

Summary
The process of excising introns from pre-mRNA complexes is directed by specific genomic DNA sequences at intron—exon borders known as splice sites. These regions contain well-conserved motifs which allow the splicing process to proceed in a regulated and structured manner. However, as well as conventional splicing, several genes have the inherent capacity to undergo alternative splicing, thus allowing synthesis of multiple gene transcripts, perhaps with different functional properties. Within the human genome, therefore, through alternative splicing, it is possible to generate over 100,000 physiological gene products from the 35,000 or so known genes. Abnormalities in normal or alternative splicing, however, account for about 15% of all inherited single gene disorders, including many with a skin phenotype. These splicing abnormalities may arise through inherited mutations in constitutive splice sites or other critical intronic or exonic regions. This review article examines the process of normal intron—exon splicing, as well as what is known about alternative splicing of human genes. The review then addresses pathological disruption of normal intron—exon splicing that leads to inherited skin diseases, either resulting from mutations in sequences that have a direct influence on splicing or that generate cryptic splice sites. Examples of aberrant splicing, especially for the COL7A1 gene in patients with dystrophic epidermolysis bullosa, are discussed and illustrated. The review also examines a number of recently introduced computational tools that can be used to predict whether genomic DNA sequences changes may affect splice site selection and how robust the influence of such mutations might be on splicing.
Keywords: Inherited skin disease, RNA, Intron, Exon, Gene mutation, Splice site, Cryptic splicing
1. Introduction
In the early 1970s, studies on the processing of eukaryotic RNAs identified major differences between the size/length of heterogeneous nuclear RNAs (hnRNAs) compared to cytoplasmic messenger RNAs (mRNAs). Following electron microscopy and other analyses, the discrepancy as to why mRNAs are much shorter was explained by the phenomenon of pre-mRNA splicing [1]. This process involves modification of the initial primary transcript (copy of the entire gene) with retention of the protein-coding exons and removal (splicing out) of the intervening introns. For an individual gene, however, this splicing reaction is not limited to a single set of splice sites and thus use of alternative sites provides a versatile means of genetic regulation. Significantly, alterations in splice site choice can have vastly different effects on the mRNA and protein products of a gene. Commonly, alternative splicing patterns may lead to the inclusion or exclusion of a portion of coding sequence in the mRNA, giving rise to protein isoforms that differ in their peptide sequences and perhaps in the chemical, physical or biological activities of the protein(s). Alternative splicing is a major contributor to protein diversity in metazoan organisms. Estimates of the minimum number of human gene products that undergo alternative splicing are as high as 60% [2]. Moreover, many gene transcripts have multiple splicing patterns and some may have thousands [3]. Understanding the complexities of gene splicing is important not only to gain insight into normal patterns of gene regulation but because sequence alterations/mutations in DNA motifs critical to the process of gene splicing may result in a spectrum of inherited human diseases. The purpose of this review article is to provide an overview of the mechanisms of normal gene splicing, how splicing can be predicted from in silico models and to show how particular naturally occurring mutations can disrupt this highly regulated system and lead to disease. In this review, the splice site mutations illustrated and discussed predominantly involve the type VII collagen gene, COL7A1, which underlie certain forms of the mechanobullous disease, dystrophic epidermolysis bullosa.
2. RNA splicing
An interrupted gene is generally characterised by several short exons interposed with relatively longer introns. In order to translate into proteins, genes are first transcribed and the ends modified, with attachment of a 5′ cap and 3′ polyadenylation sequences. Exons are then identified and joined together, with introns removed, giving rise to mature mRNAs. Mature mRNAs are then transported out of nuclei and are translated into proteins.
RNA splicing is carried out by the spliceosome–—a large macromolecular complex that assembles onto these sequences and catalyses the two trans-esterification steps of the splicing reaction. This system also requires sequence elements necessary for recognition for mRNA splicing which, as shown by deletion experiments, are limited to the immediate vicinity of splice sites; the bulk of the intron is dispensable for splicing [4], but may contain other elements that are necessary for development. This intronic functional redundancy is evidenced by sequence analysis of Fugu rubripes, commonly known as the puffy fish, which shows compactness of its genome (one-eighth of the size of that of man), although it contains a similar complement of protein-coding genes to humans. The relative compaction results from reduction in the size of the puffy fish introns and intergenic regions [5].
2.1. Sequence motifs required during RNA splicing
The excision of the introns from a pre-mRNA and the joining of the exons are directed by special sequences at the intron—exon junctions called splice sites. The conserved motifs around splice sites were deduced by systematic analyses of intron—exon junctions throughout the GENBANK databank [6]. The 5′ splice site (donor) marks the exon—intron junction at the 5′ end of the intron. This almost invariably includes a GU dinucleotide at the intron end encompassed within a larger, less conserved sequence. Occasionally, the donor splice site sequence may vary; for example, intron 103 of the COL7A1 gene has GC at this site. Indeed, information theory-based analysis of the human genome reveals ~800 of this GC type donor sites [7,8]. At the other end of the intron, the 3′ splice site (acceptor) region has three conserved sequence elements: a branchpoint, followed by a polypyrimidine (Py) tract, followed by a terminal AG at the extreme 3′ end of the intron (Fig. 1). The process of splicing starts by excision of pre-mRNA at the donor site. The spliced intron folds back to form a lariat connecting the intron 5′ G to the 2′ of A at the branchpoint site. Next, the acceptor site is then cut, releasing the ‘lariat’ intron while the downstream exon is ligated to the upstream exon, immediately followed by product release from the spliceosome.
Fig. 1.

Nuclear splicing. (a) Schematic representation of an intron, flanked by two exons (A, light blue) and (B, pink). The essential splicing signals that define the exon boundaries are relatively short and poorly-conserved sequences. Only the GT (at the 5′ end of the intron), AG (at the 3′ end) and the branchpoint adenosine at the 2′ position are always conserved (all shown in red). (b) Pre-mRNA, showing the two flanking exons, branchpoint sequence and the conserved nucleotides as in (a). (c) The spliceosome cuts the pre-mRNA at its 5′ end, and then the intron forms a lariat by joining G at the 5′ end with the branchpoint A at the 2′ position. (d) The spliceosome cuts at the 3′ end of the intron and the intron is released as a lariat. (e) Exons A and B are then joined and released from the spliceosome.
The branchpoint, sited approximately 18–40 nucleotides upstream of the acceptor splice site, provides the means by which the 3′ splice site is identified. It is highly conserved in yeasts, with the sequence UACUAAC, mutation of which prevents mRNA splicing. In higher eukaryotes, however, the branchpoint sequence is not well-conserved apart from the target A nucleotide. These relaxed constraints result in the flexibility to use related sequences in the vicinity when the authentic branchpoint is deleted.
2.2. Other sequence motifs that may impact on RNA splicing
The splice site consensus sequences are generally not sufficient in themselves to determine whether a particular site will assemble a spliceosome and lead to gene splicing. Moreover, the natural splice site can also be very different from the consensus sequence, such as U12 introns that start and end with AT–AC [9]. Thus, other information and interactions are often necessary to activate splice site selection. To add further complexity to this process, certain genes such as hprt contain intronic sequences that match splice site sequences but which merely surround intronic false exons. Indeed, these intronic false exons may often outnumber real exons by several fold [10]. These intronic components, therefore, must rely on negative regulators to prevent aberrant splicing. Commonly, spliceosomal components binding on opposite sides of an exon can interact to stimulate excision of the flanking introns. This process is called exon definition and occurs in most internal exons. On top of this process, there are many non-splice site regulatory sequences that strongly affect spliceosome assembly, these are known as splicing enhancers. Exonic splicing enhancers (ESEs) are commonly found even in constitutive exons. Intronic enhancers also occur and appear to differ from exonic enhancers. Conversely, other RNA sequences may act as splicing silencers or repressors to block spliceosome assembly and certain splicing choices.
ESEs serve as binding sites for serine/arginine-rich (SR) proteins, conserved splicing factors characterised by the presence of RNA recognition motifs and carboxy-terminal arginine/serine (RS) domains. SR proteins that are bound to ESEs can promote exon definition by directly recruiting spliceosomes and/or antagonising the action of nearby exon splice suppressors (ESSs). ESE motifs can be identified by multiple methods such as systematic evolution of ligands by exponential enrichment (SELEX) [11] and S100 complementation assay [12], although the former produces binding sites that are biased towards consensus sequences and are not representative of the population of functional sites. Interestingly, from these RNA-binding sites, it appears that, although SR proteins have distinct RNA-binding specificities, they can bind to many different sequences and the same sequence can be a binding site for multiple SR proteins. The overlapping and promiscuous RNA-binding sites, therefore, may account for their apparent redundancy (for review, see Ref. [3]).
3. Non-pathogenic variability in splice site selection
It is clear that several different exonic and intronic sequences can influence splice site selection. Moreover, the process of splicing offers cells the opportunity to customise gene products to meet specific functional requirements. This can be accomplished by alternative splicing, in which selection of different splice donor and/or acceptor sites permits multiple mRNAs to be generated from a single gene. Although difficult to predict precisely, a number of in silico databases can now be used to help predict splice site options.
3.1. Prediction of splicing in silico
As splicing machinery requires sequence elements necessary for recognition for mRNA splicing, thorough and systematic analysis of RNA splice junction sequences from GENBANK databank allowed Shapiro and Senapathy to deduce mathematical formulae capable of predicting potential acceptor and donor splice sites. This approach was based on a scoring and ranking scheme linked to nucleotide weight tables [6]. Application of this model scoring system allows for prediction of normal alternative splicing as well as the impact of mutations that significantly alter these scores and which therefore may affect splice site recognition by the spliceosome and as a consequence, disturb RNA splicing. However, this method is based on consensus sequences and is highly biased towards strong splice sites. The values produced by this method do not correlate with binding site affinities and are therefore not very useful for determining inherent differences in their propensities to be recognised and spliced [13]. Details of web-based computational tools that may help predict options and changes in splicing, based on the conservation of nucleotides at splicing sites or ESEs and the features of base composition and base correlation around these regions, are listed in Table 1. For the purposes of this review, all analyses referred to were carried out using automated splice site analyses, by which the information in bits for a splice site (Ri) is defined as the dot product of a weight matrix derived from the nucleotide frequencies at each position of the splice site from the database and the vector of a particular splice junction sequence, and hence are more accurate than Senapathy scores [14–16].
Table 1.
Web-based resources for splice site and ESE predictions
| Websites | Addresses |
|---|---|
| Automated splice site analyses | https://splice.cmh.edu/ |
| Splice site prediction by neural network | http://www.fruitfly.org/seq_tools/splice.html |
| NetGene2 server | http://www.cbs.dtu.dk/services/NetGene2/ |
| GENIO/splice | http://www.biogenio.com/splice/ |
| ESE finder | http://rulai.cshl.edu/tools/ESE/ |
3.2. Alternative splicing
Alternative splicing is a very common phenomenon. It may occur in all cell types or be limited to certain tissues. How alternative splice sites are selected, however, is only partially understood. Most alternatively, spliced exons match poorly with canonical splicing consensus sequences. Thus, variations in the amount of a limiting splicing factor may have a significant impact on whether a poorly defined exon is included in the mature mRNA or not. Within skin biology, alternative splicing of component structural proteins has not been described very often. Recently, however, Sawamura et al. [17] described the first alternative splicing in the COL7A1 gene. Amplification of overlapping cDNA from keratinocytes using reverse transcription-polymerase chain reaction identified alternative splicing within the NC-1 domain of type VII collagen, which was generated by a different exon 18 acceptor site 27 bp upstream from the common acceptor site. This transcript variant, which encodes a protein that is nine amino acids longer than the usual transcript, is found in keratinocyte biopsies from the wound edge of patients with epithelialising skin ulcers and keratinocyte cultures treated with the cytokine, transforming growth factor beta-1.
Analysis of information content of all splice acceptor/donor sites throughout the COL7A1 gene reveals that, although exon definition of exon 18 is strong, there is another acceptor site 27 bp upstream from the natural site, with a higher Ri when compared with the natural site: 11.9 bits versus 10.8 bits (red arrow, Fig. 2). This new site is approximately 50 bp downstream from the branchpoint sequence and thus should not affect lariat formation. No similar acceptor site is predicted in any other part of the COL7A1 gene from the in silico analysis.
Fig. 2.

Establishing exon definition through in silico analysis of the COL7A1 gene for sequence motifs of acceptor/donor sites. Analysis of potential donor/acceptor splice motifs around exon 18 to 20 reveals an area within intron 18 that has a higher sequence motif conservation than the natural acceptor splice site of exon 18. In silico analysis reveals that although exon definition of exon 18 is strong, there is another acceptor site 27-bp upstream to the natural site, with a higher information content when compared with the natural site (red arrow). This site has the capability to lead to cryptic splicing and an in-frame insertion of 27-bp into the mRNA, increasing the length of the translated protein by 9 amino acids. This in silico observation provides an explanation for the findings of cryptic splicing by Sawamura et al. [17]. This computational modelling also demonstrates the relatively low information content for the intron 19 donor site (blue arrow) and may help explain the non-pathogenic retention of intron 19 observations made by Whittock et al. [19].
3.3. Intron retention
The retention of introns, as a physiological event in pre-RNA splicing, has not been well recognised because such transcripts are often believed to arise erroneously from unspliced or partially spliced pre-mRNAs. However, this phenomenon is not a rare event. Indeed, it happens in almost 15% of all known genes and in fact some retained introns may participate in the coding of protein domains [18]. Introns that are retained usually have higher GC contents than non-retained introns, a similar GC distribution for their flanking exons, a lower frequency of stop codons and conservation of the same introns across species [18]. In skin biology, intron retention was first described by Whittock et al. [19], who found inclusion of all of intron 19 of COL7A1 in mRNA extracted from leucocytes of many recessive dystrophic epidermolysis bullosa patients, although the pathogenic mutations in these individuals clearly were located elsewhere. The significance of intron 19 retention and why this occurs is not known. Using the criteria set out by Galante et al. [18], the GC content of intron 19 is not particularly high (62.9%), compared with other introns of COL7A1 (average 58%) and although the GC distribution is similar in the flanking exons (exon 19: 59.9%; exon 20: 71.5%), there is a stop codon within this intron, leading to protein truncation (909 amino acids compared to the 2944 amino acids of full-length type VII collagen). Furthermore, there is no similarity of intron 19 across species. However, computer analysis of the acceptor/donor splice sites for the entire COL7A1 gene shows that exon 19 has the lowest information content of any exon (blue arrow, Fig. 2). The spliceosome has a lower affinity for these donor and acceptor sites. This means that one or both sites may fail to be recognised and the exon will not be defined, therefore, it will not be excised. Another mitigating factor is the relatively short length of the IVS18-exon19-IVS19 span (354 bp), which permits the entire interval to be included in the transcript. The distribution of internal exon lengths in the genome is quite constrained, presumably because of the requirements of the exon definition model.
On the other hand, both splice site strength and accessory regulatory sequences dictate splice site use. In the majority of alternative splices, each skipped upstream acceptor site has a lower Ri value than that of the ultimate downstream acceptor site [20]. In other words, there is an inverse relationship between splice acceptor and propensity for exon skipping. For consecutive short exon—intron intervals (such as IVS18-exon19-IVS19), this same mechanism could alternatively produce intron 19 inclusion due to failure to recognise the donor at the end of exon 18 [19].
4. Splice site mutations and inherited skin disease
A thorough survey of human mutations show that approximately 15% of point mutations that are associated with genetic diseases affect the splice signals at the ends of introns, leading to aberrant splicing patterns, typically with skipping of the neighbouring exon(s) [21]. In COL7A1, splicing mutations account for almost 17% of all mutations [19] and similar mutations have been reported in several other human collagen genes [16]. However, these data derive mainly from genomic DNA analyses and the effect of a mutation on the mRNA or protein is usually predicted from the genomic sequence, as opposed to study of the mRNA.
4.1. Intron—exon border splice site mutations
In reviewing pathogenic splicing mutations in the COL7A1 gene, most occur within either the ‘‘GT’’ at the 5′ end or the ‘‘AG’’ at the 3′ end of the introns, with approximately 69% involving the donor site [19]. These splice site mutations have mostly been assumed to result in exon skipping, but this is not always the case. Indeed, computational analyses of genomic sequences using base composition can be used to help understand and predict cryptic splicing. For example, the COL7A1 mutation IVS30-1G > A is not predicted to lead to exon skipping since the sequence change results in a reduction in information content of the acceptor splice site, thus, creating a cryptic acceptor splice site 1 bp downstream into exon 31 (Fig. 3). Direct sequencing of cDNA derived from the skin of a patient with this mutation confirms this prediction. Likewise, the mutation c.4118C > T at the end of exon 35, has been previously classified as a splice site mutation [19], but the donor Ri is only minimally reduced, from 9.5 to 9.4 bits, and therefore, the intron 35 donor splice site may not be compromised. Another example is the COL7A1 mutation IVS7-6del5, which abolishes the natural donor splice site (Ri reduced from 5.9 to −4.3 bits) and is predicted to create a cryptic donor site 13 bp downstream (Ri increased from −1.3 to 4.0 bits), this outcome is fully supported by cDNA sequence analysis [22].
Fig. 3.

In silico analysis of a splice site mutation in COL7A1 predicts a frameshift rather than exon skipping. (a) Genomic DNA wild-type sequence for the intron30/exon 31 border. (b) In contrast, genomic sequence from a patient with dystrophic epidermolysis bullosa demonstrates a heterozygous splice site mutation, IVS30-1G > A mutation (arrow). By current paradigms, this mutation should result in skipping of exon 31. (c) Analyses of information content, however, shows a reduction of Ri at the natural acceptor site from 12.0 to 4.4 bits. (d) This mutation, therefore, is also predicted to create a new cryptic donor splice site 1-bp into exon 31, with an increase in Ri from 0.5 to 8.7 bits, resulting in the deletion of the first nucleotide G in exon 31. (e) Direct sequencing of cDNA derived from the patient’s skin confirms this frameshift in exon 31 and illustrates how computer analysis can help in predicting the consequences of splice site mutations.
Once a natural donor splice site is weakened or abolished, the cryptic site activated by this mutation must reside within a few hundred nucleotides of the natural splice site, since the novel exon in restricted in length [23].
4.2. Exons: missense and nonsense mutations, deletions and insertions
The most common pathogenic missense mutations in the COL7A1 gene involve substitution of glycine codons within the X—Y-glycine repeats, destabilising the integrity of the triple helix and accounting for most cases of dominant dystrophic epidermolysis bullosa [24]. By contrast, with few exceptions, missense mutations within the non-collagenous regions are not thought to be pathological, merely indicating common or rare polymorphisms. Computational models of such sequence changes, however, can shed new light on their disease relevance. For example, the mutation 341G> T in exon 3 of COL7A1 gene is expected to convert a glycine (GGG) residue to valine (GTG) residue within the non-collagenous NC-1 domain. Analyses of information content, however, reveals that this mutation creates a new cryptic donor splice site 87 bp upstream from the consensus donor splice site, with Ri value higher than the nearest natural site (8.2 bits versus 6.6 bits). Direct sequencing of cDNA derived from the skin of a patient with this mutation (and a clinical phenotype of recessive dystrophic epidermolysis bullosa) confirms this prediction and demonstrates the pathogenic relevance of this seemingly innocuous amino acid substitution [25]. Adding further complexity to nucleotide substitutions that may appear non-pathogenic are mutations that do not change a particular amino acid, termed silent mutations. Some of these mutations, however, may have pathogenic consequences, such as 3009C > Tin exon 20 of the LAMB3 gene, which leads to a cryptic donor splice site and a clinical phenotype of junctional epidermolysis bullosa [26].
Nonsense mutations are usually predicted to result in truncated proteins and/or accelerated mRNA decay of the mutant transcripts. However, this is not always the case. For example, the mutation R185X in the COL7A1 gene is actually predicted to generate a cryptic donor splice site (Ri increases from 0.1 to 7.6 bits, compared with the natural donor site of 5.6 bits) (Fig. 4).
Fig. 4.
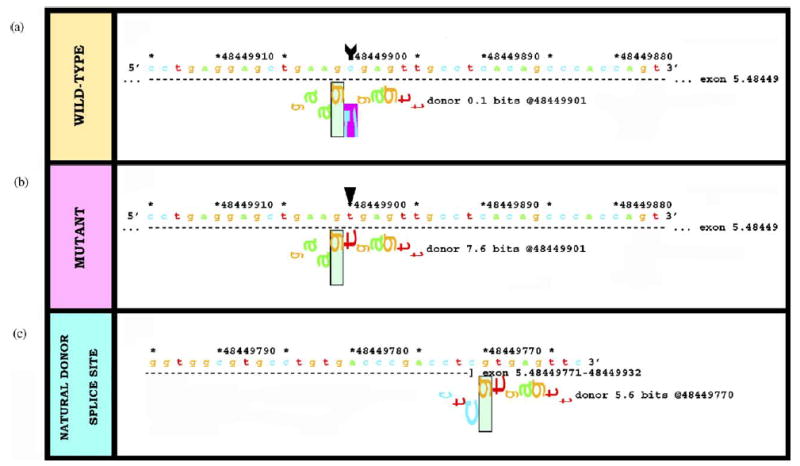
In silico analysis of a nonsense mutation, R185X, in exon 5 of the COL7A1 gene, surprisingly predicts cryptic splicing. Data analysis of (a) wild-type sequence, (b) the mutation R185X, and (c) the natural donor splice site of exon 5. The nucleotide substitution 553C > T in exon 5 of the COL7A1 gene changes an arginine codon to a termination codon, designated R185X. However, this transition raises the donor Ri content at this position from 0.1 (a) to 7.6 (b) bits, higher than the natural site at 5.6 bits (c) and is therefore predicted to result in an out-of-frame cryptic splice site 122-bp before the end of exon 5.
These examples highlight the fact that several different sequence changes can modulate splicing and without characterisation at the mRNA level, their interpretation can be difficult to predict. Indeed, a study comparing cDNA and genomic DNA in neurofibromatosis patients clearly illustrates this problem [27]. Splicing anomalies were identified in 50% of the patients in whom mutations were identified, and in at least 13% of these, the nature of the mutation would have been wrongly classified (frameshift, missense or nonsense) if only genomic DNA analysis, and not additional cDNA sequencing, been carried out.
Pathogenic mutations may also occur in sequences encoding ESE or ESS regions. This type of mutation has not been formally studied in the dermatological literature, probably because it is rare and because the mutation itself does not cause cryptic splicing—it merely enhances or suppresses splicing at natural splice sites. However, one previously published COL7A1 mutation, IVS2-3C > G, does appear, in retrospect, to compromise an ESE, resulting in the retention of the last 70 nucleotides of intron 2 [22]. Analysis of this mutation shows that it abolishes the acceptor splice site (Ri reduces from 8.3 to 2.3 bits) and creates a new site at position −2 to the natural site (Ri increases from −1.8 to 5.5 bits) (Fig. 5). Surprisingly, this site is not used for splicing. Interestingly, computer analysis also reveals that the mutation abolishes SRp40 ESE at position −3 (Ri reduces from 6.2 to 1.5 bits). Although hypothetical, it is plausible that without this enhancer, the splicing machinery fails to detect the new cryptic splice site at the −2 position and then has to resort to the nearest natural acceptor site, which is 70 bp upstream from the acceptor splice site (Ri = 5.0 bits), hence providing an explanation for the partial retention of intron 2.
Fig. 5.

In silico analysis of IVS2-3C > G in COL7A1 explains how an ESE mutation affects cryptic splicing. This mutation was previously shown to result in partial retention of intron 2 but the mechanism was not explained [22]. New insight, however, can be gained from computational analysis. In silico data for (a) wild-type sequence, and (b) the mutation IVS2-3C > G. Analysis of IVS2-3C > G in intron 2 of the COL7A1 gene shows that the natural acceptor splice site is abolished (Ri reduces from 8.3 to 2.3 bits). This mutation also creates a new cryptic acceptor splice site, 2-bp upstream into intron 2 (Ri increases from −1.8 to 5.5 bits). However, this site is not used in vivo because the same mutation also abolishes SRp40 ESE (Ri reduces from 6.2 to 1.5 bits). (c) Splicing apparatus instead uses the nearest natural acceptor site, 70-bp upstream into intron 2 (Ri = 5.0 bits).
Inferring the potential consequences of ESE mutations should not be based solely on in silico prediction, however, since the role of ESE sites are still not fully characterised and the biology has not caught up sufficiently to assert that these changes always result in cryptic splicing. For example, the mutation c.1760A > T in the middle (61 bp from the acceptor splice site) of exon 12 of the DSP gene creates a new SC35 ESE (Ri changes from 0.8 to 6.5 bits) at position −1 to a donor site (Ri = 6.8 bits, compared to the natural donor site 96 bp down-stream with Ri = 9.5 bits). It is conceivable that this might result in a cryptic splicing in the middle of this exon. Analysis of RT-PCR products, however, shows normal splicing. Thus, the in silico predictions of ESE mutations should always be substantiated by mRNA/protein work.
4.3. Introns: branchpoints, stem-loop, pseudoexon splicing
Although the branchpoint consensus sequence (BPS) in higher eukaryotes is not as well-conserved as in prokaryotes and a mutated BPS can easily be replaced by a cryptic BPS with only moderately reduced splicing efficiency in vitro [28], splice lariat BPS mutations have been reported in many inherited diseases, chiefly in dominant or X-linked disorders. These include nail patella syndrome [29] and type II Ehlers—Danlos syndrome [30]. However, in xeroderma pigmentosum, an autosomal recessive condition, splice lariat BPS mutations have also recently been reported [31]. Pathogenic mutations can either be missense changes that affect BPS information content, for example, an A > G change at the 2′ A position of the BPS (IVS3-24A>G) in the XPC gene in a case of xeroderma pigmentosum [31] that reduces Ri from 9.2 to −2.6 bits or, alternatively, intronic deletions of the BPS, for example, the mutation IVS1-37del17 in the LMX1B gene in a patient with nail patella syndrome [29]. Such mutations prevent binding of the U2 snRNP to the BPS, leading to destabilisation of U2AF and U1 snRNP and loss of the exon boundary definition of the subsequent exon, typically resulting in exon skipping.
Another disease-associated mechanism leading to aberrant splicing may involve formation of stem-loop structures at exon—intron junctions. Masking of these sites by mutations may lead to exon skipping through loss of the motif that the spliceosome normally recognises. An example of this has been noted in studies on the MAPT gene in the disorder, fronto-temporal dementia with Parkinsonism. Here, a stem-loop structure is formed at the junction between exon 10 and intron 10 that inhibits inclusion of exon 10 by interfering with U1 snRNP hybridisation to the pre-mRNA. Mutations that destabilise this structure by reducing the number of potential base pairs in the stem result in inclusion of exon 10 and consequently alter the protein isoform ratio, eventually leading to the disease phenotype [32]. This type of mutation has not been described in the skin.
Mutations found deep into the intron can also cause aberrant splicing. For example, in a case of neurofibromatosis, a NF1 gene mutation sited more than 300 bp into the intron, designated IVS30 + 332A > G, creates a new donor splice site, with the information content increasing from −4.1 to 8.7 bits. This activates a natural acceptor splice site 177 bp upstream with Ri = 2.1 bits and results in a cryptic splicing of a new exon [27]. Another example of a pathogenic intronic mutation has been reported by Pagani et al. [33], who described a 4 bp deletion in the intron-splicing processing element (ISPE) within intron 20 of the ATM gene in a patient with ataxia telangiectasia. This motif is complementary to U1 snRNA and interacts specifically with U1 snRNP particle. Abolition of this ISPE leads to the aberrant inclusion of a cryptic 65 bp exon.
Interestingly, mutation at a natural site can also activate sites that are further away inside the intron when a cryptic exon is created. For example, mutation of the first nucleotide of exon 3 of the CFTR gene, designated c.406G > A, activates a cryptic exon 183 bp in length in intron 3 (2354 nucleotides downstream of exon 3 and 19,329 nucleotides upstream of exon 4) [34]. In this instance, the cryptic exon is activated by weakening the natural acceptor site of the downstream exon. In other words, this activation depends on the comparative affinity of the cryptic versus the natural acceptor. Since intron 3 is so large, the downstream acceptor of exon 4 has evolved to be quite strong (Ri = 14.8 bits), thus precluding the activation of the cryptic exon (which notably has very weak binding sites, with Ri = 2.91 bits). Any reduction in the strength of the natural acceptor (in this case, Ri is reduced only from 14.8 to 12.6 bits) appears to activate this cryptic exon [16]. This is an interesting mutation, since this would be very difficult to predict a priori.
5. Conclusions
The process of intron removal from pre-mRNA complexes leading to mature mRNA is directed mainly by interactions between the spliceosome and well-conserved genomic DNA sequences at intron—exon borders, known as donor and acceptor splice sites. Other areas such as branchpoint sequences in the introns or exon splice enhancers/suppressors can also influence RNA splicing. Pathological disruption of normal intron—exon splicing can result from mutations in sequences that have a direct influence on splicing or from mutations that generate cryptic splice sites. Traditionally, mutation screening of genodermatoses is generally based on genomic DNA analysis and the effect of a mutation on the mRNA or protein is usually inferred from the genomic sequence, as opposed to direct evaluation by RT-PCR studies. Moreover, sequence changes in the introns not directly adjacent to natural splice sites are generally ignored as part of most mutation detection strategies. The failure to search for such mutations and the lack of appreciation of the potential for other intronic or exonic sequence variations that may affect intron splicing means that the incidence of disease-associated mutations that affect splicing is probably much higher than previously thought. To improve understanding of splice site anomalies, insight into whether changes in genomic DNA sequences might affect splicing has been gleaned from a number of computational tools. Indeed, there is now a decade of research on information theory-based analysis of splicing, which supports the use of an in silico approach to accurately predict the consequences of splicing mutations. In fact, so many splice variants have now been revealed by sequencing methods that it is impractical to comprehensively test the disease relevance of all of them in the laboratory. When no obvious coding sequence mutation is evident, therefore, information analysis can prioritise and direct subsequent laboratory workup of the remaining variants to establish which, if any, might be pathogenic.

John McGrath qualified MBBS at Guy’s Hospital Medical School in 1985, MRCP(UK) in 1988, MD (London University) in 1994 and FRCP in 1999. He trained in clinical dermatology and dermatopathology at St. John’s Institute of Dermatology in London and in molecular biology at Jefferson Medical College in Philadelphia. Since 2000, he has been Professor of molecular dermatology at St. John’s within King’s College, London. His research interests include the molecular basis of inherited skin disease and prenatal diagnosis.

Vesarat Wessagowit qualified MD at Chulalongkorn University in 1989, Diplomate Board of Dermatology (Thailand) in 1995 and PhD (London University) in 2004. He trained in dermatopathology and molecular biology at St. John’s Institute of Dermatology in London. His research interests include the molecular basis of inherited skin disease.
Acknowledgments
Funding for this work from the Dystrophic Epidermolysis Bullosa Research Association (DebRA UK) and the Royal Thai Government is gratefully acknowledged.
References
- 1.Berget SM, Berk AJ, Harrison T, Sharp PA. Spliced segments at the 5′ termini of adenovirus-2 late mRNA: a role for heterogeneous nuclear RNA in mammalian cells. Cold Spring Harb Symp Quant Biol. 1978;42(Pt 1):523–9. doi: 10.1101/sqb.1978.042.01.055. [DOI] [PubMed] [Google Scholar]
- 2.Modrek B, Lee C. A genomic view of alternative splicing. Nat Genet. 2002;30:13–9. doi: 10.1038/ng0102-13. [DOI] [PubMed] [Google Scholar]
- 3.Graveley BR. Alternative splicing: increasing diversity in the proteomic world. Trends Genet. 2001;17:100–7. doi: 10.1016/s0168-9525(00)02176-4. [DOI] [PubMed] [Google Scholar]
- 4.Wieringa B, Hofer E, Weissmann C. A minimal intron length but no specific internal sequence is required for splicing the large rabbit beta-globin intron. Cell. 1984;37:915–25. doi: 10.1016/0092-8674(84)90426-4. [DOI] [PubMed] [Google Scholar]
- 5.Aparicio S, Chapman J, Stupka E, Putnam N, Chia JM, Dehal P, et al. Whole-genome shotgun assembly and analysis of the genome of Fugu rubripes. Science. 2002;297:1301–10. doi: 10.1126/science.1072104. [DOI] [PubMed] [Google Scholar]
- 6.Shapiro MB, Senapathy P. RNA splice junctions of different classes of eukaryotes: sequence statistics and functional implications in gene expression. Nucleic Acids Res. 1987;15:7155–74. doi: 10.1093/nar/15.17.7155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nalla VK, Rogan PK. Automated splicing mutation analysis by information theory. Hum Mutat. 2005;25:334–42. doi: 10.1002/humu.20151. [DOI] [PubMed] [Google Scholar]
- 8.Rogan PK, Svojanovsky S, Leeder JS. Information theory-based analysis of CYP2C19, CYP2D6 and CYP3A5 splicing mutations. Pharmacogenetics. 2003;13:207–18. doi: 10.1097/00008571-200304000-00005. [DOI] [PubMed] [Google Scholar]
- 9.Burge CB, Padgett RA, Sharp PA. Evolutionary fates and origins of U12-type introns. Mol Cell. 1998;2:773–85. doi: 10.1016/s1097-2765(00)80292-0. [DOI] [PubMed] [Google Scholar]
- 10.Sun H, Chasin LA. Multiple splicing defects in an intronic false exon. Mol Cell Biol. 2000;20:6414–25. doi: 10.1128/mcb.20.17.6414-6425.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tuerk C, Gold L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science. 1990;249:505–10. doi: 10.1126/science.2200121. [DOI] [PubMed] [Google Scholar]
- 12.Abmayr SM, Reed R, Maniatis T. Identification of a functional mammalian spliceosome containing unspliced pre-mRNA. Proc Natl Acad Sci USA. 1988;85:7216–20. doi: 10.1073/pnas.85.19.7216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.O’Neill JP, Rogan PK, Cariello N, Nicklas JA. Mutations that alter RNA splicing of the human HPRT gene: a review of the spectrum. Mutat Res. 1998;411:179–214. doi: 10.1016/s1383-5742(98)00013-1. [DOI] [PubMed] [Google Scholar]
- 14.Schneider TD. Information content of individual genetic sequences. J Theor Biol. 1997;189:427–41. doi: 10.1006/jtbi.1997.0540. [DOI] [PubMed] [Google Scholar]
- 15.Schneider TD. Sequence walkers: a graphical method to display how binding proteins interact with DNA or RNA sequences. Nucleic Acids Res. 1997;25:4408–15. doi: 10.1093/nar/25.21.4408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rogan PK, Faux BM, Schneider TD. Information analysis of human splice site mutations. Hum Mutat. 1998;12:153–71. doi: 10.1002/(SICI)1098-1004(1998)12:3<153::AID-HUMU3>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 17.Sawamura D, Goto M, Yasukawa K, Kon A, Akiyama M, Shimizu H. Identification of COL7A1 alternative splicing inserting 9 amino acid residues into the fibronectin type III linker domain. J Invest Dermatol. 2003;120:942–8. doi: 10.1046/j.1523-1747.2003.12232.x. [DOI] [PubMed] [Google Scholar]
- 18.Galante PA, Sakabe NJ, Kirschbaum-Slager N, de Souza SJ. Detection and evaluation of intron retention events in the human transcriptome. RNA. 2004;10:757–65. doi: 10.1261/rna.5123504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Whittock NV, Ashton GH, Mohammedi R, Mellerio JE, Mathew CG, Abbs SJ, et al. Comparative mutation detection screening of the type VII collagen gene (COL7A1) using the protein truncation test, fluorescent chemical cleavage of mismatch, and conformation sensitive gel electrophoresis. J Invest Dermatol. 1999;113:673–86. doi: 10.1046/j.1523-1747.1999.00732.x. [DOI] [PubMed] [Google Scholar]
- 20.Thompson TE, Rogan PK, Risinger JI, Taylor JA. Splice variants but not mutations of DNA polymerase beta are common in bladder cancer. Cancer Res. 2002;62:3251–6. [PubMed] [Google Scholar]
- 21.Krawczak M, Reiss J, Cooper DN. The mutational spectrum of single base-pair substitutions in mRNA splice junctions of human genes: causes and consequences. Hum Genet. 1992;90:41–54. doi: 10.1007/BF00210743. [DOI] [PubMed] [Google Scholar]
- 22.Hovnanian A, Rochat A, Bodemer C, Petit E, Rivers CA, Prost C, et al. Characterization of 18 new mutations in COL7A1 in recessive dystrophic epidermolysis bullosa provides evidence for distinct molecular mechanisms underlying defective anchoring fibril formation. Am J Hum Genet. 1997;61:599–610. doi: 10.1086/515495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hawkins JD. A survey on intron and exon lengths. Nucleic Acids Res. 1988;16:9893–908. doi: 10.1093/nar/16.21.9893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Christiano AM, McGrath JA, Tan KC, Uitto J. Glycine substitutions in the triple-helical region of type VII collagen result in a spectrum of dystrophic epidermolysis bullosa phenotypes and patterns of inheritance. Am J Hum Genet. 1996;58:671–81. [PMC free article] [PubMed] [Google Scholar]
- 25.Wessagowit V, Kim S-C, Oh SW, McGrath JA. Genotype—phenotype correlation in recessive dystrophic epidermolysis bullosa: when missense doesn’t make sense. J Invest Dermatol. 2005;124:863–6. doi: 10.1111/j.0022-202X.2005.23650.x. [DOI] [PubMed] [Google Scholar]
- 26.Buchroithner B, Klausegger A, Ebschner U, Anton-Lamprecht I, Pohla-Gubo G, Lanschuetzer CM, et al. Analysis of the LAMB3 gene in a junctional epidermolysis bullosa patient reveals exonic splicing and allele-specific nonsense-mediated mRNA decay. Lab Invest. 2004;84:1279–88. doi: 10.1038/labinvest.3700164. [DOI] [PubMed] [Google Scholar]
- 27.Ars E, Serra E, Garcia J, Kruyer H, Gaona A, Lazaro C, et al. Mutations affecting mRNA splicing are the most common molecular defects in patients with neurofibromatosis type 1. Hum Mol Genet. 2000;9:237–47. doi: 10.1093/hmg/9.2.237. [DOI] [PubMed] [Google Scholar]
- 28.Reed R, Maniatis T. The role of the mammalian branchpoint sequence in pre-mRNA splicing. Genes Dev. 1988;2:1268–76. doi: 10.1101/gad.2.10.1268. [DOI] [PubMed] [Google Scholar]
- 29.Hamlington JD, Clough MV, Dunston JA, McIntosh I. Deletion of a branch-point consensus sequence in the LMX1B gene causes exon skipping in a family with nail patella syndrome. Eur J Hum Genet. 2000;8:311–4. doi: 10.1038/sj.ejhg.5200448. [DOI] [PubMed] [Google Scholar]
- 30.Burrows NP, Nicholls AC, Richards AJ, Luccarini C, Harrison JB, Yates JR, et al. A point mutation in an intronic branch site results in aberrant splicing of COL5A1 and in Ehlers—Danlos syndrome type II in two British families. Am J Hum Genet. 1998;63:390–8. doi: 10.1086/301948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Khan SG, Metin A, Gozukara E, Inui H, Shahlavi T, Muniz-Medina V, et al. Two essential splice lariat branchpoint sequences in one intron in a xeroderma pigmentosum DNA repair gene: mutations result in reduced XPC mRNA levels that correlate with cancer risk. Hum Mol Genet. 2004;13:343–52. doi: 10.1093/hmg/ddh026. [DOI] [PubMed] [Google Scholar]
- 32.D’Souza I, Poorkaj P, Hong M, Nochlin D, Lee VM, Bird TD, et al. Missense and silent tau gene mutations cause frontotemporal dementia with parkinsonism-chromosome 17 type, by affecting multiple alternative RNA splicing regulatory elements. Proc Natl Acad Sci USA. 1999;96:5598–603. doi: 10.1073/pnas.96.10.5598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pagani F, Buratti E, Stuani C, Bendix R, Dork T, Baralle FE. A new type of mutation causes a splicing defect in ATM. Nat Genet. 2002;30:426–9. doi: 10.1038/ng858. [DOI] [PubMed] [Google Scholar]
- 34.Will K, Dork T, Stuhrmann M, Meitinger T, Bertele-Harms R, Tummler B, et al. A novel exon in the cystic fibrosis trans-membrane conductance regulator gene activated by the nonsense mutation E92X in airway epithelial cells of patients with cystic fibrosis. J Clin Invest. 1994;93:1852–9. doi: 10.1172/JCI117172. [DOI] [PMC free article] [PubMed] [Google Scholar]