

Abstract
Both epidermal growth factor receptor (EGFR) and protein kinase C (PKC) play important roles in glioblastoma invasive growth, however, the interaction between EGFR and PKC is not well characterized in glioblastomas. Treatment with EGF stimulated global phosphorylation of tyrosine 845, 992, 1068 and 1045 residues of the EGFR in glioblastoma cell lines (U-1242 MG and U-87 MG). Interestingly, phorbol 12-myristate 13- acetate (PMA) stimulated phosphorylation of the EGFR only at Tyr 1068 residues in the two glioblastoma cell lines. The phosphorylation of EGFR at Tyr 1068 was not detected in normal human astrocytes treated with the phorbol ester. The PMA-induced phosphorylation of EGFR at Tyr 1068 was blocked by BIM, a PKC inhibitor, and rottlerin, a specific PKC δ inhibitor. In contrast Go 6976, an inhibitor of classical PKC isozymes had no effect on PMA-induced EGFR phosphorylation. Furthermore, gene silencing with PKC δ siRNA, siRNA against c-Src, mutant c-Src (Ser12Cys/Ser48Ala) and treatment with a c-Src inhibitor (PP2) abrogated PMA-induced EGFR phosphorylation at Y1068. PMA induced serine/threonine phosphorylation of Src which was blocked by both BIM and rottlerin. Inhibition of EGFR with AG 1478 did not significantly alter PMA-induced EGFR (Tyr 1068) phosphorylation, but completely blocked EGF-induced phosphorylation of EGFR. The effect of PMA on mitogen activated protein kinase (MAPK) phosphorylation and glioblastoma cell proliferation were reduced by BIM, rottlerin, the MEK inhibitor UO 126, PKC δ siRNA and c-Src siRNA. Taken together, our data demonstrate that, PMA transactivates EGFR and increases cell proliferation by activating PKC δ/c-Src pathway in glioblastomas.
The abbreviations used are: PMA, Phorbol myristate acetate; PKC, protein kinase C; EGF, Epidermal growth factor; EGFR, Epidermal growth factor receptor; BIM, bisindolylmaleimide; ERK, extracellular signal-regulated kinase; MEK, mitogen-activated kinase effector kinase; α-MEM, minimal essential medium- α; siRNA, small interfering ribonucleic acid; PAGE, polyacrylamide gel electrophoresis; GBM, glioblastoma multiforme
Glioblastoma multiforme (GBM) as a biologically aggressive neoplasm has an elevated, often abberant, proliferative capacity with a diffuse pattern of brain invasion. It is the most malignant astrocytic tumor, composed of poorly differentiated neoplastic astrocytes (1–3). 50–60 % and 40 % of GBMs have overexpression and amplification of the EGFR, respectively (4–7). The overexpression and amplification of EGFR contribute to the malignant phenotype of human glioblastomas (6, 8). In addition, malignant glioma cells have higher levels of PKC than nonneoplastic astrocytes (9–12). This may suggest that excessive PKC activity may significantly contribute to astrogial tumorigenicity.
The EGFR or ErbB family belongs to the subclass I of the super family of the receptor tyrosine kinase (RTKs). Receptor tyrosine kinases represent an important subclass of these transmembrane proteins with the EGFR being the most prominent representative. The EGFR controls a wide variety of biological processes such as cell proliferation, differentiation, migration and modulation of apoptosis (13). There are four members of the ErbB family: EGFR (also termed as ErbB1/HER1), ErbB2/Neu/HER2, ErbB3/HER3 and ErbB4/HER4. Members of the ErbB family are characterized by a modular structure consisting of an extracellular ligand binding domain, a single hydrophobic transmembrane region and the intracellular part habouring the highly conserved tyrosine kinase domain (14). The ErbB receptors are activated by a number of ligands known as EGF-related peptide growth factors (15). These ligands include EGF, amphiregulin and transforming growth factor α (TGFα) which bind specifically to ErbB1, betacellulin, heparin-binding EGF (HB-EGF) and epiregulin which exhibit dual specificity in that they bind ErbB1 and ErbB4. The neuregulins 1 and 2 both bind to ErbB3 and ErbB4 (16), while neuregulins 3 and 4 bind to ErbB4 and not ErbB3 (17–18). The EGF-related peptide growth factors bind the extracellular domain of the ErbB receptor leading to formation of homo-and heterodimers. Dimerization consequently stimulates the intrinsic tyrosine kinase activity of the receptors and triggers autophosphorylation of specific residues within the cytoplasmic domain. These phosphorylated residues serve as docking sites for signaling molecules involved in the regulation of intracellular cascades (19). The activation of the EGFR in a large percentage of glioblastomas suggests an important role for this receptor in astrocytic tumor development. EGFR activation may therefore contribute to tumor malignancy by promoting invasion, proliferation and metastasis (20).
PKC represents a major cellular receptor for tumor- promoting phorbol esters (21–23). PKC comprises a family of phospholipid-dependent serine threonine kinases that play important roles in signal transduction associated with a variety of cellular responses including cell growth and differentiation, gene expression, hormone secretion and membrane function (24–27). Activation of PKC leads to the phosphorylation of proteins that are involved in the regulation of cell growth, differentiation and apoptosis (28–30). PKC consists of at least 11 isoforms showing diversity in their structures, cellular distribution and biological functions and have been divided into three groups based on their structure and cofactor requirements (31). The conventional PKC isoforms α, βI, βII and γ require diacylglycerol (DAG), phosphatidylserine, and Ca2+ for activity. The novel PKC isoforms δ, ɛ, η, and θ do not require Ca2+ as a cofactor but do require DAG and phosphatidylserine. The atypical PKC isoforms ζ, ι/λ do not require Ca2+ and are not activated by PMA or DAG but do bind to phosphatidylserine when active (24,28,29,32).
The EGFR, a protein tyrosine kinase is phosphorylated by protein kinase C at threonine 654 in A431 cells (33–35). Moro et al (36) have reported that integrins induced phosphorylation of EGF receptor on tyrosine residues 845, 1068, 1086 and 1173 by complexing with p130Cas adaptor protein. Furthermore, Samet et al., (37) have shown that Zn2+, a particulate matter induces the phosphorylation of EGF receptor at tyrosine 1068, in addition to tyrosine 845 and 1173. The mechanism through which PKC transactivates EGFR is not fully understood. In this study we examined the relationship between PKC and EGFR expression in glioblastoma cell lines to identify the PKC isoform and the intermediate downstream target involved in the activation of EGFR. The data suggest that PKC δ and c-Src are involved in the transactivation of EGFR and glioblastoma mitogenic response to PMA. The existence of a cross talk between PKC activation and the EGFR tyrosine kinase receptor that is overexpressed in more than 50% of primary GBMs provides a novel signaling pathway that is altered in astrocytic tumors and may provide a useful therapeutic target.
EXPERIMENTAL PROCEDURES
Materials
PMA, EGF and tubulin antibody (DMA 1) were purchased from Sigma Chemical (St. Louis MO). The phosphospecific antibodies directed against EGFR at tyrosine 992, 845, 1045, and EGFR antibody were purchased from Cell Signaling Technology (Beverly, MA). Phosphospecific antibodies directed against EGFR at tyrosine 1068 were purchased from Biosource (Camarillo, CA) and Cell Signaling Technology (Beverly, MA). The specific PKC inhibitors, Go 6976, bisindolylmaleimide (BIM), MEK inhibitor (UO 126), Rottlerin, PP2 (4-Amino-5-(4- chlorophenyl)-7-(t-butyl) pyrazolo[3,4-d] pyrimidine, PP3 (4-Amino-7-phenylpyrazol[3,4-d] pyrimidine and AG1478 (EGFR kinase inhibitor) are products of Calbiochem (San Diego, CA). Ingenol was purchased from RBI (Natick, MA). The phosphospecific antibody to dually phosphorylated extracellular signal regulated kinase –1 and –2 (ERK1/ERK2) was obtained from Biosource (Camarillo, CA), PKC δ antibody was obtained from Transduction Laboratories (San Diego, CA), while PKCɛ and θ antibody were purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA). PKC δ siRNA and c-Src siRNA were purchased from Dharmacon (Lafayette, CO). c-Src mutant (Ser12Cys/Ser48Ala) was made in Dr. Sally Parsons laboratory.
Cell Cultures
Human U-1242 MG cell line was kindly supplied by Dr. A. J. Yates (Ohio State University), while U-87 MG was obtained from ATCC (Manassas, VA). The normal human astroctytes were obtained from Clonetics (San Diego). The cell lines were originally isolated from astrocytic tumors that were designated as glioblastomas and their characteristics were described previously by Hussaini et al (38). Cell lines were regularly determined to be free of mycoplasma with reagent from Gen-Probe Inc. (San Diego, CA). Cells were grown in minimal essential medium -α modification with 10% defined fetal bovine serum (Hyclone, Logan UH) and 20 μg/ml bovine zinc insulin (25.7 IU/mg; Sigma). The cells were cultured to 100% confluence and passage every 4–5 days, from an initial concentration of 6–8 × 103/cm2 in T-flasks or 6- or 24-well plates and cultured in astrocyte growth medium (AGM 7, containing 5% fetal bovine serum) at 37°C in 5% CO2 and 90% relative humidity. Prior to assays, cultures that were 80–100% confluent were washed three times with serum free medium before exposure to PMA (100 nM). In studies involving the pharmacological inhibitors, the inhibitors were added to the cells for 60 min before PMA. After the treatments the medium was removed and the cells were examined by Western blot analysis.
Western Blot Analysis
After the different treatments, the cells were washed in PBS containing sodium orthovanadate (0.2 mM) and extracted with 1% Triton X-100, 0.2% Nonidet P- 40, in the presence of 2 mM EDTA, 100 μM phenylmethylsulfonyl fluoride, 5 μg/ml leupeptin, and 1 μg /ml aprotinin. Cells were centrifuged at 14,000 g for 1 min at 4°C. Protein concentration of supernatant was determined by the BCA protein assay (Bio-Rad). Proteins were boiled for 5 min in SDS . polyacrylamide gel electrophoresis (PAGE) buffer. Proteins (200 μg/lane) were separated by SDS-PAGE on 8% polyacrylamide gels and then electroblotted onto nitrocellulose and reacted with monoclonal and polyclonal antibodies. Immunoblotted proteins were detected using the ECL reagents (Amersham Pharmacia Biotech) as described by the manufacturer with horseradish peroxidase-conjugated secondary antibodies (Sigma). EGFR phosphorylation was stimulated in cells that had been cultured in serum free medium for 24 h by incubation with either PMA (100 nM) or EGF (25 ng/ml) for 10–60 min. The incubation was terminated by the addition of ice-cold phosphate-buffered saline (137 mM NaCl, 8.1 mM Na2HPO4, 2.7 mM KCl, 1.5 mM KH2PO4, at pH 7.4) containing 0.2 mM sodium orthovanadate to the culture. Phosphate buffered saline was then aspirated, and cells were solubilized with 1% Triton X-100, 0.2% Nonidet P-40, in the presence of 2 mM EDTA, 100 μM phenylmethylsulfonyl fluoride, 5 μg/ml leupeptin, and 1 μg /ml aprotinin, 50 mM sodium fluoride, 5 mg/ml dithiothreitol, and 0.2 mM sodium orthovanadate. The 1% Triton-X extract was centrifuged at 14,000 × g for 1 min, elecrophoresed, and immunoblotted as described above with mouse or rabbit antibodies specific for EGFR. Densitometer and Image Quant software (Molecular Dynamics) were used to quantitate the protein bands.
siRNA Transfections
PKC δ siRNA and siRNA c-Src were synthesized and purified by Dharmacon Inc.(Lafayette, CO). siRNA PKC δ (200nM) and siRNA c-Src (400 nM) were transfected separately into both U-1242 MG and U-87 MG cells using the Amaxa Nucleofector ™(Amaxa, Gaithersburg, MD). Briefly, confluent cells were trypsinised and resuspended in amaxa Nucleofector solution V at a density of 2 X106 per 100 μl of solution, and either 200 nM of the PKC δ siRNA or siRNA c-Src (400 nM) was added. Cells were transfected using the A23 pulsing program. Immediately after the electroporation, cells were suspended in 4.9 ml of α-MEM plus 10% FBS and incubated at 37°C (39). Similarly, 5 μg mutant c-Src (ser12cys/ser48ala) was transfected into astrocytic tumor cells following similar protocol. 48 h later the cells were lysed as stated above. The cell lysates (200 μg of proteins/ lane) were separated by SDS-PAGE on 8% polyacrylamide gels. Proteins were then electroblotted onto nitrocellulose and reacted with PKC δ monoclonal antibody, PKC α polyclonal antibody and phospho EGFR (Tyr 1068). The antibodies were detected with anti-mouse or anti-rabbit peroxidase –conjugates, and final detection was carried out with ECL (Amersham Pharmacia Biotech) as described by the manufacturer.
Immunoprecipitation
Cells were allowed to grow to 70–90% confluence. After starvation for 24 h in α-MEM containing no FBS, cells were treated with EGF (25 ng/ml) or PMA 100 nM for 10 and 30 min respectively at 37°C. Other cultures were treated with PKC inhibitor (BIM 1 μM) and PKC δ inhibitor (Rottlerin 5 μM) for 60 min before the addition of PMA. Cells were washed twice with cold PBS and extracted with 1% Triton X-100, 0.2% Nonidet P-40, in the presence of 2 mM EDTA, 100 μM phenylmethylsulfonyl fluoride, 5 μg/ml leupeptin, and 1 μg /ml aprotinin. Cell lysates were then subjected to centrifugation at 14,000 g for 1 min at 4°C. After normalization of supernatant derived from untreated and treated cells by protein concentration (Biorad, Richmond, CA), 1 mg of each cell lysate was incubated with monoclonal c-Src antibody (2–17) overnight at 4°C. Immune complexes were collected with Protein-G beads and washed five times with immunoprecipation buffer containing 50 mM sodium acetate buffer (pH 5.0), 500 mM NaCl, 0.1 % SDS, 1 % NP-40. The resulting immunoprecipitate was then subjected to 8% SDS-polyacrylamide gel electrophoresis (PAGE) and then transferred onto nitrocellulose membranes followed by Western blot analysis with phospho serine/threonine and c-Src (2–17) antibodies using the ECL detection system.
Incorporation of [3H] Thymidine into DNA.
Relative rates of DNA synthesis were assessed by determination of [3H] thymidine incorporation. U-87 MG cells (4.5 X 104) were seeded into a 24 well plate. Upon serum deprivation for 24 h, cells were subjected to 60 min preincubation with BIM (1 μM) Rottlerin (5 μM), PP2 (5 μM), AG1478 (100 nM), or UO126 (10 μM), followed by treatment with PMA (100 nM) or EGF (25 ng/ml) for an additional 18 h. In another set of experiments, cells transfected with either siRNA directed against PKC δ or c-Src were also seeded into a 24-well plate at a density of 4.5 X 10−4 cells. Upon serum starvation, the transfected cells were treated with either PMA or EGF for 18 h. Cells were pulsed with 0.5 μCi of [3H] thymidine for 6 h and then washed with phosphate buffered saline. This was followed by 10 min washes with 10% trichloroacetic acid, first at 4°C and then at room temperature. Cells were then dissolved in 1 N NaOH overnight and neutralized with an equal volume of 2N HCl and placed in scintillation fluid. [3H] Thymidine incorporation was determined in a Beckman scintillation counter.
Statistical analysis
Data are expressed as mean ± SEM. Differences between two or multiple groups were tested using the analysis of variance (ANOVA) followed by a calculation of Fisher’s least significant difference. Differences with a p value of <0.05 were considered statistically significant.
RESULTS
PMA and EGF induce EGFR Tyrosine Phosphorylation in Glioblastoma Cells
Two well characterized glioblastoma cell lines (U-1242 MG and U-87 MG) were used for phosphorylation studies using EGF and PMA. The U-1242 MG cells have mutant p53 (40), while the U-87 MG cells have wildtype p53 and mutant PTEN (41). Treatment with EGF (25 ng/ml) induced EGFR phosphorylation at all tyrosine residues tested, Tyr 1068, Tyr 1045, Tyr 992 and Tyr 845. The phosphorylation induced by EGF was time-dependent (Figure 1). The two cell lines showed detectable levels of total EGFR, which was not altered by transient treatment with EGF for 10–60 min. Unlike EGF, treatment of the cells with PMA (100 nM) did not induce global phosphorylation of all the tyrosine residues. Only one of the major autophosphorylation sites of the EGFR (Tyr 1068) in U-1242 MG and U-87 MG was phosphorylated (Figure 2). Similarly, PMA did not change the level of total EGFR protein. When the cell lysates were immunoprecipitated with anti EGFR antibody and immunoblotted with phospho-specific EGFR (Tyr 1068) antibody, both PMA and EGF induced EGFR (Tyr 1068) phosphorylation (Figure 3). The specificity of antibody against EGFR (Y1068) was confirmed in NIH3T3 cells expressing mutant Y1068, where the tyrosine has been replaced with phenyalanine (data not shown). In contrast, treatment of normal human astrocytes with PMA (100 nM) for 30 min did not induce EGFR Tyr 1068 phosphorylation, even though the cells express the receptor (Figure 4).
Fig. 1.
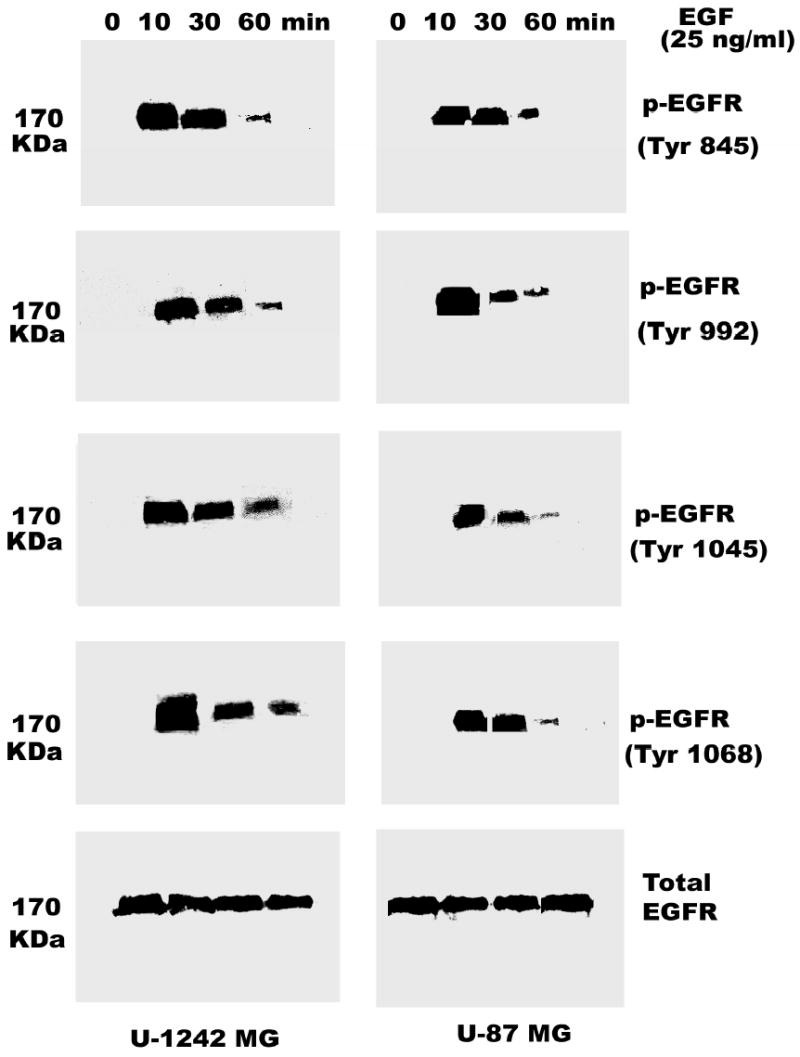
Time-dependent phosphorylation of EGFR in glioblastoma cells. Cells were serum-starved for 24 h after reaching 80–100% confluence by replacing the media with serum free α-MEM media. The cells were treated with EGF (25 ng/ml) at different time points (0, 10, 30, 60 min). Protein (200 μg/ lane) was fractionated by 8% SDS-PAGE and then transferred on to nitrocellulose. The nitrocellulose was reacted with primary antibody to EGFR (Tyr-845, Tyr-992, Tyr-1045, Tyr- 1068). Final detection was carried out using the enhanced chemuluminescence reagent (ECL). Blots were stripped and reprobed for EGFR total. The lower panel shows the total EGFR
Fig. 2.
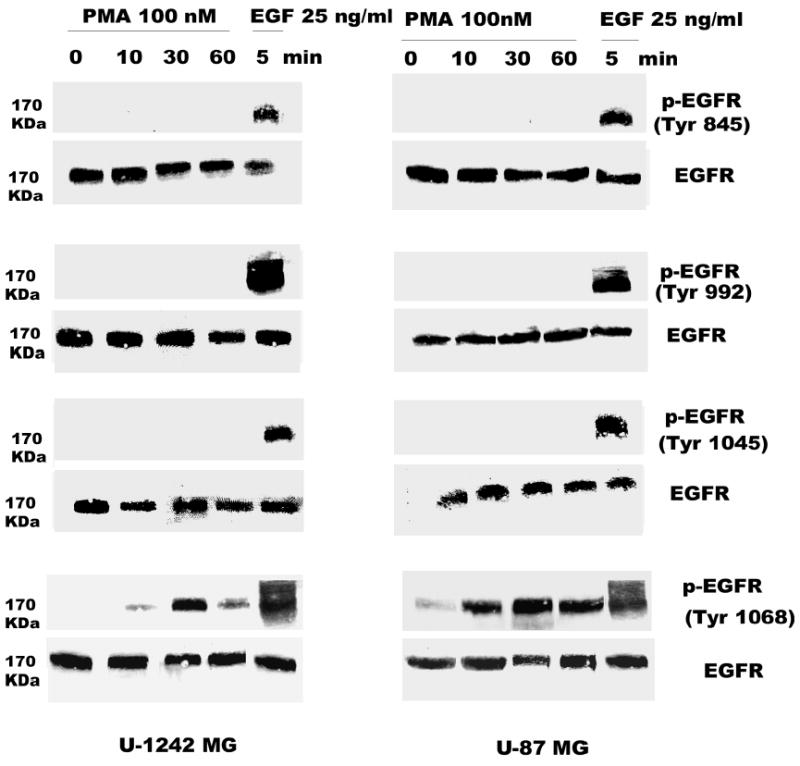
PMA induces phosphorylation of EGFR (Tyr-1068). Serum-starved U-1242 MG and U-87 MG cells were treated with PMA (100 nM) at different time points (0, 10, 30, and 60 min). Triton solubilized astrocytic tumor cell lysates (200 μg/lane) were separated using 8% polyacrylamide gels and electroblotted on to nitrocellulose. The blots were probed with antibodies to phospho-EGFR (Tyr 1068) and total EGFR.
Fig. 3.
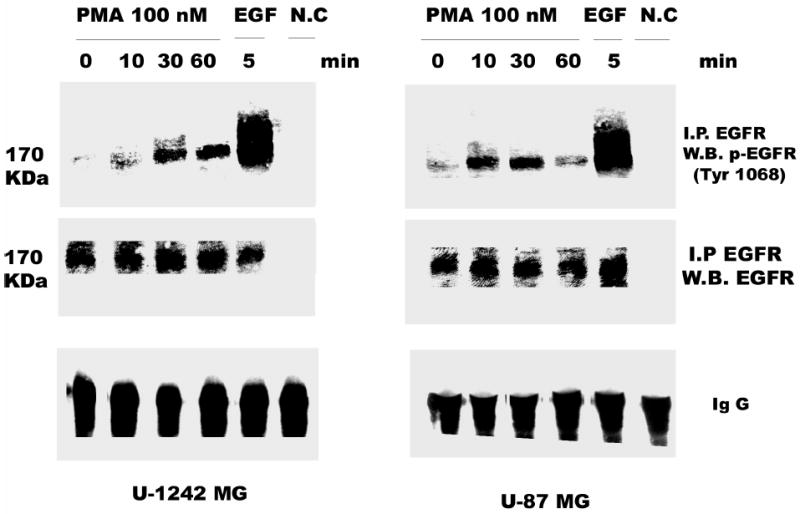
PMA stimulates tyrosine phosphorylation of EGFR (Tyr 1068). Serum- starved U-1242 MG and U-87 MG were stimulated with PMA for the indicated time periods. These cells were lysed using 1% Trition lysis buffer after washing with ice-cold PBS. Cell lysates were immunoprecipitated with anti EGFR antibody and then immunoblotted (IB) with antiphospho EGFR (Tyr 1068) or antibody to total EGFR.
Fig. 4.
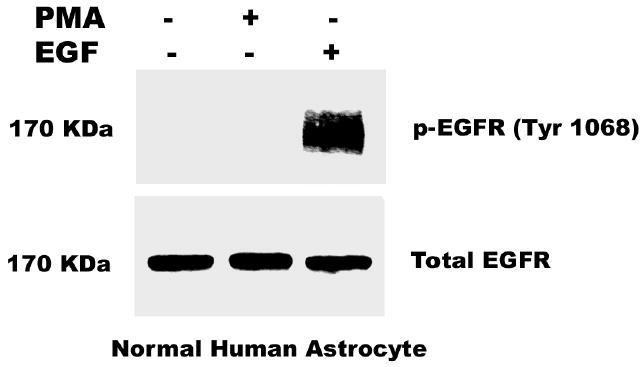
Effect of PMA and EGF on Normal Human Astrocytes (NHA). NHA cells grown to 80–100% confluence were serum-starved overnight. The cells were then treated with PMA (100nM) for 30 min and EGF (25 ng/ml) for 10 min. Protein lysates (200 μg/lane) were fractionated using 8% SDS-PAGE gels and was electroblotted onto nitrocellulose. The blots were probed with antibody to EGFR (Tyr 1068). The membrane was stripped and reprobed for total EGFR.
Effect of Pharmacological Inhibitors on PMA–induced EGFR Phosphorylation
Since PMA interacts with and activates both classical (α, βI, βII and γ) and novel (η, δ, ɛ, and θ) PKC isozymes, we designed experiments to identify the PKC isozyme(s) mediating PMA-induced phosphorylation of EGFR at Tyr 1068. Studies from our laboratory and other groups have shown that bisindolylmaleimide (BIM) inhibits the activation of both novel and classical PKCs, while Go 6976 blocks the activities of classical PKCs only. In order to determine whether classical or novel PKC isozymes were involved in PMA-induced Tyr 1068 phosphorylation, we pretreated the cells with either BIM (1 μM) or Go 6976 (10 μM) for 60 min prior to PMA addition to the cultures. BIM attenuated PMA-induced EGFR phosphorylation in both U-87 MG and U-1242 MG, while Go 6976 had no effect on PMA-induced phosphorylation of Tyr 1068 (Figure 5).
Fig. 5.
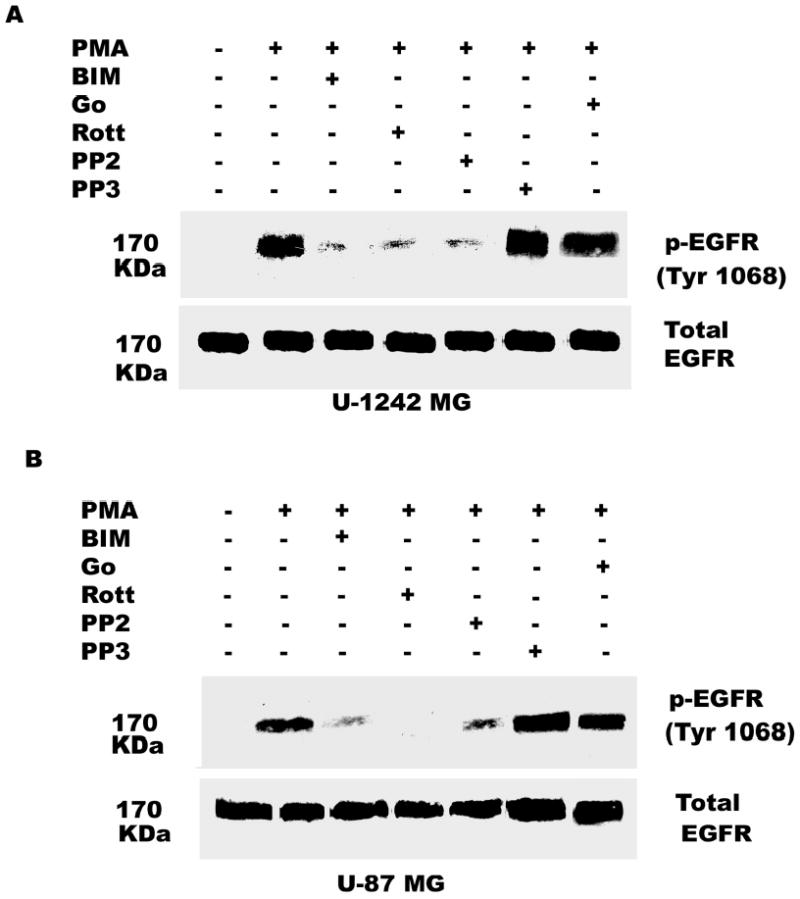
Effect of BIM (PKC inhibitor), rottlerin (PKC δ inhibitor), PP2 (Src inhibitor) and Gö 6976 on EGFR (Tyr 1068) activation by PMA. Serum-starved U-1242 MG and U-87 MG cells were incubated with inhibitors for 60 min and then treated with PMA (100 nM) for 30 min. After washing twice with ice-cold PBS, cells were solubized in 1% Triton lysis buffer, analyzed and immunoblotted for EGFR (Tyr-1068). Blots were stripped and reprobed for total EGFR.
Since BIM but not Go 6976 inhibited PMA–evoked EGFR phosphorylation, we targeted the novel PKC isozymes to identify the mediator of PMA effect. To determine whether PKC δ was involved, we used the PKC δ specific inhibitor, rottlerin (5 μM). In presence of rottlerin, the PMA–induced phosphorylation of EGFR was inhibited, suggesting that PKC δ might be mediating the response to PMA (Figure 5). Incubation of both U-1242 MG and U-87 MG cells with Ingenol (100 nM), a PKC epsilon agonist did not differentially phosphorylate EGFR (Tyr 1068) compared with untreated cells (data not shown).
Some pharmacological inhibitors may not be truly specific, and for example rottlerin apart from inhibiting PKC δ, also uncouples mitochondrial ATP production (42). Thus an alternative approach to down regulate PKC δ isozyme expression was adopted. PKC δ siRNA (200 nM) was transfected into astrocytic tumor cells for 48 h, followed by the addition of PMA (100 nM) for 30 min. Importantly, however, PKC δ gene silencing down regulated PKC δ by 90% in U-1242 MG and almost 100% in U-87 MG cells (Figure 6a, and b). PKC δ gene silencing abolished PMA-induced EGFR (Tyr 1068) phosphorylation. We also probed for the expression of PKC α to demonstrate the specificity of PKC δ siRNA. The level of PKC α was not changed nor was that of tubulin, used as loading control. Control transfections had no effect on this signaling pathway
Fig. 6.
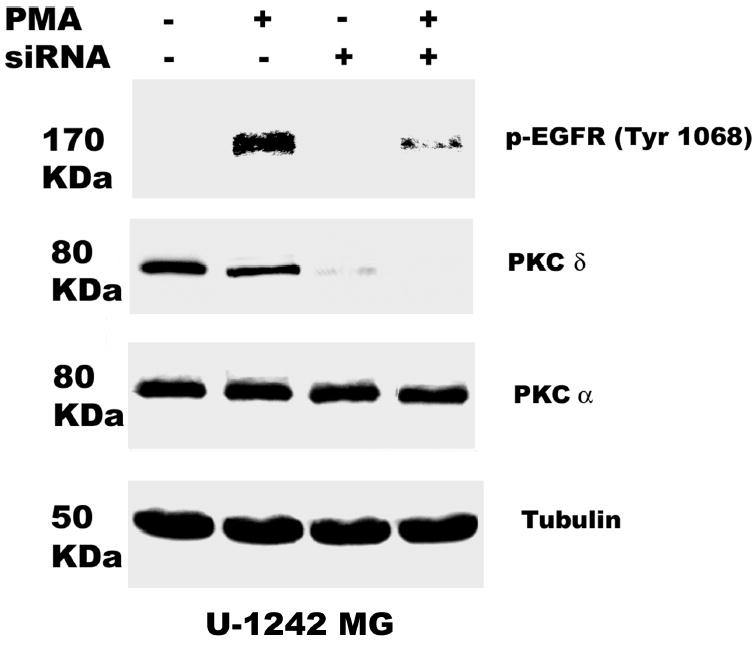
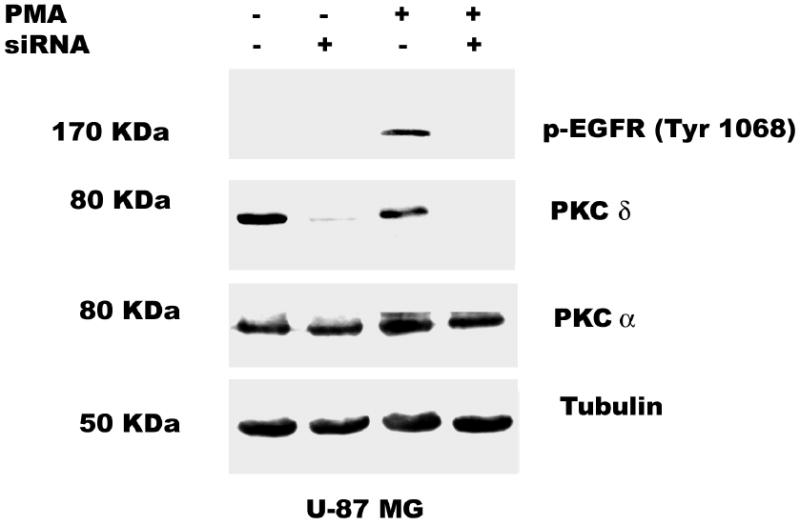
PKC δ mediates PMA-induced EGFR (Tyr 1068) phosphorylation. RNA interference with siRNA transfection was performed as described under “Experimental Procedure”. 24 h after transfection, cells were quiesced for 24 h. A, U-1242 MG cells and B, U-87 MG were transiently transfected with siRNA PKC δ and treated with or without PMA. Cells were then lysed and cell lysates of equal concentration of proteins (200 μg/lane) were separated on 8% SDS-PAGE gels and immunoblotted for phospho EGFR (Tyr 1068), PKC δ (total), anti PKCα and tubulin.
Involvement of Src in PMA Transactivation of the EGFR
The data presented above suggest that the PMA-induced EGFR tyrosine phosphorylation is mediated by PKC δ. However, PKC δ is a serine-threonine kinase and not a tyrosine kinase. What then is the intermediate kinase between PKC δ and EGFR? The non-receptor tyrosine kinase c-Src has been shown to phosphorylate EGFR at tyrosine 845 in C3H1oT1/2 murine fibroblast cell line and breast cancer cell line MDA468 (43) and tyrosine 1068 in human epidermoid A431 cells (37). We therefore determined the effect of the c-Src kinase selective inhibitor PP2 and the inactive isomer PP3 on EGFR phosphorylation in glioblastoma cell lines exposed to PMA (100 nM). As expected, addition of PMA caused phosphorylation of EGFR on tyrosine 1068. Pretreatment of glioblastoma cell lines for 60 min with PP2 (5 μM) blocked PMA-induced phosphorylation of EGFR at tyrosine 1068 by 90 %, but the inactive isomer did not attenuate this effect (Figure 5). In order to reconfirm our data on the role of c-Src in mediating the transactivation of EGFR (Y1068) phosphorylation by PMA, we used siRNA against c-Src. The c-Src siRNA (400 nM) was transfected into astrocytic tumor cells as described in “Experimental procedures”. The result showed that the siRNA knocked down the expression of c- Src and attenuated the phosphorylation of EGFR (Y1068) by PMA (Figure 7 a and b). These data suggest that PMA-induced tyrosine phosphorylation of EGF receptor is mediated directly or indirectly by Src kinase.
Fig. 7.
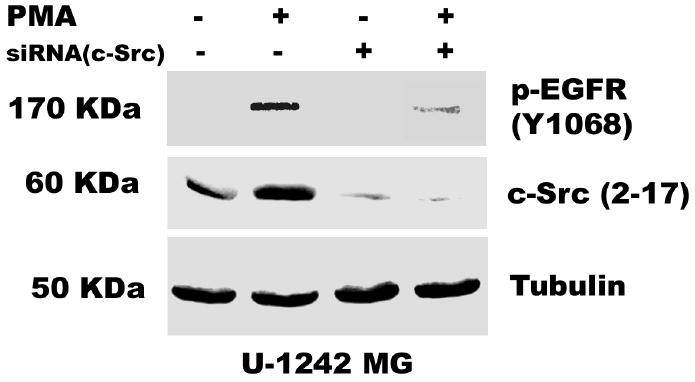
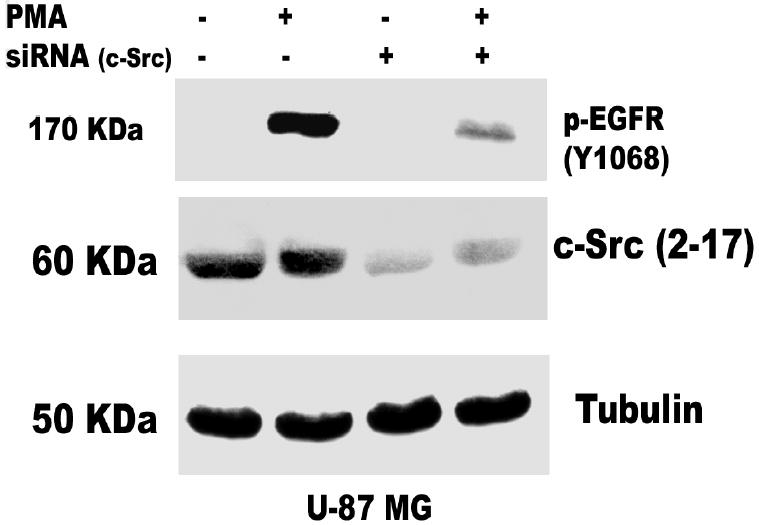
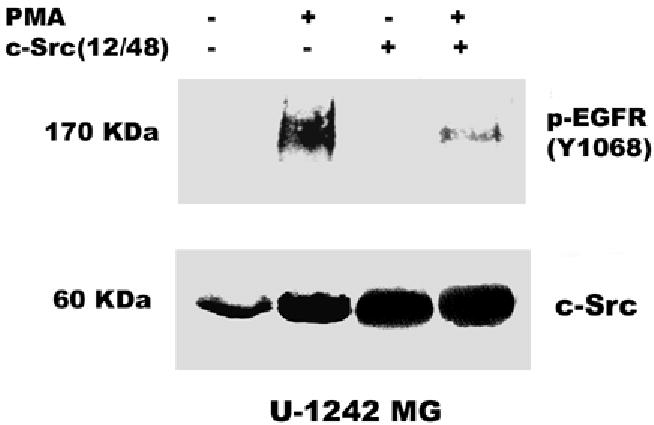
Role of c-Src in PMA-induced EGFR transactivation. siRNA against c-Src was transfected into A, U-1242 MG cells and B, U-87 MG cells and treated with PMA for 30 min. Cell were lysed using 1% Triton lysis buffer after washing with ice-cold PBS. Cell lysates were immunoblotted against EGFR (Y1068) and tubulin as loading control. C, Serum starved U-1242 MG and U-87 MG were pretreated with BIM (1 μM) and rottlerin (5 μM) for 60 min, after which PMA (100 nM) was added for 30 min. The cell lysates were immunoprecipitated with anti- c-Src (2–17) and the precipitates were immunoblotted with antibodies against phospho-serine/ threonine and total c- Src protein. D, Mutant c-Src (ser12cys/ser48ala) transfected into U-1242 MG cells were treated with PMA (100 nM) for 30 min. Cells were lysed using 1% Triton lysis buffer after washing with ice-cold PBS. Cell lysates were immunoblotted against EGFR (Y1068).
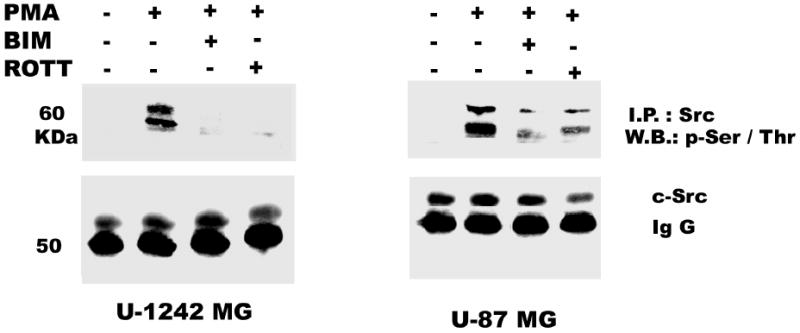
PMA Induces Phosphorylation of c-Src on Serine/Threonine residues
c-Src is a non-receptor tyrosine kinase that functions as a co-transducer of transmembrane signals (43). c-Src has a poorly conserved unique domain, that is rich in serine and threonine, that can be activated by PKC (44). In order to investigate the activation of c-Src by PMA, cell lysates were immunoprecipitated with c-Src (2–17) antibody and the immunoprecipitates were probed with phospho-specific serine/threonine antibody. The result reveals a strong activation of c-Src serine/threonine residues with PMA treatment, which was attenuated by BIM and rottlerin (Figure 7c). To further investigate the role of the amino terminus of c-Src, we generated mutants of c-src on ser 12/48, which are the phosphorylation site for PKCs, by replacing the ser12cys/ser48ala. Correct cloning and mutagenesis of all mutants were verified by sequencing. The mutant c-Src was transiently transfected into our glioblastomas cell lines and we determined its inhibitory effect on PMA-induced EGFR (Y1068) phosphorylation. The mutant c-Src (ser12cys/ser48ala) abrogated the phosphorylation of EGFR (Y1068) by about 75% (Figure 7d).
To further explore the possibility that PKC δ may associate with c-Src in our cell lines, co-immunoprecipitation experiments were performed. When U-1242 MG and U-87 MG cell lysates were immunoprecipitated with c-Src (2–17) antibody and immunoblotted with PKC δ antibody, PKC δ was detected in the c-Src immunoprecipitates. Transient treatment with PMA (30 min) did not increase the level of PKC δ pulled down with c- Src antibody (data not shown). This data demonstrates that PKC δ associate with c-Src in glioblastoma cell lines, suggesting that PKC δ activation may results in a direct c-Src activation and subsequent phosphorylation of EGFR (Tyr 1068). The result argues that PMA first interacts with PKC δ to activate or phosphorylate c-Src, which then acts as a co-transducer of EGFR (Tyr 1068) phosphorylation.
Inhibition of EGFR Kinase Activity Does not Block PMA-induced Phosphorylation of EGFR at Tyr 1068
To evaluate whether PMA activates EGF receptor kinase activity first before inducing the tyrosine phosphorylation of EGFR (Tyr 1068), we investigated the effect of AG 1478, a potent inhibitor of EGFR kinase activity. U-1242 MG and U-87 MG cells were pretreated with AG 1478 (100 nM), for 60 min and then stimulated with PMA (100 nM) or EGF (25 ng/ml) for 30 and 10 min, respectively. Western blotting demonstrated that, consistent with the blockade of EGFR kinase activity, AG 1478 effectively attenuated the robust EGFR phosphorylation of Tyr 1068 following EGF treatment. In contrast, the effective inhibition of EGFR kinase activity by AG 1478 had no discernable effect on the phosphorylation of Tyr 1068 in cells activated with PMA (Figure 8a and b). These results indicate that PMA induced phosphorylation of EGFR at Tyr 1068 first, which then lead to increase in EGFR kinase activity and the activation of Ras/Raf/MEK/MAPK pathway as shown below.
Fig. 8.
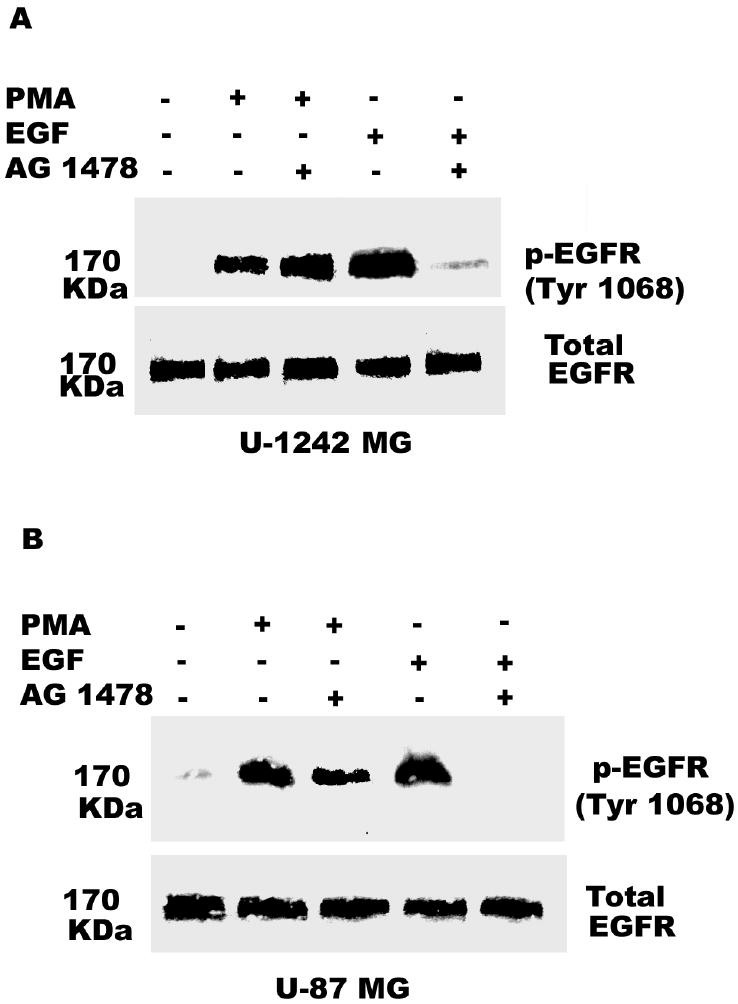
Inhibition of the epidermal growth factor receptor (EGFR) kinase activity did not affect PMA-induced phosphorylation of EGFR (Tyr 1068). Panel A, U-1242 MG cells were pretreated with EGFR kinase inhibitor AG 1478 (100 nM) for 60 min prior to treatment with media alone or EGF (25 ng/ml) or PMA (100 nM) for 10 min and 30 min, respectively. Cellular proteins were extracted and subjected to Western blotting using phospho-specific antibody against Tyr 1068. Panel B, Similarly, U-87 MG cells were pretreated with EGFR kinase inhibitor AG 1478 (100 nM) for 60 min prior to treatment with media alone, EGF (25 ng/ml) or PMA (100 nM) for 10 min and 30 min, respectively. Cellular proteins were extracted and subjected to Western blotting using phospho-specific antibody against Tyr 1068. Blots were stripped and reprobed for total EGFR.
Pharmacological Inhibition of PMA-induced ERK Activation
Activation of the ERK/MAPK pathway is a key step in the regulation of important cellular responses such as cell proliferation. In many cell types, the MAP kinase pathway has been implicated in both EGF and PMA-induced growth stimulatory responses. Our laboratory had earlier shown that PMA (100 nM) and EGF (25 ng/ml) induced the activation of ERK1/ ERK2 in glioblastoma cell lines and inhibition of MEK with UO 126 blocked PMA-induced cell proliferation (38). In the current study, we investigated the role of EGFR transactivation in PMA-induced mitogenic signaling. We found that pretreatment of the glioblastoma cell lines with BIM, Rottlerin, and AG 1478 for 60 min, before the addition of PMA (100 nM) for 30 min, partially depressed ERK1/ERK2 activation induced by PMA. The remaining MAPK phosphorylation may be as a result of EGFR-independent activation of Ras/Raf/MEK/MAPK pathway by PMA. Furthermore, pharmacological inhibition of c-Src with PP2 also decreased PMA-induced activation of MAPK. Inhibition of MEK with UO 126 (10 μM) completely abrogated both EGFR-dependent and independent MAPK phosphorylation by PMA (Figure 9a and b).
Fig. 9.
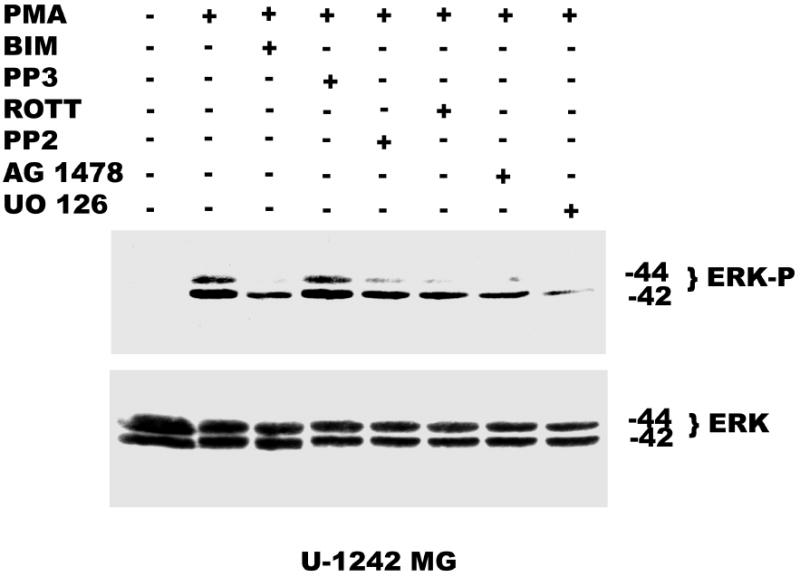
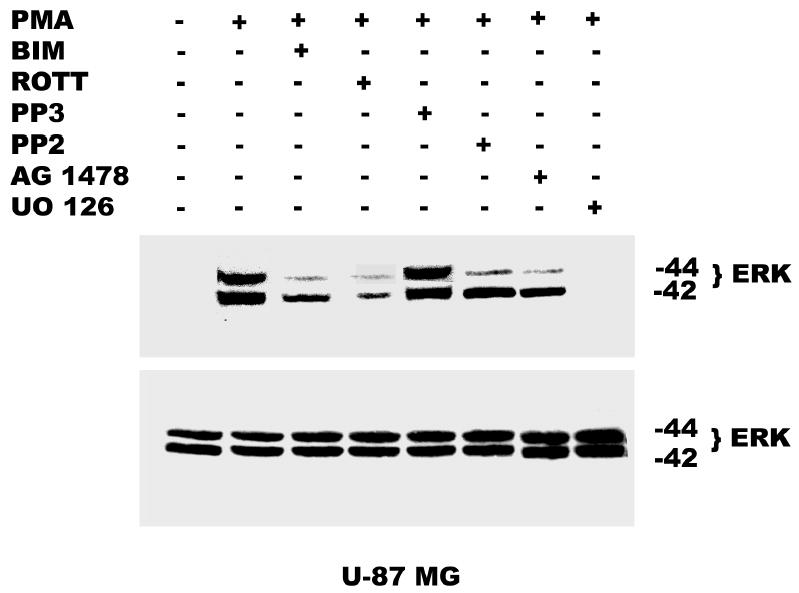
Effect of pharmacological inhibitors on ERK activation by PMA. Panel A, Serum starved U-1242 MG were pretreated with BIM (1 μM) and rottlerin (5 μM), PP2 (5 μM), AG 1478 (100 nM) or UO 126 (5 μM) for 60 min, before the addition of PMA (100 nM) for 30 min. ERK activation by PMA was attenuated in presence of different inhibitors. Panel B, Serum starved U-87 MG were pretreated with BIM (1 μM) and rottlerin (5 μM), PP2 (5 μM), AG 1478 (100 nM) and UO 126 (10 μM) for 60 min, before PMA (100 nM) addition. Proteins were subjected to Western blotting on a 10% SDS-PAGE gel and probed with phosphospecific antibody to dually phosphorylated extracellular signal regulated kinase –1 and –2 (ERK 1 / ERK 2) and subsequently reprobed for ERK 1 /ERK2 total.
Effect of PMA on [3H] Thymidine Incorporation
To determine the biological function of PMA-induced EGFR transactivation in astrocytic tumor cells, thymidine incorporation was used as measure of DNA synthesis and cell proliferation. We investigated the roles of PKC δ and c-Src in cell proliferation after stimulation with either PMA or EGF. As shown in Figure 10a, PMA increased [3H] thymidine uptake in U-87 MG cells by 1.6-fold. Pretreatment of the cells with PKC inhibitor BIM, rottlerin (PKC δ) specific inhibitor, PP2 (Src inhibitor), EGFR kinase inhibitor (AG 1478), or MEK inhibitor (UO 126), abrogated PMA effect on [3H] thymidine uptake. Similarly, EGF (25 ng/ml) increased [3H] thymidine uptake in U-87 MG cells by 1.5 fold, which was attenuated by these inhibitors (Figure 10 c). To further investigate the role of PKC δ and c-Src on PMA-induced cell proliferation, U-87 MG cells were transfected with the same concentration (200 nM) of PKC δ siRNA or (400 nM) of c-Src siRNA that completely blocked PKC δ or c-Src expression respectively and the effect of PMA on thymidine incorporation was investigated. The PKC δ siRNA and c-Src siRNA reduced PMA-induced [3H] thymidine uptake by approximately 87% and 60 %, respectively (Figure 10 b). Similarly, EGF evoked increase in thymidine incorporation was attenuated by PKC δ siRNA and c-Src siRNA by 30 and 50%, respectively (Figure 10 d).
Fig. 10.
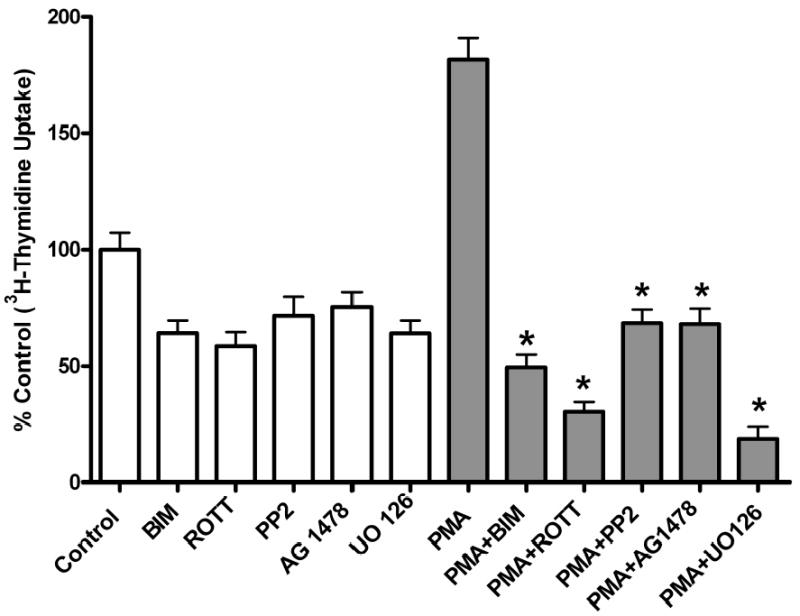
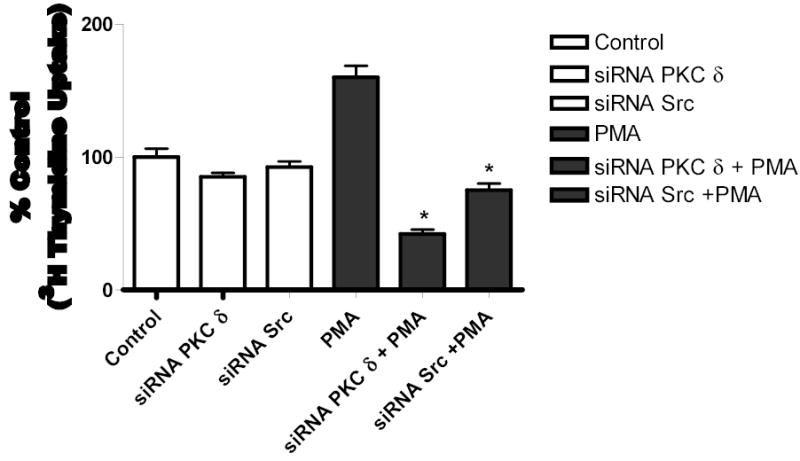
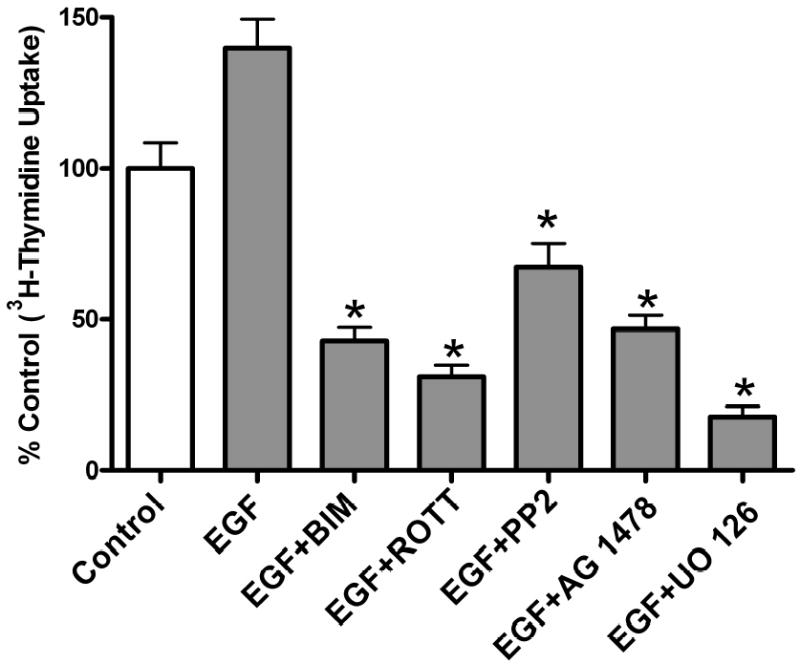
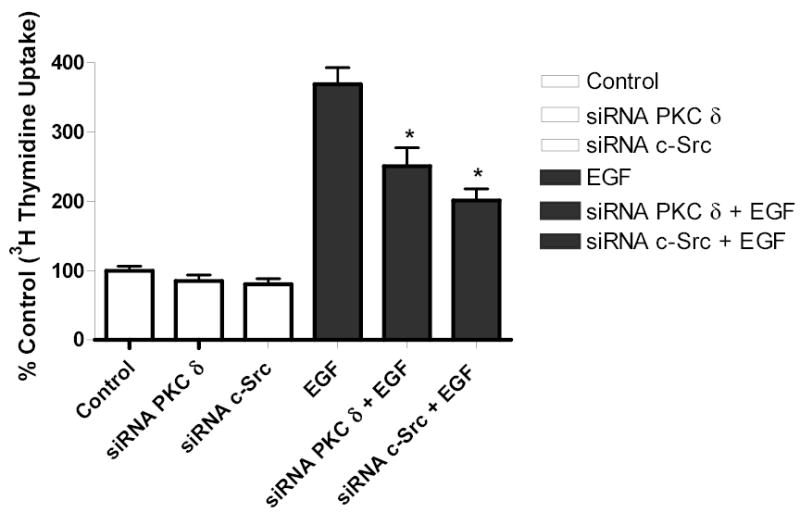
[3H] Thymidine incorporation in astroctytic tumor cells. A, Effect of PMA on cell proliferation and B, PKC δ and c-Src inhibition abrogates PMA-induced cell proliferation. Cells were transfected with PKC δ siRNA or c-Src siRNA, serum straved overnight and the incubated in the presence of PMA. C, Effect of EGF and D, PKC δ and c-Src inhibition abrogates EGF-induced [3H] thymidine incorporation in U-87 MG cells. Quiescent U-87 MG cells were preincubated with BIM (1 μM ), Rottlerin (5 μM), UO 126 (10 μM) for 60 min. Cells were incubated in the presence and absence of PMA or EGF for 18 h. Cells were then pulse-labeled with [3H] thymidine for 6 h, and thymidine incorporation was measured by Beckman scintillation counter. Quantitative analysis from three independent experiments (means ± SEM). Cell proliferation was calculated as % of control. The data in the graph are the mean ± SEM of at least 2 independent experiments with each experiment performed in quadruplicate.
DISCUSSION
Malignant gliomas are the most common adult brain tumors, are refractory to classical chemotherapy and radiotherapy and have poor prognosis (45). The EGFR is overexpressed in 50- 60% of GBM and amplified in 40% of the tumors (7), which contribute to the malignant phenotype of human glioblastomas (4,6,8). In the last ten years, the molecular mechanisms underlying astrocytic neoplastic transformation have been widely studied and a number of signaling pathways, including that of PKC, are altered in GBM (46–47). The expression and activity of PKC isozymes are highly elevated in gliomas and glioma cell lines compared with normal astrocytes (12) and PKC inhibitors markedly reduced glioma cell proliferation (38,48).
Both EGFR expression and PKC activity play a significant role in astrocytic tumor biology. Our data provide evidence for the first time that treatment of glioblastoma cell lines with PMA resulted in EGFR phosphorylation at Tyr 1068 but not at other tyrosine residues and that this phosphorylation was mediated by a PKC δ/c-Src-dependent pathway. PMA-induced phosphorylation of Tyr 1068 was blocked by BIM, an inhibitor of classical as well as novel PKC isozymes but not by Go6976, an inhibitor of classical PKC isozymes. The Tyr 1068 is a major Grb2 binding and autophosphorylation site of the EGFR (49). In contrast, EGF induced phosphorylation of the EGFR at multiple sites, including Tyr 992, 845, 1045 and 1068.
The mechanism of EGFR transactivation is not well characterized. Some studies have shown that EGFR is involved in signaling networks activated by a number of stimuli that do not interact directly with this receptor (50). These stimuli include G protein-coupled receptor agonists thrombin and lysophosphatidic acid (51), calcium (52) and UV irradiation (53). In our study prior incubation with the pharmacologic inhibitor of PKCδ, rottlerin, attenuated PMA-induced phosphorylation of EGFR (Fig. 5), which suggests a putative role for PKC δ in this process. To further confirm the role of PKC δ in mediating PMA-induced EGFR (Tyr 1068) phosphorylation, we used siRNA duplexes for PKC δ to knockdown the expression of PKC δ and to immunoblot for EGFR (Tyr 1068). Our results convincingly showed that gene silencing of PKC δ attenuated PMA-induced EGFR (Tyr 1068) phosphorylation (Figure 6 a and b). Our immunoprecipitation data reveal an association between PKC δ and c-Src in astrocytic tumor cells, as reported in other cell types. PKC δ is a widely expressed member of the novel PKCs, and this isoform has been associated with cell proliferation in a number of cell types, including NIH 3T3 fibroblast, smooth muscle cells, and human keratinocytes (54) and also shown to phosphorylate c-Src and to interact with c-Src (55–58).
Since PKC isozymes do not directly phosphorylate proteins at tyrosine residues, we examined the role of c-Src as an intermediate kinase between PKC and EGFR. Cellular Src and EGFR interact in the progression of certain human malignancies and act synergistically to induce enhanced signaling in cells that express these kinases (43). Elevated levels of Src family kinases, and mutational activation have been observed in colon carcinoma, and similar findings also have been reported in lung, breast and brain tumors (43). Evidence derived from experimental models indicates that Src activation potentiates EGF-induced mitogenesis and transformation (43). Our study shows that the c-Src inhibitor (PP2) inhibited PMA-induced EGFR phosphorylation (Fig. 5) and mitogenic response (Figure 9a and b), while the inactive form, PP3, had no effect on the phosphorylation of the receptor. To further confirm the role of c-Src in mediating PMA-induced EGFR transactivation, we used siRNA directed against c-Src to knock down the expression of c-Src. Gene silencing using the c-Src siRNA abrogated PMA-induced EGFR (Y1068) phosphorylation (Figure 7 a&b). These data imply that PMA interacts with PKC-δ, which in turn phosphorylates Src to activate EGFR. Src structurally has a poorly conserved unique domain that contains serine/threonine residues (44). The unique domain of c-Src, which is the least conserved region among Src family members, mediates protein interaction and is phosphorylated by protein kinase A, Protein kinase C, and Cdc2-cyclin complex (59–61). To test the hypothesis that PMA may be indirectly activating Src through PKCδ, we used mutant c-Src (ser12cys/ser48ala). Transient transfection of this mutant into astrocytic tumor cells abrogated the phosphorylation of EGFR (Y1068) induced by PMA (Fig 7d).
The EGFR kinase inhibitor AG 1478 did not affect PMA-induced EGFR phosphorylation at Tyr 1068 but completely abrogated EGF-induced phosphorylation of EGFR. This suggests that PMA-induced EGFR (Tyr 1068) phosphorylation through PKC δ and c-Src is upstream of EGFR kinase activity. The result agrees with similar studies that used H2O2 to transactivate EGFR (62).
The activation of the ERK/MAPK pathway is a key step in the regulation of important cellular responses such as cell proliferation (63). Extracellular regulated kinases (ERK) 1 and 2 are 44- and 42-kDa members of the MAP kinase family and are involved in the regulation of gene expression, protein synthesis, cell growth and proliferation, and in some cases cell differentiation and secretion (64). ERK phosphorylation was initially observed after ligand activation of such receptor tyrosine kinases as the EGF receptor, but many Gq-, Gi-, and Gs- coupled receptors also initiate the ERK cascade through transactivation of the EGFR (65–66). The ERK activation often involves sequential steps that include transactivation of the receptor tyrosine kinases (EGFR) in a c-Src kinase-dependent manner (51). In our study, we found that BIM (PKC inhibitor), Rottlerin (PKC δ specific inhibitor), PP2 (Src inhibitor) significantly inhibited ERK/MAPK activation by PMA (Fig.9). Similarly, AG 1478, which did not block EGFR Tyr 1068 phosphorylation and the MEK inhibitor (UO 126) inhibited PMA-evoked ERK phosphorylation (Fig.9). These data further suggest that the PMA-induced Tyr 1068 phosphorylation could lead to increase in EGFR kinase activity (blocked by AG 1478) to activate the Ras/Raf/MEK/MAPK pathway in glioblastoma cells.
Activation of PKC leads to the phosphorylation of several proteins that are involved in the regulation of cell growth, differentiation and apoptosis (28–30). Pretreament of GBM cells with BIM or rottlerin blocked PMA-induced as well as EGF-induced increase in [3H] thymidine uptake, suggesting that PKC isoforms play a critical role in glioblastoma proliferation (38). Gene silencing of PKC δ and c-Src with siRNA and pharmacological inhibition with PP2 and rottlerin also attenuated PMA-induced cell proliferation in U-87 MG cells. It is well established that following binding to EGFR, EGF increases PKC activity in a variety of cells (67- 68). In addition, PKC δ has been shown to increase transformation and metastatic progression of a number of tumors (69) and activation of PKC δ has been shown to be involved in proliferation of epithelial breast cells (70). In conclusion, our data provide evidence for the first time to demonstrate that PMA phosphorylates EGFR at Tyr 1068 through a PKC δ/Src-dependent pathway to activate MAPK and to increase cell proliferation in glioblastoma cells (Figure 11). Furthermore, our study reveals a novel pathway that may contribute to glioblastoma invasive growth.
Fig. 11.
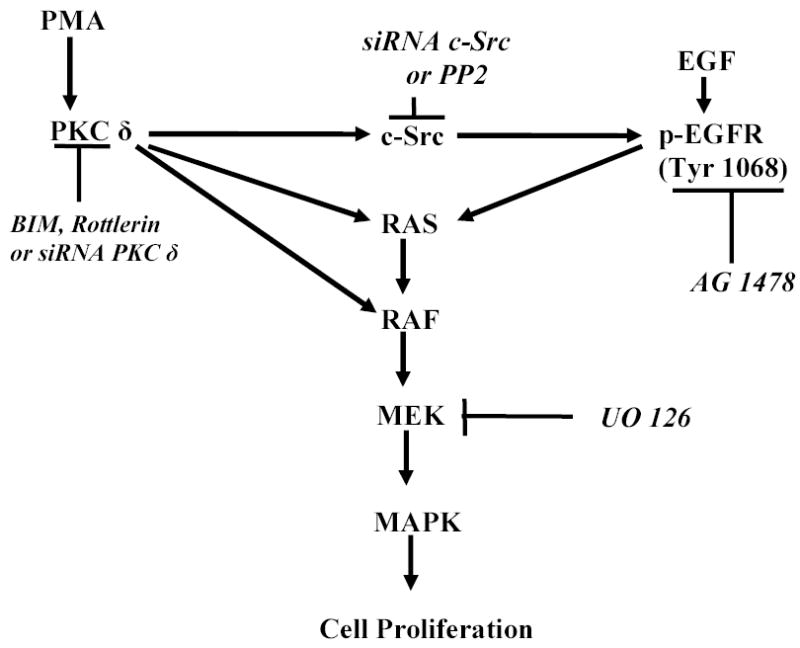
Schematic representation of the signaling pathways involved in PMA transactivation of EGFR (Tyr 1068) in glioblastoma cell lines.
Acknowledgments
This work was supported by grants NS35122 (IMH) and CA90851 (IMH) from the National Institutes of Health. The authors are grateful to Joan E. Carpenter and Gerard T. Redpath for excellent technical assistance.
References
- 1.Collins VP. Cancer Surv. 1998;32:37–51. [PubMed] [Google Scholar]
- 2.Rasheed BK, Wiltshire RN, Bigner SH, Bigner DD. Curr Opin Oncol. 1999;11:162–167. doi: 10.1097/00001622-199905000-00004. [DOI] [PubMed] [Google Scholar]
- 3.Kleihues, P., Burger, P.C., Collins V.P., Newcomb E.W., Ohgaki, H., and Cavene, W.K., (2000). Glioblastoma. Tumors of the Nervous System. Kleihues, P., and Cavene, W.K. (eds) IARC Press: Lyon, pp 29–39.
- 4.Libermann TA, Nusbaum HR, Razon N, Kris R, Lax I, Soreq H, Whittle N, Waterfield MD, Ullrich A, Schlessinger J. Nature. 1985;313:144–147. doi: 10.1038/313144a0. [DOI] [PubMed] [Google Scholar]
- 5.Ekstrand AJ, Sugawa N, James CD, Collins VP. Proc Natl Acad Sci USA. 1992;89:1169–1174. doi: 10.1073/pnas.89.10.4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wong AJ, Bigner SH, Bigner DD, Kinzler KW, Hamilton SR, Vogelstein B. Proc Natl Acad Sci USA. 1987;84:6899–6903. doi: 10.1073/pnas.84.19.6899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ohgaki H, Schauble B, Zur H, von Ammon K, Kleihues P. Virchows Arch. 1995;427:113–118. doi: 10.1007/BF00196514. [DOI] [PubMed] [Google Scholar]
- 8.Ekstrand AJ, James CD, Cevenee WK, Seliger B, Petterson RF, Collins VP. Cancer Res. 1991;51:2164–2172. [PubMed] [Google Scholar]
- 9.Todo T, Shitara N, Nakamura H, Takakura K, Ikeda K. Neurosurgery. 1991;108:11–16. doi: 10.1097/00006123-199109000-00010. [DOI] [PubMed] [Google Scholar]
- 10.Benzil DL, Finkelstein SD, Epstein MH, Finch PW. Cancer Res. 1992;52:2952–2956. [PubMed] [Google Scholar]
- 11.Couldwell WT, Uhm JH, Antel JP, Yong VW. Neurosurgery. 1991;29:880–887. doi: 10.1097/00006123-199112000-00013. [DOI] [PubMed] [Google Scholar]
- 12.Couldwell WT, Uhm JH, Antel JP, Yong VW. Neurosurgery. 1992;31:717–724. doi: 10.1227/00006123-199210000-00015. [DOI] [PubMed] [Google Scholar]
- 13.van der Geer P, Hunter T, Linberg RA. Ann Rev Cell Biol. 1994;10:251–337. doi: 10.1146/annurev.cb.10.110194.001343. [DOI] [PubMed] [Google Scholar]
- 14.Prenzel N, Fischer OM, Streit S, Hart S, Ullrich A. Endocr Relat Cancer. 2001;8:11–31. doi: 10.1677/erc.0.0080011. [DOI] [PubMed] [Google Scholar]
- 15.Riese II DJ, Stern DF. Bioessays. 1998;20:41–48. doi: 10.1002/(SICI)1521-1878(199801)20:1<41::AID-BIES7>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 16.Chang H, Riese II DJ, Gilbert W, Stern DF, McMahan UJ. Nature. 1997;387:509–512. doi: 10.1038/387509a0. [DOI] [PubMed] [Google Scholar]
- 17.Zhang D, Sliwlkowski MX, Mark M, Frantz G, Akita R, Sun Y. Hillan, K. Crowley, C. Brush, J. and, Godowski, P.J Proc Natl Acad Sci USA. 1997;94:9562–9567. doi: 10.1073/pnas.94.18.9562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harari D, Tzahar E, Romano J, Shelly M, Pierce JH, Andrews GC, Yarden Y. Oncogene. 1999;18:2681–2689. doi: 10.1038/sj.onc.1202631. [DOI] [PubMed] [Google Scholar]
- 19.Ullrich A, Schlessinger J. Signal transduction by receptors with tyrosine kinase activity. Cell. 1990;61:203–212. doi: 10.1016/0092-8674(90)90801-k. [DOI] [PubMed] [Google Scholar]
- 20.Nishikawa R, Ji XD, Harman RC, Lazar CS, Gill GN, Cavenee WK, Huang HJ. Proc Natl Acad Sci USA. 1994;91:7727–7731. doi: 10.1073/pnas.91.16.7727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Castagna M, Takai Y, Kaibuchi K, Sano K, Kikkawa V, Nishizuka Y. J Biol Chem. 1982;257:7847–7851. [PubMed] [Google Scholar]
- 22.Niedel JE, Kuhn LJ, Vanderbark GR. Proc Natl Acad Sci USA. 1983;80:36–40. doi: 10.1073/pnas.80.1.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leach KL, James ML, Blumberg PM. Proc Natl Acad Sci USA. 1983;80:4208–4212. doi: 10.1073/pnas.80.14.4208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nishizuka Y. Nature. 1984;308:693–698. doi: 10.1038/308693a0. [DOI] [PubMed] [Google Scholar]
- 25.Nishizuka Y. Nature. 1988;334:661–665. doi: 10.1038/334661a0. [DOI] [PubMed] [Google Scholar]
- 26.Basu A. Pharmacol Ther. 1993;59:257–80. doi: 10.1016/0163-7258(93)90070-t. [DOI] [PubMed] [Google Scholar]
- 27.Toker A. Front Biosci. 1998;3:D1134–1147. doi: 10.2741/a350. [DOI] [PubMed] [Google Scholar]
- 28.Newton AC. J Biol Chem. 1995;270:28495–28498. doi: 10.1074/jbc.270.48.28495. [DOI] [PubMed] [Google Scholar]
- 29.Nishizuka Y. FASEB J. 1995;9:484–496. [PubMed] [Google Scholar]
- 30.Stabel S, Parker PJ. Pharmacol Ther. 1991;51:71–95. doi: 10.1016/0163-7258(91)90042-k. [DOI] [PubMed] [Google Scholar]
- 31.Jaken S. Curr Opin Cell Biol. 1996;8:168–173. doi: 10.1016/s0955-0674(96)80062-7. [DOI] [PubMed] [Google Scholar]
- 32.Ron D, Kazanietz MG. FASEB J. 1999;13:1658–76. [PubMed] [Google Scholar]
- 33.Cochet C, Gill GN, Meisenhelder J, Cooper JA, Hunter T. J Biol Chem. 1984;259:2553–2558. [PubMed] [Google Scholar]
- 34.Hunter T, Ling N, Cooper JA. Nature. 1984;311:480–483. doi: 10.1038/311480a0. [DOI] [PubMed] [Google Scholar]
- 35.Davis RJ, Czech MP. Proc Natl Acad Sci USA. 1985;82:1974–1978. doi: 10.1073/pnas.82.7.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moro L, Dolce L, Cabodi S, Bergatto E, Erba EB, Smeriglio M, Turco E, Retta SF, Giuffrida MG, Venturino M, Godovac-Zimmermann J, Conti A, Schaefer E, Beguinot L, Tacchetti C, Gaggini P, Silengo L, Tarone G, Defilippi P. J Biol Chem. 2002;277:9405–9414. doi: 10.1074/jbc.M109101200. [DOI] [PubMed] [Google Scholar]
- 37.Samet MJ, Dewar BJ, Wu W, Graves ML. Toxicol & Appl Pharmacol. 2003;191:86–93. doi: 10.1016/s0041-008x(03)00219-9. [DOI] [PubMed] [Google Scholar]
- 38.Hussaini IM, Karns LR, Vinton G, Carpenter JE, Redpath GT, Sando JJ, VanderBerg SR. J Biol Chem. 2000;275:22348–22354. doi: 10.1074/jbc.M003203200. [DOI] [PubMed] [Google Scholar]
- 39.Maasho K, Marusina A, Reynolds NM, Coligan JE, Borrego F. J Immunol Meth. 2004;284:133–140. doi: 10.1016/j.jim.2003.10.010. [DOI] [PubMed] [Google Scholar]
- 40.Uhrbom L, Nister M, Westermark B. Oncogene. 1997;15:505–514. doi: 10.1038/sj.onc.1201227. [DOI] [PubMed] [Google Scholar]
- 41.Bostrom J, Cobbers JM, Wolter M, Tabatabai G, Weber RG, Licher P, Collins VP, Reifenberger G. Cancer Res. 1998;58:29–33. [PubMed] [Google Scholar]
- 42.Soltoff S. J Biol Chem. 2001;276:37986–37992. doi: 10.1074/jbc.M105073200. [DOI] [PubMed] [Google Scholar]
- 43.Biscardi JS, Tice DA, Parsons SJ. Adv Cancer Res. 1999;76:61–119. doi: 10.1016/s0065-230x(08)60774-5. [DOI] [PubMed] [Google Scholar]
- 44.Brown MT, Cooper JA. Biochimica et Biophysica Acta. 1996;1287:121–149. doi: 10.1016/0304-419x(96)00003-0. [DOI] [PubMed] [Google Scholar]
- 45.Prados MD, Levin V. Semin Oncol. 2000;27:1–10. [PubMed] [Google Scholar]
- 46.Baltuch GH, Yong VW. Brain Res. 1996;710:143–149. doi: 10.1016/0006-8993(95)01395-4. [DOI] [PubMed] [Google Scholar]
- 47.Bredel M, Pollack IF. Acta Neurochir. 1997;139:1000–1013. doi: 10.1007/BF01411552. [DOI] [PubMed] [Google Scholar]
- 48.Ahmad S, Mineta T, Martuza RL, Glazer RI. Neurosurgery (Baltim) 1994;35:904–908. doi: 10.1227/00006123-199411000-00015. [DOI] [PubMed] [Google Scholar]
- 49.Downward J, Parker P, Waterfield MD. Nature. 1984;311:483–485. doi: 10.1038/311483a0. [DOI] [PubMed] [Google Scholar]
- 50.Carpenter G. J Cell Biol. 1999;146:697–702. doi: 10.1083/jcb.146.4.697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Daub H, Wallasch C, Lankenau A, Herrlich A, Ullrich A. EMBO. 1997;16:7032–7044. doi: 10.1093/emboj/16.23.7032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dethlefsen SM, Raab G, Moses MA, Adam RM, Klagsbrun M, Freeman MR. J Cell Biochem. 1988;69:143–153. doi: 10.1002/(sici)1097-4644(19980501)69:2<143::aid-jcb5>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 53.Dent P, Reardon DB, Park JS, Bowers G, Logsdon C, Valerie K, Schmidt-Ullrich R. Mol Biol Cell. 1999;10:2493–2506. doi: 10.1091/mbc.10.8.2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gschwendt M. Eur J Biochem. 1999;259:555–564. doi: 10.1046/j.1432-1327.1999.00120.x. [DOI] [PubMed] [Google Scholar]
- 55.Zang Q, Lu Z, Curto M, Barile N, Shalloway D, Foster DA. J Biol Chem. 1997;272:13275–13280. doi: 10.1074/jbc.272.20.13275. [DOI] [PubMed] [Google Scholar]
- 56.Saito S, Frank GD, Mifune M, Ohba M, Utsunomiya H, Motley ED, nagami T, Eguchi S. J Biol Chem. 2002;277:44695–44700. doi: 10.1074/jbc.M208332200. [DOI] [PubMed] [Google Scholar]
- 57.Joseloff E, Cataisson C. Aamodt, H. Ocheni, H. Blumberg, P.M. Kraker, A.J. and, Yuspa, S.H J Biol Chem. 2002;277:12318–12323. doi: 10.1074/jbc.M111618200. [DOI] [PubMed] [Google Scholar]
- 58.Song JS, Swann PG, Szallasi Z, Blank U, Blumberg PM, Rivera J. Oncogene. 1998;16:3357–3368. doi: 10.1038/sj.onc.1201886. [DOI] [PubMed] [Google Scholar]
- 59.Collett MS, Erickson E, Erickson RL. J Virol. 1979;29:770–781. doi: 10.1128/jvi.29.2.770-781.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shenoy S, Chackalaparampil I, Bagrodia S, Liu PH, Shalloway D. Proc Natl Acad Sci USA. 1992;89:7237–7241. doi: 10.1073/pnas.89.15.7237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Denning MF, Dlugosz AA, Threadgill DW, Magnuson T, Yuspa SH. J Biol Chem. 1996;271:5325–5331. doi: 10.1074/jbc.271.10.5325. [DOI] [PubMed] [Google Scholar]
- 62.Schonwasser DC, Marais RM, Marshall CJ, Parker PJ. Mol Cell Biol. 1998;18:790–798. doi: 10.1128/mcb.18.2.790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gutkind JS. J Biol Chem. 1998;273:1839–1842. doi: 10.1074/jbc.273.4.1839. [DOI] [PubMed] [Google Scholar]
- 64.Hackel PO, Zwick E, Prenzel N, Ullrich A. Curr Opinion Cell Biol. 1999;11:184–189. doi: 10.1016/s0955-0674(99)80024-6. [DOI] [PubMed] [Google Scholar]
- 65.Luttrell LM, Daaka Y, Lefkowitz RJ. Curr Opinion Cell Biol. 1999;11:171–183. doi: 10.1016/s0955-0674(99)80023-4. [DOI] [PubMed] [Google Scholar]
- 66.Eguchi S, wasaki H, nagami T, Numaguchi K, Yamakawa T, Motley E.D. Owada, K.M. Marumo, F. and, Hirata, Y Hypertension. 1999;33:201–206. doi: 10.1161/01.hyp.33.1.201. [DOI] [PubMed] [Google Scholar]
- 67.Welsh J, Gill GN, Rosenfeld MG, Wells A. J Cell Biol. 1991;114:533–543. doi: 10.1083/jcb.114.3.533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Iwabu A, Smith K, Allen FD, Lauffenburger DA, Wells A. J Biol Chem. 2004;279:14551–14560. doi: 10.1074/jbc.M311981200. [DOI] [PubMed] [Google Scholar]
- 69.Li W, Jiang Y, Zhang J, Soon L, Flechner L, Kapoor V, Pierce JH, Wang L. Mol Cell Biol. 1998;18:5888–5898. doi: 10.1128/mcb.18.10.5888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Greco S, Muscella A, Elia M.G. Romano, S. Storelli, C. and, Marsigliante S. J Cell Physiol. 2004;201:84–96. doi: 10.1002/jcp.20052. [DOI] [PubMed] [Google Scholar]