

Abstract
Expression of the insulin gene is nearly exclusive to the β cells of the pancreatic islets. Whereas the sequence-specific transcription factors that regulate insulin expression have been well studied, the interrelationship between these factors, chromatin structure, and transcriptional elongation by RNA polymerase II (Pol II) has remained undefined. In this regard, recent studies have begun to establish a role for the methylation of histone H3 in the initiation or elongation of transcription by Pol II. To determine a role for the transcriptional activator Pdx-1 in the maintenance of chromatin structure and Pol II recruitment at the insulin gene, we performed small interfering RNA-mediated knockdown of Pdx-1 in βTC3 cells and subsequently studied histone modifications and Pol II recruitment by chromatin immunoprecipitation. We demonstrate here that the 50% fall in insulin transcription following knockdown of Pdx-1 is accompanied by a 60% fall in dimethylated histone H3-Lys4 at the insulin promoter. H3-Lys4 methylation at the insulin promoter may be mediated, at least partially, by the methyltransferase Set9. Immunohistochemical analysis reveals that Set9 is expressed in an islet-enriched pattern in the pancreas, similar to the pattern of Pdx-1 expression. The recruitment of Set9 to the insulin gene appears to be a consequence of its direct interaction with Pdx-1, and siRNA-mediated knockdown of Set9 attenuates insulin transcription. Pdx-1 knockdown was also associated with an overall shift in the recruitment of Pol II isoforms to the insulin gene, from an elongation isoform (Ser2-P) to an initiation isoform (Ser5-P). Our findings therefore suggest a model whereby Pdx-1 plays a novel role in linking H3-Lys4 di-methylation and Pol II elongation to insulin transcription.
During pancreatic development, endocrine and exocrine cell types differentiate from a common endodermal precursor cell via a tightly coordinated sequence of transcriptional events (1,2). The Hox-like transcription factor Pdx-1 is thought to initiate this cascade by regulating specific genes within the endodermal precursor cell (3,4). The disruption of pdx-1 expression during development, either by targeted knockout in mice (5,6) or homozygous mutations in humans (7), can be catastrophic, resulting in the failure of pancreas formation and consequent development of diabetes. Within the mature pancreas, Pdx-1 is necessary for the maintenance and function of endocrine cell types within the islets of Langerhans. In the islet β cell, Pdx-1 is believed to activate a number of critical genes involved in glucose sensing and insulin production, including glucokinase, glut2, and insulin (3,8).
Expression of the insulin gene is almost exclusive to the islet β cells. We recently demonstrated, by use of small interfering (si) RNA and chromatin immunoprecipitation (ChIP) in isolated mouse islets, that Pdx-1 is a direct activator of insulin transcription upon binding to A box DNA elements in the 5′ regulatory region of the gene (the insulin promoter) (4). In addition to A boxes, other defined regulatory elements within the promoter are believed to be bound by ubiquitous (e.g. E47) and cell-specific (e.g. NeuroD1, MafA) transcription factors (9); in sum, these factors are thought to act in concert to stabilize coactivators (e.g. CBP/p300) and the basal transcriptional proteins at the promoter. Whereas the sequence-specific transcription factors that regulate insulin expression have been well studied, the interrelationship between these factors, histone/chromatin structure, and transcriptional elongation by RNA polymerase II (Pol II) has remained undefined. In this regard, recent studies in yeast systems have begun to establish a role for the methylation of histone H3 in initiation and/or elongation of transcription by Pol II (10,11). For example, Set1 (a histone H3-Lys4 (H3-K4) methyltransferase) and Set2 (an H3-K36 methyltransferase) are found in association with initiation phosphorylated (Ser5-P) and elongation phosphorylation (Ser2-P) isoforms of Pol II, respectively (12,13). Studies in higher eukaryotic systems are also beginning to suggest that histone methylation patterns may indeed correlate with the activity of Pol II (14,15). These findings also emphasize that histone Lys methylation is not necessarily a stable epigenetic marker of active genes, but may play a more dynamic role in the acute regulation of gene transcription.
In this study, we describe a novel role for the insulin gene activator Pdx-1 in the maintenance of H3-K4 methylation at the promoter and coding regions of the mouse insulin I and II loci. We demonstrate that Pdx-1 physically associates with and recruits the H3-K4 methyltransferase Set9 (16,17) to the insulin gene. siRNA-mediated knockdown of Pdx-1 abrogates Set9 recruitment, H3-K4 methylation, Pol II elongation (but not initiation), and insulin transcription in a promoter-dependent manner. Our findings suggest a model whereby cell type-and sequence-specific transcription factors, exemplified by Pdx-1, may be central to linking H3-K4 methylation and Pol II elongation to insulin transcription.
MATERIALS AND METHODS
Antibodies and vectors
Rabbit polyclonal antiserum against Pdx-1 was provided by Dr. M. German (University of California, San Francisco). Rabbit polyclonal antiserum against Set9 and di-methyl H3-K4 were from Upstate Biotechnology. Antisera against mono- and tri-methyl H3-K4 were purchased from Abcam. Pol II C-terminal domain (CTD) and phospho-specific antisera were from Covance. Wildtype and mutant Set9 cDNA expression constructs in vector pcDNA3 were provided by Dr D. Reinberg (University of Medicine and Dentistry, New Jersey). Rabbit antiinsulin, goat anti-glucagon, and mouse mono-clonal anti-HA antibodies were purchased from Santa Cruz Biotechnology. Mouse monoclonal anti-Pdx-1 antibody was purchased from R&D Systems. Cy2-conjugated goat anti-rabbit, Cy2- conjugated donkey anti-mouse, Cy3-conjugated goat anti-rabbit, and Cy3-conjugated donkey antigoat antibodies were all purchased from Jackson Immunoresearch Laboratories.
For generation of a vector expressing siRNA specific for set9, a previously published siRNA sequence (5′-GATCTGCACCTGGACGATGACGGATTACCT TCCTGTCAGTAATCCGTCATCGTCCAGGTG CTTTTTGTCGACG-3′, ref. (18)) was subcloned into the U6 promoter-driven vector pSIREN-DNR (Clontech) according to manufacturer’s instructions. Recombinant adenoviruses encoding siRNAs specific for pdx-1 and luciferase (Ad-siPdx and Ad-siLuc) were described previously (4,19). Infectious titers of virus, as determined by endpoint dilution assay in HEK293 cells, were on the order of 1010–1011 pfu/ml.
The 5S ribosomal array fragment containing 5X-E2/A3/A4 for nucleosomal reconstitutions was generated by subcloning the 5X-E2/A3/A4-TATA-Luc fragment from vector pFoxLucPrl5FF (20) into the positioning vector pIC-2085S (21). The array fragment was excised by digestion of the resulting plasmid (pIC-5FFPrl) with NdeI and ClaI, and purified by preparative agarose gel electrophoresis. The array fragment containing 5X-Gal4-TATA-E4 was excised from vector pIC208G5E4 as described previously (22).
Protein purification and GST pulldown assays
Pdx-1 and Set9 proteins were prepared as N-terminal His6-tagged proteins overexpressed in E. coli BL21(DE3)/pLysS using the pET expression system (Novagen), and purified using Ni-nitrolotriacetic acid resin (Qiagen), as previously described (23). Glutathione S-transferase (GST) assays were performed as described previously (20). Briefly, GST fusion proteins were overexpressed in E. coli BL21(DE3)/pLysS overnight at 30°C and purified by Glutathione agarose beads (Amersham). For in vitro-translation, proteins (encoded within the T7 promoter-based vector, pBAT11) were radiolabeled with 35S-Met using the T7-TNT® in vitro transcription-translation kit (Promega). 5 μl of radiolabeled proteins were incubated in 1 ml of binding buffer (20 mM HEPES, pH 7.9, 10 mM MgCl2, 150 mM KCl, 10% glycerol, 0.12% NP-40) with GST purified proteins for 12 h at 4°C with gentle rocking. Protein complexes were washed 4–5 times with binding buffer, and subjected to electrophoresis on a 12% sodium dodecylsulfate polyacrylamide gel and visualized by fluorography.
Cell culture and transfections
The mouse insulinoma cell line βTC3 and mouse fibroblast cell line NIH3T3 were maintained in Dulbecco’s modified Eagle’s medium as previously reported (3). HEK293 cells and HeLa cells were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% newborn calf serum and 1% penicillin/streptomycin. Transient transfections were performed using 5x104 NIH3T3 or HEK293 cells or 5x105 βTC3 cells per well in a 6 well dishes. For activation studies, 1μg of reporter plasmid (pFoxLucPrl5FF or pFoxLucPrl5Gal4 (24)) were combined with 0.05 μg CMV promoter-driven Pdx-1 expression plasmids (pBAT12, pBAT12Pdx1) and/or 0.5 μg CMV-promoter driven Set9 expression plasmids (pcDNASet9 or pcDNASet9m (16)). For repression studies, 1μg of insulin promoter reporter plasmid pFoxLucSwt (23) or control reporter plasmid pFoxLuc was cotransfected with 0.5 μg of siRNA encoding plasmids (pSuper-siPdx, pMIGHTY-siGFP (25), or pSIREN-DNR-siSet9) and mixed with 3 μl of Transfast® reagent (Promega). Transfected cells were harvested 48 hours later and assayed for luciferase activity using a commercially available luminometric kit (Promega).
For adenoviral transduction of βTC3 cells, cells were cultured overnight at a density of 5x105 cells/well in 6 well tissue culture plates, followed by addition of recombinant adenoviruses at a multiplicity of infection of ~2500 pfu/cell directly to the medium. After 5 h, virus-containing medium was removed and cells were washed once with PBS and cultured in fresh medium. After 72 h, cells were processed for isolation of total RNA or whole cell extracts, as described previously (4).
HeLa cells stably expressing hemagglutinin (HA)-tagged Pdx-1 were generated by cotransfecting a vector encoding HA-Pdx-1 downstream of the CMV promoter (pBAT12- HAPdx) with a vector encoding for a resistance gene against hygromycin (pcDNA3.1-hygro). 48 h after transfection, cells stably incorporating the vectors were selected by incubating in medium containing 400ug/ml of hygromycin. Approximately 3 weeks later, all hygromycinresistant clones were pooled and replated. Expression was confirmed by immunoblot.
Real-time RT-PCR
Real-time reverse transcriptase (RT)-PCR was performed using forward and reverse primers to amplify insulin pre-mRNA (5′-GGGGAGCGTGGCTTCTTCTA- 3′ and 5′-GGGGACAGAATTCAGTGGCA-3′), or β –actin (5′-AGGTCATCACTATTGGCAACGA-3′ and 5′- CACTTCATGATGGAATTGAATGTAGTT-3′) as described in detail previously (4). PCR reactions were cycled 40 times using the following conditions: 95°C for 15 sec, 64°C for 1 min. Samples containing no reverse transcriptase were run parallel reactions to assay for genomic contamination. In all instances, samples without reverse transcriptase exhibited threshold cycle numbers greater than 40, indicating negligible amounts of genomic DNA.
Amplified products from initial PCR reactions were subcloned into the T/A cloning vector pCR2.1 (Invitrogen), and 3–4 clones were sequenced to confirm the identity of the amplified product. The threshold cycle (CT) methodology (3) was used to calculate relative quantities of mRNA products from each sample; all samples were corrected for total input RNA by normalizing CT values to the CT value of β actin message. These corrections were verified by quantitation of total RNA in each sample by fluorescence using RiboGreen® dye (Molecular Probes).
Quantitative Chromatin immunoprecipitation
ChIP assays, including quantitation of coimmunoprecipitated DNA fragments by real-time PCR, were performed as described previously (3). ChIP assays were performed on at least 3 independent occasions; for each ChIP assay, promoter samples were quantitated in triplicate. Forward and reverse primer sequences, respectively, used for PCR were: distal insulin gene : 5′-CCTCCATACAGACAAAATACTCTCC-3′ and 5′-CTCACATCCAAAGCAGAAGTTC-3′; insulin promoter : 5′-TACCTTGCTGCCTGAGTTCTGC-3′ and 5′-GCATTTTCCACATCATTCCCC-3′; insulin coding region : 5′-TGGCTTCTTCTACACACCCAAG-3′ and 5′-ACAATGCCACGCTTCTGCC-3′; myoD promoter: 5′-GCACTGCCACCGATTCATTTG-3′ and 5′- CAGGAGGTTTGGAGAGAGACTCAAG-3′.
Chromatin reconstitution and in vitro transcription
Chromatin reconstitutions of the 5S array fragments were performed using HeLa-purified histone octamers and a sequential salt dilution procedure (21). Reconstituted (chromatinized) fragments were stored at 4°C for up to 1 month. In vitro transcription was performed using 2 ng of the chromatinized array fragment (or corresponding naked DNA fragments) and incubated in the presence or absence of 100 ng Pdx-1, 100 ng Set9, and/or 2 μM S-adenosyl methionine (Ado-Met) in transcription buffer (20mM HEPES, pH 7.9, 100mM KCl, 0.2mM EDTA, and 20% Glycerol) for 30 min prior to addition of 10 μg HeLa Nuclear Extract (prepared by method of Dignam (26)) in a total volume of 25 μl. Reactions were started by addition of 0.4 mM rNTPs with or without 2 μM Ado-Met. Reactions proceeded for 60 min. at 30 °C and subsequently digested with DNaseI for 30 min, and then stopped by addition of 250 μl of stop buffer (20mM EDTA, 0.2M NaCl, 1% SDS, and 3ug/ml tRNA). Reactions were extracted with phenol:chloroform:isoamyl alcohol and ethanol precipitated. RT-PCR was performed at 37°C for 50 minutes with MMLV reverse transcriptase and 3 μg of random hexamers (both from Invitrogen) and a unique 5′ capping primer (5′-AAGCAGTGGTATCAACGCAGAGTACGCGG G-3′). The MMLV was inactivated at 70°C for 15 min. Real time PCR using forward capping primer (5′-AAGCAGTGGTATCAACGCAGAGT-3′) and reverse luciferase coding primer (for the E2/A3/A4 array) (5′-CCTCTAGAGGATAGAATGGC-3′) or reverse E4 coding primer (for the Gal4 array) (5′-AACATACAGCGCTTCCACAGCGGCAG-3′) was performed to quantitate transcription product.
Co-immunoprecipitation assays
For coimmunoprecipitation of endogenous SET9 and HA-tagged Pdx-1, nuclear lysates from HeLa cells stably expressing HA-Pdx-1 were prepared and subject to immunoprecipitation (using anti-Set9 antibody or normal rabbit serum) as described previously (27). Immunoprecipitated proteins were subjected to immunoblot analysis using monoclonal anti-HA antibody.
Immunoblot analysis
Aliquots of 5μg of whole cell or nuclear extract (prepared as described) were resolved by electrophoresis on a 12% sodium dodecylsulfate-polyacrylamide gel followed by immunoblot analysis using anti-Pdx-1, -Set9, and –dimethyl H3-K4 antisera. Immunoblots were visualized using the ECL-Plus system (Amersham).
Immunohistochemistry
Pancreata from 6–8 week old C57/BL6 mice were isolated and fixed in 4% paraformaldehyde and paraffin-embedded. 5 μm tissue sections were immunostained as described (28), and visualized using a Zeiss Axioplan fluorescence microscope. For immunofluorescence, the excitation wavelengths were selected using narrow bandpass filters 450–515 nm and 546–590 nm for Cy2 and Cy3, respectively. Images were obtained with a SPOT® CCD camera (Diagnostic Instruments) and analyzed using Advanced SPOT® Software.
RESULTS
H3-K4 methylation correlates with Pol II recruitment at the insulin gene loci in pancreatic β cells
Several studies have demonstrated the enrichment of H3-K4 methylation within the promoter and coding regions of active genes in both yeast and higher eukaryotes, and that it may serve as an important mark for recruitment of basal transcriptional machinery (10,11). Because mono-, di-, and tri-methylated forms of H3-K4 demonstrate different patterns of enrichment across a given locus (14) and may be catalyzed by different methyltransferases, we first sought to determine the correlation between different methylated forms of H3-K4 and Pol II recruitment at the insulin I and II loci1. For these studies, we utilized a mouse β cell-derived cell line, βTC3, which we and others have previously demonstrated to exhibit insulin gene regulatory characteristics similar to pancreatic islets (4,29). Fig. 1A demonstrates that mono-, di-, and tri-methylated H3-K4 species are enriched at the proximal insulin promoter and coding region relative to a distal region of the insulin locus located 4500 base pairs upstream of the transcriptional start site. The data in Fig. 1 were normalized to methylation observed at the distal region of insulin primarily to highlight changes in methylation patterns across the gene. Notably, there is strikingly greater enrichment (25-fold) of dimethylated H3-K4 at the proximal promoter compared to the other methylated species. To confirm that methylation of H3-K4 correlates with active transcription, we also analyzed methylation at the inactive myoD1 gene. As shown in Fig. 1B, substantially less enrichment of all methylated H3- K4 species is observed at the regulatory region of the myoD1 promoter in βTC3 cells. Recruitment of Pol II is observed coordinately with the H3-K4 methylation patterns across the insulin locus (Fig. 1C), with no Pol II recruitment at the inactive myoD promoter (Fig. 1D). Taken together, these data support a strong correlation between H3-K4 methylation and active transcription in βTC3 cells.
Figure 1. Relative Histone H3-K4 methylation and Pol II and Pdx-1 occupancies along the insulin and myoD1 genes in mouse βTC3 cells.
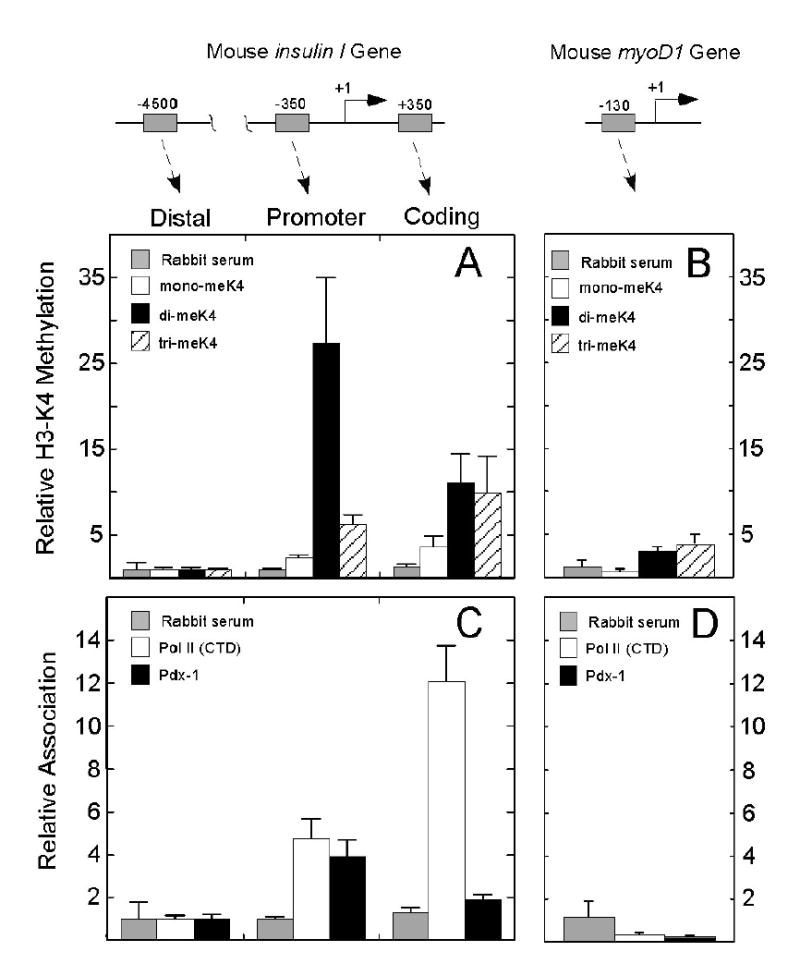
Schematic representations of the insulin and myoD1 genes are shown at the top of the figure, highlighting approximate positions of the fragments amplified by PCR (gray boxes) following chromatin immunoprecipitation. The numbers indicate positions in base pairs relative to the transcriptional start site (+1). The data shown in panels A-D are derived from Q-PCR analysis of the respective gene fragments after ChIP. All data (including those for the myoD1 gene) are expressed as fold-differences relative to methylation or factor association observed at the distal insulin gene (defined as “1”). This normalization was performed primarily to highlight the methylation and factor association patterns across the gene. Specific antibodies used in the ChIP experiments were anti-mono-, di-, and tri-methylated H3-K4 (panels A and B) or anti-Pol II (CTD) and anti-Pdx-1 (panels C and D). Approximately 0.02% of input DNA was recovered when normal rabbit serum was used instead of specific antiserum. Data represent the mean ± SEM of at least four independent ChIP experiments.
Pdx-1 is required for the maintenance of di-methylated H3-K4 at the insulin gene
To determine a potential role for Pdx-1 in the maintenance of H3-K4 methylation and Pol II recruitment, we treated βTC3 cells with an adenovirus encoding siRNA against Pdx-1 (Ad-siPdx), and subsequently monitored histone modifications by ChIP and insulin transcription by quantitative real-time RT-PCR. As demonstrated in Fig. 1D, Pdx-1 associates preferentially with the proximal insulin promoter. We do note, however, that significant Pdx-1 occupancy was observed within the coding region, suggesting either incomplete chromatin shearing (and hence primer overlap) or that Pdx-1 occupancy within this region is real and reflective of the overall “accessible” (euchromatic) state of the insulin promoter (3). Notably, however, no Pdx-1 occupancy was observed at the myoD1 promoter (Fig. 1D). As shown in Fig. 2A, treatment of βTC3 cells for 72h with Ad-siPdx results in a decrease in Pdx-1 protein levels to only 20% compared to untreated cells or cells treated with an adenovirus encoding an siRNA against luciferase (Ad-siLuc). Coordinately, we observed a decline in Pdx-1 occupancy at the insulin promoter by ChIP and a decline in insulin transcription to only 45% compared to Ad-siLuc treated cells by quantitative RT-PCR of insulin pre-mRNA levels (data not shown, but previously demonstrated in ref. (4)). The results of Pdx-1 knockdown on insulin transcription reported here are identical to what we observed in a prior study (4), and the details of the pre-mRNA assay are provided in that study. Fig. 2B demonstrates that this decline in Pdx-1 levels is associated with 60% decreases in di-methylated H3-K4 at the proximal promoter and coding regions of the insulin gene. Interestingly, no significant changes in mono- and tri-methylated H3-K4 were observed upon Ad-siPdx treatment (Fig. 2B). The decrease in di-methylated H3-K4 with Ad-siPdx was not a result of any change in the total levels of di-methylated H3-K4 in the cell, as determined by immunoblot analysis (data not shown).
Figure 2. siRNA-directed knockdown of Pdx-1 diminishes insulin transcription and reduces di-methylated H3-K4 at the insulin promoter and coding regions.
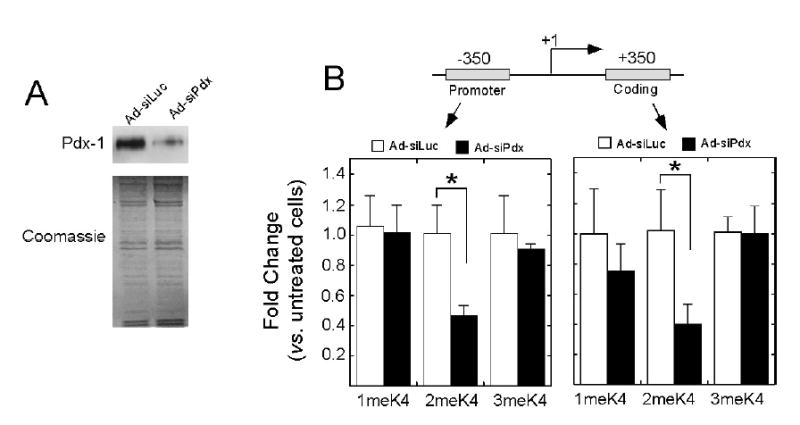
A, Representative immunoblot for Pdx-1 from whole cell extract of βTC3 cells treated with Ad-siLuc or Ad-siPdx for 72 h (top), and the corresponding Coomassie stain of the 15% polyacrylamide gel demonstrating equivalent loading of protein samples (bottom). B, Quantitative ChIP analysis of mono-, di-, and tri-methylated H3-K4 at the promoter and coding regions of the insulin gene following treatment of βTC3 cells with Ad-siLuc or Ad-siPdx for 72 h. Data are expressed as fold-changes relative to untreated cells. Data represent the mean ± SEM of least four independent ChIP experiments. “*” signifies that the value for the comparisons shown is statistically significant (p<0.05).
We next examined Pol II occupancy by ChIP. Using an antibody directed against the C-terminal domain (CTD) of Pol II, we noticed a trend to increased Pol II occupancy at the proximal insulin promoter in Ad-siPdx-treated cells compared to controls (Ad-siLuc treatment) (Fig. 3A). In contrast, and consistent with the trend in insulin transcription, Ad-siPdx treatment led to a significant 2-fold decrease in Pol II occupancy within the insulin coding region (Fig. 3A). Because differentially phosphorylated isoforms of the CTD of Pol II have been correlated with changes in histone methylation in yeast systems, we performed ChIP using antibodies against the Ser2-P and Ser5-P isoforms of Pol II. In other systems, Ser5-P has been associated with transcriptional initiation and Ser2-P has been observed with transcriptional elongation (30). As shown in Fig. 3B, we observed that the Ser5 phosphorylated isoform was enhanced 1.5-fold at the promoter region in Ad-siPdx-treated cells compared to controls, and was not observed over background in the coding region. The Ser2-P isoform, which showed no change in the promoter region, was decreased to background levels in AdsiPdx treated cells within the coding region compared to controls (Fig. 3C). These results suggest that Pdx-1 is necessary for switching the initiation isoform of Pol II (Ser5-P) to the elongation isoform (Ser2-P) at the insulin promoter. Moreover, as in other systems, our results also suggest a potential relationship between dimethylation of H3-K4 and conversion to the Ser2-P isoform.
Figure 3. Occupancies of Pol II (CTD) and its phosphorylated isoforms (Ser5-P and Ser2-P) at the insulin promoter and coding regions following Pdx-1 knockdown.
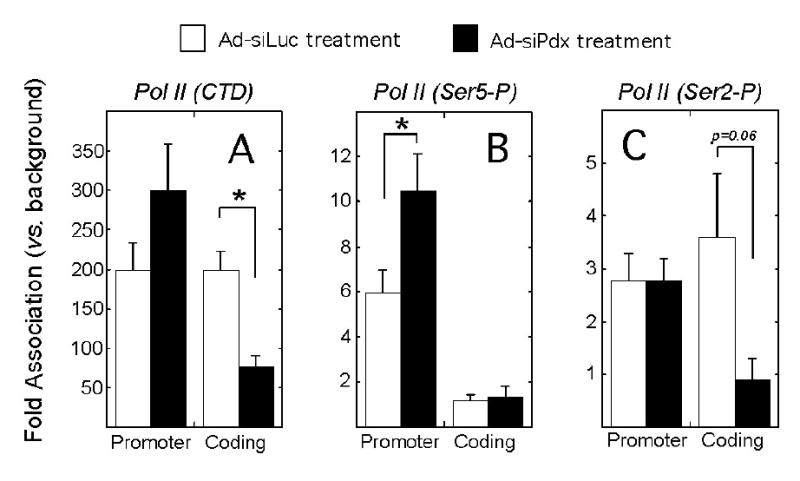
βTC3 cells were treated with Ad-siLuc or Ad-siPdx for 72 h, then subject to quantitative ChIP analysis for occupancies of Pol II (CTD) (panel A), Pol II (Ser5-P) (panel B), or Pol II (Ser2-P) (panel C) at the insulin promoter and coding regions. Data are expressed as fold-differences relative to background (in which normal rabbit serum is used instead of specific antibody). Background recovery was approximately 0.02% of input. Data represent the mean ± SEM of 4 independent ChIP experiments. “*” signifies that the value for the comparisons shown is statistically significant (p<0.05).
The H3-K4-specific methyltransferase Set9 is expressed in pancreatic islets
The SET domain protein Set9 was originally identified as an H3-K4-specific methyltransferase (16,17). In earlier studies, we demonstrated that this enzyme occupied the endogenous proximal insulin promoter in β TC3 cells in a pattern consistent with the dimethylation of H3-K4 (15). To determine if Set9 is also expressed in normal pancreatic tissue, we performed immunohistochemical localization of Set9 in fixed, serial sections of mouse pancreas. As shown in Fig. 4, islets displayed a characteristic, mantle-type (α cell) staining for glucagon (Fig. 4B), and central-type (β cell) staining for insulin (Fig. 4C). Consistent with its known distribution (6), Pdx-1 displayed primarily central-type staining, similar to insulin, with additional staining seen in duct cells (Fig. 4D). Interestingly, Set9 staining was also observed in an islet-enriched pattern (Fig. 4E), whereas staining for p300 (a ubiquitously-expressed histone acetyltransferase), occurred in a broad distribution throughout the pancreatic parenchyma. To determine if Set9 expression overlaps with that of Pdx-1, we performed dual fluorescence imaging of islets using antibodies against both Pdx-1 and Set9. Fig. 4G-I demonstrates that Set9 staining almost completely overlaps with Pdx-1 staining, suggesting that these two proteins colocalize to the same cell types (β cells) within the islet. Just as importantly, these data suggest for the first time that Set9 exhibits a cell type-specific pattern of expression.
Figure 4. Set9 exhibits an islet-enriched expression pattern in the pancreas.
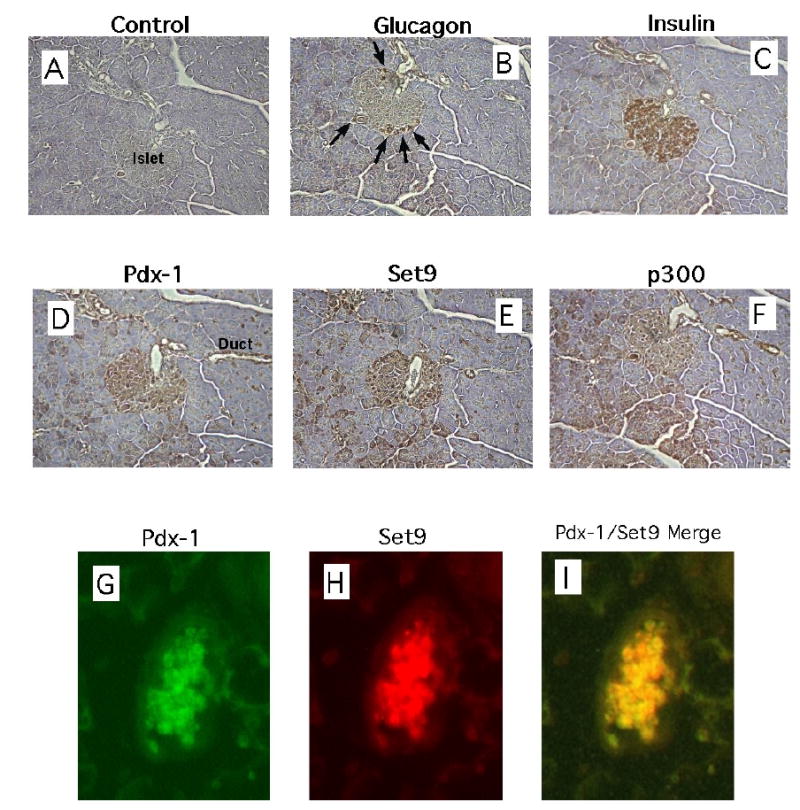
A-F, Serial, fixed pancreatic sections from C57/BL6 mice were treated with control serum (A), or antisera against glucagon (B), insulin (C), Pdx-1 (D), Set9 (E), and p300 (F), and subject to staining after using a secondary antibody linked to horseradish peroxidase. Arrows in panel B identify mantle cells of the islet staining for glucagon-producing cells. G-I, a section of C57/BL6 mouse pancreas containing the islet shown was treated with a mouse monoclonal antibody against Pdx-1 and a rabbit polyclonal antibody against Set9, then secondarily treated with antibodies fused to Cy2 or Cy3 recognizing mouse or rabbit Fc portions, respectively. Images from the green channel, representing Pdx-1 expression (G), and the red channel, representing Set9 expression (H), were merged to show co-localization of the two proteins in the same islet (I).
Pdx-1 physically interacts with and recruits the H3-K4-specific methyltransferase Set9 to the insulin promoter
To establish a possible relationship between histone methylation and Set9 recruitment to the insulin gene, we next examined the occupancy of Set9 at the insulin gene by ChIP after treatment of β TC3 cells with Ad-siPdx or control adenovirus (Ad-siLuc). As shown in Fig. 5A, Set9 occupancy at the insulin gene parallels the pattern of methylated H3-K4 (c.f. Fig. 1A). By contrast, we observed no Set9 occupancy at the distal insulin or myoD promoters (latter promoter data not shown) in β TC3 cells, consistent with the relative deficiency of methylated H3-K4 at these loci (Fig. 5A). Importantly, following Ad-siPdx treatment, Set9 occupancy at both the proximal insulin promoter and coding regions declined by approximately 40% compared to controls. These data are coincident with the 80% decline in Pdx-1 levels (Fig. 2A) and the 40–60% decline in di-methylated H3-K4 following Ad-siPdx treatment (Fig. 2B). Our findings strongly suggest that Pdx- 1 may at least partially be responsible for the recruitment of Set9 to the insulin gene, and that Set9 may be the relevant histone methyltransferase. Notably, Ad-siPdx treatment did not affect total Set9 levels in cells, as shown by the immunoblot in Fig. 5A (inset).
Figure 5. Pdx-1 recruits the H3-K4 methyltransferase Set9 to the insulin gene and directly interacts with the Set9 N-terminus in vitro.
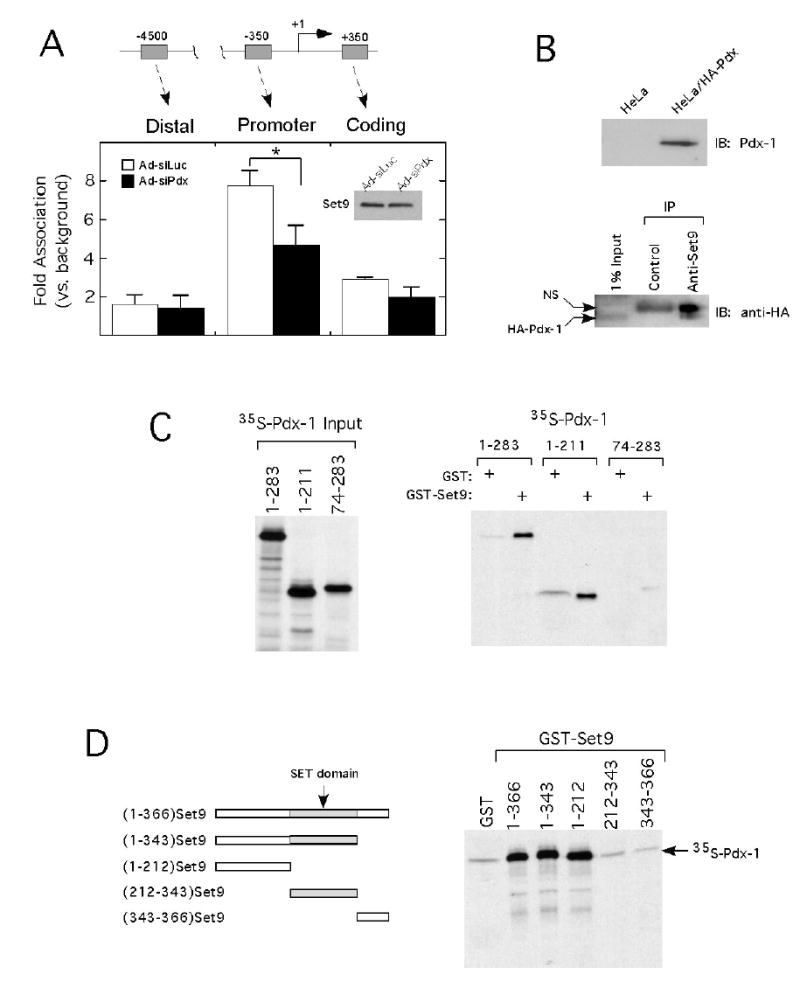
A, occupancy of Set9 along the insulin gene locus following treatment of βTC3 cells with Ad-siLuc or Ad-siPdx for 72 h. Data are derived from quantitative ChIP analysis and are expressed as fold-differences relative to background (in which normal rabbit serum is used instead of Set9 antibody). Background recovery was approximately 0.02% of input. Data represent the mean ± SEM of at least 3 independent ChIP experiments. “*” signifies that the value for the comparisons shown is statistically significant (p<0.05); inset: immunoblot showing Set9 protein levels after treatment of βTC3 cells with siLuc and siPdx. B, upper panel: immunoblot using anti-Pdx-1 antibody to demonstrate expression of HA-Pdx-1 in stably-transfected HeLa cells; lower panel: extracts from HeLa cells stably expressing HA-Pdx-1 were immunoprecipitated using the antiserum indicated, then subject to immunoblot analysis using anti-HA antibody to detect Pdx-1. IP, immunoprecipitation antibody; IB, immunoblotting antibody. C, left panel: 15% polyacrylamide gel of Pdx-1 protein fragments translated in vitro in the presence of 35S-Met that were used as input in GST pull-down experiments; right panel: 15% polyacrylamide gel to assess recovery of 35S-Pdx-1 proteins following incubation with GST-Set9, as detailed in Materials and Methods. D, Left panel: Schematic representation of GST-Set9 fragments used in these studies; right panel: 15% polyacrylamide to assess recovery of 35S-Pdx-1 following incubation with various GST-Set9 fusion proteins, as detailed in Materials and Methods.
To determine if Pdx-1 and Set9 interact physically, we first performed co-immunoprecipitation assays in HeLa cells stably expressing HA-tagged Pdx-1 protein (Fig. 5B, upper panel). As shown in Fig. 5B (lower panel), immunoprecipitation of Set9 protein using a polyclonal antibody resulted in co-precipitation of HA-Pdx-1, whereas no HA-Pdx-1 was recovered upon immunoprecipitation using control rabbit serum. To confirm the interaction in vitro, we next performed GST pull-down assays using bacterially-purified GST-Set9 proteins and 35S-labelled, in vitro translated Pdx-1 proteins (Fig. 5C, left panel). Fig. 5C (right panel) demonstrates that full-length Pdx-1 (amino acids 1-283) interacts with GST-Set9 but not GST alone (lanes 1–2); moreover, this interaction requires the N-terminal residues of Pdx-1 (amino acids 1–73, lanes 3–6). Importantly, the interaction between Set9 and Pdx-1 is only observed when the N-terminal residues of Set9 (amino acids 1–212) are fused to GST (Fig. 5D). These data suggest that Pdx-1 and Set9 physically interact via their respective N termini.
Pdx-1 and Set9 synergistically activate the insulin promoter
To determine if Pdx-1 and Set9 functionally interact at the insulin promoter to activate transcription, we performed cotransfection experiments in mammalian cells. As shown in Fig. 6A, when a cDNA encoding Pdx-1 is cotransfected into NIH3T3 cells with a reporter plasmid containing tandem copies of the E2/A3/A4 insulin mini-enhancer driving luciferase (20), 15-fold activation of luciferase activity is observed. No activation is observed, however, upon cotransfection of the reporter with either a cDNA encoding wild-type Set9 or a catalytically inactivate mutant of Set9 (H297A, Set9m, ref. (16)). Importantly, when cDNAs encoding Pdx-1 and wild-type Set9 are cotransfected, a synergistic 30-fold activation of the reporter is observed (Fig. 6A); cotransfection of Pdx-1 and Set9m leads to no synergy, suggesting that the methyltransferase activity of Set9 is necessary for synergistic reporter activation. No activation of a control reporter plasmid (5X-Gal4 UAS-luciferase) was observed with any of the cDNA constructs (Fig. 6A), emphasizing the specificity of our findings for the insulin promoter/enhancer. Protein levels of Pdx-1 were similar in all transfections, as were levels of Set9 and Set9m (Fig. 6B).
Figure 6. Pdx-1 and Set9 cooperatively activate the insulin promoter in vivo.
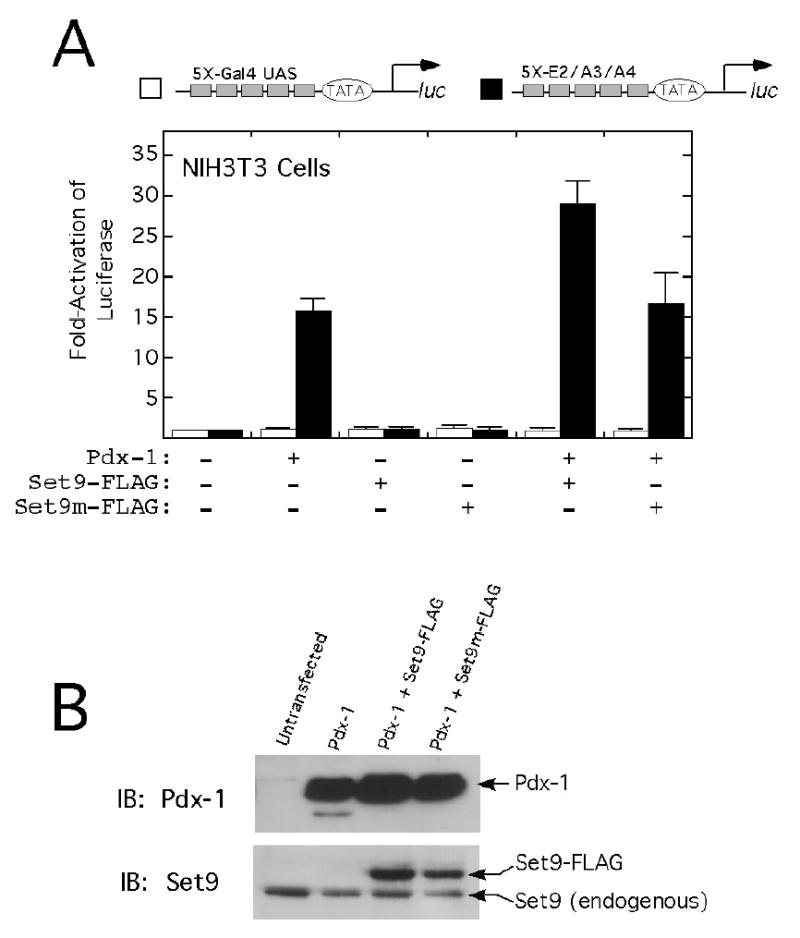
A, NIH3T3 cells were transiently co-transfected with the reporter plasmids (shown schematically at the top) and expression plasmids encoding Pdx-1, Set9-FLAG, or (H297A)Set9-FLAG (Set9m-FLAG), as detailed in Materials and Methods. Cells were harvested 48 h following transfection, and whole cell extracts were assayed for luciferase activity. Data are expressed as fold-activation relative to cells transfected with reporter only (defined as “1”). Data represent the mean ± SEM of three independent transfections performed in duplicate. B, immunoblots demonstrating levels of Pdx-1 (top) and Set9 (bottom) following transfection of NIH3T3 cells with the expression plasmids encoding the proteins indicated.
To determine if activation of the insulin mini-enhancer by Pdx-1 and Set9 requires chromatin and is accompanied by H3-K4 methylation, we performed in vitro transcription experiments using purified Pdx-1 and Set9 proteins and a “naked” or chromatin-reconstituted insulin mini-enhancer reporter (or control). Fig. 7 shows a schematic of the reporter DNA fragments used in these studies: the reporters, derived from vector pIC2085S (21), contains tandem copies of either the E2/A3/A4 mini-enhancer or Gal4 upstream activating sequences (negative control) between 5S ribosomal nucleosome positioning elements. The reporter was reconstituted with HeLa histone octamers by salt dilution (21) and demonstrated the characteristic nucleosomal laddering upon partial digestion with micrococcal nuclease (Fig. 7B). Fig. 7A (upper panel) shows that addition of Pdx-1 to naked (unreconstituted) insulin mini-enhancer DNA resulted in a 4-fold activation of the reporter, whereas no activation is observed in the absence of Pdx-1 binding sites (Gal4 control). Importantly, addition of Set9 (with or without the cofactor Ado-Met) had no independent effect upon the activation of either the insulin mini-enhancer or control reporters, consistent with the findings observed in mammalian transfection experiments (Fig. 6A). In contrast to the transfection experiments, addition of both Set9 and Pdx-1 to the naked DNA fragment resulted in no cooperative effect on transcription; this finding suggests that the cooperativity may require templates in the context of chromatin.
Figure 7. Pdx-1 and Set9 cooperatively activate only the chromatinized insulin minienhancer in vitro.
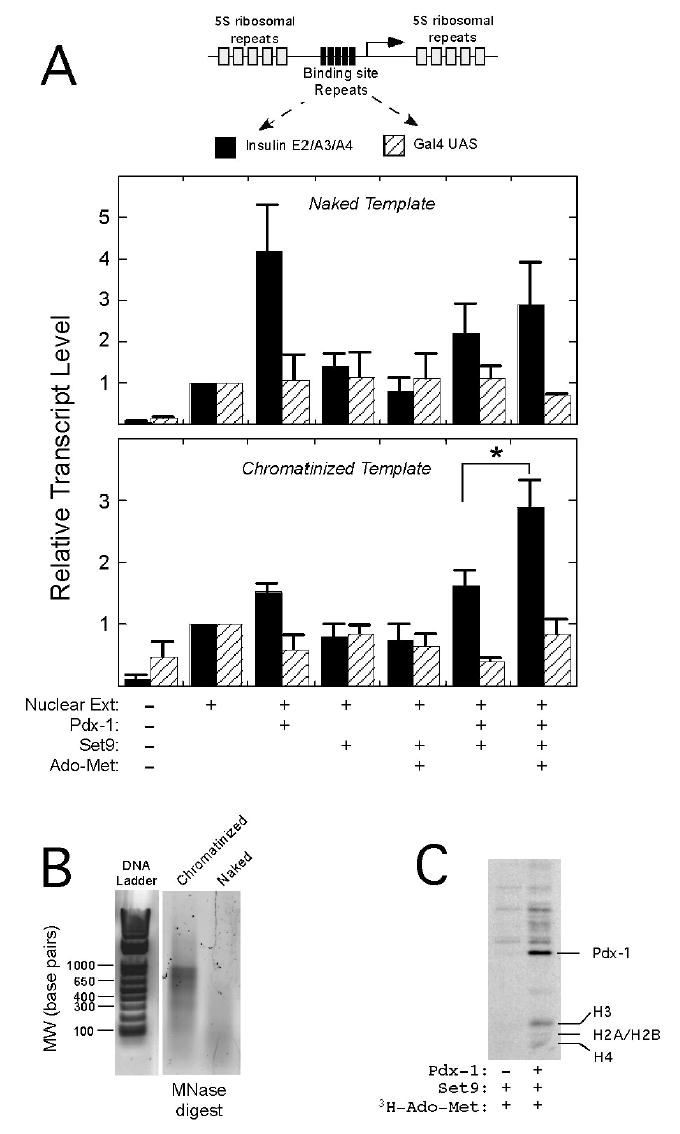
A, The reporter DNA fragments (derived from pIC2085S) containing tandem insulin E2/A3/A4 minienhancer elements or Gal4 upstream activating sequences (UASs) is shown schematically at the top. The reporter fragments were either used directly (“naked template,” upper panel) or reconstituted as chromatin with histone octamers (“chromatinized template,” lower panel) and subject to transcription reactions in vitro. The components added to the transcription reactions are indicated at the bottom of the panel. Transcription reactions were quantitated by real-time RT-PCR following a modified 5′-RACE reaction, as detailed in Materials and Methods. Data are expressed as transcript levels relative to a reaction containing only HeLa nuclear extract (defined as “1”). Data represent the mean ± SEM of three independent in vitro transcription reactions. B, 1.2% SYBR Green I-stained agarose gel analysis of chromatinized and naked reporter fragments following partial digestion with micrococcal nuclease. C, in vitro transcription reactions performed in the presence of 3H-Ado-Met and the indicated proteins were subject to electrophoresis on a 15% polyacrylamide gel and subsequent fluorography. The positions of 3H-methylated Pdx-1 and Histones H3, H2A/H2B, and H4 are indicated.
Upon chromatin-reconstitution of the insulin mini-enhancer DNA (Fig. 7A, lower panel), we observed that Set9 and Pdx-1 synergistically activate the reconstituted reporter, but only in the presence of the methylation cofactor Ado-Met. By contrast, no activation of the chromatinized control DNA was observed. These results emphasize (1) synergistic activation by Pdx-1 and Set9 requires chromatin, (2) the methyltransferase function of Set9 is necessary for transcriptional synergy, and (3) synergy between Pdx-1 and Set9 requires the presence of Pdx-1 binding sites. Polyacrylamide gel analysis of in vitro transcription reactions in the presence of 3H-Ado- Met (Fig. 7C) reveals that nucleosomal histones are not methylated by Set9 (consistent with observations by other investigators) (16,17). However, in the presence of Pdx-1 histone H3 appears to be methylated, suggesting that Pdx-1 enhances the ability of Set9 to methylate nucleosomal histones. Interestingly, Fig. 7C also suggests that Pdx-1 itself appears to be a substrate for methylation by Set9, although the significance of this methylation is unclear.
Endogenous Pdx-1 and Set9 activate the insulin promoter in β TC3 cells
We next asked whether diminishing endogenous levels of Pdx-1 and/or Set9 in β TC3 cells affect the transcription of a reporter gene driven by an intact insulin promoter. For these experiments, we generated a plasmid expressing a previously-described siRNA specific for set9 (18). This siRNA effectively knocks down endogenous Set9 levels after 48 h in transient transfections of HEK293 cells (Fig. 8A). Consistent with prior results (Fig. 2B and ref. (4)), coexpression of a plasmid expressing an siRNA against pdx-1 diminished insulin-promoter-luciferase activity in β TC3 cells by 40% (Fig. 8B). Importantly, expression of siRNA against set9 also caused ~40% decrease in reporter activity; however, the coexpression of siRNAs against both pdx-1 and set9 was no more effective in decreasing insulin reporter activity than either siRNA alone (Fig. 8B). These results suggest that Pdx-1 and Set9 contribute to activation of the insulin gene through an apparent co-dependent mechanism. Neither siRNA affected transcription of a promoterless luciferase reporter (Fig. 8B).
Figure 8. siRNA-directed knockdown of Pdx-1 and Set9 diminish insulin transcription in βTC3 cells.
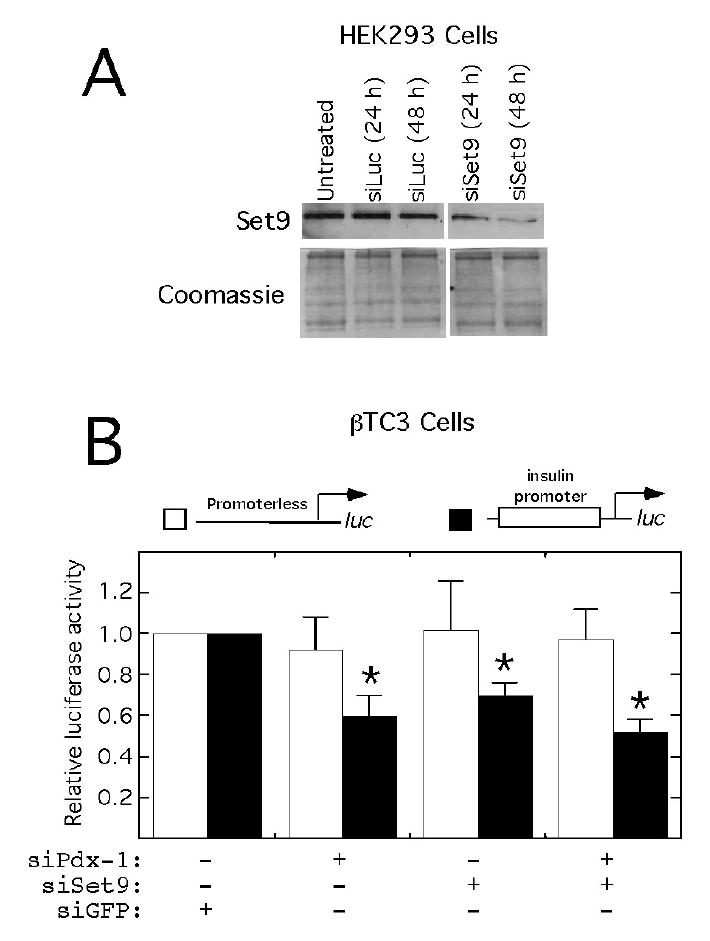
A, To demonstrate effectiveness of siRNA specific for set9, HEK 293 cells were transiently transfected with plasmids encoding a short hairpin RNA specific for either luciferase (siLuc) or set9 (siSet9), and whole cell extracts were analyzed by immunoblot for Set9 protein at the time points indicated (top panel). The corresponding 12% polyacrylamide, Coomassie-stained gel (bottom panel) demonstrates equivalent loading of protein samples. B, βTC3 cells were transiently co-transfected with the reporter plasmids shown schematically at the top and plasmids encoding siRNA specific for green fluorescent protein (GFP), pdx-1, or set9, as indicated. 48 h after transfection, cells were harvested and extracts were analyzed for luciferase activity. Data are expressed as fold-activation relative to cells transfected with reporter and siGFP only (defined as “1”). Data represent the mean ± SEM of three independent transfections performed in duplicate. “*” signifies that the value is statistically different from the activity observed upon transfection of cells with reporter and siLuc only (p<0.05).
DISCUSSION
Cell type- and sequence-specific transcription factors are well-known as crucial components in the development and function of islet β cells, however, the mechanism by which these factors regulate gene transcription is only beginning to be elucidated. In this manuscript, we describe a novel role for the insulin gene activator Pdx-1 in linking histone methylation with transcriptional elongation by RNA polymerase II. To our knowledge, this is one of the first reports implicating a cell-type specific transcription factor in the maintenance of histone H3-K4 methylation at an active mammalian gene.
Histone methylation is becoming recognized as a pivotal regulator of gene transcription. Until recently, methylation was thought to represent a stable modification, resulting in an epigenetic mark that reflected either the heterochromatic or euchromatic state of a given gene or locus. However, the recent discovery of LSD1, an H3-K4 de-methylase (31), and the recognition that differentially-modified histone variant proteins can be introduced during non-replicative phases of the cell cycle (32) suggest the possibility that histone methylation may be more dynamic than previously thought. Methylation of H3-K4 has been associated with active genes in euchromatic loci, including insulin (15). In this regard, out data show that siRNA-mediated knock-down of Pdx-1 results in both the attenuation of insulin transcription and the partial depletion of di-methylated H3-K4 at the insulin promoter and coding regions. Whether the decrease in di-methylated H3-K4 is secondary to de-methylation or histone replacement is unclear, but it does not appear that conversion of this species to mono- or tri-methylated forms is occurring, because we observed no quantitative changes in these other methylated isoforms upon knock-down of Pdx-1. The di-methyl mark of H3-K4 likely serves as a recognition site for chromodomain-containing proteins, which themselves contain chromatin remodeling activities that allow for more active gene transcription. Recently, the chromodomain-containing protein Chd1 (and its likely mouse homologs CHD1 and CHD4) was identified as a di-methyl H3-K4-binding chromatin modifying component of the yeast SAGA/SLIK complex (33).
Differential phosphorylation of Ser residues within the heptapeptide repeats of the CTD of Pol II has been linked to transcriptional initiation and elongation (10,11). Evidence for the relationship between histone methylation and CTD phosphorylation has been most compelling in yeast systems, where methylation of H3-K4 by Set1 is related to Ser5-P and initiation/early elongation, and methylation of H3-K36 by Set2 is related to Ser2-P and late elongation/termination (12,13). Our data confirm that Ser5 phosphorylation appears to be closely linked to transcriptional initiation whereas Ser2-P is linked to elongation at the insulin gene. More importantly, we have identified a unique role for the sequence-specific transcription factor Pdx-1 in the elongation by Pol II at the insulin gene. Because knock-down of Pdx-1 decreases di-methylation of H3-K4 and increases occupancy of the Ser5-P isoform of Pol II at the promoter, our data suggest a compelling relationship between histone methylation and Pol II phosphorylation that is distinctly different than that so far described in yeast. We recognize that our data are correlative at this stage and do not establish a cause-and-effect relationship between methylation and Pol II phosphorylation. We also note that because insulin transcription is attenuated (and not entirely eliminated) upon knock-down of Pdx-1, our data should be interpreted in the context of relative (rather than absolute) transcription of the insulin gene; thus di-methylation of H3-K4 may be a mechanism that causes (or reflects) changes to overall Pol II transcriptional rates.
Several models can be envisaged to explain the link between Pdx-1, histone methylation, and Pol II phosphorylation. In one such model, Pdx-1 may directly recruit a histone methyltransferase to the insulin gene, and the subsequent methyl mark imposed by this enzyme is then recognized by complexes that change Pol II phosphorylation states. In favor of this model, our data suggest that Pdx-1 is responsible for the recruitment of the H3-K4-specific histone methyltransferase Set9 to the insulin gene. In this regard, we show that (1) the two proteins exhibit islet-enriched, co-expression patterns in the pancreas, (2) the two proteins interact with each other via their N termini, (3) Set9 can potentiate transcriptional activation by Pdx-1, (4) Pdx-1 enhances methylation of nucleosomal histones by Set9, and (5) Set9 knock-down diminishes insulin gene reporter activity in β TC3 cells.
Interestingly, it appears that Set9 functions as a mono-methyltransferase in vitro (34), an observation that is seemingly inconsistent with our hypothesis that it controls histone di-methylation. However it is possible that the mono-methyltransferase specificity observed in vitro is altered in vivo, or that Set9 is closely linked to another methyltransferase that completes the dimethylation. Our data do not distinguish these possibilities; however, they are similar to the previously published observation that loss of Set9 recruitment to the β-globin locus resulted in loss of H3-K4 dimethylation (35). Set9 has also been demonstrated to methylate and functionally activate non-histone proteins (18), including the TAF10 component of TATA Binding Protein (36). We found that Set9 also appears to methylate Pdx-l. On the one hand, these findings raise the possibility that at least part of the transcriptional synergy we observed between Set9 and Pdx-1 might be related to methylation of TAF10 and/or Pdx-1 in vivo. On the other, it is clear from our in vitro transcription experiments (Fig. 7A) that synergy between Set9 and Pdx-1 occurs only in the presence of chromatin, and therefore the methylation of histones (rather than of TAF10 or Pdx-1) may be the more likely mechanism underlying the transcriptional synergy in our system.
Other models to explain the link between Pdx-1, histone methylation, and Pol II phosphorylation, however, cannot be ruled out. These would include the possibility that Pdx-1 first influences Pol II phosphorylation itself (through recruitment of a kinase), and the subsequent phosphorylated polymerase interacts with a histone methyltransferase to cause the specific methyl mark at the promoter and coding regions of the insulin gene. In this context, yeast Set1 and Set2 have been demonstrated to interact with phosphorylated Pol II at actively transcribed genes (12,37). Finally, the link between Pdx-1, histone methylation, and transcription could well represent some combination of the above models. Taken together, our findings clearly point to an interplay between chromatin modification and Pol II elongation as a mechanism by which sequence-specific factors at the insulin gene lead to transcriptional activation.
Acknowledgments
We would like to thank the UVA Diabetes and Endocrine Research Center Islet Isolation Core for assistance in immunostaining. We also wish to acknowledge Drs. C. Newgard, G. Weir, and T. Becker for provision of siRNA adenoviruses, Dr. D. Reinberg for the Set9 expression vectors, Dr. P. Grant for the pIC2085S vector, and Dr. M. German for the polyclonal Pdx-1 antibody.
Footnotes
This work was supported by grant R01 DK60581 from the National Institutes of Health and a Thomas R. Lee Career Development Award from the American Diabetes Association (both to R.G.M.)
Abbreviations: Ado-Met: S-Adenosyl Methionine; Ad-siPdx: adenovirus encoding siRNA specific for pdx-1; Ad-siLuc: adenovirus encoding siRNA specific for luciferase; ChIP: chromatin immunoprecipitation; CTD: C-terminal domain of RNA polymerase II; HA: hemagglutinin; Pol II: RNA polymerase II; RT-PCR: reverse transcriptase PCR; siRNA: small interfering RNA
The primers used in these studies equally and efficiently amplify both the mouse insulin I and insulin II genes. Hence, the reference to the insulin gene throughout the text refers to both mouse genes.
References
- 1.Chakrabarti SK, Mirmira RG. Trends Endocrinol Metab. 2003;14:78–84. doi: 10.1016/s1043-2760(02)00039-5. [DOI] [PubMed] [Google Scholar]
- 2.Habener JF, Kemp DM, Thomas MK. Endocrinology. 2005;146:1025–1034. doi: 10.1210/en.2004-1576. [DOI] [PubMed] [Google Scholar]
- 3.Chakrabarti SK, James JC, Mirmira RG. J Biol Chem. 2002;277:13286–13293. doi: 10.1074/jbc.M111857200. [DOI] [PubMed] [Google Scholar]
- 4.Iype T, Francis J, Garmey JC, Schisler JC, Nesher R, Weir GC, Becker TC, Newgard CB, Griffen SC, Mirmira RG. J Biol Chem. 2005;280:16798–16807. doi: 10.1074/jbc.M414381200. [DOI] [PubMed] [Google Scholar]
- 5.Jonsson J, Carlsson L, Edlund T, Edlund H. Nature. 1994;371:606–609. doi: 10.1038/371606a0. [DOI] [PubMed] [Google Scholar]
- 6.Offield MF, Jetton TL, Labosky PA, Ray M, Stein RW, Magnuson MA, Hogan BL, Wright CV. Development. 1996;122:983–995. doi: 10.1242/dev.122.3.983. [DOI] [PubMed] [Google Scholar]
- 7.Stoffers DA, Zinkin NT, Stanojevic V, Clarke WL, Habener JF. Nat Genet. 1997;15:106–110. doi: 10.1038/ng0197-106. [DOI] [PubMed] [Google Scholar]
- 8.Melloul D. Ann N Y Acad Sci. 2004;1014:28–37. doi: 10.1196/annals.1294.003. [DOI] [PubMed] [Google Scholar]
- 9.Ohneda K, Ee H, German M. Semin Cell Dev Biol. 2000;11:227–233. doi: 10.1006/scdb.2000.0171. [DOI] [PubMed] [Google Scholar]
- 10.Gerber M, Shilatifard A. J Biol Chem. 2003;278:26303–26306. doi: 10.1074/jbc.R300014200. [DOI] [PubMed] [Google Scholar]
- 11.Hampsey M, Reinberg D. Cell. 2003;113:429–432. doi: 10.1016/s0092-8674(03)00360-x. [DOI] [PubMed] [Google Scholar]
- 12.Ng HH, Robert F, Young RA, Struhl K. Mol Cell. 2003;11:709–719. doi: 10.1016/s1097-2765(03)00092-3. [DOI] [PubMed] [Google Scholar]
- 13.Xiao T, Hall H, Kizer KO, Shibata Y, Hall MC, Borchers CH, Strahl BD. Genes Dev. 2003;17:654–663. doi: 10.1101/gad.1055503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schneider R, Bannister AJ, Myers FA, Thorne AW, Crane-Robinson C, Kouzarides T. Nat Cell Biol. 2004;6:73–77. doi: 10.1038/ncb1076. [DOI] [PubMed] [Google Scholar]
- 15.Chakrabarti SK, Francis J, Ziesmann SM, Garmey JC, Mirmira RG. J Biol Chem. 2003;278:23617–23623. doi: 10.1074/jbc.M303423200. [DOI] [PubMed] [Google Scholar]
- 16.Nishioka K, Chuikov S, Sarma K, Erdjument-Bromage H, Allis CD, Tempst P, Reinberg D. Genes Dev. 2002;16:479–489. doi: 10.1101/gad.967202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang H, Cao R, Xia L, Erdjument-Bromage H, Borchers C, Tempst P, Zhang Y. Mol Cell. 2001;8:1207–1217. doi: 10.1016/s1097-2765(01)00405-1. [DOI] [PubMed] [Google Scholar]
- 18.Chuikov S, Kurash JK, Wilson JR, Xiao B, Justin N, Ivanov GS, McKinney K, Tempst P, Prives C, Gamblin SJ, Barlev NA, Reinberg D. Nature. 2004;432:353–360. doi: 10.1038/nature03117. [DOI] [PubMed] [Google Scholar]
- 19.Schisler JC, Jensen PB, Taylor DG, Becker TC, Knop FK, Takekawa S, German M, Weir GC, Lu D, Mirmira RG, Newgard CB. Proc Natl Acad Sci U S A. 2005;102:7297–7302. doi: 10.1073/pnas.0502168102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ohneda K, Mirmira RG, Wang J, Johnson JD, German MS. Mol Cell Biol. 2000;20:900–911. doi: 10.1128/mcb.20.3.900-911.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Steger DJ, Eberharter A, John S, Grant PA, Workman JL. Proc Natl Acad Sci U S A. 1998;95:12924–12929. doi: 10.1073/pnas.95.22.12924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Utley RT, Ikeda K, Grant PA, Cote J, Steger DJ, Eberharter A, John S, Workman JL. Nature. 1998;394:498–502. doi: 10.1038/28886. [DOI] [PubMed] [Google Scholar]
- 23.Mirmira RG, Watada H, German MS. J Biol Chem. 2000;275:14743–14751. doi: 10.1074/jbc.275.19.14743. [DOI] [PubMed] [Google Scholar]
- 24.Iype T, Taylor DG, Ziesmann SM, Garmey JC, Watada H, Mirmira RG. Mol Endocrinol. 2004;18:1363–1375. doi: 10.1210/me.2004-0006. [DOI] [PubMed] [Google Scholar]
- 25.Yoshida T, Sinha S, Dandre F, Wamhoff BR, Hoofnagle MH, Kremer BE, Wang DZ, Olson EN, Owens GK. Circ Res. 2003;92:856–864. doi: 10.1161/01.RES.0000068405.49081.09. [DOI] [PubMed] [Google Scholar]
- 26.Dignam JD, Lebovitz RM, Roeder RG. Nucleic Acids Res. 1983;11:1475–1489. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Okada Y, Feng Q, Lin Y, Jiang Q, Li Y, Coffield VM, Su L, Xu G, Zhang Y. Cell. 2005;121:167–178. doi: 10.1016/j.cell.2005.02.020. [DOI] [PubMed] [Google Scholar]
- 28.Yang Z, Chen M, Fialkow LB, Ellett JD, Wu R, Nadler JL. Pancreas. 2003;26:e99–104. doi: 10.1097/00006676-200305000-00021. [DOI] [PubMed] [Google Scholar]
- 29.Efrat S, Surana M, Fleischer N. J Biol Chem. 1991;266:11141–11143. [PubMed] [Google Scholar]
- 30.Palancade B, Bensaude O. Eur J Biochem. 2003;270:3859–3870. doi: 10.1046/j.1432-1033.2003.03794.x. [DOI] [PubMed] [Google Scholar]
- 31.Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA, Shi Y. Cell. 2004;119:941–953. doi: 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 32.Schwartz BE, Ahmad K. Genes Dev. 2005;19:804–814. doi: 10.1101/gad.1259805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pray-Grant MG, Daniel JA, Schieltz D, Yates JR, 3rd, Grant PA. Nature. 2005;433:434–438. doi: 10.1038/nature03242. [DOI] [PubMed] [Google Scholar]
- 34.Xiao B, Jing C, Wilson JR, Walker PA, Vasisht N, Kelly G, Howell S, Taylor IA, Blackburn GM, Gamblin SJ. Nature. 2003;421:652–656. doi: 10.1038/nature01378. [DOI] [PubMed] [Google Scholar]
- 35.West AG, Huang S, Gaszner M, Litt MD, Felsenfeld G. Mol Cell. 2004;16:453–463. doi: 10.1016/j.molcel.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 36.Kouskouti A, Scheer E, Staub A, Tora L, Talianidis I. Mol Cell. 2004;14:175–182. doi: 10.1016/s1097-2765(04)00182-0. [DOI] [PubMed] [Google Scholar]
- 37.Kizer KO, Phatnani HP, Shibata Y, Hall H, Greenleaf AL, Strahl BD. Mol Cell Biol. 2005;25:3305–3316. doi: 10.1128/MCB.25.8.3305-3316.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]