

Abstract
The discovery of endothelin two decades ago has now evolved into an intricate vascular endothelin (ET) system. Several ET isoforms, receptors, signaling pathways, agonists, antagonists, and clinical applications have been identified and documented in first-rate patents. The role of ET as one of the most potent endothelium-derived vasoconstricting factors is now complemented by a newly discovered role in vascular relaxation. ET synthesis is initiated by the transcription of ET genes in endothelial cells and the generation of the gene products preproET and big ET, which are further cleaved by specific ET converting enzymes into ET-1, -2, -3 and -4 isoforms. ET isoforms bind with different affinities to ETA and ETB2 receptors in vascular smooth muscle, and stimulate [Ca2+]i, protein kinase C, mitogen-activated protein kinase and other signaling mechanisms of smooth muscle contraction, growth and proliferation. ET also binds to endothelial ETB1 receptors, which mediate the release of vasodilator substances such as nitric oxide, prostacyclin and endothelium-derived hyperpolarizing factor. Endothelial ETB1 receptors may also function in ET re-uptake and clearance. Although the effects of ET on vascular function and growth are well-recognized, the role of ET and its receptors in the regulation of blood pressure and in the pathogenesis of hypertension is not clearly established. Salt-dependent hypertension in experimental animals and some forms of moderate to severe hypertension in human may show elevated levels of plasma or vascular ET; however, other forms of hypertension show normal ET levels. The currently available ET receptor antagonists reduce blood pressure in some forms of experimental hypertension. Careful examination of recent patents may identify more effective and specific modulators of the vascular ET system for clinical use in human hypertension.
Keywords: endothelium, smooth muscle, calcium, hypertension
List of abbreviations: AngII, angiotensin II; [Ca2+]i, intracellular free Ca2+ concentration; DOCA, deoxycorticosterone acetate; ECE, endothelin converting enzyme; ET-1, endothelin-1; ETA, endothelin receptor A; ETB, endothelin receptor B; MAPK, mitogen-activated protein kinase; MLC, myosin light chain; NO, nitric oxide; PGI2, prostacyclin; Phe, phenylephrine; PKC, protein kinase C; S6c, sarafotoxin 6c; SHR, spontaneously hypertensive rat; VSM, vascular smooth muscle
INTRODUCTION
Hypertension is a multifactorial disease that involves pathological changes in the neuronal, renal, hormonal and vascular control mechanisms of blood pressure. The endothelium functions as a major vascular control mechanism by releasing vasodilator substances such as nitric oxide (NO), prostacyclin (PGI2) and endothelium-derived hyperpolarizing factors (EDHFs) [1–3]. A decrease in endothelium-derived vasodilators impairs vascular relaxation and thereby contributes to the increased vascular resistance and blood pressure in hypertension.
In addition to the vasodilator factors, the endothelium releases vasoconstrictor substances such as endothelin (ET), thromboxane A2, and angiotensin II (AngII) [4,5]. Numerous studies have described the biochemistry, structure and function of ET [6,7]. Several ET isoforms and receptor subtypes have been identified in neuronal, renal and vascular tissues, and an array of ET-mediated physiological functions, such as regulation of vascular tone, sodium balance and neurotransmission have been suggested [7–9]. However, the role of ET and its receptors in the regulation of blood pressure and in the pathogenesis of hypertension is not clearly established. For instance, while the plasma or vascular ET levels are elevated in some forms of experimental and human hypertension, this is not a consistent finding in all forms of hypertension. Also, while ET receptor antagonists decrease blood pressure in some forms of experimental hypertension, their potential use in medicine has not been fully evaluated.
The purpose of this review is to provide insight into the role of ET and its receptors in the regulation of vascular tone and blood pressure, and the alterations in their amount, distribution and function in hypertension. We will first provide a brief background on ET biochemistry, synthesis pathways, plasma/tissue levels and metabolism. We will then describe the vascular ET receptor subtypes, tissue/subcellular distribution, signaling pathways, function and currently available agonists and antagonists. The changes in ET metabolism/function in human and experimental hypertension will then be described. The review will conclude with a discussion of the potential use of currently available ET receptor antagonists and recently patented modulators of the vascular ET system in experimental and human hypertension.
ET Synthesis
In the mid-1980s, careful examination of the role of endothelial cells in the vascular system led to the discovery of an endothelium-derived constricting factor. In 1988, a 21-amino-acid vasoconstricting factor was isolated from cultured porcine aortic endothelial cells and termed endothelin [6]. The ET peptide family now includes three 21-amino acid peptides ET-1, -2, and -3, 31-amino acid forms, and a more recently discovered ET-4. ET-1 is the main isoform released from the endothelium and acts in a paracrine or autocrine fashion by interacting with ET receptors in vascular smooth muscle (VSM) and endothelial cells, and thereby modifying vascular function and cell growth and proliferation [10]. ET-1 is also produced by airway epithelial cells, macrophages, fibroblasts, cardiomyocytes and neurons [9,11–13]. ET-2 is expressed by intestinal epithelial cells, while ET-3 is produced by intestinal epithelial cells, brain neurons and renal tubular epithelial cells [9,14]. ET-4 is found in the gut mucosa, lung and renal epithelial cells [15–17].
ET synthesis begins with the transcription of the preproET gene, which is regulated by c-fos and c-jun, and nuclear factor-1, AP-1 and GATA-2 [10,18,19]. The translation of preproET mRNA results in the formation of a 203-amino acid preproET peptide [10,20]. PreproETs are cleaved at dibasic sites by furin-like endopeptidase to form biologically inactive 37- to 41-amino acid intermediates, termed big ETs [4,9]. Big ET-1 and big ET-2 are cleaved at Trp21-Val22 by ET converting enzyme-1 (ECE-1) and ECE-2 to produce ET-11-21 and ET-21-21, respectively [10,21]. Mast cell chymases cleave big ET-1 and big ET-2 at Tyr31 -Gly32 to produce ET-11-31 and ET-21-31, respectively [10,23]. Big ET-3 is cleaved at Trp21-lle22 by ECE-1, -2 and -3 to produce ET-31-21, and at Gly31 -Leu32 by chymases to form ET-31-31 [10,22] (Fig. 1).
Fig. 1.
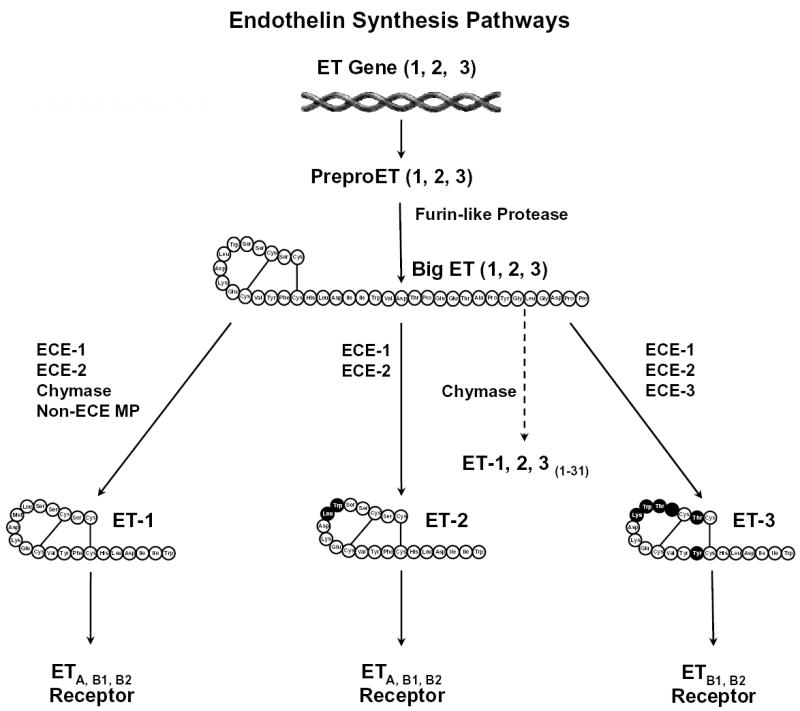
ET-1, -2, and -3 are encoded on different genes by various vasoactive and growth stimuli. The translation of preproET mRNA results in the formation of a 203-amino acid preproET, which is cleaved by a furin-like protease to form big ET-1, -2, and -3. Big ET is cleaved by ECE, metalloprotease, and chymase at amino acid-21 and -31 to produce ET-1, ET-2 and ET-3 (1–21) and (1–31). ET isoforms stimulate ET receptor subtypes with different affinities.
ECEs belong to the metalloprotease family, they are part of the neprilysin superfamily, share functional and structural similarity with neutral endopeptidases, and are partially inhibited by phosphoramidon [10,21]. ECE-1 expression is regulated by protein kinase C (PKC), ETB receptors, the transcription factor ets-1, and cytokines [10,23–26]. ECE-1 is found in a variety of cells including endothelial cells, has peak activity at neutral pH, and is processed both intracellularly and on the cell surface [9,27]. When ECE-1 cDNA is transfected into cultured cells that secrete only big ET-1, it promotes the secretion of mature ET-1. Also, in ECE-1 transfected cells, endogenous big ET-1 and exogenously supplied big ET-1 interact with ECE-1 on the cell surface. The specificity of ECE-1 provides a target for selective pharmacological intervention to alter ET-1 production in certain cardiovascular disorders [28].
ECE-2 is produced by several cell types including neurons and has peak activity at pH 5.8, which makes it a likely intracellular-processing enzyme [9,21]. Both ECE-1 and ECE-2 show preference for big ET-1 over big ET-2 or big ET-3 in vitro. Interestingly, mice lacking both ECE-1 and ECE-2 still have significant levels of mature ET peptides, suggesting that ECE-3 or other unidentified enzyme(s) may carry out the final processing step of ET [9,28].
Regulation of peptide mediators may occur at the synthesis, storage or release level. Regulation of the ET system takes place mainly at the synthesis level, particularly during transcription. ET-1 mRNA is upregulated and ET-1 synthesis is stimulated during major cardiovascular stress and in response to vasoactive agents such as AngII, norepinephrine, vasopressin and thrombin, and cytokines such as tumor necrosis factor-α, interleukins and transforming growth factor-β [6,9,29,30]. ET-1 mRNA levels are upregulated by hypocapnia and downregulated by hypoxia [31]. In endothelial cells, ET-1 mRNA initially increases then decreases by mechanical shear stress and stretch [32], but is consistently decreased in response to NO, prostacyclin and atrial natriuretic factor [9,33,34].
Endothelial cells contain elongated vesicles known as Weilbel-Palade bodies, which serve as a storage compartment for ET-1 [35]. Upon activation of endothelial cells, the Weilbel-Palade bodies relocate from the cytoplasm towards the plasma membrane, fuse with the plasma membrane and release their contents by exocytosis.
ET Plasma and Tissue Levels
Small but measurable levels of ET have been detected in the plasma and blood vessels of human and experimental animals. Studies in healthy adults have shown basal plasma levels between 0.7 and 5 pg/mL [36,37]. In normal Wistar rats the basal plasma ET-1 levels range between 0.7 and 4.9 fmol/mL [38,39]. Also, in isolated aorta of normal Sprague-Dawley rats ET levels of 120 pg/g tissue have been observed [40].
ET Metabolism/Clearance
The low basal plasma/tissue levels of ET may be related to rapid elimination of ET from the bloodstream. ET levels are controlled by continuous metabolism/clearance. In renal tissues, neutral endopeptidase restricts the turnover of ET-1, and inhibitors of this endopeptidase increase urinary ET levels [9,41]. The 24-hour urinary ET excretion has been used as a predictor of ET levels, but the measurements show significant variability. For example, in normal adults, the 24-hour urinary excretion of ET could range between 1.7 pg/mL and 6.8 ng/mL [42,43]. Other forms of ET metabolism may involve its uptake by certain endothelial ET receptors that could function as “clearance receptors” [9].
ET Receptors
There are three known ET receptors, ETA, ETB and ETC. ETA and ETB receptors are widely expressed in a partially overlapping tissue distribution [9,44]. ETA receptors mediate vasoconstriction and cell proliferation, whereas ETB receptors are important for ET clearance, endothelial cell survival, release of NO and prostacyclin and the inhibition of ECE-1 (Fig. 2). Based on their in vivo pharmacology, ETB receptors are classified into two subtypes, ETB1 and ETB2; however, the molecular basis for the existence of these subtypes is still lacking. ETA and ETB receptors share 63% amino acid identity and are encoded by distinct genes located on chromosomes 4 and 13, respectively [9]. Immunoblot analysis of vascular tissues have shown intense ETA receptor immunoreactive band with an apparent molecular mass of 59 kDa, and less dense bands at 44 and 32 kDa. Anti-ETB receptor antiserum has revealed two immunoreactive bands at 64 and 44 kDa. The information on ETC receptors is scant, and additional studies are needed to further characterize this ET receptor subtype.
Fig. 2.
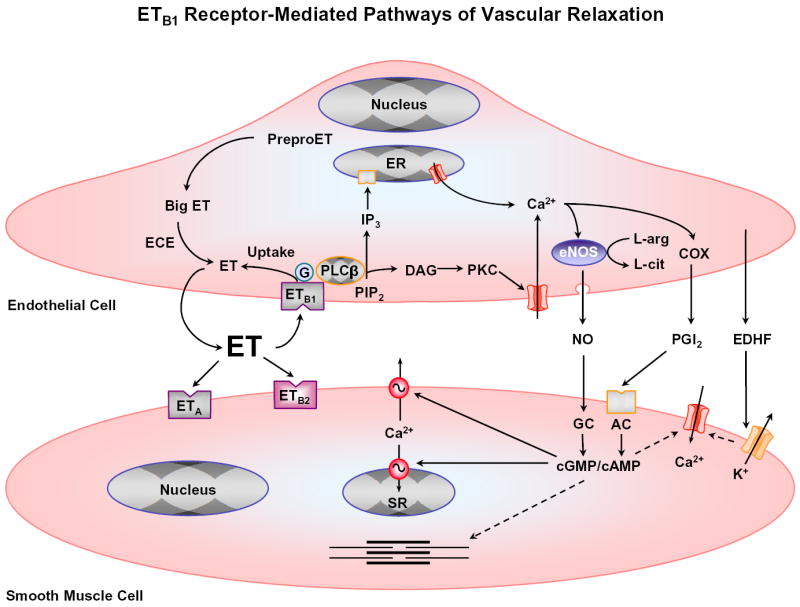
ET-induced VSM relaxation pathways. ET binds to ETB1 receptor in endothelial cells or ETA and ETB2 receptors in VSM. ETB1 may function in ET uptake. ETB1 is also coupled to activation of PLCβ, hydrolysis of PIP2, and release of IP3 and diacylglycerol (DAG). IP3 stimulates Ca2+ release from the endoplasmic reticulum (ER). Ca2+ stimulates eNOS, which converts L-arginine to L-citrulline and increases NO production. NO diffuses into VSM, stimulates guanylate cyclase (GC) and increases cGMP. cGMP causes VSM relaxation by decreasing [Ca2+]i and the myofilament sensitivity to Ca2+. ETB1 -mediated increase in endothelial Ca2+ also stimulates cyclooxygenases (COX) and prostacyclin (PGI2) production. PGI2 activates adenylate cyclase (AC) and increases cAMP, which causes VSM relaxation similar to cGMP. ETB1 also increases the release of EDHF, which activates K+ channels and causes hyperpolarization, inhibition of Ca2+ influx, and VSM relaxation. Interrupted arrows indicate inhibition.
A number of factors affect the expression of ET receptors. In VSM, ETA receptors are upregulated by insulin and NO. In endothelial cells, ETB receptors are upregulated by tumor necrosis factor-α and basic fibroblast growth factor [9].
Tissue Distribution of ET Receptors
ETA receptors are present in VSM of most blood vessels (Table 1), and in airway smooth muscle, cardiomyocytes, liver stellate cells, hepatocytes, neurons, osteoblasts, melanocytes, keratinocytes, adipocytes and various cells in the reproductive system [9,45,46]. ETB receptors predominate in endothelial cells, but are also present in VSM of some vascular beds [46,47] (Table I). ETB receptors have been identified in the aorta, mesenteric arteries, coronary arteries, and veins of different animal species, and in human mammary arteries. ETB receptors are also present in the brainstem glia and neurons, which are involved in the central control of cardiovascular function, the atrial and ventricular myocardium and the atrioventricular conducting tissue [44,46]. ETB receptors have also been localized in renal tubules and collecting duct epithelial cells, airway smooth muscle, liver hepatocytes, osteoblasts, central and peripheral neurons, multiple endocrine tissues and various cells of the reproductive tract [9,44]. Together ETA and ETB are widely distributed in vascular tissues, the central and sympathetic nervous systems, and some regions of the kidney, such as arterioles, glomerular capillaries and inner medullary collecting ducts [48,49].
Table 1.
Examples of endothelin receptor distribution, function, and signaling pathways in the vascular system
| ETA | ETB1 | ETB2 | |
|---|---|---|---|
| Molecular Weight | Mainly 59 kDa | 64/44 kDa | 64/44 kDa |
| Vascular Distribution | |||
| Endothelial Cells | + | ||
| Vascular Smooth Muscle | + | + | |
| Coronary Arteries | + | + | |
| Subcutaneous Arteries | + | + | + |
| Pulmonary Artery | + | + | + |
| Mammary Arteries | + | + | + |
| Veins | + | + | + |
| Glomerular Capillaries | + | + | + |
| Sub-Cellular Distribution | |||
| Cytosol | + | + | + |
| Nucleus | + | ||
| Sarcolemma | + | + | + |
| Function | VSM Contraction | VSM Relaxation | VSM Contraction |
| Vasoconstriction
VSM Growth |
Vasodilatation | Vasoconstriction | |
| Signaling Pathways | - Heterotrimeric G Proteins | - Intracellular Ca2+ | - G Proteins |
| - PLCβ, PLD, PLA2 | Mobilization | - PLCβ, PLD, PLA2 | |
| - Intracellular Ca2+ | - NO Synthesis | - Intracellular Ca2+ | |
| Mobilization | - PGI2 Synthesis | Mobilization | |
| - Activation of Ca2+ Channels | - EDHF Release | - Activation of Ca2+ Channels | |
| - MAPK | - MAPK | ||
Subcellular Distribution of ET Receptors
Immunohistochemical analyses have identified ETA and ETB receptors in the sarcolemma and cytosol of many cell types. ETB receptors have also been found in the nuclear envelope membranes and nucleoplasm [50] (Table I).
ET Receptor-Mediated Signaling Pathways
ET receptor activation leads to diverse cellular responses through interaction with pertussis toxin-sensitive and insensitive pathways, indicating that multiple G-proteins are involved [51–53] (Fig. 3). ETA receptors are functionally coupled to Gq/11 protein to activate phospholiphase C-β (PLC-β), and to Gi protein to inhibit adenylyl cyclase [54]. ETA receptor-mediated activation of Gq/11 protein and PLC-β result in the breakdown of phosphatidylinositol 4,5-bisphosphate, and the generation of inositol 1,4,5-trisphosphate (IP3) and diacylglycerol. IP3 acts on specific receptors on the intracellular Ca2+ stores and stimulate Ca2+ release [51]. ET also activates plasma membrane Ca2+ channels and stimulates Ca2+ influx from the extracellular space [55,56]. The ET-induced increase in diacylglycerol stimulates PKC activity [57–59]. ETA receptor stimulation could also activate phospholipase D with generation of diacylglycerol, phospholipase A2 with release of arachidonic acid, the Na+/H+ exchanger, Src-family tyrosine kinases, mitogen-activated protein kinase (MAPK), c-Jun-NH2-terminal kinase (JNK), p38 MAPK, and phosphatidylinositol 3-kinase [9,60–62].
Fig. 3.
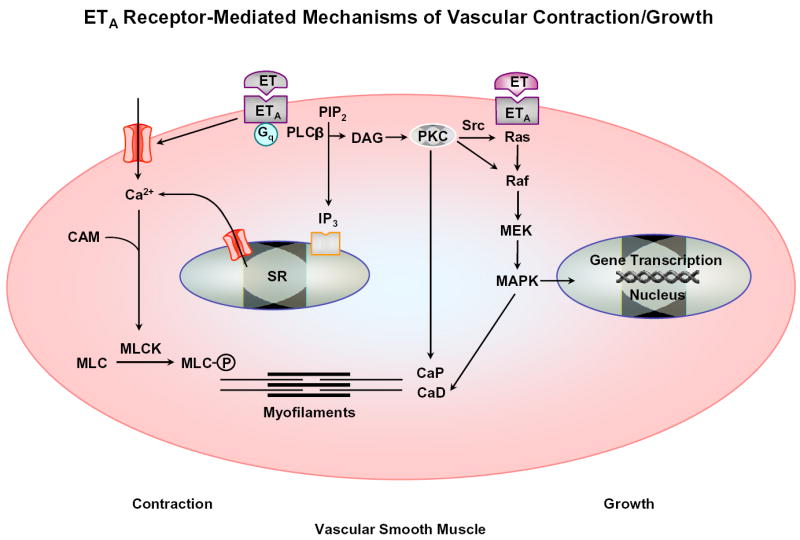
ET binds to ETA receptor, stimulates PLCβ, and increases production of IP3 and DAG. IP3 stimulates Ca2+ release from the SR. ET also stimulates Ca2+ entry through Ca2+ channels. Ca2+ binds calmodulin (CAM), activates myosin light chain (MLC) kinase, causes MLC phosphorylation, and initiates VSM contraction. DAG activates PKC. PKC could phosphorylate calponin (CaP) and/or activate a protein kinase cascade involving Raf, MAPK kinase (MEK) and MAPK, leading to phosphorylation of caldesmon (CaD) and an increase in the myofilament force sensitivity to Ca2+. ETA receptor-mediated activation of MAPK could induce gene transcription and VSM growth. ETB2 receptors could activate similar mechanisms of VSM contraction/growth.
Stimulation of endothelial ETB receptors activates signaling pathways that promote the release of relaxing factors such as NO, prostacyclin and EDHFs (Fig. 2). The ETB receptor-mediated release of NO from endothelial cells may account for the transient vasodilator action of ET-1. NO may also regulate ET-1 production, and NO donors may inhibit ET-1 release, while NOS inhibitors stimulate ET-1 release from endothelial cells. Also, the vasodilator actions of NO antagonize the constrictor actions of ET-1 on VSM [63].
The role of ETB receptors in promoting EDHF release from endothelial cells is not clear. EDHFs are important mediators of vascular relaxation, particularly in resistance arteries where they regulate tissue blood flow. The release of EDHFs is modulated by a number of factors including agonist stimulation and shear stress. The chemical identification and functional characterization of EDHFs vary depending on vascular size, vascular bed and species. Major EDHF candidates include the cytochrome P450 metabolites of arachidonic acid epoxyeicosatrienoic acids, K+ and hydrogen peroxide [2,3]. Additionally, electrical coupling through myoendothelial gap junctions serves to conduct electrical changes from the endothelium to VSM and may mediate or propagate hyperpolarization.
Functions of ET/ET Receptors
ET-1 induces vasoconstriction, is proinflammatory, promotes fibrosis and has mitogenic effects on VSM, which are important factors in the regulation of vascular tone, and in vascular injury and remodeling [9]. The ET-1 induced vascular effects are ET receptor-specific. ETA receptors are localized in VSM and produce vasoconstriction. Also, ET-1, -2 and -3 could induce vascular contraction via activation of VSM ETB2 receptors [64]. For example, both ETA and ETB receptors are present and produce vasoconstriction in the renal circulation [65]. Also, studies using selective ET receptor agonists and antagonists have suggested the presence of the constrictor ETB receptors in human subcutaneous and rat mesenteric arteries, albeit in low numbers. These data agree with the reports that ETB receptor agonists could elicit vasoconstriction in vivo and ETB receptor-mediated contraction in isolated blood vessels [66].
Although much attention has been given to the role of ETA and perhaps ETB2 receptors in the pathophysiology of cardiovascular and renal disease, recent studies suggest an equally important role for endothelial ETB1 receptors n the regulation of vascular tone, sodium balance and arterial pressure [5]. ET-1 may produce vasodilatation via activation of endothelial ETB receptors, and enhanced NOS activity and NO release [63] (Fig. 2). Endothelial ETB receptors also mediate the release of prostacyclin and produce vasodilation in numerous vascular beds. In support of a role of ETB receptors in blood pressure regulation it has been shown that ETB heterozygous (+/−) knockout mice are hypertensive, possibly due to unbalanced activation of ETA receptors by endogenous ET-1 [67]. Also, studies in ETB receptor-knockout mice have suggested that the vascular ETB receptors in vivo may play a favorable inhibitory role in vascular remodeling after injury [68]. ETB receptors are also abundant in tubular epithelium of the renal medulla. ET-1, via ETB receptors, may inhibit sodium and water reabsorption [69].
Big ET-1 may produce similar effects to ET-1 as a result of its conversion to the latter peptide by ECE. However, big ET-1 is a less potent renal vasoconstrictor than ET-1, despite the fact that both peptides produce comparable increases in arterial pressure [4]. Studies suggest that big ET-1, but not ET-1, provokes significant diuresis and natriuresis [4,70]. For example, renal cortical blood flow decreases in response to big ET-1 in rats on a normal or high salt diet. Also, big ET-1 increases medullary blood flow in rats on a high but not normal salt diet. These data demonstrate that medullary vasodilation produced by big ET-1 is more prominent in rats on a high salt diet. The data are consistent with contribution of ETB-mediated events in the natriuretic response to high salt intake, and further support a role for ET in regulating sodium excretion through activation of ETB receptors [71].
ET Clearance via ETB Receptors
ETB receptors also likely function as “clearance receptors”. For example, the ETB selective antagonist BQ788 inhibits accumulation of intravenously administered radiolabeled ET-1 in the lungs and kidneys, thereby slowing its clearance from the circulation, whereas the ETA receptor-selective antagonist BQ123 has no such effect [9]. In conditions that are associated with changes in the expression of ET receptors, the ET clearance, plasma levels and vascular responses could vary. For example, ETB receptor deficiency is associated with high plasma ET-1 and could cause a decrease in ETA-dependent contraction to ET-1 in mesenteric arteries. This effect occurs in the presence of an increase in ETA receptor protein, suggesting possible uncoupling of receptor expression and functional effects [64].
ET Receptor Agonists
ETA and ETB receptors can be distinguished by their ligand specificities (Table II). ETA receptor has subnanomolar affinities for ET-1 and ET-2 and 2-orders of magnitude lower affinity for ET-3. ETB receptor has equal subnanomolar affinities for ET-1, -2 and -3 [9,72],. The chymase-derived peptide ET-1 is a relatively selective agonist of ETA receptors [73,74].
Table 2.
Examples of Recently Published and Commercially Available ET Receptor Agonists and Antagonists and Their Selectivity to ET Receptor Subtypes
| Compound | Chemistry | Receptor Specificity | US Patent |
|---|---|---|---|
| Agonists | |||
| ET1
ET2 ET3 |
Cys-Ser-Cys-Ser-Ser-Leu-Met-Asp-Lys-Glu-Cys-Val-Tyr- Phe- Cys-His-Leu-Asp-Ile-Ile-Trp | ETA, ETB
ETA, ETB ETB |
5468623 [87]
5230999 [88] |
| IRL 1620 | Suc-Asp-Glu-Glu-Ala-Val-Tyr-Phe-Ala-His-Leu-Asp-Ile-Ile-Trp | ETB | 20050084872* [89] |
| S6c | Cys-Thr-Cys-Asn-Asp-Met-Thr-Asp-Glu-Glu-Cys-Leu-Asn-Phe- Cys-His-Gln-Asp-Val-Ile-Trp | ETB | 20050090514* [90] |
| Antagonists | |||
| A127722 | 2-(4-methoxyphenyl)-4-(1,3-benzodioxol-5-yl)-1-(((dibutyl amino)carbonyl) methyl)pyrrolidine-3-carboxylic acid | ETA | 6573285 [91] |
| ABT 627 | (2R,3R,4S)-1-[(dibutyl carbamoyl)methyl]-2-(p-methoxyphenyl)- 4[3,4-(methylenedioxy)phenyl]-3-pyrrolidinecarboxylic acid | ETA | 20050042172* [92] |
| BMS 182874 | 5-(dimethylamino)-N-(3,4-dimethyl-5-isoxazolyl)-1- naphthalenesulfonamide | ETA | |
| Bosentan | 4-t-butyl-N-(6-(2-hydroxyethoxy)-5-(2-methoxyphenoxy)-2,2′- bipyrimidin-4-yl)benzenesulfonamide | ETA, ETB | 20030176356* [93] |
| BQ 123 | Cyclo(D-α-aspartyl-L-prolyl-D-valyl-L-leucyl-D-tryptophyl) | ETA | 6573285 [91] |
| BQ 610 | Homopiperidinyl-CO-Leu-D-Trp(CHO)-D-Trp-OH | ETA | 20030176356* [93] |
| BQ 788 | N-cis-2,6-dimethylpiperidinocarbonyl-γ-methylleucyl-tryptophyl(COOMe)-norleucine | ETB | 20030176356* [93] |
| FR 139317
(PD 147953) |
N-(N-(N-((hexahydro-1H-azepin-1-yl)carbonyl)-L-leucyl)-1-methyl-D-tryptophyl)-3-(2-pyridinyl)-2-((1-(hexahydro-1H-azepinyl)carbonyl)amino-4-methyl-pentanoyl)-3-(-(1-methyl-1H-indolyl)propionyl)amino-3-(2-pyridyl)propionic acid | ETA | 20030176356* [93] |
| LU 135252 | 2-(4,6-dimethoxypyrimidin-2-yloxy)-3-methoxy-3,3-diphenylpropionic acid | ETA | 20030176356* [93] |
| PD 142893 | Ac-D-(3,3-diphenylalanine)-leu-Asp-Ile-Ile-Trp-OH | ETA, ETB | 20030176356* [93] |
| PD 145065 | Ac-D-(5H-dibenzo[a,d]cycloheptene-5-glycine)-Leu-Asp-Ile-Ile- Trp-OH | ETA, ETB | 20030176356* [93] |
| PD 156707 | Sodium 2-benzo[1,3]dioxol-5-yl-4-(4-methoxy-phenyl)-4-oxo-3-(3,4,5-trimethoxy-benzyl)-but-2-enoate | ETA | 6573285 [91] |
| RES 701–1 | L-Tryptophan, glycyl-L-asparaginyl-L-tryptophyl-L-histidylglycyl- L-threonyl-L-alanyl-L-prolyl-L-α-aspartyl-L-tryptophyl-L- phenylalanyl-L-phenylalanyl-L-asparaginyl-L-tyrosyl-L- tyrosyl-, cyclic (9–1)-peptide | ETB2 | 6855701 [94] |
| SB 209670 | 3-(2-(carboxymethoxy)-4-methoxyphenyl)-1-(3,4-(methylene dioxy)phenyl)-5-(prop-1-yloxy)indan-2-carboxylic acid | ETA, ETB1 | 6573285 [91] |
| TAK 044 | cyclo(D-alpha-aspartyl-3-((4-phenylpiperazin-1-yl)carbonyl)-L-alanyl-L-alpha-aspartyl-D-2-(2-thienyl)glycyl-L-leucyl-D-tryptophyl) disodium salt | ETA, ETB | 6573285 [91] |
US patent application number.
indicates reference number
The ETB agonist sarafotoxin 6c (S6c) induces smaller vascular contraction than ET-1 in resistance vessels isolated from human subcutaneous fat and rat mesentery. The small S6c contraction remains in the presence of the ETA antagonist BQ610. S6c-induced contraction is slightly enhanced in the presence of the NOS inhibitor L-NAME or after removal of the endothelium. These data are consistent with the notion that vasoconstrictor ETB receptors are present in resistance vessels of human subcutaneous fat and in rat’s mesentery, but their contribution to ET-mediated constriction is small. Other studies have shown that in tissues treated with L-NAME, S6c induces only a minimal contraction (less than 5%) [75]. ET-3 is considered a high affinity ETC receptor agonist.
ET Receptor Antagonists
ET receptor antagonists have been useful in determining the role of ET in various systems (Table II). ABT627 and BQ610 are selective ETA receptor antagonists [73,74,76]. BQ123 inhibits ET-1 induced contraction in the abdominal aorta [75]. Also, ET-1 induced contraction of human and rat resistance arteries is reduced by the ETA antagonists BQ123 and BQ610, suggesting that the ETA receptor is a major ET receptor in these resistance vessels [46].
ETB receptor antagonists have recently become available (Table II). Studies in rats treated with the ETB antagonist A192621 have shown significant increases in blood pressure associated with enhanced vascular contraction and reduction of endothelium-dependent vascular relaxation and NO production. The vasoconstrictive effects of A192621 were enhanced in rats on a high salt diet. These studies suggest that endothelial ETB1 receptors could play a role in the regulation of vascular tone particularly during high salt diet [77].
Role of ET in the Regulation of blood Pressure and in Hypertension
ET has been implicated in multiple cardiovascular functions and diseases, and recent evidence suggests significant involvement of ET in the regulation of blood pressure and in the pathogenesis of hypertension. Upregulation of the ET system appears to occur mainly in moderate to severe forms of human hypertension, as well as in experimental animal models of severe and salt-sensitive hypertension [66].
ET Plasma/Tissue Levels in Hypertension
The role of ET-1 in human hypertension has been investigated by measuring its plasma levels. Although some studies have demonstrated an increase in plasma levels of ET-1 in hypertensive patients, most studies show normal or slightly increased levels [46]. Increased plasma ET levels have been described in hypertensive African-Americans compared to normotensive controls. However, when African-Americans are compared with Caucasians, the plasma ET levels are not higher in individuals with similar severity of hypertension [66]. Upregulation of the ET system has also been reported in severe hypertension associated with coronary artery disease, heart failure, atherosclerosis and pulmonary hypertension. For example, the plasma ET-1 levels show dramatic increase in patients with heart failure (5.15 pg/mL) compared with control groups (0.75 pg/mL). Also, plasma levels of big ET-1 are greater in patients with heart failure (25.7 pg/mL) compared with control subjects (7.7 pg/mL) [42].
The inconsistent findings regarding plasma ET levels in hypertension are not surprising because elimination of ET-1 from the bloodstream occurs rapidly. Also, ET secretion is highly polarized from the endothelial cells to VSM, causing minimal increase in circulating plasma ET. Another approach to determine the role of ET in hypertension is to measure ET levels in vascular tissues. ET production is increased in vascular beds in some forms of hypertension [46]. For example, moderate to severe hypertensive patients show enhanced expression of ET-1 mRNA in the endothelium of subcutaneous resistance arteries. Other forms of human hypertension that exhibit increased ET-1 tissue expression include salt-sensitive hypertension, low renin hypertension, and obesity and insulin resistance-related hypertension [78].
Tissue levels of ET-1 also vary in animal models of experimental hypertension. ET-1 is overexpressed in the vascular wall of deoxycorticosterone acetate (DOCA)-salt hypertensive rats. The aorta of DOCA-salt rats show significant elevation of ET-1 (730 pg/g) compared to control tissues (120 pg/g) [40]. The increase in ET tissue levels is significantly higher in comparison to the plasma levels, supporting the contention that tissue levels of ET could be a better indicator of the hypertensive changes in the vascular ET system. ET-1 is also overexpressed in the vascular wall of other salt-dependent models of hypertension such as DOCA-salt-treated spontaneously hypertensive rats (SHR) and Dahl salt-sensitive rats, and in salt-loaded stroke-prone SHR, AngII-infused rats and 1-kidney 1-clip Goldblatt hypertensive rats, but not in SHR, 2-kidney 1-clip hypertensive rats or L-NAME-treated rats [60].
The levels of ET in urine could also show significant changes in hypertension. For example, the 24 hour urine levels of ET-1 show an increase in hypertensive patients with heart failure (17.0 ng/g UC) as compared to control subjects (1.7 ng/g UC) [42].
ET Receptor Density in Hypertension
The amount of ET receptors could vary in different forms of hypertension, and more intriguingly in various tissues isolated from subjects with the same form of hypertension. For example, ETB receptors are upregulated in the kidneys of DOCA-salt hypertensive rats, consistent with a role for ETB receptors in the renal regulation of arterial pressure [69]. However, ET receptors could also be downregulated by ET, especially under conditions in which large amounts of ET are produced in the vasculature. For instance, the ET receptor density is reduced in some vascular beds of DOCA-salt hypertensive rats, suggesting that the ET receptors could be downregulated by the increased vascular production of ET [46].
Vascular Response to ET in Hypertension
The vascular response to ET could show variability similar to that seen in the ET plasma/tissue levels in hypertension. Vascular contraction to ET-1 is increased in rat hearts during ischemia/reperfusion and in the rat pulmonary circulation in pulmonary hypertension [46]. Evidence indicates that a defect in VSM regulation of intracellular Ca2+ may play a role in the augmented vascular reactivity to ET-1 in some forms of experimental hypertension [65,79]. However, vascular contraction to ET is unchanged in the aorta of SHR and is even decreased in the mesenteric arteries of DOCA-salt hypertensive rats [46,64]. This is possibly related to the finding that vascular ET receptor density is reduced in DOCA-salt hypertensive rats as a result of the receptors being downregulated by increased vascular ET production [46].
ET, via its growth-promoting properties, could also play a role in VSM hypertrophy in hypertension. Remodeling of large and small arteries contributes to the elevation of the blood pressure and the complications of hypertension. In hypertension, large arteries may exhibit increased lumen size, thickened media, increased collagen deposition and decreased compliance, leading to elevation of systolic blood pressure and pulse pressure. In milder forms of hypertension, the VSM of resistance arteries are restructured around a smaller lumen without true hypertrophy. Remodeled resistance arteries in most hypertensive animals and in human hypertension exhibit a reduced circumference, which acts as an amplifier of pressor stimuli. This structurally based amplification may explain the enhanced vasoconstriction observed in isolated perfused vascular beds of SHR and renovascular hypertensive rats [46]. In severe forms of hypertension and in secondary hypertension, hypertrophic remodeling of VSM occurs [66]. The ET growth-promoting properties could play a role in the hypertrophy of VSM observed in severe hypertension and in DOCA-salt hypertensive rats [46].
Changes in the responsiveness of endothelial ETB receptors may also occur in hypertension. The ETB receptor-mediated vasorelaxation induced by ET-1 is greater in SHR and DOCA-salt hypertensive rats than normotensive controls. Thus, ET does not appear to release less vasorelaxant substances in hypertensive rats, and this may not be a mechanism via which ET contributes to the pathophysiology of hypertension. It has been proposed that ETA receptors may play a role in the development of DOCA-salt–induced hypertension, whereas ETB receptors may protect against vascular and renal injuries in this model [80]. However, other studies have provided opposite results, suggesting that ET may indeed release less endothelium-derived relaxing factor in SHR blood vessels [46].
Effects of ET Antagonists in Hypertension
Because the primary vasoconstrictor actions of ET are via ETA receptors, one would predict that ETA receptor antagonists would decrease the blood pressure. ETA receptor antagonists have produced variable results in normotensive and hypertensive animals. For example, chronic administration of ETA receptor antagonist in normotensive rats has no effect on arterial pressure, suggesting that ET may not play a major role in regulating basal arterial pressure [63]. Also, the ETA antagonist A127722 slightly lowers blood pressure in DOCA-salt hypertensive rats [81]. Additionally, administration of BQ123 slightly lowers the blood pressure in SHR and DOCA-salt hypertensive rats, but not in renovascular hypertension. These rather modest effects have suggested that ET involvement in hypertension is minor [66]. However, a recent study indicates that acute administration of the ETA antagonist ABT-627 to DOCA-salt rats produces significant hypotensive effect and that long-term treatment with this agent suppresses the development of hypertension [80]. Also, long-term treatment with the nonselective ETA/ETB antagonist bosentan attenuates the development of hypertension and vascular remodeling in DOCA-salt rats. Bosentan decreases the blood pressure to a degree similar to that observed with selective ETA receptor antagonist, suggesting that ETA receptors are the main receptors involved in the pathogenesis of DOCA-salt-sensitive hypertension [80].
The net benefits of ET antagonists in hypertension may depend on their ability to suppress the vasoconstrictive effects of ET or its growth promoting properties or both. In hypertensive animals that overexpress ET-1 in their blood vessels, the vasoconstrictor effect of ET-1 may contribute to blood pressure elevation, while its growth-promoting action contributes to vascular hypertrophy. In these hypertensive rats overexpressing ET-1, ETA/B and ETA-selective receptor antagonists lower blood pressure slightly, but significantly reduce vascular growth, particularly of small arteries [60]. Also, intravenous infusion of bosentan reduces the blood pressure in SHR and DOCA-salt hypertensive rats. On the other hand, the vascular hypertrophy and remodeling of resistance arteries is practically abolished by bosentan treatment, beyond what could be explained by the blood pressure lowering effect [46].
Targeting the ET system could be useful in treatment of hypertension in human, particularly by preventing target organ damage and cardiovascular complications. ETA-selective antagonists could block many of the ET-induced effects on VSM, and thereby prevent the pathophysiologic effects of ET in cardiovascular diseases such as hypertension, heart failure, atherosclerosis, coronary heart disease, restenosis after angioplasty and primary pulmonary hypertension [60]. In clinical trials, combined ETA-ETB receptor blockers produce significant blood pressure-lowering effects [78]. In a study of mild cases of essential hypertensive patients, a 4-week trial of bosentan reduced the blood pressure similar to the angiotensin converting enzyme (ACE) inhibitor enalapril. Also, in acute and chronic studies, bosentan improves hemodynamics in hypertensive patients with heart failure. ET receptor antagonists may also offer promise in primary pulmonary hypertension [60]. Thus, the ET system appears to be involved in different forms of cardiovascular disease in human, and its interruption using ET antagonists offers great promise as a therapeutic intervention in hypertension, heart failure and other cardiovascular diseases [60].
Vascular ET and Other Control Mechanisms of Blood Pressure
If ET plays a role in the regulation of blood pressure and in the pathogenesis of hypertension, then infusion of ET in experimental animals should increase the blood pressure. Although some studies have shown that ET infusion in rats causes elevation of blood pressure [82], other studies have demonstrated that ET infusion causes slight or no change [56]. The difference in the results could be related to the activity of the ET batches from different commercial sources. ET could also be rapidly metabolized leaving less ET to act on the blood vessels. Additionally, ETA mediated vascular contraction could be counterbalanced by ETB mediated increase in endothelium-derived vasodilators and promotion of vascular relaxation. Furthermore, the vascular ET system could interact with other vascular, neuronal and renal control mechanisms of blood pressure in the setting of hypertension. For example, the effects of vascular ET on the blood pressure could be influenced by endothelial NO, oxidative stress, the sympathetic nervous system, dietary salt, and the renin-angiotensin system.
ET and NO
NO plays a major role in the regulation of vascular function and a more significant role than ET-1 in the long-term maintenance of blood pressure. Inhibition of NO production causes elevation of blood pressure in experimental animals. Interestingly, ETA receptor blockade attenuates the hypertension in the early stages of chronic NOS inhibition, while investigations of the role of ET receptors in the long term have not supported ET involvement [63]. Although ET appears to contribute to the hypertension in the early stages of NOS inhibition, blockade of either ETA or both ETA and ETB receptors has only a minor effect on the hypertension beyond the initial two weeks of NOS inhibition. It appears that ET may play a role in the development of early vascular lesions associated with NOS inhibition, at least within the kidney, which may be related to AngII activity. However, the processes involved in the hypertension associated with chronic NOS inhibition appear to be complex and variability in the results in different animal species may relate to genetic factors and the choice of NOS inhibitor [63].
ET and Oxidative Stress
Increased vascular oxidative stress has been observed in DOCA-salt hypertensive rats, a rat model with elevated plasma ET levels. Also, in vivo blockade of ETA receptors in DOCA-salt hypertensive rats results in reduction in oxidative stress, supporting a role for ET-1 in the generation of reactive oxygen species. Since oxidative stress influences specific signaling pathways and redox-sensitive genes that coordinate several responses in the cardiovascular system including VSM growth and endothelial cell function, and because each of these alterations could be produced by ET-1, oxidative stress may play a role in the cardiovascular changes observed in DOCA-salt hypertension as a result of ET-1 overexpression/actions [83].
ET and the Sympathetic Nervous System
Salt-sensitive hypertensive patients often have low plasma renin activity and their plasma ET levels are dramatically increased, in association with enhanced plasma catecholamines. This suggests a relationship between the sympathetic system, sodium sensitivity and reactivity of the ET system that may contribute to blood pressure elevation in these subjects [66].
ET and High Salt
In salt-dependent hypertension, a high-salt diet intensifies the increase in blood pressure. A decrease in endothelial-derived NO may contribute to the development of salt-dependent hypertension [84]. High salt diet is also associated with increased ET production, and stimulation of both ETA mediated vascular contraction and ETB mediated vascular relaxation. Also, chronic ETB receptor blockade increases arterial pressure in normal rats and the hypertension is much greater in rats on a high-salt diet [77]. However, the increase in arterial pressure during ETB receptor blockade is larger than that predicted solely from the elevated salt intake such that there was a significant shift in the pressure-natriuresis relationship. These findings are consistent with a role for ET-1 in regulating the blood pressure during conditions of high salt intake [69].
ET and the Renin-Angiotensin System
The renin-angiotensin system plays a major role in the regulation of blood pressure. Some of the effects of AngII, particularly growth of VSM media, are mediated by ET-1 via ETA receptors. AngII may also stimulate the production of ET in SHR blood vessels to a greater extent than in normotensive control vessels [46].
Current and Future Developments
The ET system plays an intricate role in many physiological and pathological conditions. Underlying the complex physiology of the ET system is the diverse expression pattern of its components [9]. Several ET isoforms and receptor subtypes have been identified in various tissues, and many ET-mediated signaling pathways have been proposed in various cell types.
The vascular ET system functions as a modulator of vascular tone, growth, and the vascular control mechanisms of blood pressure. ET-1 could be a factor in many cardiovascular diseases, and may contribute to the increased blood pressure observed in some models of experimental hypertension and in human hypertension. ET plays a role in blood pressure elevation and vascular growth in moderate-to-severe hypertension, in salt-sensitive forms of hypertension, and in certain populations such as African-Americans. Although ET infusion may produce hypertension in some animals, not all animal models of hypertension have high ET levels, and ET infusion does not always increase the blood pressure.
There are different ways for treatment of hypertension including diuretics, β-adrenergic blockers, angiotensin-converting enzyme inhibitors, AngII receptor subtype 1 antagonists and long-acting Ca2+ channel blockers. However, in hypertension, as the blood pressure increases, endothelial damage may increase the expression of ET-1 in the blood vessels and the heart. ET could then further contribute to blood pressure elevation and the progression of vascular damage and atherosclerosis. Therefore, blocking the ET system may provide a new therapeutic approach beyond blood pressure lowering in hypertension, by contributing to the arrest of vascular damage, and thereby improving the prognosis.
One way to block the ET system is to use ET receptor antagonists and thereby reduce VSM contraction/growth. Currently available ET receptor antagonists reduce blood pressure in some forms of experimental hypertension (Table II), and could be effective disease-modifying agents if they are shown in clinical trials to blunt vascular growth and endothelial dysfunction, reduce stroke and exert the vascular protective effects already reported in experimental hypertension. ET antagonists could also reduce the long-term cardiovascular complications of hypertension [60]. Careful examination of recent patents may identify more effective/specific modulators of the vascular ET system for clinical use in human hypertension (Table III).
Table 3.
Recently Patented Compounds with Potential ET Receptor Antagonist Properties
| Compounds | Examples | Receptor Specificity | US Patent |
|---|---|---|---|
| Benzothia(oxa)diazols | 3-(2,1,3-benzothiadiazol-5-yl)-4-(3-cyclopentyloxy-4,5-dimethoxybenzyl)-5-hydroxy-5-(4 methoxyphenyl)- 5H-furan-2-one | ETA, ETB | 6017939 [95]
6197800 [96] |
| Benzothiazine dioxides | 4-(3,5-Dimethoxy-phenyl)-2-(2-trifluoromethyl-phenyl)-1,1-dioxo-1,2-dihydro -1.γ6 -benzo[e][1,2]thiazine-3-carboxylic acid | ETA | 6440962 [97] |
| Cyclopentanes | (1RS,2RS,3SR)-1,3 bis(4-methoxyphenyl) cyclopentane-2-carboxylic acid | ETA, ETB | 5929106 [98] |
| Dibenzodiazepines | 5,11-Dihydro-8-(1-naphthalenylmethoxy)-11-oxo-10H-dibenzo[b,e][1,4]diazepine-10-acetic acid | ETA, ETB | 5420123 [99] |
| Furans | (E)-3-[3-[2-(2-Carboxyphenyl)methoxy-4-methoxy] phenylfuran-2-yl]-2-[(2-meth oxy-4,5-methylene dioxy) phenylmethyl]-prop-2-enoic acid | ETA, ETB | 6051599 [100] |
| Indanes, Indenes, Indoles | (1RS,2SR,3RS)-3-(2-Carboxymethoxy-4-methoxy phenyl)-1-(3,4-methylenedioxy-phenyl)-5-(prop-1-yloxy)-indane-2-carboxylic acid | ETA, ETB | 6448260 [101]
6384070 [102] |
| Isooxazoles, Oxazoles, Imidazoles | (E)-Ethyl alpha-[[3-[4methoxy-2-[[2-(methoxycarbonyl) phenyl]methoxy]-phenyl]isoxazol -4-yl]methylene]-6-methoxy-1,3-benzodioxole-5-propanoate | ETA, ETB | 6174906 [103]
6620826 [104] |
| Ketoacids | Benzo[1,3]dioxol-5-yl-4-(4-methoxy-phenyl)-4-oxo-3-(3,4,5-trimethoxy- benzyl)-but-2-enoic acid | ETA, ETB | 6043241 [105] |
| Phenoxyphenylacetic acids | 2-[(2,6-Dipropyl-4-hydroxymethyl) phenoxy]-2-(3-methyl phenyl)acetic acid | ETA, ETB | 5565485 [106] |
| Phenylalanine derivatives | (R,S)-N-(Diphenylacetyl)-3-methoxy-2-(phenylmethoxy) phenylalanine | ETA, ETB | 5658943 [107] |
| Prostaglandins | difluoro-13,14-dihydro-15-keto-PGE1 Methyl Ester | ETA, ETB | 6197821 [108] |
| Pyrazoles, Triazoles | (E)-a-[[5-[2-[(2-carboxyphenyl)methoxy]-4-methoxy phenyl]-1-ethyl-1H-pyrazol-4-yl]methylene]-6-methoxy-1,3-benzodioxole-5-propanoic acid | ETA, ETB | 6545031 [109] |
| Pyridizinones | 2-(1,3-benzodioxol-5-yl)-2-(2,3-dihydro-4,6-dimethyl pyridazin-3-on-2-yl)N- (4-isopropylphenyl sulfonyl) acetamide | ETA, ETB | 5883090 [110] |
| Pyrimidines | 3-6-(4-t-butylphenyl-sulfonylamino)-5-(2-methoxyphen oxy)-2-morpholino-4-pyrimidinyl oxypropionic acid | ETA, ETB | 5883092 [111] |
| Pyrrolo[2,3-b]pyridines | (Methoxyphenyl)-1-(3,4-methylenedioxy-phenylmethyl) pyrrolo[2,3-b]pyridine-2-carboxylic acid | ETA, ETB | 6075037 [112] |
| Sulfanyl derivatives | Methyl 3,3-diphenyl-2-(4,6-dimethoxypyrimidine-2-sulfanyl)propionate | ETA, ETB | 6235903 [113] |
| Aryl, hetaryl, isoxazole, N-heterocyclic, phenyl, and thieno-pyridine sulfonamides | 5-(Dimethylamino)-N-(3,4-dimethyl-5-isoxazolyl)-1-naphthalenesulfonamide | ETA, ETB | 6387915 [114]
6686382 [115] 6107320 [116] 6013655 [117] |
| Benzene, biphenyl sulfonamides | N-(3,4-Dimethyl-5-isoxazolyl)-4-biphenylsulfonamide | ETB | 6541498 [118] |
indicates reference number
Another potential way to treat hypertension and prevent its cardiovascular/renal injuries is to use ECE inhibitors. Small molecular biaryl compounds are potential ECE inhibitors [85]. Quinazoline compounds also have potent ECE inhibitory effects [86]. An alternative way to treat hypertension is to take a genetic approach and knock-out a specific ET isoform or ET receptor subtype. The genetic approach has shown promising results in experimental animals, but remains to be validated in humans with cardiovascular disease [66].
Although this review has emphasized on the role of vascular ET in the control of blood pressure and the pathogenesis of hypertension, that should not minimize the role of ET on the neuronal, hormonal and renal control mechanisms of the blood pressure. Also, no disease entity including hypertension could be attributed solely to an abnormality in ET alone, and it would be unrealistic to expect that ET receptor antagonists alone could cure hypertension.
Acknowledgments
This work was supported by grants from National Heart, Lung, and Blood Institute (HL-65998, HL-70659). RA Khalil is an Established Investigator of the American Heart Association.
References
- 1.Ignarro LJ, Kadowitz PJ. The pharmacological and physiological role of cyclic GMP in vascular smooth muscle relaxation. Annu Rev Pharmacol Toxicol. 1985;25:171–91. doi: 10.1146/annurev.pa.25.040185.001131. [DOI] [PubMed] [Google Scholar]
- 2.Vanhoutte PM. Vascular biology. Old-timer makes a comeback. Nature. 1998;396:213. doi: 10.1038/24261. 215–6. [DOI] [PubMed] [Google Scholar]
- 3.Gauthier KM, Deeter C, Krishna UM, Reddy YK, Bondlela M, Falck JR, Campbell WB. 14,15-Epoxyeicosa-5(Z)-enoic acid: a selective epoxyeicosatrienoic acid antagonist that inhibits endothelium-dependent hyperpolarization and relaxation in coronary arteries. Circ Res. 2002;90(9):1028–36. doi: 10.1161/01.res.0000018162.87285.f8. [DOI] [PubMed] [Google Scholar]
- 4.Pollock DM, Opgenorth TJ. Evidence for metalloprotease involvement in the in vivo effects of big endothelin 1. Am J Physiol. 1991;261(1 Pt 2):R257–63. doi: 10.1152/ajpregu.1991.261.1.R257. [DOI] [PubMed] [Google Scholar]
- 5.Granger JP. Endothelin. Am J Physiol Regul Integr Comp Physiol. 2003;285(2):R298–301. doi: 10.1152/ajpregu.00249.2003. [DOI] [PubMed] [Google Scholar]
- 6.Yanagisawa M, Kurihara H, Kimura S, Tomobe Y, Kobayashi M, Mitsui Y, Yazaki Y, Goto K, Masaki T. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature. 1988;332(6163):411–5. doi: 10.1038/332411a0. [DOI] [PubMed] [Google Scholar]
- 7.Rubanyi GM, Polokoff MA. Endothelins: molecular biology, biochemistry, pharmacology, physiology, and pathophysiology. Pharmacol Rev. 1994;46(3):325–415. [PubMed] [Google Scholar]
- 8.Konishi F, Okada Y, Takaoka M, Gariepy CE, Yanagisawa M, Matsumura Y. Role of endothelin ET(B) receptors in the renal hemodynamic and excretory responses to big endothelin-1. Eur J Pharmacol. 2002;451(2):177–84. doi: 10.1016/s0014-2999(02)02228-8. [DOI] [PubMed] [Google Scholar]
- 9.Kedzierski RM, Yanagisawa M. Endothelin system: the double-edged sword in health and disease. Annu Rev Pharmacol Toxicol. 2001;41:851–76. doi: 10.1146/annurev.pharmtox.41.1.851. [DOI] [PubMed] [Google Scholar]
- 10.Lüscher TF, Barton M. Endothelins and endothelin receptor antagonists: therapeutic considerations for a novel class of cardiovascular drugs. Circulation. 2000;102:2434–40. doi: 10.1161/01.cir.102.19.2434. [DOI] [PubMed] [Google Scholar]
- 11.Lee ME, de la Monte SM, Ng SC, Bloch KD, Quertermous T. Expression of the potent vasoconstrictor endothelin in the human central nervous system. J Clin Invest. 1990;86(1):141–7. doi: 10.1172/JCI114677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rubanyi GM, Botelho LH. Endothelins. FASEB J. 1991;5(12):2713–20. doi: 10.1096/fasebj.5.12.1916094. [DOI] [PubMed] [Google Scholar]
- 13.Sakai S, Miyauchi T, Kobayashi M, Yamaguchi I, Goto K, Sugishita Y. Inhibition of myocardial endothelin pathway improves long-term survival in heart failure. Nature. 1996;384(6607):353–5. doi: 10.1038/384353a0. [DOI] [PubMed] [Google Scholar]
- 14.Matsumoto H, Suzuki N, Onda H, Fujino M. Abundance of endothelin-3 in rat intestine, pituitary gland and brain. Biochem Biophys Res Commun. 1989;164(1):74–80. doi: 10.1016/0006-291x(89)91684-7. [DOI] [PubMed] [Google Scholar]
- 15.Ishida N, Tsujioka K, Tomoi M, Saida K, Mitsui Y. Differential activities of two distinct endothelin family peptides on ileum and coronary artery. FEBS Lett. 1989;247(2):337–40. doi: 10.1016/0014-5793(89)81365-1. [DOI] [PubMed] [Google Scholar]
- 16.Saida K, Mitsui Y, Ishida N. A novel peptide, vasoactive intestinal contractor, of a new (endothelin) peptide family. Molecular cloning, expression, and biological activity. J Biol Chem. 1989;264(25):14613–6. [PubMed] [Google Scholar]
- 17.Cunningham ME, Huribal M, Bala RJ, McMillen MA. Endothelin-1 and endothelin-4 stimulate monocyte production of cytokines. Crit Care Med. 1997;25(6):958–64. doi: 10.1097/00003246-199706000-00011. [DOI] [PubMed] [Google Scholar]
- 18.Inoue A, Yanagisawa M, Takuwa Y, Mitsui Y, Kobayashi M, Masaki T. The human preproendothelin-1 gene. Complete nucleotide sequence and regulation of expression. J Biol Chem. 1989;264(25):14954–9. [PubMed] [Google Scholar]
- 19.Dorfman DM, Wilson DB, Bruns GA, Orkin SH. Human transcription factor GATA-2. Evidence for regulation of preproendothelin-1 gene expression in endothelial cells. J Biol Chem. 1992;267(2):1279–85. [PubMed] [Google Scholar]
- 20.Denault JB, Claing A, D’Orleans-Juste P, Sawamura T, Kido T, Masaki T, Leduc R. Processing of proendothelin-1 by human furin convertase. FEBS Lett. 1995;362:276–80. doi: 10.1016/0014-5793(95)00249-9. [DOI] [PubMed] [Google Scholar]
- 21.Emoto N, Yanagisawa M. Endothelin-converting enzyme-2 is a membrane-bound, phosphoramidon-sensitive metalloprotease with acidic pH optimum. J Biol Chem. 1995;270(25):15262–8. doi: 10.1074/jbc.270.25.15262. [DOI] [PubMed] [Google Scholar]
- 22.Hasegawa H, Hiki K, Sawamura T, Aoyama T, Okamoto Y, Miwa S, Shimohama S, Kimura J, Masaki T. Purification of a novel endothelin-converting enzyme specific for big endothelin-3. FEBS Lett. 1998;428(3):304–8. doi: 10.1016/s0014-5793(98)00554-7. [DOI] [PubMed] [Google Scholar]
- 23.Uchida K, Uchida S, Nitta K, Yumura W, Nihei H. Regulated expression of endothelin converting enzymes in glomerular endothelial cells. J Am Soc Nephrol. 1997;8(4):580–5. doi: 10.1681/ASN.V84580. [DOI] [PubMed] [Google Scholar]
- 24.Naomi S, Iwaoka T, Disashi T, Inoue J, Kanesaka Y, Tokunaga H, Tomita K. Endothelin-1 inhibits endothelin-converting enzyme-1 expression in cultured rat pulmonary endothelial cells. Circulation. 1998;97(3):234–6. doi: 10.1161/01.cir.97.3.234. [DOI] [PubMed] [Google Scholar]
- 25.Yoshioka S, Fujiwara H, Yamada S, Tatsumi K, Nakayama T, Higuchi T, Inoue T, Maeda M, Fujii S. Endothelin-converting enzyme-1 is expressed on human ovarian follicles and corpora lutea of menstrual cycle and early pregnancy. J Clin Endocrinol Metab. 1998;83(11):3943–50. doi: 10.1210/jcem.83.11.5277. [DOI] [PubMed] [Google Scholar]
- 26.Kroger, B., Seulberger, H., Meyer, T., Schmidt, M., Jacob, E., Otter, R., Subkowski, T., Hillen, H.: US6066502 (2000). [DOI] [PubMed]
- 27.Xu D, Emoto N, Giaid A, Slaughter C, Kaw S, deWit D, Yanagisawa M. ECE-1: a membrane-bound metalloprotease that catalyzes the proteolytic activation of big endothelin-1. Cell. 1994;78(3):473–85. doi: 10.1016/0092-8674(94)90425-1. [DOI] [PubMed] [Google Scholar]
- 28.Yanagisawa H, Hammer RE, Richardson JA, Emoto N, Williams SC, Takeda S, Clouthier DE, Yanagisawa M. Disruption of ECE-1 and ECE-2 reveals a role for endothelin-converting enzyme-2 in murine cardiac development. J Clin Invest. 2000;105(10):1373–82. doi: 10.1172/JCI7447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kurihara H, Yoshizumi M, Sugiyama T, Takaku F, Yanagisawa M, Masaki T, Hamaoki M, Kato H, Yazaki Y. Transforming growth factor-beta stimulates the expression of endothelin mRNA by vascular endothelial cells. Biochem Biophys Res Commun. 1989;159:1435–40. doi: 10.1016/0006-291x(89)92270-5. [DOI] [PubMed] [Google Scholar]
- 30.Emori T, Hirata Y, Imai T, Ohta K, Kanno K, Eguchi S, Marumo F. Cellular mechanism of thrombin on endothelin-1 biosynthesis and release in bovine endothelial cell. Biochem Pharmacol. 1992;44(12):2409–11. doi: 10.1016/0006-2952(92)90687-e. [DOI] [PubMed] [Google Scholar]
- 31.Yoshimoto S, Ishizaki Y, Sasaki T, Murota S. Effect of carbon dioxide and oxygen on endothelin production by cultured porcine cerebral endothelial cells. Stroke. 1991;22(3):378–83. doi: 10.1161/01.str.22.3.378. [DOI] [PubMed] [Google Scholar]
- 32.Malek AM, Zhang J, Jiang J, Alper SL, Izumo S. Endothelin-1 gene suppression by shear stress: pharmacological evaluation of the role of tyrosine kinase, intracellular calcium, cytoskeleton, and mechanosensitive channels. J Mol Cell Cardiol. 1999;31(2):387–99. doi: 10.1006/jmcc.1998.0873. [DOI] [PubMed] [Google Scholar]
- 33.Boulanger C, Luscher TF. Release of endothelin from the porcine aorta. Inhibition by endothelium-derived nitric oxide. J Clin Invest. 1990;85(2):587–90. doi: 10.1172/JCI114477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kohno M, Yokokawa K, Horio T, Yasunari K, Murakawa K, Takeda T. Atrial and brain natriuretic peptides inhibit the endothelin-1 secretory response to angiotensin II in porcine aorta. Circ Res. 1992;70(2):241–7. doi: 10.1161/01.res.70.2.241. [DOI] [PubMed] [Google Scholar]
- 35.van Mourik JA, Romani de Wit T, Voorberg J. Biogenesis and exocytosis of Weibel-Palade bodies. Histochem Cell Biol. 2002;117(2):113–22. doi: 10.1007/s00418-001-0368-9. [DOI] [PubMed] [Google Scholar]
- 36.Predel HG, Meyer-Lehnert H, Backer A, Stelkens H, Kramer HJ. Plasma concentrations of endothelin in patients with abnormal vascular reactivity. Effects of ergometric exercise and acute saline loading. Life Sci. 1990;47(20):1837–43. doi: 10.1016/0024-3205(90)90286-z. [DOI] [PubMed] [Google Scholar]
- 37.Shichiri M, Hirata Y, Ando K, Emori T, Ohta K, Kimoto S, Ogura M, Inoue A, Marumo F. Plasma endothelin levels in hypertension and chronic renal failure. Hypertension. 1990;15(5):493–6. doi: 10.1161/01.hyp.15.5.493. [DOI] [PubMed] [Google Scholar]
- 38.Grafov MA, Gavrilova SA, Rubina AYu, Masenko VP, Medvedeva NA. Active immunization against endothelin-1 is associated with a decrease in plasma endothelin-1 and changes in vascular reactivity. J Cardiovasc Pharmacol. 2000;36(5 Suppl 1):S132–4. doi: 10.1097/00005344-200036051-00042. [DOI] [PubMed] [Google Scholar]
- 39.Abdel-Sayed S, Brunner HR, Nussberger J. Volume expansion enhances plasma endothelin-1. Am J Hypertens. 2003;16(12):1057–61. doi: 10.1016/s0895-7061(03)01031-8. [DOI] [PubMed] [Google Scholar]
- 40.Zhao H, Joshua IG, Porter JP. Microvascular responses to endothelin in deoxycorticosterone acetate-salt hypertensive rats. Am J Hypertens. 2000;13(7):819–26. doi: 10.1016/s0895-7061(00)00260-0. [DOI] [PubMed] [Google Scholar]
- 41.Abassi ZA, Tate JE, Golomb E, Keiser HR. Role of neutral endopeptidase in the metabolism of endothelin. Hypertension. 1992;20(1):89–95. doi: 10.1161/01.hyp.20.1.89. [DOI] [PubMed] [Google Scholar]
- 42.Modesti PA, Cecioni I, Costoli A, Poggesi L, Galanti G, Serneri GG. Renal endothelin in heart failure and its relation to sodium excretion. Am Heart J. 2000;140(4):617–22. doi: 10.1067/mhj.2000.109917. [DOI] [PubMed] [Google Scholar]
- 43.Cantaro S, Milan Manani S, Marcon R, Bonfante L, Masiero M, D'Angelo A, Calo L. Urinary excretion of vasoactive substances in chronic renal failure. Clin Nephrol. 2001;55(5):393–9. [PubMed] [Google Scholar]
- 44.Gariepy CE, Ohuchi T, Williams SC, Richardson JA, Yanagisawa M. Salt-sensitive hypertension in endothelin-B receptor-deficient rats. J Clin Invest. 2000;105(7):925–33. doi: 10.1172/JCI8609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Arai H, Hori S, Aramori I, Ohkubo H, Nakanishi S. Cloning and expression of a cDNA encoding an endothelin receptor. Nature. 1990;348(6303):730–2. doi: 10.1038/348730a0. [DOI] [PubMed] [Google Scholar]
- 46.Schiffrin EL. Endothelin: potential role in hypertension and vascular hypertrophy. Hypertension. 1995;25(6):1135–43. doi: 10.1161/01.hyp.25.6.1135. [DOI] [PubMed] [Google Scholar]
- 47.Sakurai T, Yanagisawa M, Takuwa Y, Miyazaki H, Kimura S, Goto K, Masaki T. Cloning of a cDNA encoding a non-isopeptide-selective subtype of the endothelin receptor. Nature. 1990;348(6303):732–5. doi: 10.1038/348732a0. [DOI] [PubMed] [Google Scholar]
- 48.Dai X, Galligan JJ, Watts SW, Fink GD, Kreulen DL. Increased O2− production and upregulation of ETB receptors by sympathetic neurons in DOCA-salt hypertensive rats. Hypertension. 2004;43(5):1048–54. doi: 10.1161/01.HYP.0000126068.27125.42. [DOI] [PubMed] [Google Scholar]
- 49.Tostes RC, Touyz RM, He G, Chen X, Schiffrin EL. Contribution of endothelin-1 to renal activator protein-1 activation and macrophage infiltration in aldosterone-induced hypertension. Clin Sci (Lond) 2002;103 (Suppl 48):25S–30S. doi: 10.1042/CS103S025S. [DOI] [PubMed] [Google Scholar]
- 50.Bkaily G, Choufani S, Sader S, Jacques D, d'Orleans-Juste P, Nader M, Kurban G, Kamal M. Activation of sarcolemma and nuclear membranes ET-1 receptors regulates transcellular calcium levels in heart and vascular smooth muscle cells. Can J Physiol Pharmacol. 2003;81(6):654–62. doi: 10.1139/y03-020. [DOI] [PubMed] [Google Scholar]
- 51.Neylon CB. Vascular biology of endothelin signal transduction. Clin Exp Pharmacol Physiol. 1999;26(2):149–53. doi: 10.1046/j.1440-1681.1999.03013.x. [DOI] [PubMed] [Google Scholar]
- 52.Hilal-Dandan R, Merck DT, Lujan JP, Brunton LL. Coupling of the type A endothelin receptor to multiple responses in adult rat cardiac myocytes. Mol Pharmacol. 1994;45(6):1183–90. [PubMed] [Google Scholar]
- 53.Hilal-Dandan R, Ramirez MT, Villegas S, Gonzalez A, Endo-Mochizuki Y, Brown JH, Brunton LL. Endothelin ETA receptor regulates signaling and ANF gene expression via multiple G protein-linked pathways. Am J Physiol. 1997;272(1 Pt 2):H130–7. doi: 10.1152/ajpheart.1997.272.1.H130. [DOI] [PubMed] [Google Scholar]
- 54.Robin P, Boulven I, Desmyter C, Harbon S, Leiber D. ET-1 stimulates ERK signaling pathway through sequential activation of PKC and Src in rat myometrial cells. Am J Physiol Cell Physiol. 2002;283(1):C251–60. doi: 10.1152/ajpcell.00601.2001. [DOI] [PubMed] [Google Scholar]
- 55.Chew DK, Orshal JM, Khalil RA. Elastase-induced suppression of endothelin-mediated Ca2+ entry mechanisms of vascular contraction. Hypertension. 2003;42(4):818–24. doi: 10.1161/01.HYP.0000086200.93184.8E. [DOI] [PubMed] [Google Scholar]
- 56.Smith L, Payne JA, Sedeek MH, Granger JP, Khalil RA. Endothelin-induced increases in Ca2+ entry mechanisms of vascular contraction are enhanced during high-salt diet. Hypertension. 2003;41(3 Pt 2):787–93. doi: 10.1161/01.HYP.0000051643.05700.56. [DOI] [PubMed] [Google Scholar]
- 57.Kanashiro CA, Altirkawi KA, Khalil RA. Preconditioning of coronary artery against vasoconstriction by endothelin-1 and prostaglandin F2alpha during repeated downregulation of epsilon-protein kinase C. J Cardiovasc Pharmacol. 2000;35(3):491–501. doi: 10.1097/00005344-200003000-00021. [DOI] [PubMed] [Google Scholar]
- 58.Sirous ZN, Fleming JB, Khalil RA. Endothelin-1 enhances eicosanoids-induced coronary smooth muscle contraction by activating specific protein kinase C isoforms. Hypertension. 2001;37(2 Part 2):497–504. doi: 10.1161/01.hyp.37.2.497. [DOI] [PubMed] [Google Scholar]
- 59.McNair LL, Salamanca DA, Khalil RA. Endothelin-1 promotes Ca2+ antagonist-insensitive coronary smooth muscle contraction via activation of ε-protein kinase C. Hypertension. 2004;43(4):897–904. doi: 10.1161/01.HYP.0000118520.92686.3b. [DOI] [PubMed] [Google Scholar]
- 60.Schiffrin EL. Endothelin and endothelin antagonists in hypertension. J Hypertens. 1998;16(12 Pt 2):1891–5. doi: 10.1097/00004872-199816121-00007. [DOI] [PubMed] [Google Scholar]
- 61.Cain AE, Tanner DM, Khalil RA. Endothelin-1--induced enhancement of coronary smooth muscle contraction via MAPK-dependent and MAPK-independent [Ca2+]i sensitization pathways. Hypertension. 2002;39(2 Pt 2):543–9. doi: 10.1161/hy0202.103129. [DOI] [PubMed] [Google Scholar]
- 62.Daou GB, Srivastava AK. Reactive oxygen species mediate Endothelin-1-induced activation of ERK1/2, PKB, and Pyk2 signaling, as well as protein synthesis, in vascular smooth muscle cells. Free Radic Biol Med. 2004;37(2):208–15. doi: 10.1016/j.freeradbiomed.2004.04.018. [DOI] [PubMed] [Google Scholar]
- 63.Pollock DM. Chronic studies on the interaction between nitric oxide and endothelin in cardiovascular and renal function. Clin Exp Pharmacol Physiol. 1999;26(3):258–61. doi: 10.1046/j.1440-1681.1999.03027.x. [DOI] [PubMed] [Google Scholar]
- 64.Perry MG, Molero MM, Giulumian AD, Katakam PV, Pollock JS, Pollock DM, Fuchs LC. ETB receptor-deficient rats exhibit reduced contraction to ET-1 despite an increase in ETA receptors. Am J Physiol Heart Circ Physiol. 2001;281(6):H2680–6. doi: 10.1152/ajpheart.2001.281.6.H2680. [DOI] [PubMed] [Google Scholar]
- 65.Schroeder AC, Imig JD, LeBlanc EA, Pham BT, Pollock DM, Inscho EW. Endothelin-mediated calcium signaling in preglomerular smooth muscle cells. Hypertension. 2000;35(1 Pt 2):280–6. doi: 10.1161/01.hyp.35.1.280. [DOI] [PubMed] [Google Scholar]
- 66.Schiffrin EL. Role of endothelin-1 in hypertension and vascular disease. Am J Hypertens. 2001;14(6 Pt 2):83S–89S. doi: 10.1016/s0895-7061(01)02074-x. [DOI] [PubMed] [Google Scholar]
- 67.Berthiaume N, Yanagisawa M, D'Orleans-Juste P. Contribution of endogenous endothelin-1 and endothelin-A-receptors to the hypertensive state of endothelin-B heterozygous (+/-) knockout mice. J Cardiovasc Pharmacol. 2000;36(5 Suppl 1):S72–4. [PubMed] [Google Scholar]
- 68.Murakoshi N, Miyauchi T, Kakinuma Y, Ohuchi T, Goto K, Yanagisawa M, Yamaguchi I. Vascular endothelin-B receptor system in vivo plays a favorable inhibitory role in vascular remodeling after injury revealed by endothelin-B receptor-knockout mice. Circulation. 2002;106(15):1991–8. doi: 10.1161/01.cir.0000032004.56585.2a. [DOI] [PubMed] [Google Scholar]
- 69.Pollock DM, Pollock JS. Evidence for endothelin involvement in the response to high salt. Am J Physiol Renal Physiol. 2001;281(1):F144–50. doi: 10.1152/ajprenal.2001.281.1.F144. [DOI] [PubMed] [Google Scholar]
- 70.Hoffman A, Haramati A, Dalal I, Shuranyi E, Winaver J. Diuretic-natriuretic actions and pressor effects of big-endothelin (1–39) in phosphoramidon-treated rats. Proc Soc Exp Biol Med. 1994;205(2):168–73. doi: 10.3181/00379727-205-43693. [DOI] [PubMed] [Google Scholar]
- 71.Vassileva I, Mountain C, Pollock DM. Functional role of ETB receptors in the renal medulla. Hypertension. 2003;41(6):1359–63. doi: 10.1161/01.HYP.0000070958.39174.7E. [DOI] [PubMed] [Google Scholar]
- 72.Imura, H,: Nakao, K.: Nakanishi, S. US6821743 (2004)
- 73.Nagata N, Niwa Y, Nakaya Y. A novel 31-amino-acid-length endothelin, ET-1(1–31), can act as a biologically active peptide for vascular smooth muscle cells. Biochem Biophys Res Commun. 2000;275(2):595–600. doi: 10.1006/bbrc.2000.3292. [DOI] [PubMed] [Google Scholar]
- 74.Maguire JJ, Kuc RE, Davenport AP. Vasoconstrictor activity of novel endothelin peptide, ET-1(1 – 31), in human mammary and coronary arteries in vitro. Br J Pharmacol. 2001;134(6):1360–6. doi: 10.1038/sj.bjp.0704384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhou Y, Dirksen WP, Zweier JL, Periasamy M. Endothelin-1-induced responses in isolated mouse vessels: the expression and function of receptor types. Am J Physiol Heart Circ Physiol. 2004;287(2):H573–8. doi: 10.1152/ajpheart.01170.2003. [DOI] [PubMed] [Google Scholar]
- 76.Watts SW, Fink GD, Northcott CA, Galligan JJ. Endothelin-1-induced venous contraction is maintained in DOCA-salt hypertension; studies with receptor agonists. Br J Pharmacol. 2002;137(1):69–79. doi: 10.1038/sj.bjp.0704831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Giardina JB, Green GM, Rinewalt AN, Granger JP, Khalil RA. Role of endothelin B receptors in enhancing endothelium-dependent nitric oxide-mediated vascular relaxation during high salt diet. Hypertension. 2001;37(2 Part 2):516–23. doi: 10.1161/01.hyp.37.2.516. [DOI] [PubMed] [Google Scholar]
- 78.Touyz RM, Schiffrin EL. Role of endothelin in human hypertension. Can J Physiol Pharmacol. 2003;81(6):533–41. doi: 10.1139/y03-009. [DOI] [PubMed] [Google Scholar]
- 79.Tostes RC, Wilde DW, Bendhack LM, Webb RC. Calcium handling by vascular myocytes in hypertension. Braz J Med Biol Res. 1997;30(3):315–23. doi: 10.1590/s0100-879x1997000300004. [DOI] [PubMed] [Google Scholar]
- 80.Matsumura Y, Hashimoto N, Taira S, Kuro T, Kitano R, Ohkita M, Opgenorth TJ, Takaoka M. Different contributions of endothelin-A and endothelin-B receptors in the pathogenesis of deoxycorticosterone acetate-salt-induced hypertension in rats. Hypertension. 1999;33(2):759–65. doi: 10.1161/01.hyp.33.2.759. [DOI] [PubMed] [Google Scholar]
- 81.Larouche I, Schiffrin EL. Cardiac microvasculature in DOCA-salt hypertensive rats : effect of endothelin ET(A) receptor antagonism. Hypertension. 1999;34(4 Pt 2):795–801. doi: 10.1161/01.hyp.34.4.795. [DOI] [PubMed] [Google Scholar]
- 82.Mortensen LH, Fink GD. Captopril prevents chronic hypertension produced by infusion of endothelin-1 in rats. Hypertension. 1992;19(6 Pt 2):676–80. doi: 10.1161/01.hyp.19.6.676. [DOI] [PubMed] [Google Scholar]
- 83.Callera GE, Touyz RM, Teixeira SA, Muscara MN, Carvalho MH, Fortes ZB, Nigro D, Schiffrin EL, Tostes RC. ETA receptor blockade decreases vascular superoxide generation in DOCA-salt hypertension. Hypertension. 2003;42(4):811–7. doi: 10.1161/01.HYP.0000088363.65943.6C. [DOI] [PubMed] [Google Scholar]
- 84.Sullivan JC, Pollock DM, Pollock JS. Altered nitric oxide synthase 3 distribution in mesenteric arteries of hypertensive rats. Hypertension. 2002;39(2 Pt 2):597–602. doi: 10.1161/hy0202.103286. [DOI] [PubMed] [Google Scholar]
- 85.Cheng, X., Massa, M.A., Patt, W.C.: US5891892 (1999).
- 86.Ahn, K., Cheng, X., Doherty, A.M., Elslager, E.F., Kornberg, B., Lee, C., Leonard, D., Nikam, S., Werbel, L.M.: US5773444 (1998).
- 87.Suzuki, N., Matsumoto, H.: US5230999 (1993).
- 88.Ohwaki, T., Sakai, H.: US5468623 (1995).
- 89.Lum, P.Y., Tan, Y., Dai, H., Muise, E.S., Berger, J.P., Thompson, J.R.: US20050084872 (2005).
- 90.Reynolds, M., Polakis, P.: US20050090514 (2005).
- 91.Lebwohl, D.E.: US6573285 (2003).
- 92.Whittle, B.A.: US20050042172 (2005).
- 93.Yorio, T., Prasanna, G., Dibas, A., Stokely, M.E.: US20030176356 (2003).
- 94.Lawrence, III, J.H., Donahue, J.K.: US6855701 (2005).
- 95.Dorsch, D., Osswald, M., Mederski, W., Wilm, C., Schmitges, C., Christadler, M., Anzali, S.: US6017939 (2000). [DOI] [PubMed]
- 96.Dorsch, D., Osswald, M., Mederski, W., Wilm, C., Christadler, M., Schmitges, C.J.: US6197800 (2001).
- 97.Bunker, A.M., Cheng, X., Doherty, A.M., Edmunds, J.J., Kanter, G.D., Lee, C., Repine, J.T., Skeean, R.W.: US6440962 (2002).
- 98.Elliott, J.D., Franz, R.G., Lago, M.A., Gao, A.: US5929106 (1999).
- 99.Murugesan, N.: US5420123 (1995).
- 100.Elliott, J.D., Gao, A.: US6051599 (2000).
- 101.Cousins, R.D., Elliott, J.D., Lago, M.A., Leber, J.D., Peishoff, C.E.: US6448260 (2002).
- 102.Rawson, D.J., Dack, K.N., Dickinson, R.P., James, K.: US6384070 (2002).
- 103.Elliott, J.D., Luengo, J.I., Xiang, J.: US6174906 (2001).
- 104.Elliott, J.D., Weinstock, J., Xiang, J.: US6620826 (2003).
- 105.Cheng, X., Doherty, A.M., Hurley, T.R., Lovdahl, M.J., Patt, W.C., Repine, J.T.: US6043241 (2000).
- 106.Bagley, S.W., Broten, T.P., Chakravarty, P.K., Dhanoa, D.S., Fitch, K.J., Greenlee, W.J., Kevin, N.J., Pettibone, D.J., Rivero, R.A., Walsh, T.F., Williams, Jr., D.L., Toupence, R.B., Mathews, J.M.: US5565485 (1996).
- 107.Berryman, K.A., Cheng, X., Doherty, A.M., Edmunds, J.J., Klutchko, S.: US5658943 (1997).
- 108.Ueno, R.: US6197821 (2001).
- 109.Luengo, J.I., Elliott, J.D., Xiang, J.: US6545031(2003).
- 110.Dorsch, D., Osswald, M., Mederski, W., Wilm, C., Schmitges, C.J., Christadler, M.: US5883090 (1999). [DOI] [PubMed]
- 111.Hirata, M., Deushi, T., Takahashi, Y., Tamura, M., Ohshima, T., Oda, T., Ishikawa, T., Sonoki, H., Shiratsuchi, M.: US5883092 (1999).
- 112.Elliott, J.D., Leber, J.D.: US6075037 (2000).
- 113.Amberg, W., Jansen, R., Klinge, D.: US6235903 (2001).
- 114.Banks, B.J., Chubb, N.A.L., Eshelby, J.J., Pacey, M.S., Schulz, D.J.: US6387915 (2002).
- 115.Wu, C., Blok, N., Holland, G.W.: US6686382 (2004).
- 116.Murugesan, N., Hunt, J.T.: US6107320 (2000).
- 117.Verner, E.J.: US6013655 (2000).
- 118.Chan, M.F., Raju, B.G., Kois, A., Verner, E.J., Wu, C., Castillo, R.S., Yalamoori, V., Balaji, V.N., Ramnarayan, K.: US6541498 (2003).