

Abstract
Messenger RNA levels of phospholemman (PLM), a member of the FXYD family of small single-span membrane proteins with putative ion-transport regulatory properties, were increased in postinfarction (MI) rat myocytes. We tested the hypothesis that the previously observed reduction in Na+-K+-ATPase activity in MI rat myocytes was due to PLM overexpression. In rat hearts harvested 3 and 7 days post-MI, PLM protein expression was increased by 2- and 4-fold, respectively. To simulate increased PLM expression post-MI, PLM was overexpressed in normal adult rat myocytes by adenovirus-mediated gene transfer. PLM overexpression did not affect the relative level of phosphorylation on serine68 of PLM. Na+-K+-ATPase activity was measured as ouabain-sensitive Na+-K+ pump current (Ip). Compared to control myocytes overexpressing green fluorescent protein alone, Ip measured in myocytes overexpressing PLM was significantly (P<0.0001) lower at similar membrane voltages, pipette Na+ ([Na+]pip) and extracellular K+ concentrations ([K+]o). From −70 to +60 mV, neither [Na+]pip nor [K+]o required to attain half-maximal Ip was significantly different between control and PLM myocytes. This phenotype of decreased Vmax without appreciable changes in Km for Na+ and K+ in PLM overexpressed myocytes was similar to that observed in MI rat myocytes. Inhibition of Ip by PLM overexpression was not due to decreased Na+-K+-ATPase expression since there were no changes in either protein or messenger RNA levels of either α1 or α2 isoforms of Na+-K+-ATPase. In native rat cardiac myocytes, PLM co-immunoprecipitated with α-subunits of Na+-K+-ATPase. Inhibition of Na+-K+-ATPase by PLM overexpression, in addition to previously reported decrease in Na+-K+-ATPase expression, may explain altered Vmax but not Km of Na+-K+-ATPase in postinfarction rat myocytes.
Keywords: primary cardiac myocyte culture, patch clamp, ion transport, Western blots
INTRODUCTION
Phospholemman (PLM) is a 72-amino acid membrane phosphoprotein with a single transmembrane domain (24). It belongs to the FXYD gene family of small ion transport regulators (36). Studies in non-cardiac tissues suggest that PLM can be a channel (15), a channel subunit, or an ion transport regulator (4, 9, 21–23) and is likely involved in regulation of cell volume (7, 22, 23). In heart and skeletal muscle, PLM is a major sarcolemmal substrate for protein kinase A (PKA) and protein kinase C (PKC) (16, 24, 25). Specifically, β-adrenergic agonists phosphorylate serine68 via PKA while PKC phosphorylates both serine68 and serine63 at the C-terminus of PLM (40). Additional studies by Crambert et al. (6) and Feschenko et al. (9) demonstrated association of PLM with α-subunits of Na+-K+-ATPase in bovine cardiac sarcolemma and central nervous system. When co-expressed with α- and β-subunits of Na+-K+-ATPase in Xenopus oocytes, PLM was shown to modulate Na+-K+-ATPase activity, primarily by decreasing apparent affinities for Na+ and K+ without affecting Vmax (6). It is not known whether PLM directly affects Na+-K+-ATPase in cardiac myocytes.
In cardiac sarcolemma isolated from the uninfarcted portion of rat left ventricles 8–16 wk after myocardial infarction (MI), Na+-K+-ATPase activities were depressed primarily due to decreases in Vmax without any changes in the apparent affinities for Mg-ATP, Na+, and K+ (8). In addition, in rat hearts subjected to coronary ligation, application of cDNA microarrays (containing 86 known genes and 989 unknown cDNAs) to analyze transcript levels indicated that PLM was 1 of only 19 genes to increase after MI (29). Although reduced expression of both α1 and α2 but not α3 isoforms of Na+-K+-ATPase may account for the decreased Vmax post-MI (28, 30), increased PLM expression post-MI may also contribute to the suppression of Na+-K+-ATPase activity. The present study was undertaken to test the hypothesis that enhanced PLM expression partly explains the depressed Na+-K+-ATPase activities observed in post-MI rat hearts.
METHODS
Induction of myocardial infarction
To induce MI in male Sprague-Dawley rats (~ 250g), the left main coronary artery of each anesthetized (2% isoflurane – 98% O2), intubated, and ventilated rat was ligated 3–5 mm distal to its origin from the ascending aorta (5, 41, 45). Sham operation, except that the coronary artery was not ligated, was identical to MI. In our hands, Sham operated rats had close to 100% survival while the mortality for coronary ligation procedure was ~30% within 24h of the operation. All surviving rats (Sham, n=3; MI, n=6) received rat chow and water ad libitum and were maintained on a 12:12h light-dark cycle. Survivors typically had 36 ± 3% of myocardium infarcted as determined histologically (5). In addition, despite no overt signs of heart failure in MI rats, we observed at 1 and 3 wk postinfarction 20% lower LV systolic pressure in MI hearts when perfused in vitro (5). At 3 and 7 days post-MI, MI rats were overdosed with pentobarbital sodium (34 mg/kg body wt ip), and the left ventricles and septae were excised for immunoblotting studies. Sham-operated rat hearts were harvested at 7 days post-op for protein measurements. The protocol for induction of MI and subsequent heart excision was approved by Institutional Animal Care and Usage Committee.
Myocyte isolation and culture
Cardiac myocytes were isolated from the septum and left ventricular free wall of normal male Sprague-Dawley rats (~280g), seeded on laminin-coated coverslips and subjected to continuous pacing culture (1 Hz, [Ca2+]o = 1.8 mM) as previously described (20, 32, 33, 37, 42–44). Under continuous pacing culture conditions, we have previously demonstrated that myocyte contractility did not decline for at least 72h (33). The protocol for heart excision for myocyte isolation was approved by Institutional Animal Care and Usage Committee.
Adenoviral infection of cardiac myocytes
Recombinant, replication-deficient adenovirus (Adv) expressing either green fluorescent protein (GFP) alone or GFP and dog PLM were constructed as described previously (33). Two hours after isolation, myocytes were infected with Adv-GFP or Adv-GFP-PLM, and studied after 72h of continuous pacing culture as previously described (20, 32, 33). We have previously demonstrated that over 95% of myocytes were successfully infected (44) and that adenoviral infection of myocytes had no effects on myocyte contractility when examined after 72 h of continuous pacing culture (33). We have also shown that the effects of PLM overexpression on contractility and [Ca2+]i transients were manifest 72h after Adv-PLM infection (33). In addition, PLM overexpression did not affect action potential amplitude and morphology, sarcoplasmic reticulum (SR) Ca2+ uptake, and protein levels of Na+/Ca2+ exchanger, α-subunits of Na+-K+-ATPase, calsequestrin, and sarco(endo)plasmic reticulum Ca2+-ATPase in adult rat myocytes (32, 33, 42). For the sake of brevity, myocytes infected with Adv-GFP and Adv-GFP-PLM are referred to as GFP and PLM myocytes, respectively.
Measurement of Na+-K+-ATPase pump current (Ip)
Whole cell patch-clamp recordings were performed at 30ºC as described previously (20, 32, 37, 42, 45). Standard pipette filling solution consisted of (in mM): 70 Na-aspartate, 20 K-aspartate, 8 CsOH, 7 MgSO4, 11 EGTA, 10 TEA.Cl, 1 CaCl2, 5 HEPES, 5 Na2ATP, and 0.2 GTP, pH 7.2. The Na+ concentration in the pipette ([Na+]pip) was varied between 5 and 80 mM by equimolar substitution of Na+ with Cs+. At 5 mM [Na+]pip, K2ATP was used instead of Na2ATP. External solution contained (in mM): 137.7 NaCl, 5.4 KCl, 2.3 NaOH, 1 MgCl2, 2 BaCl2, 1 CdCl2, 5 HEPES and 10 glucose, pH 7.4. Extracellular K+ concentration ([K+]o) was varied between 1 and 18 mM.
Holding potential was switched from −70 to −40 mV (300ms) before application of a negative voltage ramp (from +60 to −120 mV, 20 mV/sec; Fig. 2B). Ouabain (1 mM) was added and the voltage-ramp stimulus protocol was repeated. Currents were filtered at 1 KHz, and data was acquired at 2 KHz. Ip was defined as the difference in currents before and after ouabain addition. To facilitate comparison of Ip among cells, Ip of each myocyte was divided by its capacitance Cm to account for variations in cell sizes (42, 45).
Figure 2. Na+-K+-ATPase pump current (Ip) in cultured adult rat ventricular myocytes.

Isolated rat cardiac myocytes were infected with adenovirus expressing GFP or PLM, and placed in continuous pacing culture (Methods). Membrane currents were measured after 72h of culture. (A): Top panel: currents measured in a GFP myocyte (held at –40 mV) before and after puffer superfusion of 0 [K+]o medium. Bottom panel: currents measured in another GFP myocyte (held at –40 mV) before and after application of 1 mM ouabain. (B): Voltage-ramp protocol applied to myocytes. (C): Currents recorded in a control GFP myocyte during the voltage ramp from +60 to −120 mV, in the absence and presence of 1 mM ouabain. (D): Current-voltage relationships of Ip, measured as the ouabain-sensitive current (5.4 mM [K+]o, 80 mM [Na+]pip, 30°C) in GFP (open circles; n=8) and PLM (filled circles; n=5) myocytes.
To determine the concentration of [Na+]pip (K0.5, Na +) required to attain half-maximal Ip at each voltage, [K+]o was set to be saturating at 18 mM while [Na+]pip was varied between 5 to 80 mM. Similarly, to determine the concentration of [K+]o (K0.5, K+) required to attain half-maximal Ip, [Na+]pip was set at the saturating level of 80 mM while [K+]o was varied between 1 and 18 mM. Mean Ip values (at each [Na+]pip or [K+]o) were used to fit the Hill equation, using the Levenberg-Marquardt algorithm (SlideWrite Plus; Advanced Graphics Software, Encinitas, CA) which minimizes the sum of square deviation (chi-squared value). Values for nh (the Hill coefficient), Imax (maximal Ip), and K0.5, Na + or K0.5, K+ were determined at each voltage from −70 to +60 mV.
PLM, calsequestrin, and Na+-K+-ATPase immunoblotting
Left ventricles and septae were excised at 3- and 7-days post-MI and homogenized separately in a buffer described previously (44). Cultured myocytes were harvested for immunoblotting on day 3 as described previously (20, 32, 33, 37, 42–44). Primary antibodies used were: for unphosphorylated PLM, C2 Ab (1:12,000) (27, 32, 33); for PLM phosphorylated at serine68, C68P Ab (1:10,000) (27, 31); and for calsequestrin, anti-calsequestrin antibody (1:2,500; Swant). Isoform-specific antibodies were used for α1 subunit (0.5 μg/ml; Cat#96-520, Upstate USA Inc., Charlottesville, VA) and α2 subunit of Na+-K+-ATPase (1:1000; polycloncal antibody gift from Dr. Robert Levenson, Pennsylvania State University).
Co-immunoaffinity purification experiments
C2 Ab was covalently linked to agarose support beads according to manufacturer’s instructions (Affi-Gel Hz immunoaffinity kit; Cat# 153-6060, Biorad)(20). Myocyte lysates were repeatedly (50 times) loaded onto the immunoaffinity column which, following washing, was eluted with 0.2M glycine-HCl, pH 2.5, as previously described (20). The 3 eluant fractions (500 μl each) with the highest protein concentrations were subjected to PAGE (10% polyacrylamide gel, 32 to 40 μl/lane), followed by protein transfer to ImmunoBlot PVDF membranes for detection of PLM and (α1 + α2)-subunits of Na+-K+-ATPase (α5 antibody, 1:250; Developmental Studies Hybridoma Bank, University of Iowa) by Western blotting.
Quantitative Real-time RT-PCR studies
RNA (18S and 28S bands) from GFP and PLM myocytes were purified (RNeasy Mini Kit; Qiagen; Valencia, CA), visualized by chromatography (Agilent 2100 Bioanalyzer; Agilent Technologies, USA) and concentrations adjusted by UV spectrophotometry. First strand cDNA was synthesized from 1.5 μg of total RNA using Oligo(dT)12–18 primers and random hexamer primers ( SuperScript III Reverse Transcription kit; InVitrogen; Carlsbad, CA). Forty-two ng of cDNA per sample was utilized as a template for real-time PCR, using a SYBR Green Master Mix (Qiagen) and gene-specific primers (GenScript Corporation; Scotch Plains, NJ). PCR amplification was performed (Sequence Detection System 7000; Applied Biosystems; Foster City, CA) with the following cycling parameters: 95°C for 10 min, followed by 45 cycles of 95°C for 15 seconds then 60°C for 1 minute. Taqman 18S rRNA primers (Eurogentec; San Diego, CA) were also used with 42 ng of cDNA and Taqman PCR Master Mix (Qiagen) under identical conditions for amplification of 18S, which was used as an internal standard. To exclude the possibility of genomic DNA contamination, control PCR reactions with no cDNA template were also performed for each gene-specific primer and probe set. Amplification data for α1 and α2 subunits of Na+-K+-ATPase were normalized for 18S within each individual PCR reaction. Duplicates of each PCR reaction were performed and the resultant data were averaged. The following primers and probes were utilized for real-time PCR amplification: for Na+-K+-ATPase α1 subunit (Atp1A1, rattus norvegicus; accession # NM_012504) forward 5′-CACGGCCTTCTTTGTCAGTA-3′, reverse 5′-TTGTTCTTCATTCCCTGCTG-3′ and probe 5′ Fam-TGCAGATGACCAAGTCAGCCCA-3′ Tamra; for Na+-K+-ATPase α2 subunit (Atp1A2, rattus norvegicus; accession # NM_012505) forward 5′-TTATCGTCACTGGCTGCTTC-3′, reverse 5′-TTCTCTCCCTCTCGGATGAC-3′ and probe 5′ Fam-ACATGGTGCCTCAGCAAGCTCTG-3′ Tamra.
Statistical Analysis
All results are expressed as means ± SE. In experiments in which Ip was measured as a function of experimental group (GFP vs. PLM), voltage and [Na+]pip or [K+]o, three-way ANOVA was performed to determine significance of difference. Student’s t-test was used to compare protein abundance between Sham and MI hearts, and between GFP and PLM myocytes. A commercial statistical analysis package (JMP 4.04) was used. In all analyses, P < 0.05 was taken to be statistically significant.
RESULTS
Effects of myocardial infarction on PLM expression in adult rat myocytes
Fig. 1 shows that at 3 and 7 days post-MI, PLM increased in MI myocytes when compared to Sham myocytes. There were no differences in PLM protein levels between septum and left ventricle and the results were combined. At 3 days post MI, PLM increased 2.4-fold in MI myocytes (241 ± 53 vs. 110 ± 35 arbitrary units)(p=0.066). At 7 days post-MI, the 4-fold increase in PLM in MI myocytes (407 ± 83 vs. 110 ± 35 arbitrary units) is statistically significant (p=0.008). Calsequestrin was used as an internal control for protein loading since its expression has been shown to be unchanged during ontogenic development, aging, cardiac hypertrophy and failing human myocardium (11). There were no differences in calsequestrin levels between sham (292 ± 40 arbitrary units) and day 3 MI hearts (326 ± 24 arbitrary units)(p=0.48); and between sham and day 7 MI hearts (306 ± 26 arbitrary units)(p=0.78).
Figure 1. Immunoblots of phospholemman (PLM) and calsequestrin in Sham and postinfarction (MI) myocytes.

Proteins in heart homogenates (50 μg/lane) were separated by gel electrophoresis and transferred to ImmunBlot PVDF membranes, and PLM (C2 Ab) and calsequestrin were identified by immunoblotting (METHODS). Numbers on left refer to apparent molecular mass in kDa.
Effects of PLM overexpression on Ip in adult rat myocytes
Fig. 2A shows that in experiments in which GFP myocytes were voltage-clamped at −40 mV, either reducing [K+]o to zero in one myocyte (top panel) or adding 1 mM ouabain to another myocyte (bottom panel) resulted in decreases in steady-state membrane current, indicating that the [K+]o- or ouabain-sensitive currents represented Ip under our experimental conditions. The apparent differences in Ip measured under 0 [K+]o conditions and after ouabain addition were due to differences in cell sizes. Figure 2B shows the voltage-ramp protocol applied to myocytes at 30ºC. Fig. 2C shows ramp currents measured in a control GFP myocyte (with [Na+]pip and [K+]o at 80 and 5.4 mM, respectively) in the absence and presence of 1 mM ouabain. The ouabain-sensitive currents, i.e., Ip, are displayed in Fig. 2D. It is clear that Ip was voltage-dependent in that its amplitude was small at negative membrane potentials and progressively increased with depolarization, reaching a maximum at ~ 0 mV. In addition, the absolute magnitudes of Ip were generally lower in PLM than GFP myocytes (Fig. 2D). Two-way ANOVA confirmed a significant group (GFP vs. PLM) effect (P<0.0001). In both GFP and PLM myocytes, depolarization to more positive potentials increased the absolute magnitude of Ip (voltage effect, P<0.0001).
The relationship between Ip and [Na+]pip (with [K+]o saturating at 18 mM) measured at −10 mV is shown in Fig. 3A. The fitted values for K0.5, Na+ are 9.95 ± 0.52 and 9.96 ± 0.73 mM; for Imax are 1.08 ± 0.04 and 0.71 ± 0.03 pA/pF; and for nh are 3.45 ± 0.74 and 2.97 ± 0.70; for GFP and PLM myocytes, respectively. The coefficient of determination (r2) describes the variability in y (Ip) due to variation in x ([Na+]pip). The closer r2 is to 1, the better is the curve fit. For GFP myocytes, r2 is 0.9913 and it is 0.9842 for PLM myocytes at −10 mV.
Figure 3. Dependence of Ip on [Na+]pip and voltage dependence of K0.5, Na+ and Imax in GFP and PLM myocytes.

Ip at 18 mM [K+]o and 30°C was measured in GFP and PLM myocytes (Methods). [Na+]pip was varied between 5 to 80 mM. (A): Ip (mean ± SE; measured at −10 mV) as a function of [Na+]pip in control GFP (open circles) and PLM (filled circles) myocytes are shown. Ip was measured in 4 – 8 myocytes for each [Na+]pip. At 5 mM [Na+]pip, data obtained from GFP and PLM myocytes overlap such that only a filled circle is shown. Mean Ip values are fitted to the Hill equation, and the fitted curves are shown for GFP (broken line) and PLM (solid line) myocytes, from which nh, Imax, and K0.5, Na + are obtained. Fitted values of K0.5, Na+ (B) and Imax (C) derived from data relating mean Ip and [Na+]pip from −70 to +60 mV are shown for control GFP (open circles) and PLM (filled circles) myocytes.
When Ip was analyzed as a function of group (GFP vs. PLM), voltage, and [Na+]pip (with [K+]o fixed at 18 mM), 3-way ANOVA (n=826) indicated significant group (P<0.0001), voltage (P<0.0001) and [Na+]pip (P<0.0001) main effects. In addition, the group x voltage (P=0.0005) and group x [Na+]pip (P<0.0001) interaction effects were highly significant, indicating the inherent differences in Ip between GFP and PLM myocytes were amplified at more positive voltages and higher [Na+]pip. There were no other significant interaction effects.
The voltage dependence of K0.5, Na+ and Imax of GFP and PLM myocytes are shown in Figs. 3B and 3C, respectively. There were no apparent differences in K0.5, Na+ between GFP and PLM myocytes although Imax was clearly suppressed with PLM overexpression. Across the physiological voltage range from −70 to + 60 mV, fitted values of nh varied between 2.74 to 3.77 (mean 3.27 ± 0.09) for GFP myocytes, and between 2.66 to 4.13 (mean 3.22 ± 0.11) for PLM myocytes. The coefficient of determination r2 was ≥ 0.9632 for GFP myocytes and ≥ 0.9596 for PLM myocytes across the voltage range examined.
The relationship between Ip and [K+]o (with [Na+]pip saturating at 80 mM) measured at −10 mV is shown in Fig. 4A. Fitted values for K0.5, K+ are 2.69 ± 0.32 and 3.18 ± 0.36 mM; for Imax are 1.17 ± 0.08 and 0.73 ± 0.06 pA/pF; and for nh are 1.96 ± 0.23 and 1.95 ± 0.77; for GFP and PLM myocytes, respectively. The coefficient of determination r2 is 0.9774 and 0.9686, for GFP and PLM myocytes, respectively.
Figure 4. Dependence of Ip on [K+]o and voltage dependence of K0.5, K+ and Imax in GFP and PLM myocytes.

Ip at 80 mM [Na+]pip and 30ºC was measured in GFP and PLM myocytes. [K+]o was varied between 1 to 18 mM. (A): Ip (measured at −10 mV) as a function of [K+]o in control GFP (open circles) and PLM (filled circles) myocytes are shown. Each point represents mean ± SE of 4 – 10 myocytes. Mean Ip values are fitted to the Hill equation, and the fitted curves are shown for GFP (broken line) and PLM (solid line) myocytes, from which nh, Imax, and K0.5, K+ are obtained. Fitted values of K0.5, K+ (B) and Imax (C) derived from data relating mean Ip and [K+]o from −70 to +60 mV are shown for GFP (open circles) and PLM (filled circles) myocytes.
When Ip was analyzed as a function of group (GFP vs. PLM), voltage, and [K+]o (with [Na+]pip fixed at 80 mM), 3-way ANOVA (n=987) indicated significant group (P<0.0001), voltage (P<0.0001) and [K+]o (P<0.0001) main effects. In addition, the group x [K+]o (P<0.0001) interaction effects were highly significant, indicating the inherent differences in Ip between GFP and PLM myocytes were amplified at higher [K+]o. There were no other significant interaction effects.
The voltage dependence of K0.5, K+ and Imax of GFP and PLM myocytes are shown in Figs. 4B and 4C, respectively. Similar to K0.5, Na + (Fig. 3B), we did not detect a large effect of PLM overexpression on K0.5, K+. Again, PLM overexpression clearly depressed Imax. From −70 to + 60 mV, fitted values of nh varied between 1.46 to 2.50 (mean 1.80 ± 0.07) for GFP myocytes, and between 1.34 to 2.63 (mean 1.87 ± 0.12) for PLM myocytes. Across the voltage range examined, r2 was ≥0.9591 and ≥0.9466 for GFP and PLM myocytes, respectively.
Effects of PLM overexpression on Na+-K+-ATPase protein and messenger RNA levels in adult rat myocytes
We investigated whether PLM overexpression affected transcription of α-subunits of Na+-K+-ATPase by performing real-time RT-PCR studies. Real-time RT-PCR analysis demonstrated amplification of each of the primer sets specific to each of the Na+-K+-ATPase α1 and α2 isoforms, no amplification of the no template control samples, and the heat dissociation curve confirmed amplification of only a single gene transcript for each primer set (data not shown). These results indicate the presence of gene transcripts of the Na+-K+-ATPase α1 and α2 isoforms in GFP and PLM myocytes. For α1-isoform of Na+-K+-ATPase, the range of relative messenger RNA levels for GFP and PLM myocytes are 0.78–1.27 and 0.89–0.97, respectively. For α2-isoform of Na+-K+-ATPase, the range of relative messenger RNA levels for GFP and PLM myocytes are 0.87–1.15 and 0.81–1.01, respectively. Over-expression of PLM in adult rat myocytes did not result in detectable changes in messenger RNA levels of either α1 or α2 isoforms of Na+-K+-ATPase (Fig. 5A).
Figure 5. Effects of PLM overexpression on mRNA and protein levels of α-subunits of Na+-K+-ATPase.

(A): cDNA was synthesized from total RNA isolated from GFP and PLM myocytes, and used in real-time RT-PCR studies with appropriate primers for α1- and α2-isoforms of Na+-K+-ATPase (Methods). Messenger RNA levels (normalized to 18S) relative to control GFP myocytes for α1- and α2-subunits of Na+-K+-ATPase are shown. Messenger RNA levels were determined twice with similar results. (B): Antibodies specific for α1- or α2- subunits of Na+-K+-ATPase were used in immunoblotting of GFP and PLM myocytes lysates. Protein band signal intensities are given in Results.
To confirm our RT-PCR results, we next measured α1- and α2-subunits of Na+-K+-ATPase, using α1- and α2-specific antibodies. We did not detect significant differences in either α1- (GFP: 7.8 ± 1.5, PLM: 11.8 ± 1.9 arbitrary units; p<0.17) or α2-subunits (GFP: 7.5 ± 1.1, PLM: 7.1 ± 1.0 arbitrary units; p<0.79) of Na+-K+-ATPase (n=6)(Fig. 5B). Equal protein loading in the lanes was verified by similar calsequestrin levels (data not shown).
Association of PLM with Na+-K+-ATPase in adult rat myocytes
Inhibition of Ip by PLM overexpression suggests that PLM may directly interact with Na+-K+-ATPase. We next sought to examine possible association between PLM and Na+-K+-ATPase. Figure 6 demonstrates that C2 Ab, but not pre-immune serum (obtained from the same rabbit from which C2 Ab was raised), when covalently linked to agarose support beads, was able to immunoaffinity purify both PLM and α-subunits of Na+-K+-ATPase present in native adult rat myocyte lysates.
Figure 6. Association of PLM with α-subunits of Na+-K+-ATPase in native adult rat myocytes.
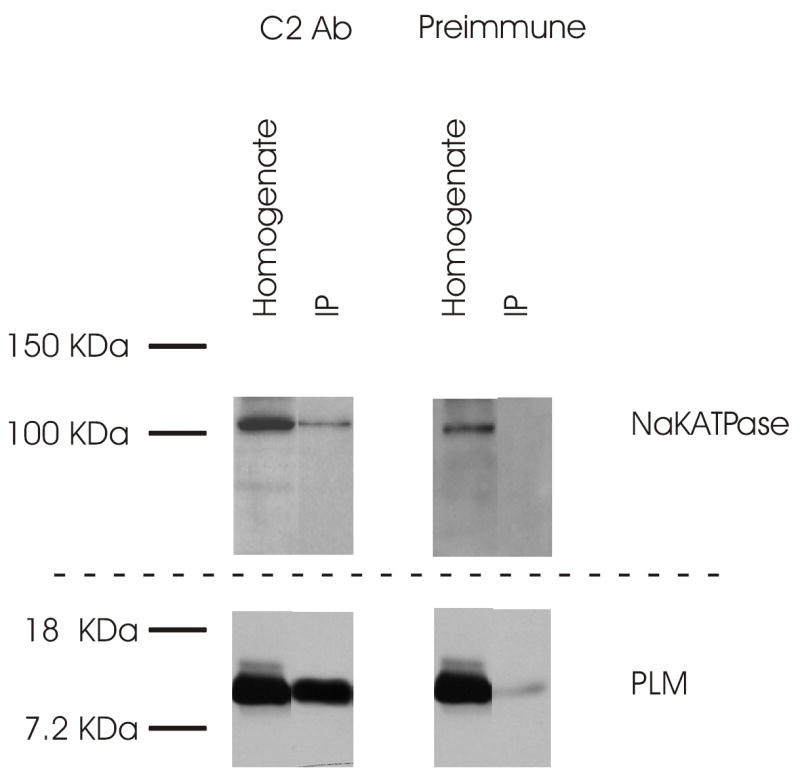
Myocyte homogenates prepared from freshly isolated adult rat myocytes were loaded repeatedly onto agarose support columns (Affi-Gel Hz immunoaffinity kit) whose beads were covalently linked with either C2 Ab or the preimmune serum (Preimmune) obtained from the same rabbit from which C2 Ab was raised (Methods). After elution, the 3 eluant fractions (only 1 is shown) with the highest protein concentrations were applied to 10% SDS-PAGE. PLM and α-subunits of Na+-K+-ATPase were detected with C2 Ab and α5 antibody, respectively. IP is immunoprecipitate. This experiment was repeated 2 times with similar results.
PLM overexpression did not affect relative level of PLM phosphorylation at serine68
Using C68P Ab, an antibody which is specific for PLM phosphorylated at serine68 (27, 31), we demonstrated that in rat cardiac myocytes, phosphorylated PLM detected by C68P Ab increased with phorbol 12-myristate-13-acetate or isoproterenol treatment, while the unphosphorylated PLM form detected by C2Ab showed the corresponding decrease (Fig. 7A). Using a 3rd PLM antibody which was engineered to recognize the N-terminus of PLM (and therefore recognizes both phosphorylated and unphosphorylated forms of PLM), we showed that protein loading was similar in all 3 lanes (Fig. 7A). Assuming either PKA or PKC stimulation resulted in 100% phosphorylation at serine68 under our experimental conditions, we estimated that 41 ± 9 % of serine68 in PLM was phosphorylated in native rat cardiac myocytes under basal conditions (n=4). To further demonstrate that C2 Ab and C68P Ab can detect changes in unphosphorylated and phosphorylated forms of PLM, respectively, we treated lysates prepared from PMA-stimulated myocytes with bacterial alkaline phosphatase. As shown in Fig. 7B, alkaline phosphatase treatment of PMA-treated myocyte lysates resulted in decrease in signal intensity detected by C68P Ab but increase in signal intensity detected by C2 Ab.
Figure 7. Serine68 phosphorylation in PLM.

(A): Freshly isolated rat myocytes were treated with phorbol 12-myristate-13-acetate (PMA, 0.5 μM), isoproterenol (Iso, 0.5 μM), or dimethylsulfoxide (Control, 0.55 μg/ml) at 37°C for 10 min. Myocyte homogenates were prepared and immunoblotting performed with C2 Ab, C68P Ab, and a polyclonal antibody that recognizes the N-terminus of PLM (to control for PLM loading). This experiment was repeated 3 times with similar results. (B): Myocytes treated with either dimethylsulfoxide (Control) or PMA were scraped into 500 μl of lysis buffer without phosphatase inhibitor cocktail, Na+ vanadate and NaF. A portion of myocyte lysate (100 μl) was treated with bacterial alkaline phosphatase (ALKP, 21.6 μg) at 37°C for 30 min. Phosphatase inhibitor cocktail (4 μl) was added to the remaining 400 μl of myocyte lysate. Phosphorylated and unphosphorylated PLM in the myocyte lysate were analyzed with C2 Ab and C68P Ab, respectively. (C): Immunoblotting was performed on cell lysates prepared from GFP and PLM myocytes (n=6 each) with either C2 Ab or C68P Ab. Protein signal band intensities are given in Results.
In PLM overexpressed myocytes, the unphosphorylated PLM (as detected by C2 Ab) was 108.6 ± 3.0 arbitrary units as compared to 6.5 ± 2.1 arbitrary units in GFP myocytes (n=6; P<0.0001)(Fig. 7C). The increase in unphosphorylated PLM in PLM-overexpressed myocytes was ~16.7-fold as compared with GFP myocytes. By comparison, PLM phosphorylated at serine68 (as detected by C68P Ab) was 3.7 ± 0.5 arbitrary units in GFP and 41.8 ± 3.9 arbitrary units in PLM myocytes (n=6; P<0.0001)(Fig. 7C). The mean increase in phosphorylated PLM in PLM myocytes was ~11.3 fold as compared with GFP myocytes. Given the similar fold increase in both unphosphorylated and phosphorylated PLM in PLM overexpressed myocytes, PLM overexpression did not grossly affect the relative degree of serine68 phosphorylation in rat myocytes.
DISCUSSION
PLM (FXYD1) belongs to the FXYD gene family of small ion transport regulators (36). Interestingly, the γ-subunit of the Na+-K+-ATPase (FXYD2) is also a member of the FXYD family and is only present in membranes from kidney tubules (39). In the actively transporting nephron, the γ-subunit regulates Na+-K+-ATPase activity by stabilizing the E1 conformation of the enzyme (26, 38, 39). A 15-kDa homologue of phospholemman isolated from shark rectal glands, PLMS, also associated with the α-subunits of Na+-K+-ATPase (18). Phosphorylation of PLMS by PKA or PKC resulted in partial dissociation of PLMS from the α-subunit of Na+-K+-ATPase, with subsequent activation of the enzyme (18). In addition, recent studies indicate that CHIF (channel inducing factor; FXYD4) (2) and FXYD7 (3) were also regulators of Na+-K+-ATPase. Thus within the FXYD family, at least four members (γ-subunit of Na+-K+-ATPase, CHIF, FXYD7 and PLMS) other than PLM (6) associated with the α-subunit of Na+-K+-ATPase or regulated its activity.
The first major finding of the present study is that we demonstrated that PLM protein levels were significantly elevated in postinfarction rat hearts (Fig. 1). Our findings are in agreement with microarray data indicating that messenger RNA levels of PLM were increased postinfarction (29).
In our experiments, Ip reached plateau amplitude at ~ 0 mV (Fig. 2D): a characteristic of the pump current that was first described by Gadsby et al. (12) and confirmed by others (34). The fitted amplitude of Imax (1.39 ± 0.02 pA/pF) in control rat myocytes measured at +60mV and 80 mM [Na+]pip (Fig. 4C) was similar to that previously reported by other investigators (1.8 – 2.0 pA/pF at +50 mV, 50 mM [Na+]pip and 5.4 mM [K+]o; Ref. 34). In addition, values for K0.5, Na + (8.9–12.6 mM; Fig. 3B) and K0.5, K+ (2.5 – 3.1 mM; Fig. 4B) were similar to those reported for K0.5, Na+ (7.8 – 40 mM) and K0.5, K+ (0.9 – 3.5 mM) for rat ventricular myocytes (for review, see Ref. 13). These observations indicate that the ouabain-sensitive current measured under our experimental conditions was indeed the Na+-K+-ATPase pump current.
The second major finding of the present study is that overexpression of PLM inhibited Ip by as much as 27 – 40% (Figs. 2–4). Inhibition of Ip by PLM was not due to sequestration of Na+-K+-ATPase by the overexpressed PLM molecules in subcellular organelles. This is because we have previously demonstrated by indirect immunofluorescence that the overexpressed PLM was correctly targeted to sarcolemma, t-tubules and intercalated discs, with little-to-no PLM signal detected in the cytosol (32, 42). Inhibition of Ip by overexpressed PLM was also not due to decreased expression of Na+-K+-ATPase, as demonstrated by no decreases in messenger RNA and protein levels of α1 and α2-subunits of Na+-K+-ATPase (Fig. 5). In contrast to the results derived from co-overexpression of PLM and α1-β1 Na+-K+-ATPase isoenzymes in Xenopus oocytes (6), in its native environment of cardiac sarcolemma, PLM overexpression decreased maximum Ip with no large effects on K0.5, Na+ or K0.5, K+ (Figs. 3 & 4). Our data agree well with a recent report which demonstrated that the Vmax of sarcolemmal Na+-K+-ATPase was increased 3-fold after acute cardiac ischemia, presumably due to less inhibition of Na+-K+-ATPase by PLM whose phosphorylation status was enhanced by >300% (10). Our present results indicate that in addition to regulation of Na+/Ca2+ exchanger activity (1, 20, 32, 42), another potential function of PLM in cardiac tissues is to modulate Na+-K+-ATPase activity.
In adult rat myocardium, both α1 and α2 subunits of Na+-K+-ATPase are expressed in the sarcolemma and t-tubules of cardiac myocytes, with the α1 subunit perhaps more abundant in the t-tubules (19, 35). Expression of α3 subunit of Na+-K+-ATPase in adult rat hearts has been reported by some (28, 30) but not by other investigators (19, 35). In addition, about 75% of the Na+ pump αβ heterodimers in the adult rat heart consisted of the fairly ubiquitous but relatively ouabain-resistant α1 isoforms (19, 35). In our experiments, we used ouabain at 1 mM, which is much higher than the Ki for the α1 isoform and thus should result in inhibition of both the α1 and α2 isoforms of Na+-K+-ATPase. Our experimental conditions, therefore, could not differentiate whether PLM modulated α1 and/or α2 isoforms of Na+-K+-ATPase to account for the observed inhibition of Ip with PLM overexpression.
The third finding is that PLM co-associates with the α-subunits of Na+-K+-ATPase (Fig. 6), in agreement with the observations by other investigators (6, 10, 31). The α5 antibody that we used to detect the α-subunits of Na+-K+-ATPase does not distinguish between α1 and α2 isoforms of the enzyme. Association of the α-subunits of Na+-K+-ATPase with PLM constitutes another line of evidence that PLM modulates Na+-K+-ATPase activity. Recent studies using immunoprecipitation and indirect immunofluorescence localization techniques indicated that PLM associated with α1 but not the α2 subunit of Na+-K+-ATPase in guinea pig myocytes (10, 31). Taken together, it is likely that PLM regulates the activity of α1 subunit of cardiac Na+-K+-ATPase.
A fourth major finding is that by assuming that either PKA or PKC stimulation resulted in 100% phosphorylation of serine68, and that C68P Ab and C2 Ab detected only the phosphorylated and unphosphorylated forms of PLM, respectively (27, 31), we estimated that ~41% of serine68 in PLM was phosphorylated in rat myocytes under the basal state. This estimate agrees reasonably well with the ~46% phosphorylation of serine68 of PLM in resting rat myocytes, based on comparing the effects of wild-type PLM and its serine68 mutants on Na+/Ca2+ exchange current (32). Using C68P Ab, Silverman et al. (31) showed that forskolin treatment of guinea pig myocytes resulted in ~4-fold increase in serine68 phosphorylation. Assuming forskolin induced 100% phosphorylation of serine68, basal serine68 phosphorylation can be estimated to be ~25% in guinea pig myocytes.
Based on analogy of phospholamban inhibition of SERCA2 and experimental observations on the effects of PLMS on shark Na+-K+-ATPase (18), the current working model is that the Na+ pump is inhibited by unphosphorylated PLM. On phosphorylation of PLM, inhibition of Na+-K+-ATPase is relieved. This hypothesis has been given indirect support by the observation that the Vmax of sarcolemmal Na+-K+-ATPase was increased 3-fold after acute cardiac ischemia, in association with increased PLM phosphorylation by >300% (10). It is at present not clear whether dissociation of the phosphorylated PLM from Na+-K+-ATPase is required to relieve its inhibition on the Na + pump (10, 18, 31). An important finding of the present study is that PLM overexpression did not grossly distort the relative level of serine68 phosphorylation. This indicates that suppression of Ip in PLM overexpressed myocytes was due to increased unphosphorylated PLM forms.
The characteristic phenotype observed in adult rat myocytes 3–9 weeks post-MI is that when compared with sham myocytes, at 0.6 mM extracellular Ca2+ concentration ([Ca2+]o), both contraction and [Ca2+]i transient amplitudes were higher. At 5 mM [Ca2+]o, both contraction and [Ca2+]i transient amplitudes were lower in MI than sham myocytes (5, 41, 43). This reduced dynamic range of contraction and [Ca2+]i transient amplitudes in response to increasing [Ca2+]o was also observed in rat myocytes in which Na+/Ca2+ exchanger was downregulated (37). In post-MI rat myocytes, both Na+/Ca2+ exchanger expression (30) and its activity (45) were depressed. In addition, contractile and [Ca2+]i transient abnormalities in post-MI rat myocytes were rescued by overexpressing the Na+/Ca2+ exchanger (43). Taken together, these observations suggest that the contractile and [Ca2+]i homeostatic abnormalities in post-MI rat myocytes were in large part due to decreased Na+/Ca2+ exchange activity. In this context, it is interesting to note that PLM, in addition to its effects on Na+-K+-ATPase, also inhibits the cardiac Na+/Ca2+ exchanger (1, 32, 42). In addition, overexpression of PLM in normal rat myocytes resulted in reduced dynamic range of contraction and [Ca2+]i transient amplitudes in response to increasing [Ca2+]o (33), similar to the contractile phenotype observed in post-MI rat myocytes (5, 41, 43). Thus in post-MI rat myocytes, contractile and [Ca2+]i transient abnormalities were largely due to decreased expression of Na+/Ca2+ exchanger (30) and PLM overexpression with its attendant inhibitory effects of Na+/Ca2+ exchange activity (1, 32, 42). On the other hand, inhibition of Na+-K+-ATPase by PLM overexpression (Figs. 2 – 4) or ouabain (14) would be expected to elevate [Na+]i, resulting in changes in thermodynamic driving force for Na+/Ca2+ exchange. This would reduce Ca2+ extrusion by forward Na+/Ca2+ exchange but increase Ca2+ influx by reverse Na+/Ca2+ exchange. The net result would be increased contraction and [Ca2+]i transient amplitudes in PLM overexpressed or ouabain-treated myocytes under both low and high [Ca2+]o conditions: a prediction not consistent with our observation in PLM overexpressed or post-MI myocytes. Viewed in this simplistic context, decreased Na+-K+-ATPase activity is unlikely to be a major factor to account for altered contractility in post-MI myocytes. Na+-K+-ATPase, however, is intimately involved with cell volume regulation (17) and affects automaticity, resting and action potentials (13). Inhibition of Na+-K+-ATPase by PLM, therefore, may have profound consequences during cardiac ischemia when disordered cell volume regulation and electrical instability are present.
There are limitations to the present study. The first is that we did not address the stoichiometry of interaction between PLM and Na+-K+-ATPase since the amounts of α1 subunits of Na+-K+-ATPase, PLM and NCX1 present in a cardiac cell are at present unknown. In addition, there may be as yet unknown partners that PLM interacts with. Furthermore, PLM may form dimers in the sarcolemma (J. Y. Cheung, unpublished observations). Therefore, precise quantification of the stoichiometry of interaction between PLM and Na+-K+-ATPase in the cardiac cell is exceedingly complex and beyond the scope of the present study. Nevertheless, qualitative mechanisms by which the overexpressed unphosphorylated PLM decreases the Vmax of Na+-K+-ATPase without affecting the α-subunit content can be speculated. The first is that there exists a pool of Na+ pump that is not associated with PLM in normal cardiac myocytes and that the overexpressed unphosphorylated PLM interacts with it. Another possibility is that the overexpressed unphosphorylated PLM successfully displaces the phosphorylated PLM from the Na+ pump. The second limitation is that the level of PLM overexpression by adenovirus infection varied between 1.4-fold (33), 2.6- to 3.5-fold (42), and 11- to 17-fold (present study), depending on the batch of adenovirus constructs and the multiplicity of infection (2 to 5) used. The range of PLM increase with adenovirus-mediated gene delivery observed in our previous (33, 42) and current study, however, is within the range of PLM increase observed in postinfarction myocytes.
In summary, we have demonstrated elevated PLM protein levels in postinfarction rat myocytes. PLM overexpression in normal rat myocytes resulted in depressed Na+-K+-ATPase pump current that was primarily due to decreases in Vmax with no significant changes in apparent affinities for Na+ and K+: changes similar to those observed in postinfarction rat myocytes. PLM overexpression did not affect messenger RNA and protein levels of α1 or α2-subunits of Na+-K+-ATPase, nor did it affect the relative phosphorylation level of serine68 of PLM. Immunoaffinity purification experiments demonstrated association of α-subunits of Na+-K+-ATPase with PLM. We conclude that PLM regulates Na+-K+-ATPase activity in the heart. We speculate that in addition to reduced expression of Na+-K+-ATPase (28, 30), overexpression of PLM may also account for depressed Na+-K+-ATPase activities observed in postinfarction rat myocytes.
A preliminary report of the present work was presented at the 48th Biophysical Society Meeting in Baltimore MD, February 14–18, 2004; and published in abstract form (Zhang XQ et al., Biophys. J. 86:192a, 2004).
Acknowledgments
We thank Kristin Gaul for assistance in the preparation of the manuscript. We acknowledge Rob Brucklacher of the Functional Genomics Core Facility, Section of Research Resources, Pennsylvania State University College of Medicine, for his expert technical assistance in real-time RT-PCR studies.
This study was supported in part by National Institutes of Health Grants HL-58672 and HL-74854 (JY Cheung), DK-46678 (JY Cheung, co-investigator), GM-46691 (LI Rothblum), HL-70548 and GM-64640 (JR Moorman), HL-69074 (AL Tucker), NS-21925, NS-37716, and NS-41363 (DJ Carey); American Heart Association Pennsylvania Affiliate Grants-in-Aid 0265426U (XQ Zhang) and 0355744U (JY Cheung); American Heart Association Pennsylvania Affiliate Post-Doctoral Fellowship 0425319U (BA Ahlers); and by grants from the Geisinger Foundation (to JY Cheung, LI Rothblum, DJ Carey and Y Chan).
References
- 1.Ahlers BA, Zhang XQ, Moorman JR, Rothblum LI, Carl LL, Song J, Wang J, Geddis LM, Tucker AL, Mounsey JP, Cheung JY. Identification of an endogenous inhibitor of the cardiac Na+/Ca2+ exchanger, phospholemman. J Biol Chem. 2005;280:19875–19882. doi: 10.1074/jbc.M414703200. [DOI] [PubMed] [Google Scholar]
- 2.Beguin P, Crambert G, Guennoun S, Garty H, Horisberger JD, Geering K. CHIF, a member of the FXYD protein family, is a regulator of Na,K-ATPase distinct from the gamma subunit. EMBO J. 20:3993–4002. doi: 10.1093/emboj/20.15.3993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beguin P, Crambert G, Monnet-Tschudi F, Uldry M, Horisberger JD, Garty H, Geering K. FXYD7 is a brain-specific regulator of Na,K-ATPase alpha 1-beta isozymes. EMBO J. 2002;21:3264–3273. doi: 10.1093/emboj/cdf330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen Z, Jones LR, O’Brian JJ, Moorman JR, Cala SE. Structural domains in phospholemman: a possible role for the carboxyl terminus in channel inactivation. Circ Res. 1998;82:367–374. doi: 10.1161/01.res.82.3.367. [DOI] [PubMed] [Google Scholar]
- 5.Cheung JY, Musch TI, Misawa H, Semanchick A, Elensky M, Yelamarty RV, Moore RL. Impaired cardiac function in rats with healed myocardial infarction: cellular vs. myocardial mechanisms. Am J Physiol Cell Physiol. 1994;266:C29–C36. doi: 10.1152/ajpcell.1994.266.1.C29. [DOI] [PubMed] [Google Scholar]
- 6.Crambert G, Fuzesi M, Garty H, Karlish S, Geering K. Phospholemman (FXYD1) associates with Na, K-ATPase and regulates its transport properties. Proc Nat’l Acad Sci U S A. 2002;99:11476–11481. doi: 10.1073/pnas.182267299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davis CE, Patel MK, Miller JR, John JE, Jones LR, Tucker AL, Mounsey JP, Moorman JR. Effects of phospholemman expression on swelling-activated ion currents and volume regulation in embryonic kidney cells. Neurochemical Research. 2004;29:177–187. doi: 10.1023/b:nere.0000010447.24128.ac. [DOI] [PubMed] [Google Scholar]
- 8.Dixon IMC, Hata T, Dhalla NS. Sarcolemmal Na+ - K+ - ATPase activity in congestive heart failure due to myocardial infarction. Am J Physiol Cell Physiol. 1992;262:C664–C671. doi: 10.1152/ajpcell.1992.262.3.C664. [DOI] [PubMed] [Google Scholar]
- 9.Feschenko MS, Donnet C, Wetzel RK, Asinovski NK, Jones LR, Sweadner KJ. Phospholemman, a single-span membrane protein, is an accessory protein of Na,K-ATPase in cerebellum and choroid plexus. J Neurosci. 2003;23:2161–2169. doi: 10.1523/JNEUROSCI.23-06-02161.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fuller W, Eaton P, Bell JR, Shattock MJ. Ischemia-induced phosphorylation of phospholemman directly activates rat cardiac Na/K-ATPase. FASEB J. 2004;18:197–199. doi: 10.1096/fj.03-0213fje. [DOI] [PubMed] [Google Scholar]
- 11.Hasenfuss G. Alterations of calcium regulatory proteins in heart failure. Cardiovasc Res. 1998;37:279–289. doi: 10.1016/s0008-6363(97)00277-0. [DOI] [PubMed] [Google Scholar]
- 12.Gadsby DC, Kimura J, Noma A. Voltage dependence of Na/K pump current in isolated heart cells. Nature. 1985;315:63–65. doi: 10.1038/315063a0. [DOI] [PubMed] [Google Scholar]
- 13.Glitsch HG. Electrophysiology of the sodium-potassium-ATPase in cardiac cells. Physiol Rev. 2001;81:1791–1826. doi: 10.1152/physrev.2001.81.4.1791. [DOI] [PubMed] [Google Scholar]
- 14.Grupp I, Im W-B, Lee CO, Lee S-W, Pecker MS, Schwartz A. Relation of sodium pump inhibition to positive inotropy at low concentration of ouabain in rat heart muscle. J Physiol (Lond) 1985;360:149–160. doi: 10.1113/jphysiol.1985.sp015609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kowdley GC, Ackerman SJ, Chen Z, Szabo G, Jones LR, Moorman JR. Anion, cation, and zwitterion-selectivity of phospholemman channel molecules. Biophys J. 1997;72:141–145. doi: 10.1016/S0006-3495(97)78653-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lindemann JP. α-adrenergic stimulation of sarcolemmal protein phosphorylation and slow responses in intact myocardium. J Biol Chem. 1985;261:4860–4867. [PubMed] [Google Scholar]
- 17.Macknight ADC, Leaf A. Regulation of cellular volume. Physiol Rev. 1977;57:510–573. doi: 10.1152/physrev.1977.57.3.510. [DOI] [PubMed] [Google Scholar]
- 18.Mahmmoud YA, Vorum H, Cornelius F. Purification of a phospholemman-like protein from shark rectal glands. J Biol Chem. 2000;275:25969–25977. doi: 10.1074/jbc.M005168200. [DOI] [PubMed] [Google Scholar]
- 19.McDonough AA, Zhang Y, Shin V, Frank JS. Subcellular distribution of sodium pump isoform subunits in mammalian cardiac myocytes. Am J Physiol Cell Physiol. 1996;270:C1221–C1227. doi: 10.1152/ajpcell.1996.270.4.C1221. [DOI] [PubMed] [Google Scholar]
- 20.Mirza MA, Zhang XQ, Ahlers BA, Qureshi A, Carl LL, Song J, Tucker AL, Mounsey JP, Moorman JR, Rothblum LI, Zhang TS, Cheung JY. Effects of phospholemman downregulation on contractility and [Ca2+]i transients in adult rat cardiac myocytes. Am J Physiol Heart Circ Physiol. 2004;286:H1322–H1330. doi: 10.1152/ajpheart.00997.2003. [DOI] [PubMed] [Google Scholar]
- 21.Moorman JR, Ackerman SJ, Kowdley GC, Griffin MP, Mounsey JP, Chen Z, Cala SE, O’Brian JJ, Szabo G, Jones LR. Unitary anion currents through phospholemman channel molecules. Nature. 1995;377:737–740. doi: 10.1038/377737a0. [DOI] [PubMed] [Google Scholar]
- 22.Morales-Mulia M, Pasantes-Morales H, Moran J. Volume sensitive efflux of taurine in HEK 293 cells overexpressing phospholemman. Biochim Biophys Acta. 2000;1496:252–260. doi: 10.1016/s0167-4889(00)00023-9. [DOI] [PubMed] [Google Scholar]
- 23.Moran J, Morales-Mulia M, Pasantes-Morales H. Reduction of phospholemman expression decreases osmosensitive taurine efflux in astrocytes. Biochim Biophys Acta. 2001;1538:313–320. doi: 10.1016/s0167-4889(01)00082-9. [DOI] [PubMed] [Google Scholar]
- 24.Palmer CJ, Scott BT, Jones LR. Purification and complete sequence determination of the major plasma membrane substrate for cAMP-dependent proteins kinase and protein kinase C in myocardium. J Biol Chem. 1991;266:11126–11130. [PubMed] [Google Scholar]
- 25.Presti CF, Jones LR, Lindemann JP. Isoproterenol-induced phosphorylation of a 15-kilodalton sarcolemmal protein in intact myocardium. J Biol Chem. 1985;260:3860–3867. [PubMed] [Google Scholar]
- 26.Pu HX, Cluzeaud F, Goldshleger R, Karlish SJ, Farman N, Blostein R. Functional role and immunocytochemical localization of the γa and γb forms of the Na,K-ATPase γ subunit. J Biol Chem. 2001;276:20370–20378. doi: 10.1074/jbc.M010836200. [DOI] [PubMed] [Google Scholar]
- 27.Rembold CM, Ripley ML, Meeks MK, Geddis LM, Kutchai HC, Marassi FM, Cheung JY and Moorman JR. Serine 68 phospholemman phosphorylation during forskolin-induced swine carotid artery relaxation. J. Vasc. Res., in press. [DOI] [PMC free article] [PubMed]
- 28.Ren B, Shao Q, Ganguly PK, Tappia PS, Takeda N, Dhalla NS. Influence of long-term treatment of imidapril on mortality, cardiac function, and gene expression in congestive heart failure due to myocardial infarction. Can J Physiol Pharmacol. 2004;82:1118–1127. doi: 10.1139/y04-115. [DOI] [PubMed] [Google Scholar]
- 29.Sehl PD, Tai JTN, Hillan KJ, Brown LA, Goddard A, Yang R, Jin H, Lowe DG. Application of cDNA microarrays in determining molecular phenotype in cardiac growth, development, and response to inquiry. Circulation. 2000;101:1990–1999. doi: 10.1161/01.cir.101.16.1990. [DOI] [PubMed] [Google Scholar]
- 30.Shao Q, Ren B, Elimban V, Tappia PS, Takeda N, Dhalla NS. Modification of sarcolemmal Na+-K+-ATPase and Na+/Ca2+ exchanger expression in heart failure by blockade of renin-angiotensin system. Am J Physiol Heart Circ Physiol. 2005;288:H2637–H2646. doi: 10.1152/ajpheart.01304.2004. [DOI] [PubMed] [Google Scholar]
- 31.Silverman BD, Fuller W, Eaton P, Deng J, Moorman JR, Cheung JY, James AF, Shattock MJ. Serine 68 phosphorylation of phospholemman: acute isoform-specific activation of cardiac Na/K ATPase. Cardiovasc Res. 2005;65:93–103. doi: 10.1016/j.cardiores.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 32.Song J, Zhang XQ, Ahlers BA, Carl LL, Wang J, Rothblum LI, Stahl RC, Mounsey JP, Tucker AL, Moorman JR, Cheung JY. Serine 68 of phospholemman is critical in modulation of contractility, [Ca2+]i transients and Na+/Ca2+ exchange in adult rat cardiac myocytes. Am J Physiol Heart Circ Physiol. 2005;288:H2342–H2354. doi: 10.1152/ajpheart.01133.2004. [DOI] [PubMed] [Google Scholar]
- 33.Song J, Zhang XQ, Carl LL, Qureshi A, Rothblum LI, Cheung JY. Overexpression of phospholemman alters contractility and [Ca2+]i transients in adult rat myocytes. Am J Physiol Heart Circ Physiol. 2002;283:H576–H583. doi: 10.1152/ajpheart.00197.2002. [DOI] [PubMed] [Google Scholar]
- 34.Stimers JR, Liu S, Kinard TA. Effect of Nai on activity and voltage dependence of the Na/K pump in adult rat cardiac myocytes. J Memb Biol. 1993;135:39–47. doi: 10.1007/BF00234650. [DOI] [PubMed] [Google Scholar]
- 35.Sweadner KJ, Herrera VLM, Amato S, Moellmann A, Gibbons DK, Repke KRH. Immunologic identification of Na+, K+-ATPase isoform in myocardium. Circ Res. 1996;74:669–678. doi: 10.1161/01.res.74.4.669. [DOI] [PubMed] [Google Scholar]
- 36.Sweadner KJ, Rael E. The FXYD gene family of small ion transport regulators or channels: cDNA sequence, protein signature sequence, and expression. Genomics. 2000;68:41–56. doi: 10.1006/geno.2000.6274. [DOI] [PubMed] [Google Scholar]
- 37.Tadros GM, Zhang XQ, Song J, Carl LL, Rothblum LI, Tian Q, Dunn J, Lytton J, Cheung JY. Effects of Na+/Ca2+ exchanger downregulation on contractility and [Ca2+]i transients in adult rat myocytes. Am J Physiol Heart Circ Physiol. 2002;283:H1616–H1626. doi: 10.1152/ajpheart.00186.2002. [DOI] [PubMed] [Google Scholar]
- 38.Therien AG, Blostein R. Mechanisms of sodium pump regulation. Am J Physiol Cell Physiol. 2000;279:C541–C566. doi: 10.1152/ajpcell.2000.279.3.C541. [DOI] [PubMed] [Google Scholar]
- 39.Therien AG, Goldshleger R, Karlish SJ, Blostein R. Tissue-specific distribution and modulatory role of the gamma subunit of the Na, K-ATPase. J Biol Chem. 1997;272:32628–32634. doi: 10.1074/jbc.272.51.32628. [DOI] [PubMed] [Google Scholar]
- 40.Walaas SI, Czernik AJ, Olstad OK, Sletten K, Walaas O. Protein kinase C and cyclic AMP-dependent protein kinase phosphorylate phospholemman, an insulin and adrenaline-regulated membrane phosphoprotein, at specific sites in the carboxyl terminal domain. Biochem J. 1994;304:635–640. doi: 10.1042/bj3040635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang XQ, Musch TI, Zelis R, Cheung JY. Effects of impaired Ca2+ homeostasis on contraction in postinfarction myocytes. J Appl Physiol. 1999;86:943–950. doi: 10.1152/jappl.1999.86.3.943. [DOI] [PubMed] [Google Scholar]
- 42.Zhang XQ, Qureshi A, Song J, Carl LL, Tian Q, Stahl RC, Carey DJ, Rothblum LI, Cheung JY. Phospholemman modulates Na+/Ca2+ exchange in adult rat myocytes. Am J Physiol Heart Circ Physiol. 2003;284:H225–H233. doi: 10.1152/ajpheart.00698.2002. [DOI] [PubMed] [Google Scholar]
- 43.Zhang XQ, Song J, Qureshi A, Rothblum LI, Carl LL, Tian Q, Cheung JY. Rescue of contractile abnormalities by Na+/Ca2+ exchanger overexpression in postinfarction rat myocytes. J Appl Physiol. 2002;93:1925–1931. doi: 10.1152/japplphysiol.00583.2002. [DOI] [PubMed] [Google Scholar]
- 44.Zhang XQ, Song J, Rothblum LI, Lun M, Wang X, Ding F, Dunn J, Lytton J, McDermott PJ, Cheung JY. Overexpression of Na+/Ca2+ exchanger alters contractility and SR Ca2+ content in adult rat myocytes. Am J Physiol Heart Circ Physiol. 2001;281:H2079–H2088. doi: 10.1152/ajpheart.2001.281.5.H2079. [DOI] [PubMed] [Google Scholar]
- 45.Zhang XQ, Tillotson DL, Moore RL, Zelis R, Cheung JY. Na+/Ca2+ exchange currents and SR Ca2+ contents in postinfarction myocytes. Am J Physiol Cell Phyiol. 1996;271:C1800–C1807. doi: 10.1152/ajpcell.1996.271.6.C1800. [DOI] [PubMed] [Google Scholar]