

Abstract
To determine the effects of expression of mutant Ki-ras on lung tumorigenesis, we developed a bitransgenic mouse model that expresses the human Ki-rasG12C allele in alveolar type II and/or Clara cells in a tetracycline-inducible, lung-specific manner. Expression of Ki-rasG12C caused multiple, small lung tumors over a 12-month time period. Although tumor multiplicity increased upon continued Ki-ras expression, most lung lesions were hyperplasias or well-differentiated adenomas. This is in contrast to the more severe phenotypes observed in other transgenic mouse models in which different mutant Ki-ras alleles were expressed in the lung. Expression of Ki-rasG12C was associated with a 2-fold increase in the activation of the Ras and Ral signaling pathways and increased phosphorylation of Ras downstream effectors, including Erk, p90 ribosomal S6 kinase, ribosomal S6 protein, p38 and MAPKAPK-2. In contrast, expression of the transgene had no effect on the activation of the JNK and Akt signaling pathways. Withdrawal of doxycycline for 1 month resulted in almost a complete absence of proliferative pulmonary lesions, suggesting tumor regression in the absence of Ki-ras expression. Mutant Ki-rasG12C expression was sufficient for initial lung tumor transformation, required for maintenance of tumor phenotype, and induced transformation of lung epithelial cells by the activation of multiple effector pathways. These results describe a novel mouse lung tumor model demonstrating benign tumor development in the absence of tumor progression, which will provide a new tool for understanding the early stages of lung tumor pathogenesis.
Introduction
Worldwide, lung cancer is the leading cause of cancer-related deaths in both men and women (1). Lung adenocarcinoma (AC), a type of non-small cell lung cancer, is the most common histological variant of lung cancer in the United States, and its incidence is increasing (1,2). Studies by several groups have documented mutations in the Ki-ras gene in human lung cancer, particularly in ACs found in smokers, ranging in incidence from 30-50% of the tumors examined (3-5). The high incidence of mutant forms of the Ki-ras gene observed in both human and animal lung ACs suggests that damage to this genetic locus may play an important role in the pathogenesis of lung tumors (3,5-12). Several groups have shown an association between the presence of a mutated Ki-ras gene and patient prognosis, demonstrating decreased survival in patients harboring the mutated Ki-ras gene (7,9,12-16).
Numerous mouse lung tumor models have been utilized to try to mimic the effects of oncogenic Ki-ras in human lung tumors with either the use of chemical carcinogens or transgenes. Until recently, alterations in the Ki-ras oncogene and p16Ink4a and Retinoblastoma tumor suppressor genes were the most widely studied genes between human and mouse lung tumors (17). Technological advances have allowed the extension of these studies into the analysis of gene expression across the entire genome in both human and rodent tumors. Recently, Sweet-Cordero et al. (18) demonstrated that mouse lung tumors initiated in mice by a mutant Ki-ras allele could be used to classify human samples as either normal or tumor with 97% accuracy using upregulated or downregulated gene sets obtained from gene-expression profiling. Another group, also using gene-expression analysis, showed a similarity between chemically-induced mouse lung tumors and human lung tumors (19). These results suggest that mouse models are a useful tool in the study of lung tumorigenesis.
A previous mouse model from our laboratory demonstrated that, following in utero exposure to the polycyclic aromatic hydrocarbon, 3-methylcholanthrene, lung tumors induced in the offspring exhibited a high incidence of mutations (84%) in the Ki-ras gene (8,20). A high incidence of Ki-ras mutations were found in hyperplastic lung tissue (60%) as well, suggesting that activation of Ki-ras may be an early event in lung cancer formation. One of the most interesting findings from this (8) and a subsequent study (21) was that the type of mutation present in the lesions appeared to influence tumor development. Lesions harboring VAL12, ASP12 or ARG13 mutations were more likely to progress to ACs than were lesions with CYS12 mutations (8,21), suggesting that different mutant Ras alleles may have different oncogenic potentials. These results and others have demonstrated that mutation to the Ki-ras gene locus is a critical target in the initiation and progression of lung tumor development.
Although mutations of Ki-ras are considered an early event in the pathogenesis of lung tumors, direct evidence for this hypothesis has only recently been obtained. Both human and chemically-induced rodent tumors contain a number of genetic lesions in addition to mutated Ki-ras, making it difficult to identify the role for mutant Ki-ras in lung tumor initiation. Mouse lung tumorigenesis protocols usually involve treatment with chemical carcinogens, which can alter any one of a number of gene loci in addition to Ki-ras. Koera et al. (22) demonstrated that expression of the Ki-ras gene is critical for normal development in mouse embryos, as deletion of the Ki-, but not the Ha- or N-ras genes, resulted in embryonic lethality. A number of investigators have confirmed the importance of Ki-ras for normal lung development and shown that expression of Ki-ras in the lung increases during gestation, reaching peak levels in the adult, suggesting that Ki-ras plays a role in normal lung morphogenesis (23,24).
Recent studies by several laboratories have utilized inducible transgenic mouse systems to express various mutant alleles of Ki-ras in a timed and/or tissue-specific manner to more directly determine the effects of Ki-ras on cancer initiation and progression (6,25-29). In these studies, expression of mutant Ki-ras was shown to be a potent oncogenic stimulus for lung epithelial cells, as expression of a mouse or human Ki-ras transgene resulted in a high tumor burden and formation of progressive ACs within 2 to 10 months.
Therefore, in an effort to understand the effects of activated Ki-rasG12C in lung tumorigenesis, we have utilized a bitransgenic mouse model that expresses the mutant human Ki-rasG12C allele in a lung-specific and tetracycline (tet)-inducible manner. In contrast to previous murine lung tumor models expressing mutant Ki-ras transgenes, expression of the CYS12 mutant allele allowed survival to an age of at least 12 months and resulted primarily in proliferative pulmonary lesions morphologically diagnosed as bronchioalveolar hyperplasias and adenomas (ADs). Activation of Ras downstream effector pathways, including the Raf/Mek/MAPK, p38, and Ral pathways was observed. The present study provides evidence for a novel lung tumor mouse model exhibiting little progression past the AD stage.
Materials and methods
Generation of transgenic mice
The Ki-rasG12C allele was cloned from the H358 human lung bronchioalveolar cell line (American Type Culture Collection, Manassas, VA) by RT-PCR. RNA samples were reverse transcribed with the Superscript Preamplification System for first strand cDNA synthesis (Life Technologies, Gaithersburg, MD). Primers to the entire Ki-ras coding sequence were used to amplify a 567 bp product by PCR, which was then subcloned into the pCR2. 1 vector using the TA Cloning Kit (Invitrogen, San Diego, CA). This plasmid was then sequenced to confirm the identity of the cloned gene by the DNA sequencing core laboratory of the Wake Forest University School of Medicine. In addition, an aliquot of the TA-cloned pCR2.1-ras plasmid was used for an in vitro translation reaction, utilizing the TNT Quick Coupled Transcription/Translation System (Promega, Madison, WI), to confirm that the cloned cDNA produced the correct sized protein.
The Ki-ras transgene was constructed using standard molecular biological methods. Twenty μg of the pCR2. 1-ras plasmid was linearized by digestion with Spe1 and blunt-ended with T4 DNA polymerase. The fragment was gel-purified and cut with Not1 to release the Ki-ras insert. The tetO7/CMV/Zeor expression vector was double digested with PmeI, which created a blunt end, and Not1 and then treated with shrimp alkaline phosphatase for 1 h at 37°C to prevent self-annealing. The linearized vector and the Ki-ras insert were then directionally cloned and ligated with 24 U of T4 DNA ligase, transformed into XL Blue cells, streaked onto an agar plate containing ampicillin and X-gal, and grown overnight at 37°C. Colonies were picked and expanded, then tested for the presence and orientation of the insertion by restriction enzyme digestion. The orientation of the inserted sequence relative to the CMV promoter was confirmed by gene sequencing.
The Ki-ras monotransgenic mouse was constructed at the Transgenic Mouse Facility at the University of Cincinnati, as described previously (30-32). The tetO-CMV-Ki-ras insert was excised from the plasmid by digestion with SpeI and PvuII and purified by CsCl gradient centrifugation. The DNA was then microinjected into donor eggs obtained from superovulated FVB/N mice. The eggs were implanted into pseudopregnant mice and founder mice containing the transgene were established. These are referred to as monotransgenic Ki-rasG12C mice.
Genotyping
DNA was extracted from mouse tails using the Wizard Genomic DNA Purification Kit (Promega) according to the manufacturer’s protocol. Genotyping was performed by PCR with Amplitaq Gold (Applied Biosystems, Foster City, CA). To identify Ki-rasG12C mice, an upstream primer in the CMV minimal promoter (5′-CCATCCACGCTGTTTTGACCTC-3′) and a downstream primer in the human Ki-ras coding sequence (5′-TACTCCTCTTGACCTGCTGTGTCG-3′) were used. Primers for genotyping mice for the presence of the CCSP-rtTA construct were 5′-ACTGCCCATTGCCCAAACAC-3′ (forward) and 5′-AAAATCTTGCCAGCTTTCCCC-3′ (reverse), and for the SPC-rtTA construct 5′-GACACATATAAGACCCTGGTCA-3′ (forward) and 5′-AAAATCTTGCCAGCTTTCCCC-3′ (reverse). Samples were amplified following denaturation at 94°C for 2 min by 40 cycles of denaturation at 94°C for 30 s, annealing at 57°C (Ras) or 61°C (CCSP and SP-C) for 1 min, and extension at 72°C for 1 min, with a final extension for 7 min at 72°C (Bio-Rad iCycler, Hercules, VA).
Tumor studies
Eight week old CCSP/Ki-ras and SP-C/Ki-ras bitransgenic mice, as well as wild-type FVB/N and monotransgenic Ki-ras mice, were either untreated or given 500 μg/ml doxycycline (DOX) (Sigma, St Louis, MO) in the drinking water for 12 days, 1.5, 3, 6, 9 or 12 months. Mice were killed by CO2 asphyxiation/exsanguination, and macroscopic lung lesions were counted and measured prior to fixation. The lungs were then inflated and fixed in 10% phosphate-buffered formalin. At the 12-month time point, liver, spleen, kidney, pancreas, heart and either testis and prostate or ovaries were examined macroscopically and then removed and placed in 10% phosphate-buffered formalin. After 24 h, the formalin solution was replaced with 70% ethanol to avoid excessive aldehyde cross-linking for immunohistochemistry. Tissues were embedded in paraffin and 5 mm sections prepared by microtomy. The cut slides were stained with hematoxylin and eosin and examined by veterinary pathologists (J.E. and N.D.K.). Proliferative lesions were evaluated using established morphological criteria for mice (33).
Ras and Ral activation
CCSP/Ki-ras and SP-C/Ki-ras bitransgenic mice were either untreated or given 500 μg/ml DOX in the drinking water for 72 h. Lungs were removed and protein was extracted using modified RIPA buffer (50 mM Tris-HCl (pH 7.4), 150 mM NaCl (pH 7.4), 1 mM EDTA, 1 mM PMSF, 1 μg/ml of aprotinin, leupeptin, pepstatin and 1 mM NaF) without detergents. Whole lung was homogenized using a Polytron homogenizer (Kinematica, Crystal Lake, IL) on setting 6 for 2 pulses of 30 s duration. Protein concentration was determined with the Bio-Rad protein assay kit according to the manufacturer’s instructions (Bio-Rad, Hercules, CA). Activation of Ras and Ral was determined from the protein lysates using kits from Upstate Technologies (Charlottesville, VA). For each assay, 15 μl of agarose beads conjugated to either the Ras binding domain (RBD) of Raf or Ral binding protein (RBP) were added to 10 mg of lysate. The samples were rocked for 1 h at 4°C. The beads were washed and denatured with 50 μl loading dye containing 2-mercaptoethanol and boiled for 5 min. An aliquot of 20 μl of each sample was loaded onto a 10% SDS-PAGE gel and transferred onto a nitrocellulose membrane. The membranes were incubated with either 1 μg/ml anti-Ras (clone Ras10) monoclonal antibody diluted in phosphate buffered saline containing 5% dry milk and 0.05% Tween-20 or 1 μg/ml anti-Ral A monoclonal antibody diluted in Tris buffered saline containing 5% dry milk and 0.05% Tween-20. Membranes were then incubated with a goat anti-mouse horseradish peroxidase-conjugated secondary antibody diluted 1:1000 in either phosphate-buffered saline or Tris buffered saline containing 5% dry milk and 0.05% Tween-20. Protein bands were visualized with Bio-Rad’s Immun-Star Substrate detection kit by autoradiography.
Immunohistochemisry (Ras effectors)
Formalin fixed, paraffin embedded mouse lungs sectioned 5 μm thick were placed on Super Frost slides and air-dried. Slides were baked for 30 min at 58°C prior to deparaffinization. Baked slides were deparaffinized in xylene 3× 5 min, rehydrated in 100% ethanol 2× 10 min, 95% ethanol 2× 10 min, and water for 2× 5 min. Antigen retrieval was performed by heating slides to 100°C for 15 min in 10 mM sodium citrate (pH 6.0), slides were then cooled to room temperature before 2× water rinse. Endogenous peroxidase activity was quenched by a 10 min incubation in 3% peroxide followed by 2 water rinses of 5 min each. Slides were blocked with TBST [25 μm Tris-HCl (pH 7.2), 100 mM NaCl and 0.1% Tween 20] containing 5% goat serum (Sigma) for 1 h. Slides were then incubated overnight at 4°C with the following primary antibodies (Cell Signaling Technology, Beverly, MA) diluted in blocking buffer: anti-phospho Erk (THR202/TYR204) rabbit monoclonal 1:300; anti-phospho p38 (THR180/TYR182) rabbit monoclonal 12F8 IHC specific 1:100; anti-phospho MAPKAPK-2 (THR319) rabbit polyclonal 1:100; anti-phospho p90Rsk (THR359/SER363) rabbit polyclonal 1:100; anti-phospho S6 (SER235/236) polyclonal 1:100; anti-phospho JNK (THR183/TYR185) rabbit polyclonal 1:50; anti-phospho Akt (SER473) rabbit polyclonal 1:50. Slides were washed 3× in TBST for 5 min then incubated with biotinylated goat anti-rabbit antibody (Vector Laboratories, Burlingame, CA) 1:1000 in TBST for 30 min, washed 3 × 5 min with TBST, then incubated with avidin-biotin complex (Vector Laboratories) for 1 h. Negative controls consisting of tissue and secondary antibody only were performed to account for non-specific binding. Nova Red (Vector Laboratories) was used as per the manufacturer’s instructions for 1 min before quenching in water. Slides were dehydrated in 2 × 95% ethanol followed by 2 × 100% ethanol for 10 s, then rinsed 3x for 10 s with xylene prior to mounting with Vectamount (Vector Laboratories). Slides were imaged with a Nikon Eclipse TE300 microscope fitted with an AxioCam digital camera.
Immunohistochemistry (CCSP, SP-C, Ki-67 and cleaved caspase 3)
Slides were deparaffinized with 3 × 5 min washes with xylene. The slides were hydrated through graded alcohols 3 × 1 min each. They were then placed in distilled water for 5 min. Antigen retrieval was performed using 10 mM citrate buffer (pH 6.0) for 35 min using a steamer. The slides were cooled for 20 min and placed in distilled water for 5 min. Primary antibodies were added: anti-CCSP 1:1000 (34); anti-SP-C 1:2000 (35-38); anti-Ki-67 1:25 (Abcam, Cambridge, MA), and anti-cleaved caspase 3 1:50 (Cell Signaling Technology). Samples as well as negative controls were incubated overnight at 4°C. Slides were then washed 5× with 1× Tris buffer (Biomeda, Foster City, CA) containing 0.5% casein. A secondary biotinylated anti-rabbit antibody (BioGenex, San Ramon, CA) was added at a dilution of 1:20 for 30 min at 32°C. Slides were washed 5× in Tris buffer followed by a 1:20 dilution of Streptavidin-alkaline phosphatase conjugate (BioGenex) for 30 min at 32°C. Slides were washed 5× in Tris buffer and Vector Red substrate (Vector Laboratories) was added for 5 min. Slides were then washed 2x in 0.1 M Tris-HCl buffer (pH 8.2-8.4) and 5× with distilled water. Slides were counterstained with Mayer’s hematoxylin for 5 min, then washed under running water for 5 min. Slides were dehydrated through graded alcohol and cleared through several changes of p-xylene.
Expression of transgenic and endogenous Ki-ras RNA by real-time fluorescent PCR
Eight week old CCSP/Ki-ras and SP-C/Ki-ras bitransgenic mice, as well as monotransgenic Ki-ras mice, were either untreated or given 500 μg/ml DOX in the drinking water for 24 h, 48 h, 7 or 14 days. Thirty mg of whole lung tissue was homogenized with a Polytron homogenizer in RLT lysis buffer supplied in the RNeasy Mini Kit at speed 6. Total RNA was extracted using the RNeasy Mini Kit (Qiagen, Valencia, CA). In addition, RNA was also isolated from the liver, spleen, thymus, pancreas, kidneys, heart, intestines and either testis and prostate or ovaries at 14 days of treatment. For RT-PCR, cDNA was initially generated from 1 μg of RNA using the iScript cDNA Synthesis Kit (Bio-Rad). One tenth of the cDNA (2 μl) was used to amplify the Ki-ras transgene, the endogenous murine Ki-ras gene, and GAPDH using the iCycler (Bio-Rad) for 40 cycles with SYBR Green Supermix (Bio-Rad).
To generate a standard curve, each PCR product (Ki-ras transgene, endogenous Ki-ras, and GAPDH) was first cloned into a pCR2.1 TA cloning vector. The plasmids were then utilized to generate the standard curves for each of the three genes using the same primers to be used for the unknown samples in order to quantify the copy number of each gene by the relative standard curve method (39-41). Both the Ki-ras transgene copy number and endogenous Ki-ras copy number were normalized to GAPDH copy number. PCR efficiencies and R2 values for the standard curves ranged from 99 to 103% and 0.991-0.998, respectively.
The primers used to amplify the Ki-rasG12C transgene were 5′-CTGCAGAATTCGCCCTTATGACTGA-3′ (forward) and 5′-TAGCTGTATCGTCAAGGCACTCTTGC-3′ (reverse); for the endogenous murine Ki-ras gene were 5′-GCAGGGTTGGGCCTTACAT-3′ (forward) and 5′-ATGCGTCGCCACATTGAAT-3′ (reverse); and for GAPDH were 5′-TCTCCCTCACAATTTCCATCCCAG-3′ (forward) and 5′-GGGTGCAGCGAACTTTATTGATGG-3′ (reverse). Cycling conditions included an initial denaturation step at 94°C for 3 min followed by 40 cycles of denaturation at 94°C for 30 s, 66.5°C (transgene and GAPDH) or 57°C (endogenous Ki-ras) for 1 min, and 72°C for 1 min. A melt curve was determined following each real-time PCR run to ensure the presence of a single product.
For values reported in Table II, the iCycler software determined a copy number for the Ki-ras transgene, endogenous Ki-ras, and GAPDH for each unknown sample based on the gene-specific standard curve. Both transgene and endogenous Ki-ras copy numbers were normalized to GAPDH by dividing each sample by their respective GAPDH copy numbers. These normalized values were averaged and standard deviations calculated. The numbers in parentheses were determined by dividing all of the normalized averages by the monotransgenic normalized endogenous Ki-ras value.
Table II.
Ki-rasG12C transgene and endogenous Ki-ras expression normalized to GAPDH in CCSP/Ki-ras mice
| Transgene copy # GAPDH copy # | Endogenous copy # GAPDH copy # | |
|---|---|---|
| Monotransgenic | 1.05 × 10−7a (0.000007) | 1.58 × 10−2 ± 6.44 × 10−3 (1.000) |
| Bitransgenic, no Dox | 2.63 × 10−6a (0.0002) | 8.56 × 10−3 ± 3.25 × 10−3 (0.542) |
| 24 h | 2.37 × 10−4 ± 9.33 × 10−5 (0.015) | 1.24 × 10−2 ± 5.61 × 10−3 (0.785) |
| 48 h | 2.53 × 10−4 ± 1.94 × 10−4 (0.016) | 8.37 × 10−3 ± 7.54 × 10−4 (0.530) |
| 7 days | 4.54 × 10−4 ± 2.03 × 10−4 (0.029) | 1.08 × 10−2 ± 9.55 × 10−4 (0.684) |
| 14 days | 4.62 × 10−4 ± 1.56 × 10−4 (0.029) | 8.57 × 10−3 ± 2.22 × 10−3 (0.542) |
| 2 months | 2.50 × 10−3 ± 1.27 × 10−3 (0.158) | 9.92 × 10−4 ± 3.92 × 10−4 (0.063) |
Numbers in parentheses represent the levels of transgene and endogenous Ki-ras expression relative to the levels of expression of endogenous Ki-ras in monotransgenic mice (value set to 1.000).
These samples were at the limits of detection.
To compare transgene expression of Ki-rasG12C to Ki-rasG12D, RNA samples from the CCSP/Ki-rasG12D mice were obtained from the University of Cincinnati Transgenic Mouse Facility for each of the following treatment groups, no DOX treatment, 24 h DOX treatment, and 7 days DOX treatment. Each treatment group was provided in triplicate. A 2 months DOX-treated lung sample was also provided by Dr Harold Varmus from the Memorial Sloan-Kettering Cancer Center. An additional time point of 2 months was included for CCSP/Ki-rasG12C mice as a comparison with this CCSP/Ki-rasG12D sample. Expression of the murine Ki-rasG12D transgene, endogenous Ki-ras, and GAPDH genes were determined by real-time PCR as described above, except that the primers for the mouse transgene used in their construct (26) were: 5′-CAAGGACAAGGTGTACAGTTATGTGACT-3′ (forward) and 5′-GGCATCTGCTCCTGCTTTTG-3′ (reverse).
Statistical methods
Continuous variables were compared among groups with t-tests. Logistic regression adjusting for sex, time and type of experiment was used to estimate adjusted means. χ2-test was used to evaluate adjusted means and generated P-values.
Results
Construction and characterization of the bitransgenic Ki-rasG12C mice
Previous studies have shown that expression of the normal Ki-ras gene is critical for normal mouse development and survival (22-24). To overcome the problems associated with expression of a strong mitogenic signal during lung development, we have utilized the inducible ‘tet-on’ system. This gene construct uses a reverse tet trans-activator (rtTA) protein that requires the presence of the ligand, DOX, in order for the rtTA gene product (consisting of the mutant tet repressor linked to the VP16 activation domain) to recognize the tetO sequence and thus stimulate gene transcription. In this approach, the gene of interest (in this case, the cDNA of mutated Ki-ras) is cloned into the tetO7-CMV plasmid, which places the Ki-ras gene downstream of a tet-inducible promoter. Founder mice established with this construct will be unable to express the Ki-ras gene because they lack the tet rtTA protein. These mice are then crossed with a second transgenic mouse line that contains the tetracycline rtTA protein linked to either the surfactant protein C (SP-C) or Clara cell secretory protein (CCSP) promoters, directing lung-specific expression of the rtTA protein. The SP-C promoter specifically targets expression to the alveolar type II cells of the lung (30,31,42,43), which has been suggested as the putative cell of origin for lung ACs in both humans and rodents (17), while the CCSP promoter appears to target expression to both the Clara and alveolar type II cells (42,44,45). Construction, generation and characterization of SP-C-rtTA and CCSP-rtTA mice have been described previously (46). The SP-C-rtTA (line 74) mice used in this study are a distinct line from the SP-C-rtTA line previously used although it contains an identical transgene construct (43). This line has very low background levels of transgene expression in the absence of DOX compared with the previously described line (unpublished data). Previous studies have shown that treatment with DOX results in very high levels of induction within 12-24 h whereas after withdrawal of DOX, expression of the transgene returns to untreated levels within 24-48 h (42,46,47).
To generate mice containing the Ki-rasG12C transgene, the insert was prepared as described in the Materials and methods section and microinjected into donor eggs obtained from superovulated FVB/N mice. The eggs were implanted into pseudopregnant mice and founder mice containing the transgene were established. To screen the pups for the presence of the transgene, genomic DNA was isolated from a small piece of the tail and then probed for the presence of the tetO and Ki-ras sequence using PCR, producing a 264 bp product that extended from the 3′ region of the tetO7-CMV expression vector through to exon 2 of the Ki-ras gene.
Initially, five Ki-rasG12C founders were identified by PCR and bred to non-transgenic littermates to establish germ line transmission. One founder did not transmit the transgene to offspring. Animals from each of the remaining four lines were bred to CCSP-rtTA or SP-C-rtTA activator mice to obtain bitransgenic mice that were then treated with DOX for 7 days to induce transgene expression. By RT-PCR analysis, one line had robust transgene expression, two had low expression, and one was not induced; however, expression was not quantified. The uninduced line was not characterized further. Bitransgenic mice from the two lines expressing low levels of Ki-ras RNA were treated with DOX from 6 weeks to 6 months. Upon killing, analysis of lung histology from these animals revealed no proliferative lesions and these lines were terminated. The remaining line developed lung adenomas upon induction of oncogenic Ki-ras expression and was used for our studies. Twenty five μg of liver DNA from the Ki-rasG12C mouse line was restricted with EcoRI and analysed by Southern blotting with a 73 bp exon 1 probe to determine the copy number of the inserted transgene. By densitometric analysis, the ratio of the 430 bp transgenic fragment to the 700 bp endogenous fragment demonstrated that there were ~22 copies of the transgene present in the mouse genome.
Treatment with DOX induces morphological changes in the lung
Bitransgenic CCSP/Ki-ras and SP-C/Ki-ras, monotransgenic Ki-ras, and control wild-type FVB/N mice were either untreated or treated with 500 μg/ml of DOX in the drinking water for 12 days, 1.5, 3, 6, 9 and 12 months. At each time point, mice were killed, macroscopic lesions counted, and the lung tissues were evaluated microscopically for the presence of proliferative changes. DOX-treated bitransgenic mice on either the CCSP or SP-C background exhibited small, hyperplastic lung foci after only 12 days of DOX treatment (Figure 1B). By 5 weeks of treatment, extensive epithelial hyperplasia of the alveolar region of the lung tissue could be seen. Lung morphology in untreated bitransgenic mice(Figure 1A) or DOX-treated single transgenic or control mice was normal.
Fig. 1.
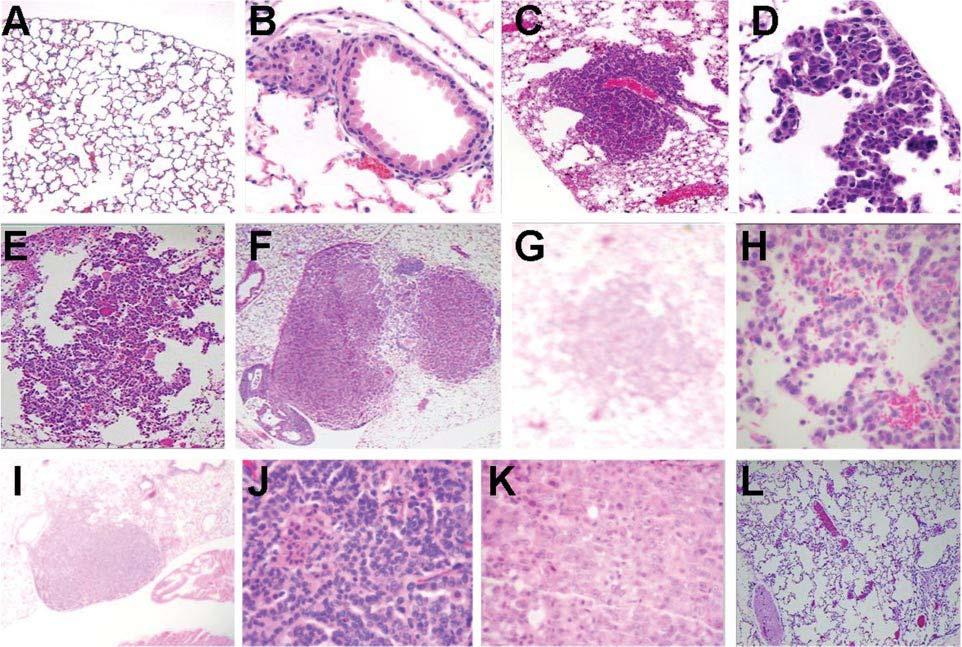
CCSP/Ki-ras lung morphology following DOX treatment. Hematoxylin and eosin stained lung tissue following (A) no DOX treatment, 10; (B) 12 days of DOX treatment, 20x, focal hyperplasia; (C) 3 months of DOX treatment, 10×, focal hyperplasia; (D) 3 months of DOX treatment, 40×, focal hyperplasia; (E) 6 months of DOX treatment, 10×, focal hyperplasia with regular cuboidal cells lining alveolar septa. The small amount of hemorrhage into the alveolar lumina is incidental; (F) 6 months of DOX treatment, 10×, solid adenoma; (G) 9 months of DOX treatment, 4, pneumocyte hyperplasia; (H) 9 months of DOX treatment, 40×, focal hyperplasia; (I) 9 months of DOX treatment, 4×, solid AD; (J) 9 months of DOX treatment, 40, solid AD with regular closely aligned pneumocytes arranged in a ribbon pattern. A small cluster have small, condensed nuclei and hypereosinophilic cytoplasm (necrosis); (K) 9 months of DOX treatment, 40×, solid AC exhibiting a solid sheet of atypical epithelial cells with pale pleomorphic nuclei, some with prominent nucleoli, and indistinct cytoplasmic borders; (L) 9 months of DOX treatment followed by 1 month of withdrawal, 10×.
Macroscopically visible lesions were first detected following 3 months of DOX treatment. CCSP/Ki-ras mice had a tumor incidence of 73% with a multiplicity of 3.5 ± 4.7 tumors per mouse while SP-C/Ki-ras mice had a tumor incidence of 55% with a multiplicity of 1.7 ± 2.0 tumors per mouse (Table I). At 3 months, all lesions were <1 mm in size. With continued exposure to DOX, the macroscopic lesions increased in number, with CCSP/Ki-ras mice displaying a higher tumor multiplicity than SP-C/Ki-ras mice at 6 and 9 months (P < 0.0001). By 6 and 9 months of DOX treatment, both CCSP/Ki-ras mice and SP-C/Ki-ras mice exhibited a high incidence of lung tumors with a tumor multiplicity of 28.8 ± 8.7 in CCSP/Ki-ras mice and 10.2 ± 7.2 in SP-C/Ki-ras mice 9 months after the initiation of DOX treatment. Tumor multiplicity increased in both lines of mice as the time of DOX treatment increased (P < 0.0001), resulting in a tumor multiplicity of 34.0 ± 8.4 in CCSP/Ki-ras mice and 12.5 ± 8.2 in SP-C/Ki-ras mice by 12 months. There was a low tumor incidence found in monotransgenic mice. These tumors were all 1 mm in size and could be attributed to either the low basal expression (Table II) of the transgene or spontaneous tumor formation often found in the FVB/N strain.
Table I.
Lung tumor incidence and tumor multiplicity
| Period in months |
||||||
|---|---|---|---|---|---|---|
| 3 (%) | 6 (%) | 9 (%) | 12 (%) | 9 a (%) | 12 a (%) | |
| Tumor incidence (mice with tumors/total mice) | ||||||
| SP-C/Ki-ras | 10/18 (55) | 14/16 (87) | 13/15 (87) | 7/7 (100) | 2/20 (10) | 0/5 (0) |
| CCSP/Ki-ras | 11/15 (73) | 18/18 (100) | 18/18 (100) | 5/5 (100) | 2/10 (20) | 0/3 (0) |
| Monotrans. +DOX | — | — | 2/20 (10) | — | 2/20 (10) | 2/5 (40) |
| FVB/N | — | — | — | — | — | 0/10 (0) |
| FVB/N + DOX | — | — | — | — | — | 0/9 (0) |
| Tumor multiplicity (number of tumors per mouse) | ||||||
| SP-C/Ki-ras | 1.7 ± 2.0 | 3.9 ± 2.4 | 10.2 ± 7.2 | 12.6 ± 8.2 | 0.2 ± 0.4 | 0.0 |
| CCSP/Ki-ras | 3.5 ± 4.7 | 13.3 ± 3.9 | 28.8 ± 8.7 | 34.0 ± 8.4 | 0.2 ± 0.4 | 0.0 |
| Monotrans. + DOX | — | — | 0.6 ± 0.3 | — | 0.1 ± 0.3 | 0.4 ± 0.4 |
| FVB/N | — | — | — | — | — | 0.0 |
| FVB/N + DOX | — | — | — | — | — | 0.0 |
Mice were treated with 500 μg/ml of DOX for the times indicated.
Untreated (DOX naive) control mice.
The majority of visible lesions detected at 3 months were morphologically diagnosed as hyperplastic lesions characterized by regular cuboidal cells lining alveolar septa (Figure 1C and D). A single adenoma was identified in one CCSP/Ki-ras mouse at this time. With continued DOX treatment for 6, 9 and 12 months, the incidence of adenomas increased (Figure 1E-J shows lesions after various time points and at different magnifications). Interestingly, by 12 months time, only two lesions were identified as low-grade carcinomas characterized by regular, but closely aligned pneumocytes arranged in a ribbon pattern. These small clusters had small, condensed nuclei and a hypereosinophilic cytoplasm (necrosis) (Figure 1K); otherwise, none of the tumors had progressed past the adenoma stage. Although there was a significant difference between SP-C/Ki-ras and CCSP/Ki-ras bitransgenic mice in tumor formation (Table I), the morphology of the proliferative lesions in both lines of bitransgenic mice were similar in being diagnosed as hyperplasias and adenomas. Immunohistochemical staining with Ki-67 demonstrated scattered nuclear staining throughout the lung lesions (Figure 2A). Comparison of the lesion morphology on the two activator backgrounds revealed that the CCSP/Ki-ras mice developed a higher incidence of proliferative changes with diminished latency. The early lesions in these animals appear to rise with characteristics of bronchiolar origin, whereas the early hyperplastic lesions in SP-C/Ki-ras mice look to be of alveologenic origin. Alveolar lipoproteinosis is found regularly in the SP-C/Ki-ras mice, indicative of abnormal surfactant metabolism. These results are particularly interesting since Fisher et al. (26), using the identical tet-on bitransgenic system but with a mutant murine Ki-rasG12D allele, found that Ki-rasG12D mice developed aggressive ACs after only 2 months of DOX. Taken together, these results suggest the development of a novel lung tumor mouse model exhibiting a less aggressive tumor type.
Fig. 2.
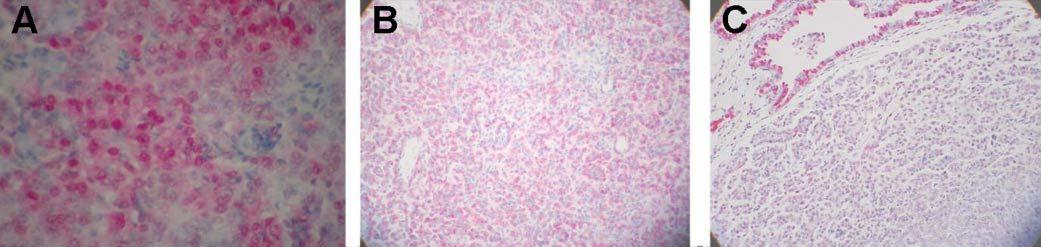
Ki-67 (A) and secretory protein (B and C) expression in lung lesions from CCSP/Ki-ras mice. Immunohistochemical analysis of lung tissue following 9 months of DOX treatment with (A) anti-SP-C, 20x, solid AD, (B) anti-CCSP, 20x, solid AD and (C) Ki-67, 20x, solid adenoma with scattered nuclear staining.
Identification of the progenitor cells of lung tumors as either Clara cells or alveolar type II cells has been an ongoing controversy, and has recently been addressed in mouse models (26,27,29,48). Several of these studies have suggested that the majority of murine lung lesions appear to arise in alveolar type II cells, while a few groups have identified cells positive for both CCSP and SP-C. Recently, cells have been identified in the bronchioalveolar junction that stain for both proteins and have been suggested to be the putative bronchioalveolar stem cells that may serve both as the stem cell population for alveolar type II and Clara cells, as well as the cell of origin for peripheral lung tumors (49). Therefore, to determine in which cell type the lung tumors in the Ki-rasG12C expressing mice arise, immunohistochemical analysis was performed using polyclonal antibodies to both the SP-C and CCSP proteins. All of the lung lesions showed moderate staining for SP-C but only very faint or no staining for CCSP in proliferative cells, which is shown for CCSP/Ki-ras mice in Figure 2B and C. While these results could be interpreted as implicating alveolar type II cells as the origin of these lesions, they would also be consistent with the presence of a common progenitor cell line as suggested by Kim et al. (49).
Ki-ras is required for tumor maintenance
Recent studies by Fisher et al. (26), utilizing a similar bitransgenic mouse system, reported that, following induction of mutant murine Ki-rasG12D expression, removal of DOX resulted in regression of the lung tumors. In order to demonstrate that the Ki-rasG12C allele is similarly important for tumor maintenance, bitransgenic CCSP/Ki-ras and SP-C/Ki-ras mice were treated with 500 μg/ml of DOX for 9 months and then withdrawn from DOX treatment for up to 1 month. Two weeks after DOX withdrawal, there were only four lung tumors still visible on the surface of the lungs in both CCSP/Ki-ras and SP-C/Ki-ras mice. By 1 month of withdrawal, 4 tumors were still visible on the surface of the lung of CCSP/Ki-ras mice, whereas no lesions were seen in SP-C/Ki-ras mice. Unfortunately, the macroscopic lesions were too small for further analysis; however, the persistent lesions could possibly be attributed to a second genetic alteration inhibiting tumor regression. Microscopically, both SP-C/Ki-ras and CCSP/Ki-ras mice exhibited few lesions after 1 month of DOX withdrawal, with only minimal hyperplastic foci generally present (Figure 1L). The remaining foci following 2 weeks of DOX withdrawal showed scattered positive nuclear staining for Ki-67, while the foci remaining following 1 month of DOX withdrawal were negative. None of the lesions were positive for cleaved caspase 3, indicating that these lesions were not regressing through an apoptotic mechanism. These data indicate that expression of Ki-rasG12C is necessary for maintenance of lung lesions and regression occurs through a mechanism alternative to apoptosis.
Expression of Ki-ras transgene
Real-time PCR was used to quantitate expression of the human Ki-rasG12C transgene in the lungs following 24 and 48 h, and 7 and 14 days of DOX treatment. Liver, spleen, pancreas, intestines, kidneys, heart, thymus and either testis and prostate or ovaries were screened for transgene expression after 14 days of antibiotic treatment. In the lung, transgene expression was shown to increase by at least 3 logs following 24 h of DOX treatment, and increased an additional 2-fold by 7 days in both lines of bitransgenic mice (Table II). Expression of the transgene in CCSP/Ki-ras mice was ~2-fold higher at every time point than in SP-C/Ki-ras mice, although this difference was not statistically significant (P < 0.9). This difference could be due to the fact that in CCSP/Ki-ras mice, both Clara cells and alveolar type II cells express the Ki-ras transgene, whereas in SP-C/Ki-ras mice only alveolar type II cells express the transgene. Except in testis and prostate, there was no detectable level of transgene expression in any of the other organs examined (Figure 3A). Since the rtTA protein should not be expressed in these two tissues, we tested the monotransgenic Ki-ras mice for transgene expression and found that the level of expression in the testis was ~6.3-fold higher than in the DOX-treated, bitransgenic lungs of SP-C/Ki-ras mice (Figure 3B). Interestingly, the testis exhibited no morphological changes even after 12 months of DOX treatment, consistent with the fact that testicular cancers do not have a high incidence of mutations in any of the Ras genes (50). The expression of the transgene in the prostate was ~8-fold higher than the lung in the same monotransgenic mice and, similar to the testis, displayed no alterations in normal histology. These results suggest that the transgene may have integrated downstream of a region of the chromosome that is regulated by male-specific hormones.
Fig. 3.
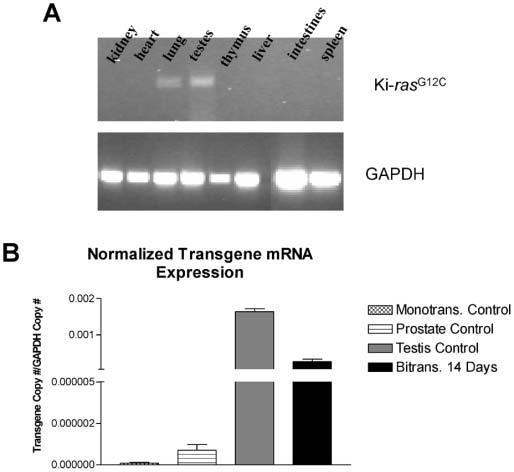
Ki-ras transgene expression in multiple organs of bitransgenic mice. (A) Total RNA was extracted from kidney, heart, lung, testis, liver, thymus, intestines and spleen tissue of CCSP/Ki-ras mice ± treatment with 500 μg/ml of DOX. RT-PCR was performed to detect Ki-ras expression (top) and GAPDH (bottom). (B) Histogram displaying transgene mRNA expression in monotransgenic control mouse lung, prostate, testis and lungs from SP-C/Ki-ras mice treated with DOX for 14 days.
We also quantified endogenous murine Ki-ras expression following 24 and 48 h, and 7 and 14 days, and 2 months of DOX treatment for both the CCSP/Ki-ras and SP-C/Ki-ras mice. The level of endogenous Ki-ras expression was identical between both lines of bitransgenic mice (data not shown). Similar to the results obtained by Fisher et al. (26), the expression levels of endogenous murine Ki-ras declined upon extended exposure to elevated levels of transgene expression (shown for CCSP/Ki-ras mice in Table II). The levels of endogenous Ki-ras expression decreased 1.5-fold after 7 days of DOX, and remained at reduced levels at 14 days (P < 0.0001). At 7 and 14 days, the level of expression of the endogenous gene was ~20-fold higher than the transgene; however, following 2 months of DOX treatment, expression of the endogenous gene was comparable with that of the transgene (Table II). These results suggest that the total levels of Ki-ras expression are strictly regulated in pulmonary epithelial cells.
The Ki-rasG12C mice used in this study demonstrated a much less severe phenotype than reported by several other laboratories utilizing transgenic mouse lines (6,26-29). In particular, the mice described by Fisher et al. (26) utilized the same vector constructs, tetO7-CMV and CCSP-rtTA, and expressed the murine Ki-rasG12D mutant allele in the same genetic background (FVB/N). Our previous data had suggested that the Ki-rasG12D mutation would be highly oncogenic, while the Ki-rasG12C mutation used in this study would be less oncogenic (8,21). To compare the levels of transgene expression, we obtained RNA from Ki-rasG12D mice either not treated or treated for 24 h and 7 days with DOX. In addition, we also isolated RNA from a sample of lung tumor tissue from Ki-rasG12D mice (a generous gift from Dr Varmus’ laboratory). In an individual experiment with newly generated standard curves, we compared these samples with RNA isolated from lung tissue in Ki-rasG12C mice following exposure to DOX for each time point. The results showed that murine Ki-rasG12D transgene expression for 24 h and 7 days was 4.31 × 10-4 ± 2.11 × 10-4 and 1.46 × 10-3 ± 1.24 × 10-3 compared with 6.83 × 10-4 ± 6.83 × 10-4 and 8.90 × 10-4 ± 1.25 × 10-3, respectively, in Ki-rasG12C mice, neither time point displaying any statistically significant difference between the two lines of mice (P = 0.5758 and 0.6022). There was a large difference in transgene expression between mice, accounting for the large standard deviations. However, this variation was not seen in our housekeeping gene, GAPDH, indicating that induction of transgene expression varied between mice. At 2 months, expression of the Ki-rasG12D transgene was ~2.5 times higher than the Ki-rasG12C allele in CCSP/Ki-ras mice. The tumor mass and lung weights were also two times higher in the Ki-rasG12D mice (26), which accounts for this difference in the RNA levels observed at 2 months between the two transgenic mouse lines. These results suggest that differences in phenotype between Ki-rasG12D and Ki-rasG12C mice were not due to differences in expression of their respective transgenes.
Activation of downstream signaling pathways
Mutations in Ki-ras result in constitutive activation of the Ras protein and increased signaling to downstream effectors. Therefore, the activation of Ras and one of its downstream effectors, Ral, was assayed. Ras and Ral activation were detected with pull down assays that detected binding of the activated protein to either the RBD of Raf or RBP, respectively. The activated proteins were then detected by western blot using antibodies for either the Ras or Ral proteins. In both bitransgenic mouse lines, Ras and Ral activation increased following treatment of the mice with DOX for 72 h. Ras activation was shown to be elevated 1.4- and 2.2-fold in DOX-treated SP-C/Ki-ras and CCSP/Ki-ras mice, respectively, compared with untreated mice (Figure 4A). Similarly, Ral activation was increased 1.4- and 1.8-fold in DOX-treated SP-C/Ki-ras and CCSP/Ki-ras mice, respectively, compared with untreated mice (Figure 4B). Thus, expression of the Ki-rasG12C allele increased signaling to downstream effectors of Ras. It should be emphasized that lysates used in the binding assays were tissue preparations from whole lung after 72 h of DOX treatment—given the large number of cell types in the lung and the restricted expression of the transgene to Clara and/or alveolar type II cells, it is likely that the activation assays have probably underestimated the actual amount of enhanced activation.
Fig. 4.
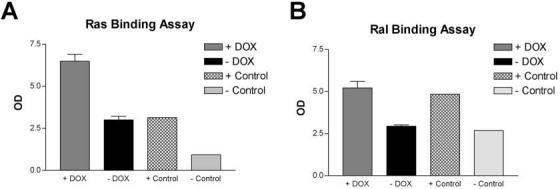
Ras and Ral activation in the lungs of CCSP/Ki-rasG12C bitransgenic mice. Total protein was extracted from whole lungs of bitransgenic mice ± treatment with 500 μg/ml of DOX. (A) Active Ras was pulled down from cell lysates with agarose beads conjugated to the RBD of Raf; (B) Active Ral was pulled down from cell lysates with agarose beads conjugated to the RBP. Activated Ras and Ral were detected by western blotting with antibodies specific for the two proteins. Controls were provided with the kit and consist of GTP (+) and GDP (-) loaded Ras or Ral proteins. The expected 2-3-fold difference in bound GTP-loaded versus GDP-loaded proteins was observed as shown in the figure.
Immunohistochemical analyses of lung tissues from CCSP/Ki-ras mice demonstrated increased phosphorylation of downstream Ras effectors after exposure to DOX. Increased phosphorylation was detected in Erk, with the highest level of activation in the nucleus and intermediate activation in the cytoplasm of hyperplastic tissue, adenomatous tissue, and normal bronchiolar epithelium (Figure 5A). Staining in lung tumors showed a relatively homogenous pattern, with scattered staining throughout the lung lesions. Downstream targets of activated Erk were also identified, including p90 Rsk which showed scattered, highly active nuclear and intermediately active cytoplasmic protein (Figure 5B) and activated ribosomal protein S6, identified by scattered cytoplasmic activation (Figure 5C). These effectors were active in the same cell types showing Erk activation. In addition, elevated phosphorylation of p38 was seen in the cytoplasm of proliferative cells (Figure 5D) and its downstream target, MAPKAPK-2, showed activation in both the nucleus and cytoplasm of proliferative cells (Figure 5E). Interestingly, phosphorylation of Akt and Jun kinase (JNK) was not increased (Figure 5F and G). These results suggest that mutant Ras-mediated activation of the Raf-Mek-Erk and Ral pathways may play a role in the early stages of lung tumorigenesis.
Fig. 5.
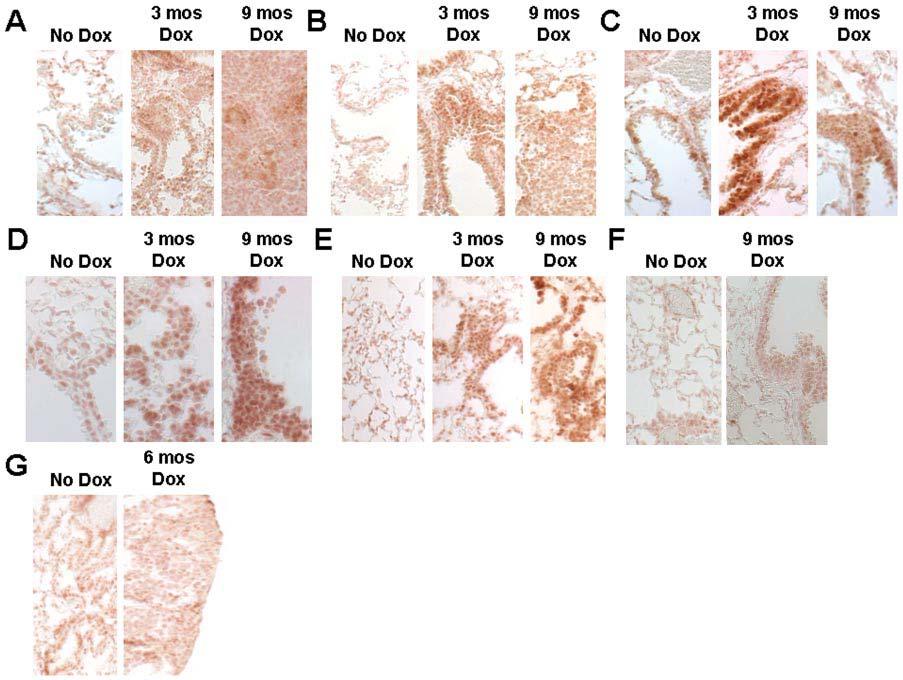
Phosphorylation of Ras downstream effectors in CCSP/Ki-rasG12C mice ± treatment with 500 μg/ml of DOX for 3, 6 or 9 months. Lung tissue was removed, embedded in paraffin, and 5 μm sections were cut for immunohistochemistry with phospho-specific antibodies. (A) Anti-phospho-ERK, 400×; (B) anti-phospho-p90 RSK, 400×; (C) anti-phospho-pS6, 400×; (D) anti-phospho-p38, 400; (E) anti-phospho-MAPKAPK-2, 400; (F) anti-phospho-JNK, 400×; (G) anti-phospho-Akt, 400×.
Discussion
Several laboratories, utilizing transgenic mouse lines, have demonstrated that expression of a mutant Ki-ras transgene results in the formation of lung tumors that rapidly progressed to ACs, with mice succumbing to their lung tumor burden in 3-10 months (6,26-29). The stark contrast in the phenotype induced in the mice reported here as compared with these other studies is particularly relevant to the results reported by Fisher et al. (26), who utilized the same tet-inducible system in the same mouse strain and the same line of CCSP-rtTA mice as employed in these studies. Fisher et al. (26) reported that expression of murine Ki-rasG12D resulted in lung ACs after only 2 months of DOX treatment. In comparison, although expression of the human Ki-rasG12C allele initially caused the appearance of visible macroscopic lesions after 3 months of DOX treatment, mice expressing the Ki-rasG12C allele for 12 months never displayed enough of a tumor burden to affect survival. The overwhelming majority of lung lesions were hyperplasias and ADs. Thus, mice with the Ki-rasG12D allele exhibited more advanced tumors than mice with the Ki-rasG12C allele; however, transgene expression in both of these mice was similar at 24 h and 7 days.
There are three possible explanations for the phenotypic differences seen between the Ki-rasG12C and Ki-rasG12D mice. First, previous studies from this laboratory provided evidence that different Ki-ras mutations influenced tumor progression, with lesions harboring the more potent VAL12 and ARG12 mutations more likely to progress to a malignant phenotype than lesions containing wild-type sequences or a CYS12 mutation (8,21). Recent studies have suggested that the correlation of mutated Ki-ras with decreased patient survival may depend on the actual base substitution, with the VAL12 and ARG12 mutations showing a trend with poor patient outcome (7,13,16). Thus, the results presented in this study, combined with those cited above, may explain the severe phenotypic differences and could be the first to demonstrate in vivo that the type of mutation induced in Ki-ras may determine the rate of lung tumor progression and the severity of the lesion produced.
Second, the Ki-rasG12C allele is a human allele while the Ki-rasG12D is a murine allele, both expressed in identical murine backgrounds. While the Ki-ras transcript and protein sequences are highly conserved between mouse and human, demonstrating 93% mRNA and 98% protein homology, there is the possibility that the human protein is signaling differently than the murine protein. Anytime there is cross-species expression, this must be considered.
Third, these transgenic mice were made through a random insertion event. Since the insertion site of neither transgene is known, there is the possibility that the phenotype is an effect of differential insertion. This problem can generally be addressed with multiple founder mice; however, we were unable to obtain a second founder line with comparable transgene expression. Further research needs to be done to distinguish between these potential mechanisms.
The benign phenotype of this model prompted us to further characterize the effects of the Ki-rasG12C transgene at the molecular level, thus we examined the activation of downstream Ras effectors. Activated Ras is thought to induce tumorigenicity by turning on multiple downstream effector pathways that increase cell proliferation and inhibit the induction of apoptosis (51). To determine effector activation, Ras and Ral activation assays as well as immunohistochemistry with phospho-specific antibodies to detect activation of downstream effectors were used. Both Ras and Ral activity were elevated ~2-fold in Ki-rasG12C expressing mice relative to untreated controls. Immunohistochemical analysis revealed that Erk and its downstream effectors, p90Rsk and pS6, were highly phosphorylated in lung tumor cells expressing mutant Ki-ras compared with non-tumor tissue or untreated, DOX naive mice. However, while we detected no increase in the activity of the JNK or Akt pathways, increased phosphorylation of p38 and MAPKAPK-2 were detected. Interestingly, Tuveson et al. (25) failed to detect phosphorylated Erk1/2 by immunohistochemistry in murine lung hyperplasias induced by expression of the Ki-rasG12D allele, although they detected elevated levels of cyclin D1. The reason for these contrasting observations is not known, but could be due to differences in the Ras alleles used in the transgene constructs, differences in levels of expression of the different Ki-ras transgenes, or differences in the antibodies or staining methods used.
Several studies have shown that stimulation of Ras activity in primary cells induces both an increase in cell proliferation and also activates a defensive mechanism to enhance apoptosis or cell cycle arrest in response to inappropriate mitogenic signals (52,53). The tumors in this model demonstrated scattered positive staining for Ki-67 but were negative when stained with an antibody specific for the cleaved fragment of caspase 3. The lack of apoptosis in both the proliferative lung lesions and in the tissue undergoing regression following DOX withdrawal suggests that there may be another regulatory mechanism accounting for the benign phenotype in this tumor model. Whereas signaling through JNK and Akt have been suggested to inhibit apoptosis (54,55), the role of p38 in the regulation of cell growth, differentiation, and cell death remains somewhat controversial (56). While an increase in Ras-mediated p38 signaling has been shown by some groups to inhibit proliferation or increase apoptosis in a number of different cell types (57-59), elevated levels of p38 have been observed in human lung tumor samples (60), suggesting that activation of p38 in some cell contexts could have a mitogenic effect. It is likely that the Ki-rasG12C allele activates a specific subset of downstream effector pathways relative to other Ki-ras mutants, and that this could determine how quickly tumors progress to more severe phenotypes.
Until recently, there has been much controversy regarding the putative progenitor cells of peripheral lung tumors. Many late-stage lung tumors express both SP-C and CCSP, making it difficult to distinguish the cell of origin. Recent transgenic mouse models have demonstrated that the majority of peripheral lung tumors stain positively for SP-C only whereas none of the lesions demonstrated staining for CCSP alone, and only a few lung tumors stain positively for both SP-C and CCSP (26,27,29). Recent studies by Kim et al. (49) have identified a potential common stem cell in the lung, which they have termed bronchioalveolar stem cells, that was identified in the bronchioalveolar junction. The authors provided evidence that these cell types may be the progenitor cells of both Clara and alveolar type II cells, and in addition may be the cells of origin for peripheral lung tumors. In the mouse model described in the present study, the Ki-rasG12C transgene is expressed in both Clara and alveolar type II cells when expressed in the CCSP-rtTA activator line, but immunohistochemical analysis demonstrated positive staining for SP-C and very little or no staining for CCSP in the proliferative cells. These data provide evidence that alveolar type II cells may serve as progenitors of murine lung tumors, but are also consistent with the possibility that tumors arise from a common progenitor cell for both Clara and alveolar type II cells.
One of the advantages of the Tet-on system is the ability to turn off transgene expression and thus determine the role of activated Ki-ras in tumor maintenance and progression. Results obtained in this study, as well as those reported by Fisher et al. (26), demonstrated that, following removal of DOX for 1 month, the number of proliferative lesions was reduced, consistent with regression of the tumors and the requirement for oncogenic Ki-ras expression for maintenance of the tumor phenotype. Using a nude mouse assay, Roth’s group transfected antisense RNA constructs of Ki-ras into human tumor cell lines and demonstrated a strong inhibition of tumor cell growth in mice, demonstrating the importance of the mutated ras allele in stimulating tumor cell proliferation and maintenance of the transformed phenotype in human cells as well (61,62). Interestingly, although the tumors that developed in Ki-rasG12C mice were not as severe, 1 month following DOX withdrawal a small number of lesions were still present when transgene expression was driven from the CCSP promoter. The remaining lesions stained positive for Ki-67 following 2 weeks of DOX withdrawal and negative following 1 month of withdrawal. However, all of the lesions were negative for cleaved caspase 3, suggesting that once the proliferative stimulus of elevated mutant Ki-rasG12C expression is removed, either tissue remodeling or the initiation of a non-apoptotic cell death pathway (63,64) may mediate the reversion of the benign lesions to normal-looking pulmonary tissue. It is possible that the longer latency of tumor development in Ki-rasG12C mice allowed a second genetic mutation to occur in some lesions that interfered with the ability of some lesions to regress. However, we also cannot rule out the possibility that due to the leakiness of transgene expression (Table II), low levels of transgene expression may prevent tumor regression in a small number of lesions.
Our results support the development of a novel mouse model for lung tumorigenesis where the tumors do not progress past the adenoma stage, in contrast to other models using Ki-ras transgenes. This long latency will allow the tumors to be monitored for an extended time period without the complications encountered by a high tumor burden or early death in the mice. With an understanding of specific activation pathways of Ras effectors, more effective targeting of novel anti-cancer agents to the specific molecules activated by specific-mutant Ras alleles could be developed, allowing therapies to be tailored to the specific molecular lesions of the tumor. Thus, these results strongly confirm the current strategy of targeting Ras proteins or members of the Ras signaling pathway in the development of novel anti-neoplastic and chemopreventive agents.
Acknowledgements
The authors thank Mr Joseph E.Moore for his technical assistance with animal husbandry, Ms Jean Clark of the University of Cincinnati Transgenic Mouse Facility for her expert technical assistance with construction of the transgenic mice, Drs Harold Varmus and William Pao of the Memorial Sloan-Kettering Cancer Center for tissue from their bitransgenic mouse model, Ms Amy Webb for assistance with isolating tail snip DNA, Hermina Bogerink for assistance with immunohistochemical staining, and Thomas McCoy for statistical analysis. Our research was supported by an initial pilot grant from the Kulynych Interdisciplinary Cancer Research Funds of Wake Forest University (to M.S.M.) and grants RO1 CA91909 (to M.S.M.) and Cancer Center Support Grant P30 CA12197 from the National Cancer Institute, training grant T32-ES07331 (for H.S.F.) from the National Institute of Environmental Health Sciences, and funding from the Francis Families Foundation (to J.T.). Partial support for Ms Webb was obtained through the Wake Forest University Summer Research Opportunities Program. Preliminary reports of this work were presented at the 41st, 42nd and 43rd annual meetings of the Society of Toxicology. This work is in partial fulfillment for the requirements of a PhD degree in the Department of Cancer Biology at the Wake Forest University Graduate School of Arts and Sciences.
Footnotes
- AC
- adenocarcinoma
- AD
- adenoma
- CCSP
- Clara cell secretory protein
- DOX
- doxycycline
- Erk
- extracellular regulated kinase
- JNK
- Jun kinase
- MAPK
- mitogen activated protein kinase
- Mek
- mitogen activated Erk kinase
- RBD
- Ras binding domain
- RBP
- Ral binding protein
- rtTA
- reverse tetracycline trans-activator
- SP-C
- surfactant protein C
- tet
- tetracycline
Conflict of Interest Statement: None declared.
References
- 1.Jemal A. Cancer Statistics. CA Cancer J. Clin. 2005;55:30. doi: 10.3322/canjclin.55.1.10. [DOI] [PubMed] [Google Scholar]
- 2.Hoffman PC, Mauer AM, Vokes EE. Lung cancer. Lancet. 2000;355:479–485. doi: 10.1016/S0140-6736(00)82038-3. [DOI] [PubMed] [Google Scholar]
- 3.Mills NE, Fishman CL, Rom WN, Dubin N, Jacobson DR. Increased prevalence of K-ras oncogene mutations in lung adenocarcinoma. Cancer Res. 1995;55:1444–1447. [PubMed] [Google Scholar]
- 4.Mitsudomi T, Viallet J, Mulshine JL, Linnoila RI, Minna JD, Gazdar AF. Mutations of ras genes distinguish a subset of non-small-cell lung cancer cell lines from small-cell lung cancer cell lines. Oncogene. 2004;6:1353–1362. [PubMed] [Google Scholar]
- 5.Reynolds SH, Anna CK, Brown KC, Wiest JS, Beattie EJ, Pero RW, Iglehart JD, Anderson MW. Activated protooncogenes in human lung tumors from smokers. Proc. Natl Acad. Sci. USA. 1991;88:1085–1089. doi: 10.1073/pnas.88.4.1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Johnson L, Mercer K, Greenbaum D, Bronson RT, Crowley D, Tuveson DA, Jacks T. Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature. 2001;410:1111–1116. doi: 10.1038/35074129. [DOI] [PubMed] [Google Scholar]
- 7.Keohavong P, DeMichele MA, Melacrinos AC, Landreneau RJ, Weyant RJ, Siegfried JM. Detection of K-ras mutations in lung carcinomas: relationship to prognosis. Clin Cancer Res. 1996;2:411–418. [PubMed] [Google Scholar]
- 8.Leone-Kabler S, Wessner LL, McEntee MF, D’Agostino RB, Jr, Miller MS. Ki-ras mutations are an early event and correlate with tumor stage in transplacentally-induced murine lung tumors. Carcinogenesis. 1997;18:1163–1168. doi: 10.1093/carcin/18.6.1163. [DOI] [PubMed] [Google Scholar]
- 9.Mitsudomi T, Steinberg SM, Oie HK, Mulshine JL, Phelps R, Viallet J, Pass H, Minna JD, Gazdar AF. ras gene mutations in non-small cell lung cancers are associated with shortened survival irrespective of treatment intent. Cancer Res. 1991;51:4999–5002. [PubMed] [Google Scholar]
- 10.Nuzum EO, Malkinson AM, Beer DG. Specific Ki-ras codon 61 mutations may determine the development of ure hane-induced mouse lung adenomas or adenocarinomas. Mol. Carcinogen. 1990;3:287–295. doi: 10.1002/mc.2940030509. [DOI] [PubMed] [Google Scholar]
- 11.Rodenhuis S, van de Wetering ML, Mooi WJ, Evers SG, van Zandwijk N, Bos JL. Mutational activation of the K-ras oncogene. A possible pathogenetic factor in adenocarcinoma of the lung. N. Engl. J. Med. 1987;317:929–935. doi: 10.1056/NEJM198710083171504. [DOI] [PubMed] [Google Scholar]
- 12.Rodenhuis S, Slebos RJ, Boot AJ, Evers SG, Mooi WJ, Wagenaar SS, van Bodegom PC, Bos JL. Incidence and possible clinical significance of K-ras oncogene activation in adenocarcinoma of the human lung. Cancer Res. 1988;48:5738–5741. [PubMed] [Google Scholar]
- 13.Rosell R, Li S, Skacel Z, Mate JL, Maestre J, Canela M, Tolosa E, Armengol P, Barnadas A, Ariza A. Prognostic impact of mutated K-ras gene in surgically resected non-small cell lung cancer patients. Oncogene. 1993;8:2407–2412. [PubMed] [Google Scholar]
- 14.Schneider PM, Praeuer HW, Stoeltzing O, Boehm J, Manning J, Metzger R, Fink U, Wegerer S, Hoelscher AH, Roth JA. Multiple molecular marker testing (p53, c-Ki-ras, c-erbB-2) improves estimation of prognosis in potentially curative resected non-small cell lung cancer. Br. J. Cancer. 2000;83:473–479. doi: 10.1054/bjoc.2000.1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Slebos RJ, Hruban RH, Dalesio O, Mooi WJ, Offerhaus GJ, Rodenhuis S. Relationship between K-ras oncogene activation and smoking in adenocarcinoma of the human lung. J. Natl Cancer Inst. 1991;83:1024–1027. doi: 10.1093/jnci/83.14.1024. [DOI] [PubMed] [Google Scholar]
- 16.Sugio K, Ishida T, Yokoyama H, Inoue T, Sugimachi K, Sasazuki T. ras gene mutations as a prognostic marker in adenocarcinoma of the human lung without lymph node metastasis. Cancer Res. 1992;52:2903–2906. [PubMed] [Google Scholar]
- 17.Malkinson AM. Molecular comparison of human and mouse pulmonary adenocarcinomas. Exp. Lung Res. 1998;24:541–555. doi: 10.3109/01902149809087385. [DOI] [PubMed] [Google Scholar]
- 18.Sweet-Cordero A, Mukherjee S, Subramanian A, You H, Roix JJ, Ladd-Acosta C, Mesirov J, Golub TR, Jacks T. An oncogenic Kras2 expression signature identified by cross-species gene-expression analysis. Nat. Genet. 2005;37:48–55. doi: 10.1038/ng1490. [DOI] [PubMed] [Google Scholar]
- 19.Bonner AE, Lemon W, Devereux TR, Lubet RA, You M. Molecular profiling of mouse lung tumors: association with tumor progression, lung development, and human lung adenocarcinomas. Oncogene. 2004;23:1166–1176. doi: 10.1038/sj.onc.1207234. [DOI] [PubMed] [Google Scholar]
- 20.Wessner LL, Fan M, Schaeffer DO, McEntee MF, Miller MS. Mouse lung tumors exhibit specific Ki-ras mutations following transplacental exposure to 3-methylcholanthrene. Carcinogenesis. 1996;17:1519–1526. doi: 10.1093/carcin/17.7.1519. [DOI] [PubMed] [Google Scholar]
- 21.Gressani KM, Leone-Kabler S, O’Sullivan MG, Case LD, Malkinson AM, Miller MS. Strain-dependent lung tumor formation in mice transplacentally exposed to 3-methylcholanthrene and post-natally exposed to butylated hydroxytoluene. Carcinogenesis. 1999;20:2159–2165. doi: 10.1093/carcin/20.11.2159. [DOI] [PubMed] [Google Scholar]
- 22.Koera K, Nakamura K, Nakao K, Miyoshi J, Toyoshima K, Hatta T, Otani H, Katsuki M. K-ras is essential for the development of the mouse embryo. Oncogene. 1997;15:1151–1159. doi: 10.1038/sj.onc.1201284. [DOI] [PubMed] [Google Scholar]
- 23.Pells S, Divjak M, Romanowski P, Impey H, Hawkins NJ, Clarke AR, Hooper ML, Williamson DJ. Developmentally-regulated expression of murine K-ras isoforms. Oncogene. 1997;15:1781–1786. doi: 10.1038/sj.onc.1201354. [DOI] [PubMed] [Google Scholar]
- 24.Thrane EV, Becher R, Lag M, Refsnes M, Huitfel HS, Schwarze PE. Differential distribution and increased levels of ras proteins during lung development. Exp. Lung Res. 1997;23:35–49. doi: 10.3109/01902149709046046. [DOI] [PubMed] [Google Scholar]
- 25.Tuveson DA, Shaw AT, Willis NA, Silver DP, Jackson EL, Chang S, Mercer KL, Grochow R, Hock H, Crowley D. Endogenous oncogenic K-rasG12D stimulates proliferation and wide-spread neoplastic and developmental defects. Cancer Cell. 2004;5:375–387. doi: 10.1016/s1535-6108(04)00085-6. [DOI] [PubMed] [Google Scholar]
- 26.Fisher GH, Wellen SL, Klimstra D, Lenczowski JM, Tichelaar JW, Lizak MJ, Whitsett JA, Koretsky A, Varmus HE. Induction and apoptotic regression of lung adenocarcinomas by regulation of a K-ras transgene in the presence and absence of tumor suppressor genes. Genes Dev. 2001;15:3249–3262. doi: 10.1101/gad.947701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, Montoya R, Jacks T, Tuveson DA. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 2001;15:3243–3248. doi: 10.1101/gad.943001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Meuwissen R, Linn SC, van der Valk M, Mooi WJ, Berns A. Mouse model for lung tumorigenesis through Cre/Lox controlled sporadic activation of the K-ras Oncogene. Oncogene. 2001;20:6551–6558. doi: 10.1038/sj.onc.1204837. [DOI] [PubMed] [Google Scholar]
- 29.Guerra C, Mijimolle N, Dhawahir A, Dubus P, Barradas M, Serrano M, Campuzano V, Barbacid M. Tumor induction by an endogenous K-ras oncogene is highly dependent on cellular context. Cancer Cell. 2003;4:111–120. doi: 10.1016/s1535-6108(03)00191-0. [DOI] [PubMed] [Google Scholar]
- 30.Glasser SW, Korfhagen TR, Wert SE, Bruno MD, McWilliams KM, Vorbroker DK, Whitsett JA. Genetic element from human surfactant protein SP-C gene confers bronchiolar-alveolar cell specificity in transgenic mice. Am. J. Physiol. Lung Cell Mol. Physiol. 1991;261:L349–L356. doi: 10.1152/ajplung.1991.261.4.L349. [DOI] [PubMed] [Google Scholar]
- 31.Wert SE, Glasser SW, Korfhagen TR, Whitsett JA. Transcriptional elements from the human SP-C gene direct expression in the primordial respiratory epithelium of transgenic mice. Dev. Biol. 1993;156:426–443. doi: 10.1006/dbio.1993.1090. [DOI] [PubMed] [Google Scholar]
- 32.Wikenheiser KA, Clark JC, Linnoila RI, Stahlman MT, Whitsett JA. Simian virus 40 large T antigen directed by transcriptional elements of the human surfactant protein C gene produces pulmonary adenocarcinomas in transgenic mice. Cancer Res. 1992;52:5342–5352. [PubMed] [Google Scholar]
- 33.Nikitin AY, Alcaraz A, Anver MR, et al. Classification of proliferative pulmonary lesions of the mouse: recommendations of the mouse models of human cancers consortium. Cancer Res. 2004;64:2307–2316. doi: 10.1158/0008-5472.can-03-3376. [DOI] [PubMed] [Google Scholar]
- 34.Hayashida S, Harrod KS, Whitsett JA. Regulation and function of CCSP during pulmonary Pseudomonas aeruginosa infection in vivo. Am. J. Physiol. Lung Cell Mol. Physiol. 2000;279:L452–L459. doi: 10.1152/ajplung.2000.279.3.L452. [DOI] [PubMed] [Google Scholar]
- 35.Wan H, Dingle S, Xu Y, Besnard V, Kaestner KH, Ang SL, Wert S, Stahlman MT, Whitsett JA. Compensatory roles of Foxa1 and Foxa2 during lung morphogenesis. J. Biol. Chem. 2005;280:13809–13816. doi: 10.1074/jbc.M414122200. [DOI] [PubMed] [Google Scholar]
- 36.Miller LA, Wert SE, Clark JC, Xu Y, Perl AK, Whitsett JA. Role of Sonic hedgehog in patterning of tracheal-bronchial cartilage and the peripheral lung. Dev. Dyn. 2004;231:57–71. doi: 10.1002/dvdy.20105. [DOI] [PubMed] [Google Scholar]
- 37.Wan H, Kaestner KH, Ang SL, Ikegami M, Finkelman FD, Stahlman MT, Fulkerson PC, Rothenberg ME, Whitsett JA. Foxa2 regulates alveolarization and goblet cell hyperplasia. Development. 2004;131:953–964. doi: 10.1242/dev.00966. [DOI] [PubMed] [Google Scholar]
- 38.Mucenski ML, Wert SE, Nation JM, Loudy DE, Huelsken J, Birchmeier W, Morrisey EE, Whitsett JA. beta-Catenin is required for specification of proximal/distal cell fate during lung morphogenesis. J. Biol. Chem. 2003;278:40231–8. doi: 10.1074/jbc.M305892200. [DOI] [PubMed] [Google Scholar]
- 39.Overbergh L, Valckx D, Waer M, Mathieu C. Quantification of murine cytokine mRNAs using real time quantitative reverse transcripatase PCR. Cytokine. 1999;11:305–312. doi: 10.1006/cyto.1998.0426. [DOI] [PubMed] [Google Scholar]
- 40.Orlando C, Pinzani P, Pazzagli M. Developments in quantitative PCR. Clin. Chem. Lab. Med. 1998;38:255–269. doi: 10.1515/CCLM.1998.045. [DOI] [PubMed] [Google Scholar]
- 41.Xu M, Miller MS. Determination of murine fetal Cyp1a1 and 1b1 expression by real-time fluorescence reverse transcription-polymerase chain reaction. Tox. Appl. Pharm. 2004;201:295–302. doi: 10.1016/j.taap.2004.05.011. [DOI] [PubMed] [Google Scholar]
- 42.Tichelaar JW, Lu W, Whitsett JA. Conditional expression of fibroblast growth factor-7 in the developing and mature lung. J. Biol. Chem. 2000;275:11858–11864. doi: 10.1074/jbc.275.16.11858. [DOI] [PubMed] [Google Scholar]
- 43.Whitsett JA, Clark JC, Picard L, Tichelaar JW, Wert SE, Itoh N, Perl AK, Stahlman MT. Fibroblast growth factor 18 influences proximal programming during lung morphogenesis. J. Biol. Chem. 2002;277:22743–22749. doi: 10.1074/jbc.M202253200. [DOI] [PubMed] [Google Scholar]
- 44.Ray MK, Magdaleno SW, Finegold MJ, DeMayo FJ. cis-Acting elements involved in the regulation of mouse Clara cell-specific 10-kDa protein. J. Biol. Chem. 1995;270:2689–2694. doi: 10.1074/jbc.270.6.2689. [DOI] [PubMed] [Google Scholar]
- 45.Stripp BR, Sawaya PL, Luse DS, Wikenheiser KA, Wert SE, Huffman JA, Lattier DL, Singh G, Katyal SL, Whitsett JA. cis-acting elements that confer lung epithelial cell expression of the CC10 gene. J. Biol. Chem. 1992;267:14703–14712. [PubMed] [Google Scholar]
- 46.Perl AK, Wert SE, Nagy A, Lobe CG, Whitsett JA. Early restriction of peripheral and proximal cell lineages during formation of the lung. Proc. Natl Acad. Sci. USA. 2002;99:10482–10487. doi: 10.1073/pnas.152238499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Perl AK, Tichelaar JW, Whitsett JA. Conditional gene expression in the respiratory epithelium of the mouse. Transgenic Res. 2002;11:23–29. doi: 10.1023/a:1013986627504. [DOI] [PubMed] [Google Scholar]
- 48.Otto WR. Lung epithelial stem cells. J. Pathol. 2002;197:527–535. doi: 10.1002/path.1160. [DOI] [PubMed] [Google Scholar]
- 49.Kim CFB, Jackson EL, Woolfenden AE, Lawrence S, Babar I, Vogel S, Crowley D, Bronson RT, Jacks T. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell. 2005;121:823–835. doi: 10.1016/j.cell.2005.03.032. [DOI] [PubMed] [Google Scholar]
- 50.Olie RA, Looijenga LH, Boerriger L, Top B, Rodenhuis S, Langeveld A, Mulder MP, Oosterhui JW. N- and K-ras mutations in primary testicular germ cell tumor: incidence and possible biological implications. Gen. Chrom. Cancer. 1995;12:110–116. doi: 10.1002/gcc.2870120205. [DOI] [PubMed] [Google Scholar]
- 51.Shields JM, Pruitt K, McFall A, Shaub A, Der CJ. Understanding ras: ‘it ain’t over ‘til it’s over’. Trends Cell Biol. 2000;10:147–154. doi: 10.1016/s0962-8924(00)01740-2. [DOI] [PubMed] [Google Scholar]
- 52.Fan J, Bertino JR. K-ras modulates the cell cycle via both positive and negative regulatory pathways. Oncogene. 1997;14:2595–2607. doi: 10.1038/sj.onc.1201105. [DOI] [PubMed] [Google Scholar]
- 53.Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- 54.Pruitt K, Pruitt WM, Bilter GK, Westwick JK, Der CJ. Raf-independent deregulation of p38 and JNK mitogen-activated protein kinases are critical for ras transformation. J. Biol. Chem. 2002;277:31808–31817. doi: 10.1074/jbc.M203964200. [DOI] [PubMed] [Google Scholar]
- 55.Rennefahrt UEE, Illert B, Kerkhoff E, Troppmair J, Rapp UR. Constitutive JNK activation in NIH 3T3 fibroblasts induces a partially transformed phenotype. J. Biol. Chem. 2002;277:29510–29518. doi: 10.1074/jbc.M203010200. [DOI] [PubMed] [Google Scholar]
- 56.Cohen P. The search for physiological substrates of MAP and SAP kinases in mammalian cells. Trends Cell Biol. 1997;7:353–361. doi: 10.1016/S0962-8924(97)01105-7. [DOI] [PubMed] [Google Scholar]
- 57.Chen G, Hitomi M, Han J, Stacey DW. The p38 pathway provides negative feedback for ras proliferative signaling. J. Biol. Chem. 2000;275:38973–38980. doi: 10.1074/jbc.M002856200. [DOI] [PubMed] [Google Scholar]
- 58.Choi JA, Park MT, Kang CM, et al. Opposite effects of Ha-ras and Ki-ras on radiation-induced apoptosis via differential activation of PI3K/Akt and Rac/p38 mitogen-activated protein kinase signaling pathways. Oncogene. 2004;23:9–20. doi: 10.1038/sj.onc.1206982. [DOI] [PubMed] [Google Scholar]
- 59.Deng Q, Liao R, Wu BL, Sun P. High intensity ras signaling induces premature senescence by activating p38 pathway in primary human fibroblasts. J. Biol. Chem. 2004;279:1050–1059. doi: 10.1074/jbc.M308644200. [DOI] [PubMed] [Google Scholar]
- 60.Greenberg AK, Basu S, Hu J, Yie TA, Tchou-Wong KM, Rom WN, Lee TC. Selective p38 activation in human non-small cell lung cancer. Am. J. Respir. Cell Mol. Biol. 2002;26:558–564. doi: 10.1165/ajrcmb.26.5.4689. [DOI] [PubMed] [Google Scholar]
- 61.Georges RN, Mukhopadhyay T, Zhang Y, Yen N, Roth JA. Prevention of orthotopic human lung cancer growth by intratracheal instillation of a retroviral antisense K-ras construct. Cancer Res. 1993;53:1743–1746. [PubMed] [Google Scholar]
- 62.Mukhopadhyay T, Tainsky M, Cavender AC, Roth JA. Specific inhibition of K-ras expression and tumorigenicity of lung cancer cells by antisense RNA. Cancer Res. 1991;51:1744–1748. [PubMed] [Google Scholar]
- 63.Broker LE, Kruyt FA, Giaccone G. Cell death independent of caspases: a review. Clin. Cancer Res. 2005;11:3155–3162. doi: 10.1158/1078-0432.CCR-04-2223. [DOI] [PubMed] [Google Scholar]
- 64.Chi S, Kitanaka C, Noguchi K, et al. Oncogenic ras triggers cell suicide through the activation of a caspase-independent cell death program in human cancer cells. Oncogene. 1999;18:2281–2290. doi: 10.1038/sj.onc.1202538. [DOI] [PubMed] [Google Scholar]