

Abstract
Marginal maternal biotin deficiency reduces hepatic activity of biotin-dependent carboxylases and causes high rates of fetal birth defects in mice. We tested the hypothesis that the decreased carboxylase activity observed in deficient dams and their offspring is mediated by decreased abundance of biotinylated carboxylases, decreased expression of their mRNAs, or both. During gestation, CD-1 mice were fed a diet that induced biotin deficiency or a biotin-sufficient diet. On gestational d 17, gravid uteri were removed, and each live fetus was examined grossly for defects. The expected high incidence of cleft palate (83%) in offspring was observed. In maternal and fetal liver, acetyl-CoA carboxylase, pyruvate carboxylase, propionyl-CoA carboxylase, and β-methylcrotonyl-CoA carboxylase abundances were determined by Western blotting; the content of mRNAs for most of these enzymes and holocarboxylase synthetase was determined by real-time RT-PCR. Biotin deficiency significantly reduced the abundance of the carboxylases in maternal and fetal liver; neither the content of mRNAs for the carboxylases nor holocarboxylase synthetase changed. This study provides evidence that the decrease in carboxylase activities is attributable to a decrease in the abundance of biotinylated carboxylases; further, this effect is more severe in fetuses than dams.
Keywords: biotin, biotin-dependent carboxylases, CD-1 mice, mRNA
Previous research in mice demonstrated that a maternal diet containing 25% egg white solids produces high rates of cleft palate, microglossia, and micromelia in offspring; the cause is severe biotin deficiency induced by egg white binding and preventing the absorption of biotin (1–3). Diets containing as little as 3% egg white solids were shown previously to produce marginal biotin deficiency in pregnant mice. Criteria for marginal biotin deficiency in that study were the absence of maternal signs of deficiency despite reduced activity of propionyl-Co A carboxylase (PCC)5 and increased urinary excretion of the leucine catabolite 3-hydroxyisovaleric acid (3HIA). Increased excretion of 3HIA indicates reduced activity of the biotin-dependent methylcrotonyl-CoA carboxylase (MCC) (3). A similar level of marginal biotin deficiency apparently occurs in about one third of human pregnancies. We and others are concerned that this marginal deficiency may be teratogenic (4).
In addition to demonstrating that marginal biotin deficiency is highly teratogenic in CD-1 mice, we further demonstrated that marginal maternal biotin deficiency resulted in fetal biotin deficiency as judged by the strong correlations between reduced maternal and fetal hepatic biotin content as well as between maternal and fetal hepatic PCC activity (3). Maternal biotin deficiency appears to produce a more severe degree of biotin deficiency in the fetuses based on the observation that a reduction in maternal hepatic PCC activity to 70% of the control level resulted in fetal hepatic PCC activity that was only 14% of that of control mice (3).
Our previous studies did not address the mechanism mediating the decrease in enzyme activity in the dam and fetus. Lewis et al. (5) observed decreased levels of hepatic MCC, PCC, pyruvate carboxylase (PC), acetyl-CoA carboxylase (ACC)-1 and ACC-2 in young male rats consuming an egg white– containing diet for 3 wk. Rodriguez-Melendez et al. (6) fed a biotin-deficient diet to young male rats for 8 wk and observed decreases in the abundance of liver PCC and MCC, but not PC; mRNA levels for PCC and PC did not differ between biotin-deficient and biotin-sufficient rats. In a subsequent study, mRNA levels for PC and PCC were not affected by biotin deficiency in kidney, muscle, brain, and liver (7). However, Rodriquez-Melendez and co-workers did observe a decrease in holocarboxylase synthetase (HCS) mRNA level in these same tissues. Leon-Del Rio et al. showed that this regulation of the enzymes involved in biotin utilization and metabolism is organ specific (8) and may spare biotin in critical organs such as the brain (9).
In a broader context, exciting new observations were made concerning additional mechanisms by which biotin can affect gene expression. Zempleni and co-workers observed that biotinylation of histones depends on the cell cycle (10,11), is associated with double-stranded DNA breaks (12), and is specific for particular lysine residues in certain histones (13). Gravel and co-workers identified HCS as the enzyme that catalyzes biotinylation of histones in the nucleus (14) and speculated that tyrosine phosphorylation may control intracellular distribution. Moreover, expression of a large number of genes appears to be influenced by biotin status (15,16). Thus, multiple mechanisms are potentially involved in teratogenesis caused by biotin deficiency.
In the study reported here, we tested the hypothesis that the decreased carboxylase activity observed in marginally biotin-deficient CD-1 maternal and fetal mice is due to reduced amounts of the biotinylated carboxylases. We investigated whether the change in carboxylase mass is caused by decreased carboxylase mRNA, decreased HCS mRNA, or both.
MATERIALS AND METHODS
Diets
Defined diets containing egg white were purchased from Teklad (Harlan Teklad). All diets were formulated to be nutritionally adequate for all components except biotin as previously described (3). In these diets, casein is replaced isonitrogenously with egg white. Experimental diets contained 5% egg white solids (biotin deficient) or 5% egg white solids with 0.08 mg biotin/kg diet added back (biotin sufficient). This amount of biotin is sufficient to saturate twice the capacity of avidin in 5% egg white to bind biotin, and provides enough biotin to meet the rodent dietary requirement of 0.2 mg biotin/kg diet (0.82 μmol biotin/kg diet) (17).
In a previous study (3), no differences in the amount of diet consumed by mice during gestation were observed for egg white content ranging from 0 to 25%; no significant differences in dam weight at term were observed (3). Food intake was also monitored in a pilot study for this study; food intake did not differ between the 2 diet groups (biotin deficient, 5.02 ± 0.7 g/d vs. biotin sufficient, 5.20 ± 0.8 g/d). Food intake was not monitored in the full study. Dam weight gain did not differ between the 2 groups. The protein content and composition were identical between the 2 diets in this study. Thus, equal diet intake also implies equal protein intake.
Animals and animal husbandry
Animal rooms were maintained under a 12-h light:dark cycle (lights on at 1030 h). Three to 4 females were housed together in plastic shoebox cages, and males were housed individually. For breeding, 2 female mice were placed with a proven breeder male for the final 3 h of the dark cycle (0730–1030 h). Those females with a copulation plug were weighed, rank ordered, and randomly assigned to one of the experimental diets, denoted gestation d 0. All animal care procedures followed those in the NRC guidelines (18) and were approved by the Animal Care and Use Committee of the National Center for Toxicological Research.
Developmental evaluations
On gestation d 17, dams were killed by ether inhalation. Maternal livers were removed by dissection, frozen immediately in liquid nitrogen, and stored at −80°C until analysis. Gravid uteri were removed and weighed. Each live fetus was weighed, examined for anatomic defects as previously described (3), and killed by decapitation. Fetal livers were removed, pooled by litter, immediately frozen on dry ice, and stored at −80°C until analysis.
Streptavidin blot analysis of proteins
Abundances of the biotinylated proteins PC, PCC, MCC, and ACC in maternal and fetal liver were determined by Western blotting as follows for both diet groups. ACC exists as 2 isotypes (ACC-1 and ACC-2) that chromatograph as a single band under the conditions used in these studies; “ACC” denotes the combined abundance. For protein extraction, a frozen liver portion (~500 mg) was thawed and homogenized in 5 volumes of ice-cold homogenization buffer [30 mmol/L HEPES, pH 7.2, 1 mmol/L EDTA, 300 mmol/L mannitol, and protease inhibitor cocktail (Sigma-Aldrich)]. The homogenates were sonicated and centrifuged at 110,000 × g for 30 min at 4°C. Protein concentration of homogenates was determined by the bicinchoninic acid assay (Pierce Biotechnology).
Biotinylated carboxylases were separated by gel electrophoresis using an adaptation of the method of Lewis et al. (19). For PC, PCC, and MCC, homogenate aliquots containing 5 μg (dams) or 10 μg (fetal pools) of protein were loaded onto a 4–12% Bis-Tris gel (Invitrogen). For ACC, 10 μg of homogenate protein was loaded onto a 3– 8% Tris Acetate gel (Invitrogen). Gels electrophoresis voltage was constant at 200 V and 115–70 mA for 50 min. Proteins were electroblotted to polyvinyldifluoride membranes for 1 h at 30 V. Membranes were blocked in 0.05% Tween-20 in PBS at room temperature for 1 h. For detection of biotinylated carboxylases, membranes were incubated at room temperature for 1 h while shaking in 0.05 μg/L avidin-alkaline phosphatase dissolved in blocking buffer. Membranes were then washed in 3 changes of wash buffer (0.05% Tween-20 in PBS). To detect biotinylated proteins labeled with avidin, membranes were incubated with 1 mL ECF substrate (Amersham Biosciences) in a sheet protector for 5 min at room temperature. Fluorescence of the bands was quantitated using a Storm 840 optical scanner (Molecular Dynamics, Amersham Biosciences); relative intensity was estimated by Image Quant software (Molecular Dynamics, Amersham Biosciences).
RT-PCR
To determine mRNA levels, a semiquantitative real-time RT-PCR assay was used. Total hepatic RNA was extracted from frozen liver using the Tri reagent (Molecular Research Center) according to the manufacturer’s instructions. RNA was quantitated spectrophotometrically using a NanoDrop ND-1000 (NanoDrop Technologies). RNA was treated with DNA-free (Ambion) according to the manufacturer’s protocol to remove DNA. Reverse transcription of 1 μg of total RNA was accomplished using the iSCRIPT cDNA synthesis kit (Bio-Rad Laboratories) according to the manufacturer’s protocol in a total volume of 20 μL using an MJ Research PTC-200 DNA Engine (MJ Research).
Primer pairs for each gene are as follows. For 18S rRNA, the forward primer was TGA CTC AAC ACG GGA AAC C, and the reverse primer was TCG CTC CAC CAA CTA GAA C. For MCC, the forward primer was TGG CTG CTG CTG GAG TTC, and the reverse primer was CCA CCA CGG ACT GCT TTG. For the α chain of PCC, the forward primer was GAA TCT CGG GTT TAT GCT GAG, and the reverse primer was AGA TGC TGA TGT CAC TTC CTG. For the β chain of PCC, the forward primer was CAG GCA GAG TAT GTG GAG AAG, and the reverse primer was GCA TAT CCG AGC ACG AGT AG. For HCS, the forward primer was CCG TGG AAG AAC AAA GGA GAG, and the reverse primer was TGG GCA GCG ATG GGT ATG. Primer pairs for ACC were those of Yang et al. (20).
Relative quantitation of mRNA levels for ACC, MCC, PCC-α, PCC-β, and HCS was determined by real-time PCR using an iCycler iQ Multi-Color Real-Time PCR Detection System (Bio-Rad Laboratories) with SYBR Green detection. PCR conditions for all genes were the same and were as follows: 95°C for 90 s; 95° for 30 s, 60°C for 30 s, 72°C for 30 s (40 cycles). A melt curve was performed for each sample after amplification by increasing the temperature to 95°C for 1 min and then lowering it to 55°C and adding 0.5°C at 10-s intervals for 80 cycles. Each well contained a total volume of 20 μL consisting of iQ SYBR green supermix (Bio-Rad Laboratories), forward and reverse primers (each at 200 nmol/L final concentration; Sigma-Genosys), cDNA equivalent to 500 pg of total RNA, and DEPC-treated water. Quantitation was performed by determining the threshold concentration (Ct) of each sample in the linear part of the curve using the PCR baseline subtracted feature of the iCycler software and manually setting the threshold at 300 arbitrary fluorescence units. Each sample was assayed in triplicate; means between each gene of interest and a housekeeping gene (18S rRNA) were compared. Briefly, for each sample, the Ct of the gene of interest (ACC-1, MCC, PCC, or HCS) was normalized to the Ct of the housekeeping gene (18S rRNA), and the mean for each group was calculated. The ratios between the means of the biotin-deficient and biotin-sufficient groups were then determined.
Using a cDNA from a pooled sample of fetal liver, standard curves were performed with each pair of primers. All genes amplified with approximately the same efficiency (85–90%); quantitation was linear over at least 4 logarithmic units. Specificity of amplification was determined by checking the melt curves for all PCR reactions; melt curves were consistent with a single gene product in each. The difference between biotin-deficient and biotin-sufficient livers for the housekeeping gene was determined by Student’s t test, and did not differ for either the maternal or fetal samples; therefore, biotin deficiency had no effect on mRNA expression of 18S rRNA.
Statistical analysis
The litter was used as the experimental unit for all fetal analyses. For continuous variables such as fetal weight and PCR relative quantitation, an F-test was used to determine whether variances were equal. An appropriate Student’s t test was used (either equal or unequal variances) to test the significance of differences between diet groups. For binomial data such as the proportion of offspring with cleft palate, the data were transformed using the arcsine transformation before analysis by t test. Differences were considered significant if the P-value was < 0.05.
RESULTS
Feeding a 5% egg white diet was shown previously to cause marginal biotin deficiency in pregnant mice as judged by significantly decreased biotin in dam liver, decreased activity of PCC in maternal liver, and increased urinary excretion of 3HIA (3); substantial rates of teratogenesis also occurred (3). In the current study, a 5% egg white diet produced the expected high incidence (83%) of cleft palate in these CD-1 mouse fetuses. The addition of 0.08 mg biotin/kg diet to the maternal diet decreased the rate of fetal clefting to the spontaneous rate (< 3%; 2 fetuses in 1 litter). For each biotin-dependent carboxylase, the holocarboxylase mass in dam livers was significantly decreased by marginal maternal biotin deficiency ( Fig. 1A, 2A). In dams fed the biotin-deficient diet, the mean hepatic masses of PC, PCC, MCC, and ACC were reduced to 42, 48, 55, and 50%, respectively, of dams fed the biotin-sufficient diet.
FIGURE 1.
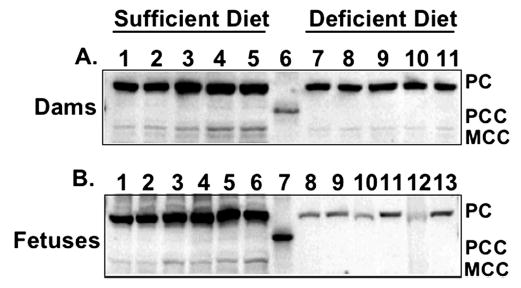
Effect of maternal diet on biotinylated carboxylases of livers of CD-1 mouse dams fed a diet that induced biotin deficiency or a biotin-sufficient diet (Panel A) and of fetal livers (Panel B). (Panel A) Gel lanes 1–5 depict individual dam liver homogenates of biotin-sufficient dams. Gel lane 6 depicts a molecular weight marker. Gel lanes 7–11 depict individual dam liver homogenates of biotin-deficient dams. PC, PCC, and MCC are noted to right of figure. (Panel B) Gel lanes 1–6 depict litter-pooled fetal liver homogenates from biotin-sufficient fetuses. Gel lane 7 depicts a molecular weight marker. Gel lanes 8–13 depict litter-pooled fetal liver homogenates from biotin-deficient fetuses. PC, PCC, and MCC are noted to the right of figure.
Holocarboxylase masses in fetal liver were also significantly decreased by marginal maternal biotin deficiency (Fig. 1B, 2B). In fetuses of dams fed the biotin-deficient diet, mean hepatic masses of PC, PCC, and MCC in fetal liver were 7, 6, and 7%, respectively, of fetuses whose dams were fed the biotin-sufficient diet. ACC was not detectable in livers from either sufficient or deficient fetuses.
Marginal maternal biotin deficiency did not affect mRNA expression for any of the genes examined. PCC, MCC, and ACC-1 mRNA expression levels in livers of deficient dams and fetuses did not differ from expression levels in sufficient controls ( Table 1). Similarly, marginal maternal biotin deficiency did not affect HCS mRNA expression in maternal or fetal liver (Table 1).
TABLE 1.
Ratio of gene expression data from biotin-deficient mice to biotin-sufficient mice normalized to 18s rRNA expression12
| Gene | Maternal liver | Fetal liver |
|---|---|---|
| ACC | 0.98 ± 0.15 | 0.99 ± 0.15 |
| MCC | 0.95 ± 0.23 | 1.00 ± 0.17 |
| PCC-α | 1.00 ± 0.21 | 1.02 ± 0.14 |
| PCC-β | 0.98 ± 0.18 | 1.05 ± 0.17 |
| HCS | 0.99 ± 0.22 | 1.03 ± 0.22 |
Values are ratios of the mean of the biotin-deficient group, n = 6, to the mean of the biotin-sufficient group, n = 6. The SD is calculated based on normalized values from both groups.
The biotin-deficient and -sufficient groups did not differ for any variable.
DISCUSSION
The studies reported here provide evidence that marginal maternal biotin deficiency in CD-1 mice altered holocarboxylase levels of ACC, PC, PCC, and MCC in mouse dam and fetal liver. Although the maternal biotin deficiency produced no overt signs of deficiency in the dams, biotinylated carboxylase mass was only half that of control mice. Marginal maternal biotin deficiency had even greater effects on biotinylated enzyme mass in the fetuses. The mass of biotinylated fetal carboxylases was < 10% of the biotin-sufficient controls. In contrast, the amounts of mRNA coding for the hepatic carboxylases in the deficient dams and fetuses were not different from the biotin sufficient controls.
Previous research by Mock et al. (3) demonstrated that feeding 5% egg white significantly decreases hepatic biotin and PCC activity of pregnant dams with none of the classic signs of biotin deficiency such as hair loss, dermatitis, or kangaroo gait. Yet marginal maternal biotin deficiency produced severe reductions in fetal carboxylase activities (10% activity compared with sufficient fetuses) and high rates of teratogenesis. Enzyme mass data from the present study provide confirmation that fetal contents of several holocarboxylases are reduced. Further, the reduction in activity and biotinylated enzyme masses are proportional, suggesting that the holocarboxylases present in the deficient fetuses are normally active. The reductions in biotinylated carboxylase activity and mass with normal gene expression for the carboxylases support the hypothesis that maternal biotin deficiency results in a lack of adequate biotin to biotinylate apocarboxylases in the fetus. Further, the relative preservation of maternal carboxylase activities suggests that the limited amount of biotin available to biotinylate proteins is sequestered in the dam liver. The fetal biotin deficiency may have been exacerbated by transport issues; the biotin transport system across the mouse placenta may not favor transport to the fetus (21). By analogy, chances of human teratogenesis due to marginal maternal biotin deficiency may be more likely because transport of biotin across the human placenta is not particularly active (22,23).
What is the relevance of this mouse model to human teratogenic events related to biotin deficiency? Watanabe and Endo (21) observed that egg white diets that produced high rates of teratogenesis in mice were not as teratogenic in rats. These investigators also observed differences in rates of teratogenesis among mice. The mechanism(s) of these differences are not known. We speculate that species differences in embryonic susceptibility to biotin deficiency, differences in biotin transport among species, or differences in the timing of onset or severity of biotin deficiency might account for species and strain differences. For example, we demonstrated previously that pregnant ICR mice exhibit significantly decreased hepatic biotin content and increased urinary excretion of 3HIA after only 7 d of consuming an egg white diet (3). In contrast, increased 3-HIA excretion and significantly decreased hepatic content of biotin are typically not present in rats consuming a similar egg white content diet until they have consumed the diet for ~2 wk (24,25). A slower onset of increased 3-HIA excretion was observed in humans (26,27); however, there are important similarities between this mouse model and human pregnancy. A significantly elevated 3-HIA excretion in response to leucine load is observed as early as d 7 in humans (26). The proportional increase of urinary 3-HIA in biotin-deficient, pregnant mice relative to normal is similar to the proportional increase observed during the first trimester of human pregnancy (3,28). Thus, the relevance of this mouse model to human teratogenesis remains to be clarified. Comparison with human pregnancy should be done with care because this mouse model seems particularly sensitive to biotin deficiency. Indeed, this model was chosen because of the high and reproducible rates of teratogenesis at marginal degrees of biotin deficiency.
No previous study has reported the effect of marginal biotin deficiency on enzyme mass and mRNA expression in mouse dams or their fetuses. However, similar trends in the abundance of biotinylated protein and mRNA were reported in biotin-deficient male rats. Lewis et al. (5) reported that moderate biotin deficiency reduced the abundance of PC, PCC, and ACC in rat liver. Rodriguez-Melendez et al. (6,7) reported that severe biotin deficiency in rats decreased the activity and abundance of total PC (apocarboxylase + holocarboxylase) and decreased the activity of PCC and abundance of total PC + MCC (apocarboxylase + holocarboxylase of both enzymes); severe biotin deficiency did not affect hepatic mRNA for PC or PCC. Rathman et al. (29) demonstrated that carbamazepine-induced biotin deficiency in rats resulted in reduced abundance of biotinylated PC, PCC, MCC, and ACC. However, there are differences between this study and the report of Rodriguez-Melendez et al. (7). Those investigators reported that severe biotin deficiency reduced holocarboxylase synthetase mRNA content of liver by more than half. Differences in the severity of biotin deficiency between the studies likely contributed to the different results; other potential sources of differences include species, sex of animals, pregnancy-induced changes, and stage of development (adult vs. fetus).
Unlike other studies that quantitated apocarboxylase + holocarboxylase mass by in vitro biotinylation before electrophoresis, this study did not directly quantitate apocarboxylase mass. Thus, the possibility remains that maternal biotin status may reduce apocarboxylase synthesis, accelerate the degradation of apocarboxylases or holocarboxylases, or both. Against these alternate mechanisms is the observation that in biotin-deficient rats, large pools of apocarboxylases are present that are immediately available for biotinylation upon the return of biotin to the system (6). Moreover, in the present study, HCS mRNA expression was not affected in biotin-deficient fetuses. We hypothesize that normal amounts of HCS mRNA code for synthesis of normal amounts and activity of HCS; normal or increased amounts of apocarboxylases are present and available for biotinylation, but deficient amounts of biotin (and hence biotinyl AMP) to biotinylate the apocarboxylases lead to decreased levels of holocarboxylases.
We did not quantitate the effects of biotin deficiency on fetal synthesis of mRNA for other proteins nor did we quantitate the resultant mass of other proteins. Because mRNA content for the proteins examined did not change, we speculate that biotin deficiency does not globally affect protein synthesis in the fetus. Consistent with that speculation, we observed no significant differences in either the weight of the biotin-deficient fetuses at d 17 nor in the number of biotin-deficient fetuses per litter. In summary, the observations presented here provide evidence that marginal biotin deficiency is relatively specific for its effect on holocarboxylases. Although the rates of teratogenesis were quite high, the anatomic effects observed exhibited a similar high degree of specificity rather than producing global anatomic disruptions. In that sense, this mouse model appears to be promising for the investigation of the pathogenesis of birth defects related to biotin deficiency.
Reduced biotin-dependent carboxylase activity during fetal development was implicated as a mechanism by which biotin deficiency might induce teratogenesis (3). Although additional research is clearly warranted to elucidate the role of biotin-dependent carboxylases in fetal development, knockout mice offer interesting observations on the effects of individual carboxylase deficiencies. Neither a knockout mouse nor isolated human inborn deficiency of ACC-1 has been reported; deficiency of ACC-1 is thought to be fatal in utero. Isolated inborn deficiency of ACC-2 in mice results in normal growth and development and an insatiable appetite without weight gain; this is thought to arise from increased fatty acid oxidation rates (30). In contrast, disruption of the α-subunit gene of PCC resulted in mice that died 24 –36 h after birth. Mice with a postnatal liver-specific PCC expression at a highly reduced level compared with wild-type mice survived the newborn and early infant period but required greater transgene expression after the late infant period to continue to grow normally (31). Thus, no isolated carboxylase deficiency recapitulates the ability of in utero biotin deficiency to cause cleft palate and limb shortening, and the teratogenic mechanism remains to be elucidated. These teratogenic events may arise from more global effects on gene expression or metabolism (11,12), from less severe combinations of carboxylase deficiencies, or from the effects of carboxylase deficiencies appearing at critical times in fetal development.
FIGURE 2.
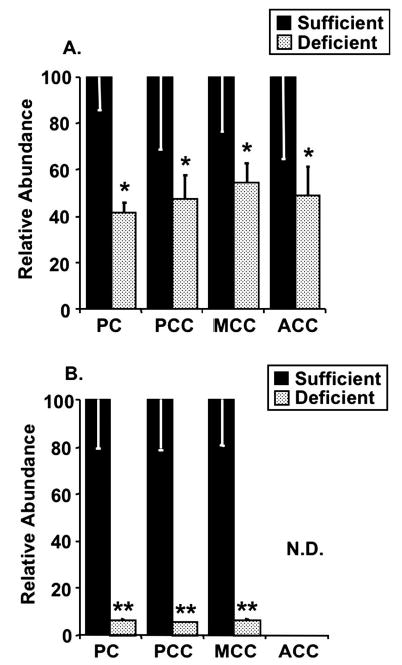
Effect of maternal diet on biotinylated carboxylase abundance in CD-1 mouse dams fed a diet that induced biotin deficiency or a biotin-sufficient diet (Panel A) and in fetuses (Panel B). (Panel A) Dams. Values are means ± SD, n = 5 for biotin-sufficient diet and n = 5 for biotin-deficient diet. *Different from sufficient, P < 0.02. (Panel B) Fetuses. Values are means ± SD (SD bars are too small to be visible in several samples), n = 6 for biotin-sufficient diet and n = 6 for biotin-deficient diet. **Different from sufficient, P < 0.002. ND, not detectable.
Acknowledgments
We thank Ms. Kelly Wall and Gene White for assistance with the fetal examinations.
Footnotes
Presented at Experimental Biology ’04, April 2004, Washington, DC [Stratton, S. L., Sealey, W. M., Hansen, D. K. & Mock, D. M. (2004) Marginal maternal biotin deficiency in CD-1 mice decreases the abundance of the biotin-dependent carboxylases in fetal liver. FASEB J. 18: A176].
Supported by the National Institutes of Health, DK-36823 (D.M.M.).
Abbreviations used: ACC, acetyl-CoA carboxylase; HCS, holocarboxylase synthetase; 3HIA, 3-hydroxyisovaleric acid; MCC, methylcrotonyl-CoA carboxylase; PC, pyruvate carboxylase; PCC, propionyl-Co A carboxylase.
References
- 1.Watanabe T. Teratogenic effects of biotin deficiency in mice. J Nutr. 1983;113:574–581. doi: 10.1093/jn/113.3.574. [DOI] [PubMed] [Google Scholar]
- 2.Watanabe T, Endo A. Teratogenic effects of avidin-induced biotin deficiency in mice. Teratology. 1984;30:91–94. doi: 10.1002/tera.1420300112. [DOI] [PubMed] [Google Scholar]
- 3.Mock DM, Mock NI, Stewart CW, LaBorde JB, Hansen DK. Marginal biotin deficiency is teratogenic in ICR mice. J Nutr. 2003;133:2519–2525. doi: 10.1093/jn/133.8.2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zempleni J, Mock DM. Marginal biotin deficiency is teratogenic. Proc Soc Exp Biol Med. 2000;223:14–21. doi: 10.1046/j.1525-1373.2000.22303.x. [DOI] [PubMed] [Google Scholar]
- 5.Lewis B, Rathman S, McMahon RJ. Dietary biotin intake modulates the pool of free and protein-bound biotin in rat liver. J Nutr. 2001;131:2310–2315. doi: 10.1093/jn/131.9.2310. [DOI] [PubMed] [Google Scholar]
- 6.Rodriguez-Melendez R, Perez-Andrade ME, Diaz A, Deolarte A, Camacho-Arroyo I, Ciceron I, Ibarra I, Velazquez A. Differential effects of biotin deficiency and replenishment on rat liver pyruvate and propionyl-CoA carboxylases and on their mRNAs. Mol Genet Metab. 1999;66:16–23. doi: 10.1006/mgme.1998.2777. [DOI] [PubMed] [Google Scholar]
- 7.Rodriguez-Melendez R, Cano S, Mendez ST, Velazquez A. Biotin regulates the genetic expression of holocarboxylase synthetase and mitochondrial carboxylases in rats. J Nutr. 2001;131:1909–1913. doi: 10.1093/jn/131.7.1909. [DOI] [PubMed] [Google Scholar]
- 8.Solorzano-Vargas RS, Pacheco-Alvarez D, Leon-Del-Rio A. Holocarboxylase synthetase is an obligate participant in biotin-mediated regulation of its own expression and of biotin-dependent carboxylases mRNA levels in human cells. Proc Natl Acad Sci USA. 2002;99:5325–5330. doi: 10.1073/pnas.082097699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pacheco-Alvarez D, Solorzano-Vargas RS, Gravel RA, Cervantes-Roldan R, Velazquez A, Leon-Del-Rio A. Paradoxical regulation of biotin utilization in brain and liver and implications for inherited multiple carboxylase deficiency. J Biol Chem. 2004;279:52312–52318. doi: 10.1074/jbc.M407056200. [DOI] [PubMed] [Google Scholar]
- 10.Stanley JS, Griffin JB, Zempleni J. Biotinylation of histones in human cells: effects of cell proliferation. Eur J Biochem. 2001;268:5424–5429. doi: 10.1046/j.0014-2956.2001.02481.x. [DOI] [PubMed] [Google Scholar]
- 11.Zempleni, J. (2003) Biotinylation of histones. In: Molecular Nutrition (Zempleni, J. & Daniel, H., eds.), pp. 267–275. CABI Press, Wallingford, UK.
- 12.Rodriguez-Melendez R, Griffin JB, Zempleni J. Biotin supplementation increases expression of the cytochrome P450 1B1 gene in Jurkat cells, increasing the occurrence of single-stranded DNA breaks. J Nutr. 2004;134:2222–2228. doi: 10.1093/jn/134.9.2222. [DOI] [PubMed] [Google Scholar]
- 13.Camporeale G, Shubert SS, Sarath G, Cerny R, Jempleni J. K8 and K12 are biotinylated in human histone H4. Eur J Biochem. 2004;271:2257–2263. doi: 10.1111/j.1432-1033.2004.04167.x. [DOI] [PubMed] [Google Scholar]
- 14.Narang MA, Dumas R, Ayer LM, Gravel RA. Reduced histone biotinylation in multiple carboxylase deficiency patients: a nuclear role for holocarboxylase synthetase. Hum Mol Genet. 2004;13:15–23. doi: 10.1093/hmg/ddh006. [DOI] [PubMed] [Google Scholar]
- 15.Zempleni J, Mock DM. Biotin homeostasis through the cell cycle. Nutr Res Rev. 2001;14:45–63. doi: 10.1079/NRR200117. [DOI] [PubMed] [Google Scholar]
- 16.Rogriguez-Melendez R, Zempleni J. Regulation of gene expression by biotin. J Nutr Biochem. 2003;14:680–690. doi: 10.1016/j.jnutbio.2003.07.001. [DOI] [PubMed] [Google Scholar]
- 17.Fenton PF, Cowgill GR, Stone MA, Justice DH. Nutrition of the mouse. VIII Studies on pantothenic acid, biotin, inositol, and p-aminobenzoic acid. J Nutr. 1950;42:257–269. doi: 10.1093/jn/42.2.257. [DOI] [PubMed] [Google Scholar]
- 18.National Research Council (1996) Guide for the Care and Use of Laboratory Animals. Institute for Laboratory Animal Resources, Washington, DC.
- 19.Lewis B, Rathman S, McMahon RJ. Detection and quantification of biotinylated proteins using the STORM 840 optical scanner. J Nutr Biochem. 2003;14:196–202. doi: 10.1016/s0955-2863(02)00283-8. [DOI] [PubMed] [Google Scholar]
- 20.Yang J, Goldstein JL, Hammer RE, Moon YA, Brown MS, Horton JD. Decreased lipid synthesis in livers of mice with disrupted Site-1 protease gene. Proc Natl Acad Sci USA. 2001;98:13607–13612. doi: 10.1073/pnas.201524598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Watanabe T, Endo A. Species and strain differences in teratogenic effects of biotin deficiency in rodents. J Nutr. 1989;119:255–261. doi: 10.1093/jn/119.2.255. [DOI] [PubMed] [Google Scholar]
- 22.Schenker S, Hu Z, Johnson RF, Yang Y, Frosto T, Elliott BD, Henderson GI, Mock DM. Human placental biotin transport: normal characteristics and effect of ethanol. Alcohol Clin Exp Res. 1993;17:566–575. doi: 10.1111/j.1530-0277.1993.tb00801.x. [DOI] [PubMed] [Google Scholar]
- 23.Hu ZQ, Henderson GI, Mock DM, Schenker S. Biotin uptake by basolateral membrane of human placenta: normal characteristics and role of ethanol. Proc Soc Biol Exp Med. 1994;206:404–408. doi: 10.3181/00379727-206-43778. [DOI] [PubMed] [Google Scholar]
- 24.Mock DM, Mock NI, Weintraub S. Abnormal organic aciduria in biotin deficiency: the rat is similar to the human. J Lab Clin Med. 1988;112:240–247. [PubMed] [Google Scholar]
- 25.Mock DM, Mock NI. Lymphocyte propionyl-CoA carboxylase is an early and sensitive indicator of biotin deficiency in rats, but urinary excretion of 3-hydroxypropionic acid is not. J Nutr. 2002;132:1945–1950. doi: 10.1093/jn/132.7.1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mock NI, Malik MI, Stumbo PJ, Bishop WP, Mock DM. Increased urinary excretion of 3-hydroxyisovaleric acid and decreased urinary excretion of biotin are sensitive early indicators of decreased biotin status in experimental biotin deficiency. Am J Clin Nutr. 1997;65:951–958. doi: 10.1093/ajcn/65.4.951. [DOI] [PubMed] [Google Scholar]
- 27.Mock DM, Henrich CL, Carnell N, Mock NI. Indicators of marginal biotin deficiency and repletion in humans: validation of 3-hydroxyisovaleric acid excretion and a leucine challenge. Am J Clin Nutr. 2002;76:1061–1068. doi: 10.1093/ajcn/76.5.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mock DM, Quirk JG, Mock NI. Marginal biotin deficiency during normal pregnancy. Am J Clin Nutr. 2002;75:295–299. doi: 10.1093/ajcn/75.2.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rathman SC, Gregory JF, 3rd, McMahon RJ. Pharmacological biotin supplementation maintains biotin status and function in rats administered dietary carbamazepine. J Nutr. 2003;133:2857–2862. doi: 10.1093/jn/133.9.2857. [DOI] [PubMed] [Google Scholar]
- 30.Abu-Elheiga L, Matzuk MM, Abo-Hashema KA, Wakil SJ. Continuous fatty acid oxidation and reduced fat storage in mice lacking acetyl-CoA carboxylase 2. Science (Washington, DC) 2001;291:2613–2616. doi: 10.1126/science.1056843. [DOI] [PubMed] [Google Scholar]
- 31.Miyazaki T, Ohura T, Kobayashi M, Shigematsu Y, Yamaguchi S, Suzuki Y, Hata I, Aoki Y, Yang X, Minjares C, Haruta I, Uto H, Ito Y, Muller U. Fatal propionic acidemia in mice lacking propionyl-CoA carboxylase and its rescue by postnatal, liver-specific supplementation via a transgene. J Biol Chem. 2001;276:35995–35999. doi: 10.1074/jbc.M105467200. [DOI] [PubMed] [Google Scholar]