison of dissected prostate tumors from each shRNA treatment group. Each bar represents the mean tumor weight ± SD of six animals per group. Significant differences from control groups (i.e., mock or EV/SV-treated) are represented by asterisks * (P <0.05).
C. Protein samples extracted from PC3 prostate tumors of six animals per group were analyzed using immunoblotting for uPA and uPAR expression levels. GAPDH was included as a loading control.
D. Representative hematoxylin and eosin sections of the orthotopic PC3 prostate mouse tumors. Primary prostate tumors were harvested from each treatment group at the conclusion of the experiment. Tumors were fixed in formalin and embedded in paraffin. Tissue sections (5 μm) were prepared and stained with H&E for histopathological analysis.
E. DNA fragEL staining of microdissected paraffin sections from established prostate tumors from the indicated treatment groups. DNA fragment end labeling assays were performed as described in Materials and Methods. Results are shown at a 40X magnification except for the box, which is at a 200X magnification.
F. Bar diagram showing quantitative data of DNA fragEL-labeled cells from six random fields per treatment group. Significant differences from control groups are indicated by asterisks * (P <0.05).
(Results are representative of three separate experiments.)
The results of the in vitro DNA laddering analysis and an in vivo DNA fragment end-labeling assay show that the simultaneous knockdown of uPA and uPAR by shRNA-based RNAi induces apoptosis. It is clear that uPA-uPAR-mediated downstream signaling is an excellent target for the treatment of hormone-independent prostate cancer. These data strongly suggest that uPA-uPAR-mediated downstream signaling is required for cell invasion, survival and proliferation in the prostate cancer cell line PC3.
DISCUSSION
The acquisition of tumor cell invasiveness is an important aspect of tumor progression and a principal factor of prostate cancer morbidity and mortality (51). Among the many proteases involved in invasion, uPA and uPAR are of great importance (15,16,19,20). These proteins are overexpressed in variety of malignancies including breast, ovarian, glioma and prostate cancers, and have been demonstrated to be essential in the maintenance of invasive and metastatic phenotypes (16). Several studies using in vitro and in vivo animal models have shown that either neutralizing uPA and/or uPAR function or blocking expression of these molecules significantly inhibits tumor invasion and metastasis in many cancers (5,33–36). While both uPA and uPAR are widely implicated in prostate tumor progression, the extent of their interaction in relation to the tumorigenicity of prostate cancer is not well understood. These earlier studies prompted us to investigate whether the uPA-uPAR system is important for prostate cancer invasion and metastasis. Although few studies have shown the relationship between uPA/uPAR expression and invasive potential in prostate cancer cell lines, there are some discrepancies in the literature (45–48). Hoosein et al., (45) reported an extremely low invasive capacity for LNCaP cells as compared with the more aggressive PC3 cell line. In contrast, Landiado et al., (46) showed a narrow range of variation between these two prostate cancer cell lines, whereas Dahiya et al., (47) reported a higher invasive capacity for LNCaP cells. Keer et al., (48) also reported that PC3 cells had a considerably higher invasive capacity as compared with DU145 cells, which is in agreement with the present study. Explanations for these differences include variations within cell lines and/or the loss of adhesion molecules over time and with passage number as well as differences in the expression of uPA activity as shown in the present study.
Our objective for testing the three prostate cancer cell lines in the cell invasion assay was to investigate whether differences in uPA and uPAR protein levels as revealed in the present study lead to differences in the invasive potential of the cells. In this study, PC3 and DU145 cell lines were more invasive than LNCaP cells, consistent with their known metastatic potentials. We found a strong correlation between the expression patterns of uPA and uPAR and the invasive potential of prostate cancer cell lines used (Fig. 1). Thus, these results show that enhanced uPA and uPAR expression in prostate cancer cell lines is associated with increased invasiveness and metastatic potential.
RNAi is a strong tool for silencing the function of specific genes (35, 37, 38). Several earlier studies have used chemical inhibitors to investigate the role of uPA system in progress of metastasis (33,35,52,53). Chemical inhibitors, however, have the intrinsic disadvantage that they often evoke nonspecific side effects (53). Sequence-based approaches, such as conventional antisense technologies, provide an attractive alternative, but usually offer only transient and partial suppression of the gene of interest (16,37,54). The development of 21-nucleotide siRNAs specifically recognizing homologous mRNA sequences provides new powerful reagents to selectively downregulate gene expression and holds great potential not only for the analysis of gene function, but also for the development of gene-specific therapeutic agents (16,40,54). Several laboratories are currently testing the feasibility of RNAi-mediated gene silencing as a novel tool for arresting tumor growth and killing cancer cells, and initial results are promising (35,40, 54,55). Our recent studies demonstrate the potential of simultaneous inhibition of two genes using a plasmid-based siRNA system (40,57). In this study, we have used plasmid-based RNAi to target uPA and uPAR genes, which are markedly overexpressed in a wide variety of human cancers including prostate cancer, singly and simultaneously. Gene-specific RNAi quite effectively downregulated uPA and uPAR mRNA as well as protein expression in the prostate cancer cell line PC3 (Fig. 2B and 2C). Although there is a little variation in the degree of gene silencing among the constructs, all these gene-specific RNAi plasmids reduced uPA and uPAR expression substantially compared to mock or EV/SV-transfected cells. Confirming these results, our immunohistochemical staining data showed that mock or cells transfected with EV/SV revealed positive staining for uPA and uPAR, while the cells transfected with sh-uPAuPAR were barely stained, with the exception of DAPI nuclear staining, suggesting the knockdown of uPA and uPAR protein expression (Fig. 2D). Similarly, the intensity of immunostaining for uPA and uPAR was reduced by gene-specific RNAi.
Aberrant expression of uPA and uPAR was found to be one of the most frequent alterations in advanced stage prostate cancer. The fact that uPA and uPAR are overexpressed only in the advanced stage of prostate cancer suggests that uPA and uPAR affect the functional pathways that are crucial in determining the phenotypes of advanced stage of cancers, such as increased proliferation and invasion. Invasion through the extracellular matrix is an important step in tumor metastasis. Therefore, understanding uPA-uPAR downstream signaling pathways will increase understanding of fundamental molecular processes responsible for cancer progression. Abrogation of either uPA or uPAR expression to suppress tumorigenesis has been achieved using several different approaches. For example, several early studies using in vitro and in vivo animals showed that either neutralizing uPA and/or uPAR function or blocking expression of these molecules significantly inhibited breast cancer invasion and metastasis (5,14). D’Alessio et al., (38) reported that reducing uPAR levels using antisense oligonucleotides inhibited tumor growth, invasion and metastasis in some cancers. Boyd et al., (5) showed that urokinase-derived peptides inhibited primary tumors and metastasis in orthotopic prostate tumors in nude mice. However, other studies on colon cancer metastasis have shown that antisense uPAR mRNA could only partially control metastasis. In our recent study with antisense uPA and uPAR bicistronic adenoviral vector targeting uPA and uPAR, we reported a reversal of the malignancy of glioma cells and an inhibition of tumor growth, invasion and angiogenesis (16). Coupling of uPA with uPAR orchestrates several different signaling molecules that form a unique network of several different types of biological responses, such as proliferation, migration, invasion, angiogenesis and metastasis. These biological responses to uPA-uPAR binding seem to be highly specific to cell-type, the nature of the downstream signaling molecule and the level of its expression. Gonias et al. (58) demonstrated that binding of uPA with uPAR activates ERK 1 and 2 and that this induced ERK activity is required for uPA-induced MCF-7 breast cancer cell migration. They further showed that a signaling cascade including FAK, Src and Shc is responsible for uPA-induced ERK activation and cell migration (59). In contrast, uPA-induced vascular smooth muscle cells (VSMC) migration and proliferation required activation of Stat pathway (60). A recent study in human breast cancer cells showed that uPA-induced mitogenic activity requires activation of both Stat and ERK pathways (61). Prior work from our laboratory has demonstrated that antisense uPA inhibited PI3K/Akt signaling and sensitized cells to apoptosis by staurosporine in the glioblastoma cell line SNB 19 (62). These findings provide evidence that binding of uPA with uPAR activates signaling cascades in order to regulate cell migration, invasion, proliferation and survival. In the context of uPA-uPAR signaling, however, direct molecular targeting against both uPA and uPAR would be a more straightforward and robust way to prove its biological contribution.
In the present study, RNAi for uPA-uPAR in PC3 cells showed remarkable suppression of invasion and proliferation as well as induction of apoptosis (Fig. 3–4). Suppression of the uPA-uPAR system and downstream signaling molecules (ERK and Stat 3) was observed in sh-uPAuPAR-transfected PC3 cells but not in mock or EV/SV-transfected cells (Fig.5). This suggests that all of the observed phenotypic changes in these cells were mediated by suppressing the uPA-uPAR interaction and the phosphorylation status of its downstream molecules. It has been demonstrated that uPA-uPAR signaling stimulates the both the Stat and ERK pathways and protects cancer cells from death. Several lines of evidence have shown that both ERK and Stat 3 pathways are capable of protecting cells from apoptotic cell death (15, 38). Transfection with either sh-uPA or sh-uPAR did not trigger apoptosis in PC3 cells (Fig. 4). We suspect that this because blocking either uPA or uPAR alone may not sufficiently affect the downstream signaling molecules ERK and Stat 3.
Since uPA-uPAR signaling modulates ERK and Stat 3 expression, simultaneous inhibition of uPA and uPAR may impair these pathways, leading to growth inhibition and induction of apoptosis. We thus reasoned that the uPA-uPAR system functions as a positive regulator of cell survival by facilitating cell proliferation and survival, the two hallmarks of cancer. Therefore, when overexpressed in cancers, uPA and uPAR would endow a cancer cell with increased proliferative and/or increased resistance to apoptosis. In contrast, knockdown of uPA-uPAR expression or function should inhibit cancer cell growth and induce apoptosis. Early studies by Gonias et al. (27) have demonstrated that the blocking uPA/uPAR expression using uPA- or uPAR-specific antibody, uPA antisense oligonucleotides or ERK pathway inhibitor PD098059, inhibited cell growth and promoted apoptosis in human breast cancer cells MDA-MB-231. These findings corroborate well with our results indicating that intercepting uPA-uPAR mediated signaling via knockdown of uPA and uPAR simultaneously inhibited cancer cell growth and induced apoptosis. Of note, uPA-uPAR RNAi worked in the hormone-resistant prostate cancer cell line PC3. This suggests that the knockdown of uPA-uPAR expression by RNAi might be an important strategy to inhibit hormone-resistant prostate tumor growth and survival.
Previous studies in a number of human cancer cell types have demonstrated that the orthotopic implantation of cells in nude mice more closely resembles the biological behaviors of these cells in humans, particularly in regards to the development of metastases. This has proven particularly true for human prostate cancer cells, which form primary tumors and metastases with much lower efficiency when implanted ectopically in nude mice. Given that uPA and uPAR are frequently overexpressed in many cancers, uPA is considered a very important target molecule for cancer therapy (16,33,34). Recently, in vivo therapies that target uPA or uPAR in various cancers have been reported by several research groups (5, 37, 38). One approach is the use of a DNA methylating agent (63). Other investigators have used antisense oligonucleotides (37), siRNA oligonucleotides (39–41), or Ad uPA-uPAR viruses (16) as targeting tools. These reports demonstrate suppression of tumor growth. Here, we show that a shRNA-based RNAi plasmid system represents an alternative strategy that can effectively suppress uPA-uPAR expression in orthotopic prostate tumors as determined by immunoblot analysis (Fig. 6C). Furthermore, the in vivo treatment of pre-established orthotopic tumors with sh-uPAuPAR-directed RNAi demonstrated a near total inhibition of tumor growth, whereas only partial reduction was observed with either sh-uPA or sh-uPAR RNAi (Fig. 6B). In addition, the co-treatment of pre-established orthotopic tumors with sh-uPA and sh-uPAR also almost completely inhibited the tumor growth (Fig. 7). No deleterious effects were noted in RNAi-treated animal groups as compared with mock or EV/SV-treated groups. Moreover, this approach can target a wide variety of tumor types and inhibit uPA-uPAR-dependent malignant phenotypes in vitro as well in vivo. Therefore, this RNAi system should provide a powerful new tool to analyze uPA-uPAR downstream signaling pathways as well as offer a new opportunity for cancer intervention. A comprehensive understanding of uPA-uPAR-mediated signaling events responsible for prostate cancer progression should lead to the development of novel therapeutic targets with clinical relevance. Furthermore, RNAi provides a novel, convenient and selective way to interfere with uPA-uPAR expression and to study the biological significance of their signaling in cancer biology.
Despite advances in understanding of the molecular mechanisms of human cancer, developing therapeutic approaches for the clinical treatment of human malignancies remains a major challenge. Knockdown of uPA-uPAR expression significantly inhibited the growth of PC3 cells in vitro as well as in vivo and ultimately resulted in apoptotic cell death. The mechanisms by which uPA-uPAR silencing reduces growth and provokes apoptosis in tumor cells certainly merit further investigation. Here, we have shown distinct target genes (ERK and Stat 3) that were regulated downstream of the uPA-uPAR signal. In our study, PC3 cells demonstrated very low Stat 3 phosphorylation and ERK phosphorylation was totally abolished when transfected with sh-uPAuPAR. Perhaps of more significance is that RNAi for uPA-uPAR induced cell death in PC3 cells in vitro as well as in vivo.
Acknowledgments
The authors are grateful to Dr. Gallick of the Department of Cancer Biology at the University of Texas M. D. Anderson Cancer Center for kindly sharing his technical expertise in orthotopic implantation procedures. We thank Shellee Abraham for preparing the manuscript and Diana Meister and Sushma Jasti for manuscript review. We also thank Noorjehan Ali for technical assistance.
Footnotes
This research was supported by National Cancer Institute Grant CA 75557, CA 85216, CA 92393, CA 95058 and N.I.N.D.S. NS47699 and The William E. McElroy Foundation and Caterpillar, Inc., OSF Saint Francis Medical Center, Peoria, IL (to J.S.R.).
References
- 1.Zhang X, Jin TG, Yang H, DeWolf WC, Khosravi-Far R, Olumi AF. Cancer Res. 2004;64:7086–7091. doi: 10.1158/0008-5472.CAN-04-1498. [DOI] [PubMed] [Google Scholar]
- 2.Shah RB, Mehra R, Chinnaiyan AM, Shen R, Ghosh D, Zhou M, Macvicar GR, Varambally S, Harwood J, Bismar TA, Kim R, Rubin MA, Pienta KJ. Cancer Res. 2004;64:9209–9216. doi: 10.1158/0008-5472.CAN-04-2442. [DOI] [PubMed] [Google Scholar]
- 3.Jemal A, Murray T, Samuels A, Ghafoor A, Ward E, Thun MJ. CA Cancer J Clin. 2003;53:5–26. doi: 10.3322/canjclin.53.1.5. [DOI] [PubMed] [Google Scholar]
- 4.Zhang Z, Li M, Wang H, Agrawal S, Zhang R. Proc Natl Acad Sci U S A. 2003;100:11636–11641. doi: 10.1073/pnas.1934692100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boyd DD, Kim SJ, Wang H, Jones TR, Gallick GE. Am J Pathol. 2003;162:619–626. doi: 10.1016/S0002-9440(10)63855-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim SJ, Uehara H, Yazici S, Langley RR, He J, Tsan R, Fan D, Killion JJ, Fidler IJ. Cancer Res. 2004;64:4201–4208. doi: 10.1158/0008-5472.CAN-03-3763. [DOI] [PubMed] [Google Scholar]
- 7.Kim SJ, Johnson M, Koterba K, Herynk MH, Uehara H, Gallick GE. Clin Cancer Res. 2003;9:5161–5170. [PubMed] [Google Scholar]
- 8.Patel P, Ashdown D, James N. Prostate Cancer Prostatic Dis. 2004;7:S14–S19. doi: 10.1038/sj.pcan.4500743. [DOI] [PubMed] [Google Scholar]
- 9.Hegeman RB, Liu G, Wilding G, McNeel DG. Clin Prostate Cancer. 2004;3:150–156. doi: 10.3816/cgc.2004.n.025. [DOI] [PubMed] [Google Scholar]
- 10.Pinthus JH, Waks T, Malina V, Kaufman-Francis K, Harmelin A, Aizenberg I, Kanety H, Ramon J, Eshhar Z. J Clin Invest. 2004;114:1774–1781. doi: 10.1172/JCI22284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Margheri F, D'Alessio S, Serrati S, Pucci M, Annunziato F, Cosmi L, Liotta F, Angeli R, Angelucci A, Gravina GL, Rucci N, Bologna M, Teti A, Monia B, Fibbi G, Del Rosso M. Gene Ther. 2005;12:702–714. doi: 10.1038/sj.gt.3302456. [DOI] [PubMed] [Google Scholar]
- 12.Desrosiers RR, Cusson MH, Turcotte S, Beliveau R. Int J Cancer. 2005;114:702–712. doi: 10.1002/ijc.20807. [DOI] [PubMed] [Google Scholar]
- 13.Singh S, Singh UP, Stiles JK, Grizzle WE, Lillard JW., Jr Clin Cancer Res. 2004;10:8743–8750. doi: 10.1158/1078-0432.CCR-04-0266. [DOI] [PubMed] [Google Scholar]
- 14.Naldini L, Tamagnone L, Vigna E, Sachs M, Hartmann G, Birchmeier W, Daikuhara Y, Tsubouchi H, Blasi F, Comoglio PM. Eur Mol Biol Org. 1992;11:4825–4833. doi: 10.1002/j.1460-2075.1992.tb05588.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rao JS. Nat Rev Cancer. 2003;3:489–501. doi: 10.1038/nrc1121. [DOI] [PubMed] [Google Scholar]
- 16.Gondi CS, Lakka SS, Yanamandra N, Siddique K, Dinh DH, Olivero WC, Gujrati M, Rao JS. Oncogene. 2003;22:5967–5975. doi: 10.1038/sj.onc.1206535. [DOI] [PubMed] [Google Scholar]
- 17.Usher PA, Thomsen OF, Iversen P, Johnsen M, Brunner N, Hoyer-Hansen G, Andreasen P, Dano K, Nielsen BS. Int J Cancer. 2005;113:870–880. doi: 10.1002/ijc.20665. [DOI] [PubMed] [Google Scholar]
- 18.Blasi F, Carmeliet P. Nat Rev Mol Cell Biol. 2002;3:932–943. doi: 10.1038/nrm977. [DOI] [PubMed] [Google Scholar]
- 19.Mamoune A, Kassis J, Kharait S, Kloeker S, Manos E, Jones DA, Wells A. Exp Cell Res. 2004;299:91–100. doi: 10.1016/j.yexcr.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 20.Nishimura K, Matsumiya K, Miura H, Tsujimura A, Nonomura N, Matsumoto K, Nakamura T, Okuyama A. Int J Androl. 2003;26:175–179. doi: 10.1046/j.1365-2605.2003.00413.x. [DOI] [PubMed] [Google Scholar]
- 21.Behren A, Binder K, Vucelic G, Herberhold S, Hirt B, Loewenheim H, Preyer S, Zenner HP, Simon C. Exp Cell Res. 2005;303:321–330. doi: 10.1016/j.yexcr.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 22.Resnati M, Pallavicini I, Wang JM, Oppenheim J, Serhan CN, Romano M, Blasi F. Proc Natl Acad Sci U S A. 2002;99:1359–1364. doi: 10.1073/pnas.022652999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dumler I, Weis A, Mayboroda OA, Maasch C, Jerke U, Haller H, Gulba DC. J Biol Chem. 1998;273:315–321. doi: 10.1074/jbc.273.1.315. [DOI] [PubMed] [Google Scholar]
- 24.Carlin SM, Resink TJ, Tamm M, Roth M. FASEB J. 2005;19:195–202. doi: 10.1096/fj.04-1644com. [DOI] [PubMed] [Google Scholar]
- 25.Tarui T, Andronicos N, Czekay RP, Mazar AP, Bdeir K, Parry GC, Kuo A, Loskutoff DJ, Cines DB, Takada Y. J Biol Chem. 2003;278:29863–29872. doi: 10.1074/jbc.M304694200. [DOI] [PubMed] [Google Scholar]
- 26.Aguirre-Ghiso JA, Estrada Y, Liu D, Ossowski L. Cancer Res (2003) 2003;63:1684–1695. [PubMed] [Google Scholar]
- 27.Ma Z, Webb DJ, Jo M, Gonias SL. J Cell Sci. 2001;114:3387–3396. doi: 10.1242/jcs.114.18.3387. [DOI] [PubMed] [Google Scholar]
- 28.Hendry L, John S. Eur J Biochem. 2004;271:4613–20. doi: 10.1111/j.1432-1033.2004.04424.x. [DOI] [PubMed] [Google Scholar]
- 29.Mora LB, Buettner R, Seigne J, Diaz J, Ahmad N, Garcia R, Bowman T, Falcone R, Fairclough R, Cantor A, Muro-Cacho C, Livingston S, Karras J, Pow-Sang J, Jove R. Cancer Res. 2002;62:6659–6666. [PubMed] [Google Scholar]
- 30.Yuan ZL, Guan YJ, Chatterjee D, Chin YE. Science. 2005;307:269–273. doi: 10.1126/science.1105166. [DOI] [PubMed] [Google Scholar]
- 31.Legrand C, Polette M, Tournier JM, de Bentzmann S, Huet E, Monteau M, Birembaut P. Exp Cell Res. 2001;264:326–336. doi: 10.1006/excr.2000.5125. [DOI] [PubMed] [Google Scholar]
- 32.Stewart DA, Cooper CR, Sikes RA. Reprod Biol Endocrinol. 2004;2:2. doi: 10.1186/1477-7827-2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rabbani SA, Gladu J. Cancer Res. 2002;62:2390–2397. [PubMed] [Google Scholar]
- 34.Schuh T, Besch R, Braungart E, Flaig MJ, Douwes K, Sander CA, Magdolen V, Probst C, Wosikowski K, Degitz K. Biol Chem. 2003;384:311–315. doi: 10.1515/BC.2003.035. [DOI] [PubMed] [Google Scholar]
- 35.Schweinitz A, Steinmetzer T, Banke IJ, Arlt MJ, Sturzebecher A, Schuster O, Geissler A, Giersiefen H, Zeslawska E, Jacob U, Kruger A, Sturzebecher J. J Biol Chem. 2004;279:33613–33622. doi: 10.1074/jbc.M314151200. [DOI] [PubMed] [Google Scholar]
- 36.Sato S, Kopitz C, Schmalix WA, Muehlenweg B, Kessler H, Schmitt M, Kruger A, Magdolen V. FEBS Lett. 2002;528:212–216. doi: 10.1016/s0014-5793(02)03311-2. [DOI] [PubMed] [Google Scholar]
- 37.Arens N, Gandhari M, Bleyl U, Hildenbrand R. Int J Oncol. 2005;26:113–119. [PubMed] [Google Scholar]
- 38.D'Alessio S, Margheri F, Pucci M, Del Rosso A, Monia BP, Bologna M, Leonetti C, Scarsella M, Zupi G, Fibbi G, Del Rosso M. Int J Cancer. 2004;110:125–133. doi: 10.1002/ijc.20077. [DOI] [PubMed] [Google Scholar]
- 39.Salvi A, Arici B, De Petro G, Barlati S. Mol Cancer Ther. 2004;3:671–678. [PubMed] [Google Scholar]
- 40.Gondi CS, Lakka SS, Dinh DH, Olivero WC, Gujrati M, Rao JS. Oncogene. 2004;23:8486–8496. doi: 10.1038/sj.onc.1207879. [DOI] [PubMed] [Google Scholar]
- 41.Jiang M, Rubbi CP, Milner J. Oligonucleotides. 2004;14:239–248. doi: 10.1089/oli.2004.14.239. [DOI] [PubMed] [Google Scholar]
- 42.Rye PD, Stigbrand T. Tumour Biol. 2004;25:329–336. doi: 10.1159/000081403. [DOI] [PubMed] [Google Scholar]
- 43.Woessmann W, Damm-Welk C, Fuchs U, Borkhardt A. Rev Clin Exp Hematol. 2003;7:270–291. [PubMed] [Google Scholar]
- 44.Sontheimer EJ. Nat Rev Mol Cell Biol. 2005;6:127–138. doi: 10.1038/nrm1568. [DOI] [PubMed] [Google Scholar]
- 45.Hoosein NM, Boyd DD, Hollas WJ, Mazar A, Henkin J, Chung LW. Cancer Commun. 1991;3:255–264. doi: 10.3727/095535491820873146. [DOI] [PubMed] [Google Scholar]
- 46.Laniado ME, Lalani EN, Fraser SP, Grimes JA, Bhangal G, Djamgoz MB, Abel PD. Am J Pathol. 1997;150:1213–1221. [PMC free article] [PubMed] [Google Scholar]
- 47.Dahiya R, Park HD, Cusick J, Vessella RL, Fournier G, Narayan P. Int J Cancer. 1994;59:126–132. doi: 10.1002/ijc.2910590122. [DOI] [PubMed] [Google Scholar]
- 48.Keer HN, Gaylis FD, Kozlowski JM, Kwaan HC, Bauer KD, Sinha AA, Wilson MJ. Prostate. 1991;18:201–14. doi: 10.1002/pros.2990180303. [DOI] [PubMed] [Google Scholar]
- 49.Ahmed N, Oliva K, Wang Y, Quinn M, Rice G. Br J Cancer. 2003;89:374–384. doi: 10.1038/sj.bjc.6601098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Aguirre Ghiso JA, Kovalski K, Ossowski L. J Cell Biol. 1999;147:89–104. doi: 10.1083/jcb.147.1.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pakneshan P, Xing RH, Rabbani SA. FASEB J. 2003;17:1081–1088. doi: 10.1096/fj.02-0973com. [DOI] [PubMed] [Google Scholar]
- 52.Joossens J, Van der Veken P, Lambeir AM, Augustyns K, Haemers A. J Med Chem. 2004;47:2411–2413. doi: 10.1021/jm0499209. [DOI] [PubMed] [Google Scholar]
- 53.Fabbrini MS, Carpani D, Bello-Rivero I, Soria MR. FASEB J. 1997;11:1169–1176. doi: 10.1096/fasebj.11.13.9367352. [DOI] [PubMed] [Google Scholar]
- 54.Miyagishi M, Hayashi M, Taira K. Antisense Nucleic Acid Drug Dev. 2003;13:1–7. doi: 10.1089/108729003764097296. [DOI] [PubMed] [Google Scholar]
- 55.Zhang Y, Zhang YF, Bryant J, Charles A, Boado RJ, Pardridge WM. Clin Cancer Res. 2004;10:3667–3677. doi: 10.1158/1078-0432.CCR-03-0740. [DOI] [PubMed] [Google Scholar]
- 56.Campbell TN, Choy FY. Curr Issues Mol Biol. 2005;7:1–6. [PubMed] [Google Scholar]
- 57.Lakka SS, Gondi CS, Yanamandra N, Olivero WC, Dinh DH, Gujrati M, Rao JS. Oncogene. 2004;23:4681–4689. doi: 10.1038/sj.onc.1207616. [DOI] [PubMed] [Google Scholar]
- 58.Nguyen DHD, Hussaini IM, Gonias SL. J Biol Chem. 1998;273:8502–8507. doi: 10.1074/jbc.273.14.8502. [DOI] [PubMed] [Google Scholar]
- 59.Nguyen DHD, Webb DJ, Catling AD, Song Q, Dhakephalkar A, Weber MJ, Ravichandran KS, Gonias SL. J Biol Chem. 2000;275:19382–19388. doi: 10.1074/jbc.M909575199. [DOI] [PubMed] [Google Scholar]
- 60.Kiyan J, Kiyan R, Haller H, Dumler I. EMBO. 2005;24:1787–1797. doi: 10.1038/sj.emboj.7600669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jo M, Thomas KS, Marozkina N, Amin TJ, Silva CM, Parsons SJ, Gonias SL. J Biol Chem. 2005;280:17449–17457. doi: 10.1074/jbc.M413141200. [DOI] [PubMed] [Google Scholar]
- 62.Chandrasekar N, Mohanam S, Gujrati M, Olivero WC, Dinh DH, Rao JS. Oncogene. 2003;22:392–400. doi: 10.1038/sj.onc.1206164. [DOI] [PubMed] [Google Scholar]
- 63.Pakneshan P, Szyf M, Farias-Eisner R, Rabbani SA. J Biol Chem. 2004;279:31735–31744. doi: 10.1074/jbc.M401669200. [DOI] [PubMed] [Google Scholar]