

Abstract
CD2 is a cell surface protein belonging to the immunoglobulin superfamily (IgSF) that plays a key role in mediating adhesion between human T-lymphocytes and target cells. The interaction between cell-adhesion molecules CD2 and CD58 is critical for immune response. Modulation or inhibition of these interactions has been shown to be therapeutically useful. Synthetic 12-mer linear and cyclic peptides and cyclic hexapeptides from the β-turn and β-strand region (hot spot) of human CD2 protein were designed to modulate CD2–CD58 interaction. The 12-amino acid synthetic cyclic peptides effectively blocked the interaction between CD2 and CD58 proteins as demonstrated by E-rosetting and heterotypic adhesion assays. NMR and molecular modeling studies indicated that these cyclic peptides exhibit β-turn structure in solution and closely mimic the β-turn structure of the surface epitopes of CD2 protein. The designed cyclic peptides with β-turn structure have the ability to modulate CD2–CD58 interaction.
Introduction
CD2 is a cell surface protein belonging to the immunoglobulin superfamily (IgSF) that plays a key role in mediating adhesion between human T-lymphocytes and target cells. Its ligand, leukocyte function-associated antigen-3 (LFA-3, CD58), also belongs to the IgSF on the target cell. The interaction between CD2 and CD58 (LFA-3) is believed to augment the adhesion in the TCR–MHC complex.1,2 The adhesion causes a costimulatory signal in the T-cell that amplifies the primary signal caused by the TCR–MHC complex; thus, the affinity of the TCR–MHC complex increases by 30–50-fold, and this can enhance the T-cell antigen interaction. However, the binding of CD2 to CD58 has a low affinity and very rapid koff rates (koff > 1 s−1).3 Inhibition of the CD2–CD58 interaction has important implications in controlling immune responses in autoimmune diseases.4,5 Therefore, molecules designed to inhibit CD2–CD58 interaction may function as immunosuppressants. It has been shown that blockage of the CD2–CD58 interaction3–8 and/or modulation of the CD2 costimulatory pathway can result in prolonged tolerance toward allografts.9–13 The soluble CD58–IgG fusion protein Amevive (LFA3TIP) has been used to treat psoriasis.4 The humanized versions of antibodies BTI-32214 and MEDI-50715 have been tested for the treatment of acute organ rejection and graft-versus-host disease. Furthermore, MEDI-507 has also been investigated for the treatment of autoimmune and other inflammatory diseases.16 However, therapeutic antibodies are huge protein molecules and non-human in origin. These often elicit significant side effects attributed to their immunogenicity. They are also susceptible to enzymatic degradation. To circumvent these problems, one approach is to design short peptides or small molecular mimics that will bind to critical areas in target proteins and, like antibodies, interfere with their activity.
Therefore, this study was undertaken to design peptides from CD2 to modulate CD2–CD58 interaction. In our earlier report,17 we showed that peptides designed from rat CD2 could inhibit CD2–CD58 interactions. On the basis of the structure of the CD2–CD58 complex18 and previously reported mutagenesis studies,3 we have designed two types of epitopes in the peptides: those that are derived from the β-turn region of CD2 and those from the β-strand region of CD2. In this paper, we report the design of linear and cyclic peptides (Table 1) from the β-turn and β-strand regions (hot spot) of human CD2 protein (Figures 1 and 2). To investigate if peptides designed from human CD2 protein could interfere with the interaction between cell surface-expressed CD2 and CD58 proteins, two cellular assays were performed. To understand the structure–function relationship of peptides, we also carried out detailed NMR and molecular modeling studies of the designed peptides. Our results indicate that the designed peptides are useful for inhibition of the T-cell adhesion mechanism.
Table 1.
Peptides Used in This Study that Are Derived from the Human CD2 Protein
| sequencea | origin | theoretical mass | mass found | purity (%) | |
|---|---|---|---|---|---|
| lAQ | Pen(Acm)AQFRKEKETFC(Acm)-OH | C′C′′ loop | 1659.8 | 1659.4 | >95 |
| cAQ | cyclo(1,12)PenAQFRKEKETFC-OH | 1515.8 | 1515.6 | >90 | |
| cFE | cyclo(1,6)FRKEKE | 818.0 | 818.5 | >90 | |
| lIL | SIYDTKGKNVLE-NH2 | FG loop | 1366.5 | 1366.5 | >90 |
| cIL | cyclo(1,12)PenIYDTKGKNVLC-OH | 1382.6 | 1382.1 | >90 | |
| cYK | cyclo(1,6)YDTKGK | 693.4 | 693.4 | >90 | |
| DS | DDIDDIKWEKTS-NH2 | C strand | 1464.5 | 1464.5 | >95 |
| SE | SDKKKIAQFRKE-NH2 | C′ strand | 1477.7 | 1476.9 | >95 |
| IK | IYKVSIYDTGK-NH2 | F strand | 1414.7 | 1414.5 | >95 |
| control | KGKTDAISVKAI-NH2 | 1230.5 | 1229.5 | >95 |
Underlined regions are the β-turn or β-strand structures in the proteins.
Figure 1.

Sequence alignment of rat CD2 and human CD2 (hCD2) (ClustalW alignment). Only 150 amino acids corresponding to domains 1 and 2 are shown. Pound signs denote the interface contact residues in the hCD2–hCD58 structure. Asterisks mark the residues in the interface. β-Turn regions are in bold letters. Amino acid residues corresponding to the designed peptide sequences are underlined.
Figure 2.
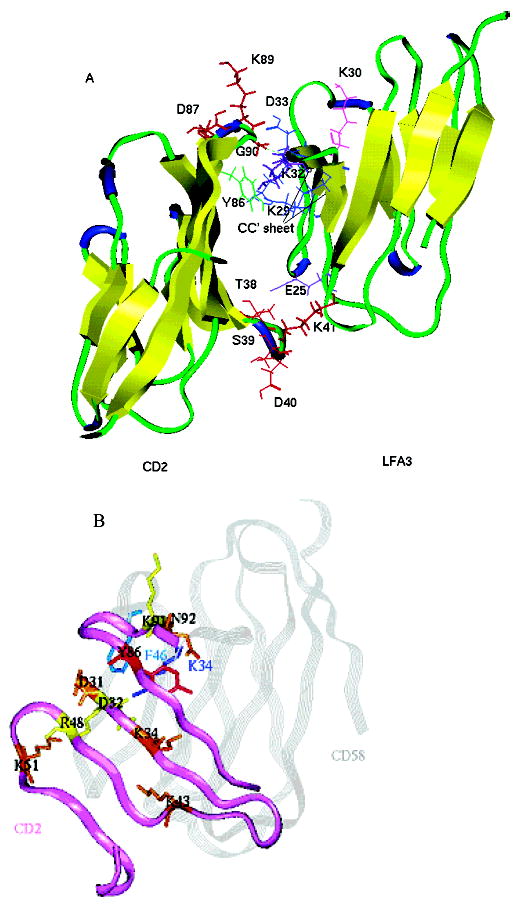
(A) Ribbon diagram of crystal structure of the CD2–CD58 (LFA-3) complex. The residues of hCD2 that are in β-turn regions are shown as red sticks. Tyr86 from CD2 is colored green. Residues from CD58 that are important in the CD2–CD58 interaction are colored: Lys32 and Glu25 in purple, Asp33, Lys29, and Glu37 in blue, and Lys30 in magenta. (B) Superimposition of the CD2 interface (purple ribbon) and CD58 (gray ribbon). The CD2 residues involved in the CD58 binding are labeled and colored on the basis of mutation results and their effects on CD2–CD58 interaction. Mutations D31A, K34A, K43A, K51A, and N92A (brown) had a stronger effect, reducing the level of adhesion by more than 50% and CD58 binding by more than 1 order of magnitude; mutations D32A, R48A, and K91A (yellow) abolished adhesion completely and manifested a 47–127-fold decrease in CD58 binding affinity. The Y86A (red) mutation resulted in a loss of binding to CD58 of more than 1000-fold. CD58 key residues K34 and F46 are labeled.
Results
On the basis of the structures of the CD2 N-terminal adhesion domain and the bimolecular complex between the N-terminal adhesion domains of CD2 and CD58,18–26 peptides were designed from the CD2 protein sequence. To identify the “minimal” structural parts of CD2-derived peptides that are responsible for CD58 binding, 12-residue peptides DS, SE, IK, lIL, and lAQ, which were derived from the sequences of C, C′, and F strands and FG and C′C′′ loops in human CD2, respectively, were designed (Figures 1 and 2 and Table 1). The peptides generated from the β-turn regions were cyclized by disulfide bonds with the introduction of amino acid penicillamine (Pen) and cysteine at the two termini of the linear peptide sequence (peptides cIL and cAQ in Table 1). Penicillamine is used in position 1 to induce conformational rigidity of the cyclic peptide.27
Cyclic 12-mer Peptides Derived from β-Turn Regions Exhibited Higher Activities than the Linear Counterparts, but Truncated Cyclic Peptides Exhibited Reduced Activities.
In the first method, E-rosetting was carried out to test the biological activity of peptides.17 E-Rosetting is the most widely used method of identifying T-cells showing CD2–CD58 interaction. Sheep red blood cells express sheep CD58 protein, while Jurkat leukemic T-cells express CD2 protein on their surface (confirmed by flow-cytometry assay, data not shown). Binding of Jurkat cells to sheep red blood cells due to CD2 and CD58 interaction results in the formation of E-rosettes. The ability of each of the designed CD2 peptides to inhibit CD2–CD58 interaction was evaluated by inhibition of E-rosette formation between Jurkat cells and sheep red blood cells.
Preincubation of sheep red blood cells with T-cells and human CD2-derived peptides, but not with a control peptide, resulted in a concentration-dependent inhibition of E-rosette formation (Figure 3). At the highest concentration that was tested (200 μM), 12-amino acid residue cyclic peptides cAQ and cIL exhibited nearly 35% inhibition of E-rossette formation, whereas the linear versions of these peptides, lAQ and lIL, respectively, exhibited 5–15% inhibition. A control peptide showed less than 5% inhibition. As a positive control, antibody to CD58 was used. The antibody showed 100% inhibition at ≤5 μM. Thus, linear peptides did not show significant inhibition. The truncated peptide cFE (hexapeptide) exhibited nearly 25% inhibition at 200 μM, whereas the hexapeptide cYK exhibited only 10% inhibition (Figure 3). This clearly suggests that truncated forms of peptides were less effective compared to 12-amino acid residue cyclic peptides.
Figure 3.
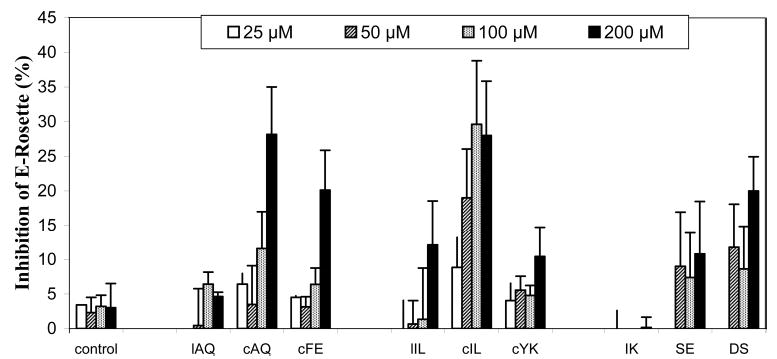
Inhibition of E-rosette formation by synthetic peptides derived from CD2 protein. Peptides were added to AET-treated sheep red blood cells (expressing CD58 protein) first, and then an equal amount of Jurkat cells (expressing CD2 protein) was added later. The cells were pelleted by centrifugation and incubated at 4 °C. The cell pellet was gently resuspended before the E-rosettes were counted. Cells with three or more SRBCs bound were counted as rosettes. At least 200 cells were counted to determine the percentage of E-rosette cells. Values are the percent inhibition of peptide-treated cells and expressed as the mean of three independent experiments.
As a second method, inhibition of adhesion between Caco-2 cells and Jurkat cells was used to evaluate the biological activity of the designed peptides. Caco-2 cells express human CD58, while Jurkat cells express human CD2 protein; thus, the inhibitory activity observed between Caco-2 cells and Jurkat cells provides evidence that the peptides designed from CD2 can inhibit the heterotypic cell adhesion by the hCD2–hCD58 pathway. By using fluorescently labeled Jurkat cells, the inhibitory activities of designed CD2 peptides were measured and the fluorescence change was calculated with and without addition of peptides. This heterotypic adhesion assay is much more sensitive than E-rosetting because of the measurement of fluorescence.28 The activities of the peptides from CD2 in the heterotypic cell adhesion assay are shown in Figure 4 along with that of a control peptide. The trends in the inhibitory activity of β-turn-based peptides were similar to those in the E-rosetting assay. Twelve-amino acid cyclic peptides cAQ and cIL exhibited nearly 50% inhibition, whereas the linear counterparts of the same peptides exhibit 25% inhibition at 90 μM. Truncated hexapeptide cFE showed 25% activity, but hexapeptide cYK showed only 20% inhibition activity in the lymphocyte epithelial assay (Figure 4).
Figure 4.
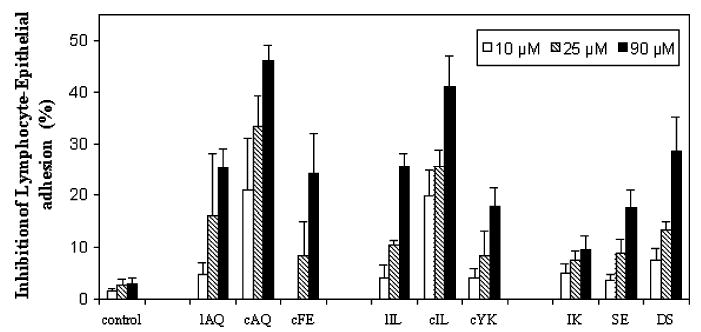
Inhibition of lymphocyte–epithelial adhesion by synthetic peptides derived from CD2 protein. CD58 and CD2 expressed in Caco-2 and Jurkat cells, respectively, were pre-examined. Peptides were added to the confluent Caco-2 monolayer, and then the BCECF-labeled Jurkat cells were added to the mixture. After incubation for 45 min at 37 °C, nonadherent Jurkat cells were removed by washing with PBS and the monolayer-associated Jurkat cells were lysed with a Triton X-100 solution. Soluble lysates were transferred to 96-well plates for reading in a microplate fluorescence analyzer. Values are showed as the percent inhibition of peptide-treated cells and expressed as the mean of three independent experiments.
Peptides Derived from β-Strand Regions Exhibited Low Inhibition Activity.
The peptides from the β-strand region were from the hot-spot region where the amino acids in this fragment are involved in CD2–CD58 interaction. Surprisingly, the three peptides derived from C, C′, and F strands, peptides DS, SE, and IK, respectively, exhibited very low activity in the two assays (~0% in the E-rosetting assay and 10% in the lymphocyte–epithelial adhesion assay). Although peptide SE, which is derived from the C′ strand with important residues Lys43 and Arg48 in CD2 binding to CD58, shared the sequence of Ala45-Gln46-Phe47-Arg48-Lys49-Glu50 with cyclic peptide cAQ (Figure 3), less than 20% inhibitory activity was observed (Figure 4). The activity of peptide DS (from the C strand with important residues Asp31, Asp32, and Lys34) showed ~20% activity in the E-rosetting assay and 30% activity in the lymphocyte–epithelial adhesion assay. This is higher than those of the other two linear peptides generated from CD2 β-sheet regions but still lower than those of the cyclic peptides from β-turn regions (30 and 45% in the two assays, respectively).
Statistical analysis of the results from the cell adhesion assay was carried out using the Mann Whitney U test (n= 4). Peptides were grouped into three categories for the analysis: group 1, lAQ, cAQ, and cFE; group 2, lIL, cIL, and cYK; and group 3, SE, IK, and DS. Comparison was made for each peptide with the negative control as well as the peptides within group. The results showed that in the heterotypic adhesion assay, almost all the peptides at all three concentrations that were tested displayed significant differences (except for cFE at 25 μM and cIL at 10 μM) with the control peptide (p < 0.05). For the E-rosetting assay, due to the limitation of the assay (low sensitivity compared to that of the heterotypic adhesion assay), the designed peptides did not show significant differences at lower concentrations (<100 μM). While comparisons are made within the group, peptide cAQ differed significantly from peptides lIL and cFE (for example, p ) 0.029 at 90 μM when comparing cAQ with lAQ and cFE). The same was found for peptide cIL (significant difference from peptides lIL and cYK). These data indicate that cyclization of 12-amino acid peptides enhanced the activity, while truncation of peptides (hexapeptide) resulted in the loss of activity. Linear peptides designed from β-strand regions (SE, IK, and DS) also displayed significant differences in activity in the E-rosetting assay at 100 and 200 μM, and in the heterotypic adhesion assay at 25 and 90 μM.
Cell Viability Assay.
The MTT assay, an index of cell viability and cell growth, is based on the ability of viable cells, not dead cells, to reduce MTT from a yellow water-soluble dye to a dark blue insoluble formazan product by mitochondrial enzymes associated with metabolic activity.29 The assay has been widely used in cell proliferation assays, cytotoxicity analysis, and apoptosis screening. To examine the toxicity of the designed peptides, the ability of the peptides to inhibit the growth of human Caco-2 cells and Jurkat cells was assessed in this assay. The results showed that the Caco-2 monolayers or Jurkat cells incubated with the highest dose of peptides for fixed periods, which were used in the lymphocyte–epithelial adhesion assay, did not display any significant inhibition of the cell viabilities (Figure 5). Therefore, the peptides used in this study were not toxic to the cells, and inhibition effects observed in the two assays carried out were presumably due to the inhibition of adhesion molecules on the surface of cells.
Figure 5.
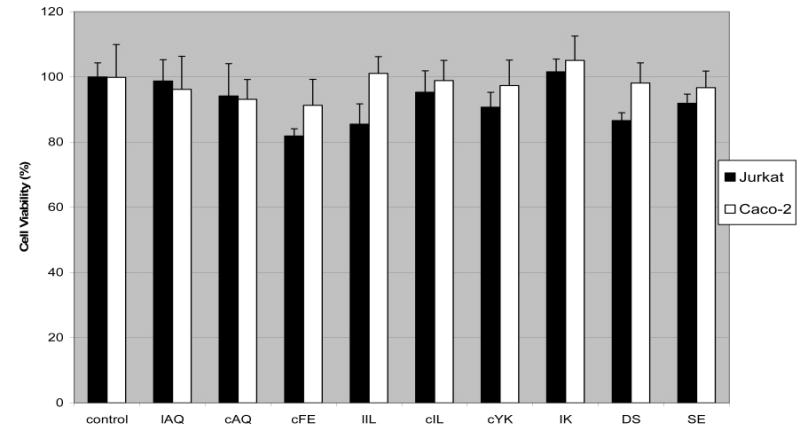
Cell viabilities of Caco-2 monolayers and Jurkat cells treated with synthetic peptides. The MTT assay was applied to examine the cytotoxicity of peptides to the Caco-2 and Jurkat cells during the lymphocyte–epithelial adhesion assay. The highest concentration of peptide used in the adhesion assay was added to the cells and incubated for the time of exposure of Caco-2 and Jurkat cells during the adherence assay. The cell viabilities were validated by incubating the cells with 5 mg/mL MTT for 3 h at 37 °C. The MTT-labeled cells were then solubilized in DMSO before the color of the reaction product was quantified using a plate reader. All samples were assayed in four independent assays, and the mean for each experiment was calculated.
Structural Studies.
CD spectra exhibited by cyclic peptides displayed β-turn structure, while linear peptides displayed random structure. As seen in Figure 6A, linear peptide lAQ showed a negative band below 200 nm, which suggests the possibility of an open or unordered conformation. The cyclic version of the same peptide cAQ exhibits a CD negative band around 202 nm, indicating the possibility of a small percentage of type I β-turn conformation (Figure 6A). The cyclic hexapeptide cFE exhibits a negative band at 203 nm, and the shift of the negative band away from 200 nm suggests a more stable conformation for this peptide.
Figure 6.
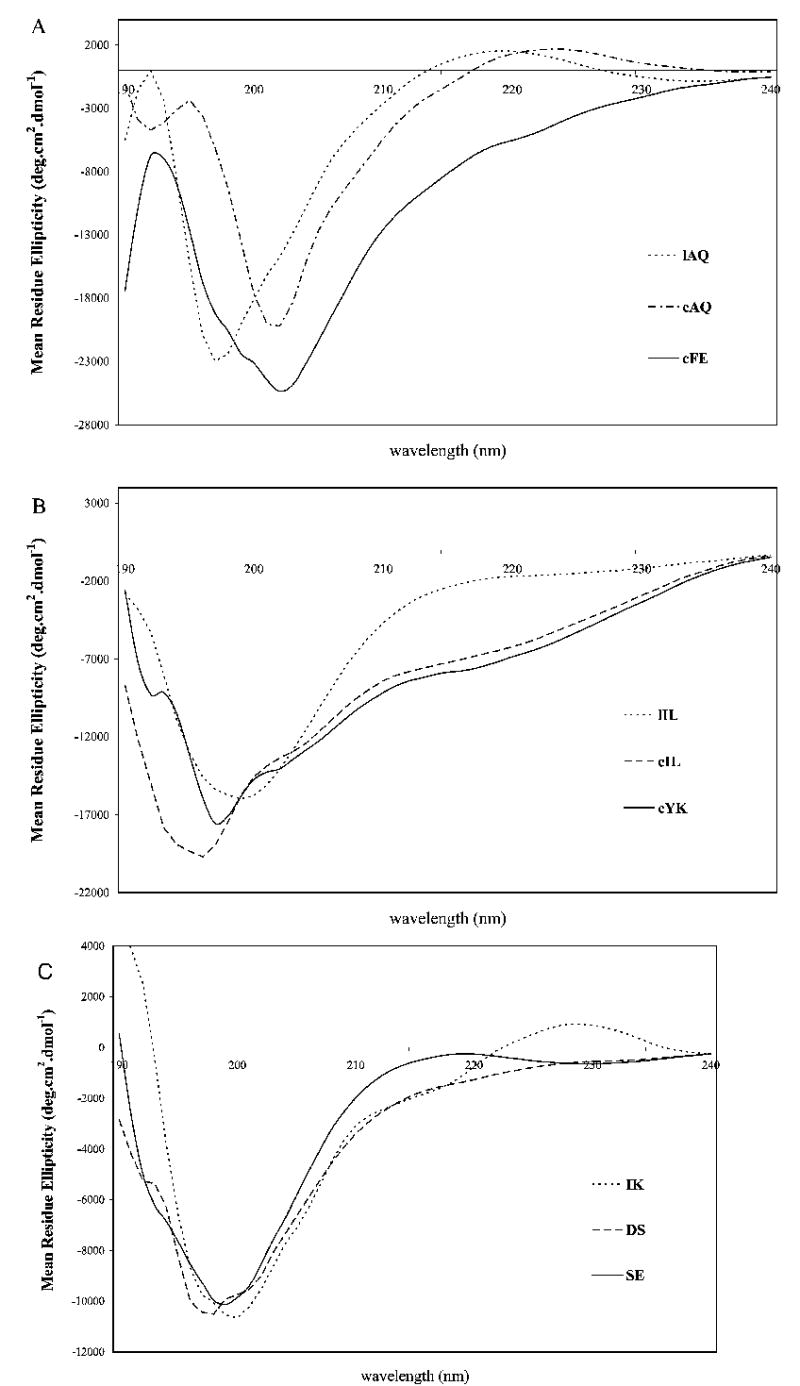
CD spectra of peptides in water. The concentration of the peptide used was in the range of 0.5–0.7 mM.
In peptide lIL, a negative band was observed below 200 nm (Figure 6B), which indicated that this linear peptide has a random structure. Compared with linear peptide lIL, the cyclic peptide cIL displayed a β-turn structure with a negative shoulder in the region of 203–205 nm. The three linear peptides DS, SE, and IK designed from the C, C′, and F β-strands, respectively, displayed mainly open/unordered structure (Figure 6C) and a small percentage of β-turn structure, characterized by a negative band around 202 nm.30,31
Structure in Solution Determined by NMR.
In a series of peptides derived from the β-turn and β-strand regions of CD2, the cyclic 12-mer peptides were identified as being more potent in inhibiting the adhesion of T-cells to sheep red blood cells or human epithelial cells than their linear counterparts. However, the smaller hexapeptides cFE and cYK were found to have lower inhibitory activities than the larger ring (12 residues) peptides cAQ and cIL. To understand the structure and biological activity relationships, the structures of the cyclic 12-mer peptides and hexapeptides cAQ, cIL, cFE, and cYK were determined by NMR. The one-dimensional (1D) 1H NMR spectrum of the peptides showed good dispersion of the chemical shifts and the coupling patterns, indicative of a stable major conformer at the experimental temperature.
12-mer Peptides.
The amide region of the 1D NMR spectrum of the cAQ peptide showed good dispersion of chemical shifts over a range of 1 ppm, indicating a well-defined conformation of the peptide in solution. At 298 K, the Lys6 and Lys8 amide resonances overlapped. However, at 293 K, the Lys6 and Lys8 amide resonances were distinguishable. The amide region of the ROESY spectrum of the cAQ peptide showed cross-peaks between the NH group of Gln3 and the NH group of Phe4, the NH group of Arg5 and the NH group of Lys6, and the NH group of Lys6 and the NH group of Glu7, suggesting the possibility of folded structure or β-turn structure around these residues. The coupling constant values of Ala2 and Lys6 were less than 6 Hz, whereas the coupling constants of all other residues were in the range of 6–8 Hz, indicating the possibility of a rapidly interconverting conformation of the peptide on the NMR time scale (Figure 7A). However, the chemical shift deviations (CSDs) of Hα 32 of all the residues deviated from random coil values and were negative. This may indicate that the cAQ peptide acquires a preferred conformation in solution. A total of 130 ROE cross-peaks were observed in the ROESY spectra which could clearly define the conformation of the peptide in solution.
Figure 7.

Summary of ROEs/NOEs for (A) cAQ, (B) cIL, (C) cYK, and (D) cFE. The thickness of the bars indicates the intensity of the cross-peaks, which were assigned as strong, medium, and weak. Temperature coefficients of chemical shifts are given in units of parts per billion per kelvin. 3JHNα are shown as arrows (down arrow if <6 Hz, up arrow if >6 Hz). Asterisks denote coupling constants that could not be measured with accuracy.
Twelve superimposed structures that satisfy the NMR data based on NMR restrained MD simulations are shown in Figure 8A. The average rms deviation of the backbone atoms was 0.70 Å, indicating the convergence nature of the energy-minimized structure. The backbone structure of the peptide is characterized by a β-turn consisting of Arg5, Lys6, Glu7, and Lys8. The existence of a β-turn is supported by NH–NH ROEs between the NH group of Arg5 and the NH group of Lys6 as well as the NH group of Lys6 and the NH group of Glu7. Furthermore, the coupling constant (3JNHα) of Lys6 was <6 Hz. The dihedral angles around Lys6 and Glu7 indicated that the β-turn is a type I turn.33 The β-turn is not stabilized by an intramolecular hydrogen bond. This is indicated in the temperature dependence of amide protons which showed δΔ/δT in the range of –4.5 to –12.0 ppb/K. In addition to this β-turn, the structure of the peptide also indicated a possibility of a β-turn consisting of Ala2, Gln3, Phe4, and Arg5. This β-turn was supported by NH–NH ROEs between the NH group of Gln3 and the NH group of Phe4 as well as observed ROE cross-peak dβN(i,i+2) between Ala2 and Phe4. However, the dihedral angle ψ of Phe4 deviated from the ideal geometry of standard β-turn dihedral angle33 (ψ of Phe4 is ~94°). Thus, the overall structure of the cAQ peptide was characterized by a type I β-turn consisting of Arg5, Lys6, Glu7, and Lys8. An average structure from the family of 12 structures was used to compare the β-turn in the structure with the crystal structure of CD2 protein. Comparison of the β-turn structure of peptide cAQ with the crystal structure of hCD2, hCD2 in complex with CD58, and rat CD2 indicated that the β-turn structure is retained in the peptide as in the native protein structure. However, the β-turn structure in the human CD2 crystal structure18–22,34 was a type II β-turn, while in the hCD2–CD58 complex crystal structure, the β-turn did not have an ideal geometry compared to the standard values.33 An overlay of the β-turn structure of the cAQ peptide, the β-turn of hCD2, and the rat CD2 β-turn region is shown in Figure 8C.
Figure 8.
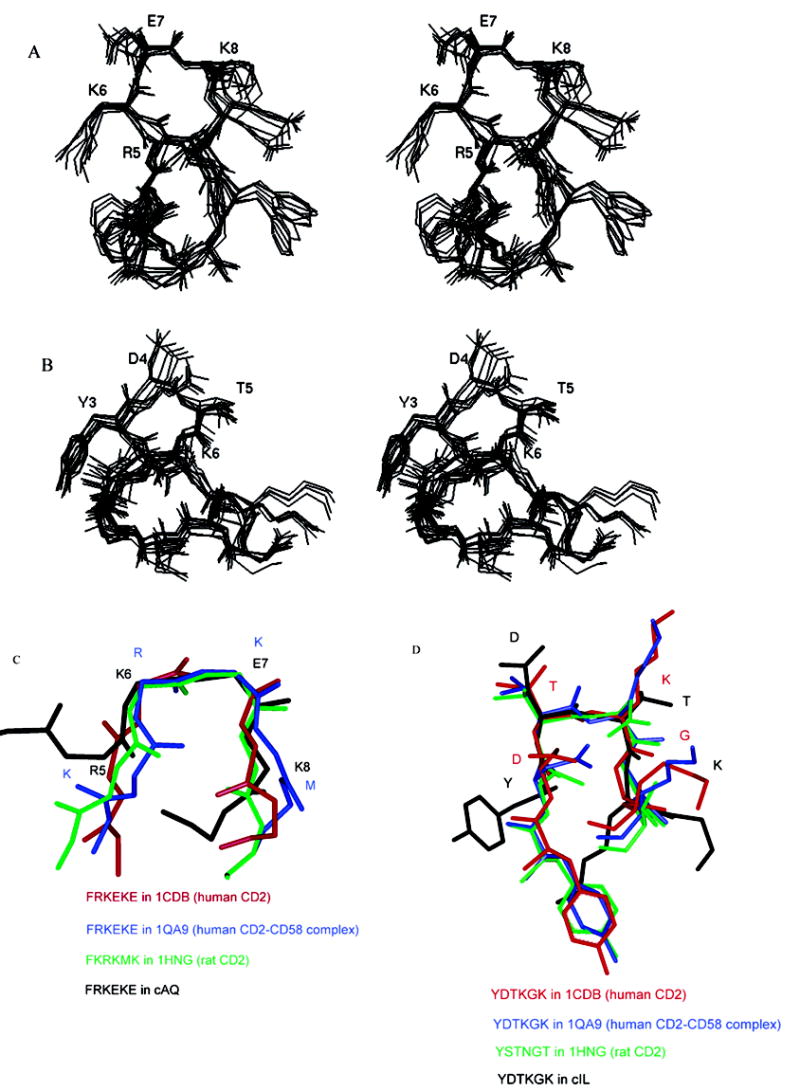
Stereoview of a family of superimposed structures from NMR-restrained MD simulations for cyclic peptides (A) cAQ and (B) cIL. The residues which are involved in the β-turn conformation are labeled. (C) Overlay of the β-turn region of cAQ (R5K6E7K8) with the β-turn region of the crystal structure of hCD2. (D) Overlay of the β-turn region (Y3-D4-T5-K6) of cIL with the β-turn region (D87-T88-K89-G90) of the crystal structure of hCD2.
The NMR spectra of the cIL peptide were well dispersed at 293 K, and all the amide resonances could be assigned without ambiguity. The chemical shift spread of amide resonances was nearly 0.7 ppm, indicating a flexible conformation for the peptide. The Gly7 diastereotopic protons were separated by greater than 1 ppm, suggesting a stable conformation around the Gly7 residue. The coupling constants of most of the amide resonances were in the range of 6–8 Hz, indicating rapid conformational interconversion on the NMR time scale (Figure 7B). The CSDs for the Hα protons deviated from random coil values32 and were positive. This may indicate that the peptide may not have a completely random structure. The amide–amide region of the ROESY spectrum indicated an ROE between the NH group of Thr5 and the NH group of Lys6, suggesting a possible turn or folded structure around these residues. There were long-range ROEs observed between the NH group of Tyr3 and the NH group of Thr5, as well as between the NH group of Ile2 and the NH group of Cys12. The ROE between the NH groups of Tyr3 and Thr5 may indicate a β-turn structure around the Tyr3-Asp4-Thr5-Lys6 sequence. The temperature coefficient of the chemical shift of amide resonances was greater than –4.5 ppb/K, which clearly suggests the absence of intramolecular hydrogen bonding in the peptide structure in solution. A total of 85 ROE cross-peaks were observed in the ROESY spectra of the peptide in solution which were used to define the structure of peptide cIL in solution.
A family of 10 superimposed structures of the cIL peptide that satisfy the NMR data is shown in Figure 8B. The average rms deviation of the backbone atoms in the family compared to the average structure in the family was 0.84 Å. The structure of the cIL peptide was characterized by a β-turn consisting of Tyr3, Asp4, Thr5, and Lys6 (Figure 8B). The existence of a β-turn was supported by NH–NH ROEs between the NH group of Thr5 and the NH group of Lys6 as well as between the NH group of Tyr3 and the NH group of Thr5. The dihedral angles around Asp4 and Thr5 indicated the β-turn was type I. Comparison of an average structure in the family with the crystal structure of the protein CD2 indicated that in the CD2 protein the β-turn is around Asp87, Thr88, Lys89, and Gly90 rather than Tyr, Asp, Thr, and Lys as observed in the peptide. Thus, in peptide cIL the β-turn is shifted by one residue (Figure 8D).
Cyclic Hexapeptides.
The NMR data of the cYK peptide were indicative of the flexible nature of the peptide, which was characterized by a degenerate chemical shift of the Gly5 Hα enantiotopic protons. The NH–NH region of the NOESY data showed connectivity between the amides of the Tyr1-Asp2 and Thr3-Lys4 sequences which are suggestive of a folded structure in the peptide and the proximity of amide protons due to the compact nature of the structure. The temperature coefficients of chemical shifts of amides of Thr3 and Lys6 were –2.4 and –4.2 ppb/K, respectively (Figure 7C), which may be due to intramolecular hydrogen bonding or solvent-shielded amide protons of these residues. However, the coupling constants of most of the amide protons were in the range of 6–9 Hz, suggestive of rapidly interconverting conformers that coexist in solution.35
From NOE-restrained MD simulations at a high temperature (900 K), eight families of conformers that satisfied the NMR data were obtained. To check for convergence, the structures in each family were superimposed on the average structure of each family. All structures presented rectangular structures with corner residues Tyr1, Asp2, Lys4, and Gly5 (Figure 9A). An average structure was taken from each family and subjected to low-temperature dynamics at 300 K with NMR constraints. These structures were subjected to energy minimization. An average structure was taken from each family. The average rms deviation of the backbone atoms of eight structures compared to the lowest-energy structure was 0.91 Å. The structure of cYK exhibits hydrogen bonding between the NH group of Thr3 and the carbonyl carbon of Tyr1, which was consistent with the low value of the temperature coefficient of chemical shifts of the amide of Thr3. An additional hydrogen bond was observed between the Lys6 NH group and the Thr3 C=O and Lys4 C=O groups in the model proposed. However, a lack of convergence was observed in the backbone and side chains except for the two corner residues of Asp2 and Gly5. An overlay of the backbone and side chains of peptide cYK to the respective fragment in human CD2 crystal structure (data not shown) showed that the overlapped structure has a relatively large rmsd compared to human CD2 (rmsd of the backbone was 2 Å), especially for the backbone of Asp2 (Asp87 in human CD2).
Figure 9.
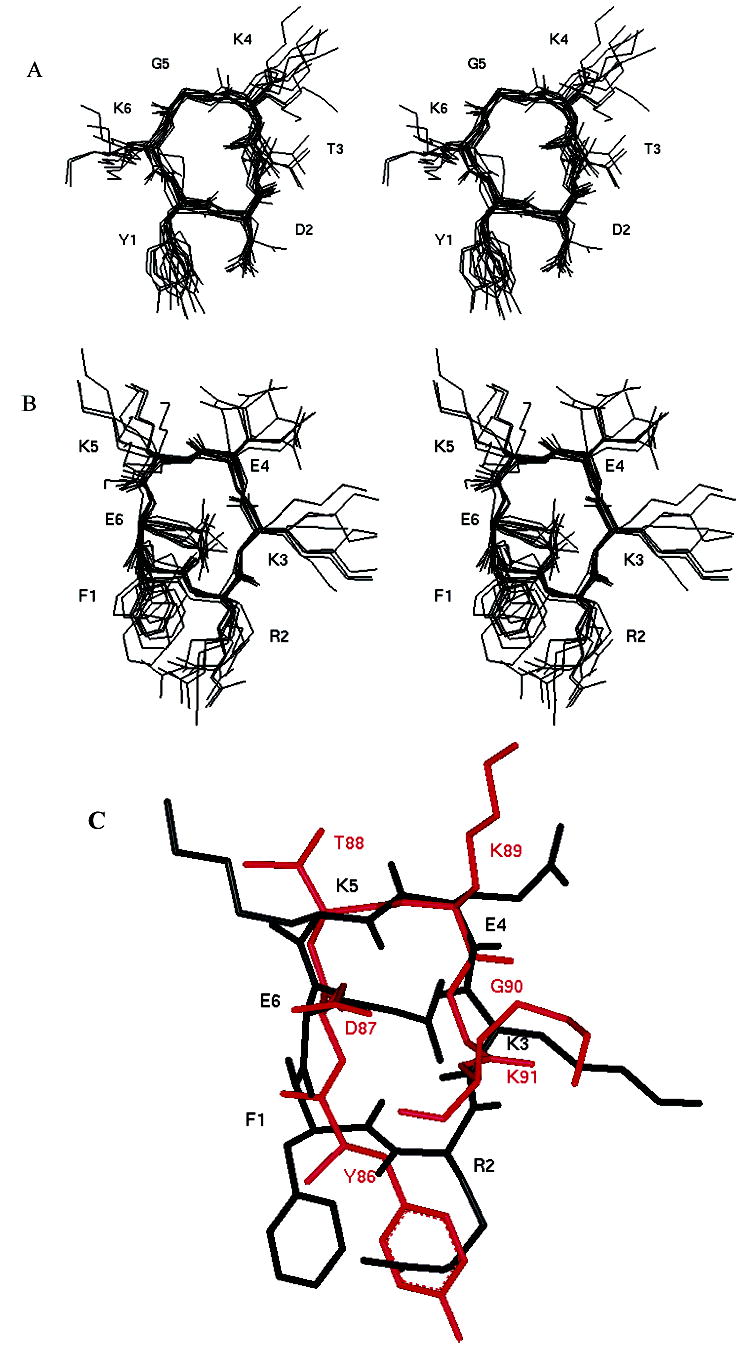
Stereoview of a family of superimposed structures from NMR-restrained MD simulations for cyclic peptides (A) cYK and (B) cFE. Only heavy atoms are shown for clarity. (C) Overlay of an average structure of peptide cFE with human CD2 (Y86–K91 fragment). Red denotes the CD2 fragment, and black denotes peptide cFE.
The NMR data for the cFE hexapeptide showed the dNN(i,i+1) cross-peaks and the stronger dαN(i,i+1) cross-peaks between Lys3 and Glu4, and dαN(i,i+2) cross-peaks between Lys3 and Lys5 suggest close contact at Lys3, Glu4, and Lys5 (Figure 7D) in the peptide. In addition, the temperature-dependent coefficients for the NH groups of Phe1, Lys3, and Glu6 were no greater than –3.0 ppb/K, which suggests that these amide protons may be intramolecularly hydrogen bonded or solvent shielded.35 Molecular modeling studies resulted in a family of conformers for the cFE peptide that best fit the NOE data. A family of 12 superimposed structures of cFE peptide is shown in Figure 9B. The average rms deviation of the backbone atoms in the family compared to the average structure was 0.75 Å. The overall structure of the cFE peptide consists of two β-turns spanning the Glu6-Phe1-Arg2-Lys3 and Lys3-Glu4-Lys5-Glu6 sequences (Figure 9B). All the structures in the family suggested a hydrogen bond between the amide proton of Lys3 and the carbonyl oxygen of Phe1, which is consistent with the low temperature coefficient of the chemical shift of the amide of Lys3. In addition, a hydrogen bond between the amide proton of Lys5 and the carbonyl oxygen of Lys3 was observed. Comparison of the cFE conformation with the Phe47-Arg48-Lys49-Glu50-Lys51-Glu52 fragment in the crystal structure of human CD2 suggested that the β-turn in the human CD2 crystal structure is at the Arg48-Lys49-Glu50-Lys51 sequence, whereas in the cFE peptide, β-turns span the Glu6-Phe1-Arg2-Lys3 and Lys3-Glu4-Lys5-Glu6 sequences. When we compared the hot-spot region of the crystal structure, Tyr86–Lys91, there was a good overlap in the backbone of the cFE peptide with the β-turn of the Asp87-Thr88-Lys89-Gly90 sequence of the crystal structure (Figure 9C). In addition, some of the hydrophobic and charged residue positions could be overlapped in both structures. For example, Tyr86 of the CD2 fragment overlapped with Phe1, Asp87 with Glu6, and Lys91 with Lys3 in the peptide.
Discussion
Design Strategy.
The CD58 (LFA-3) binding ability of CD2 is known to reside in domain 1 of the CD2 protein. CD2 peptide mapping and mutagenesis indicated that the binding surface of CD2 consists of a β-sheet formed by strands G, F, C, C′, and C′′.18 The crystal structure of CD2 (Figure 2B) indicated that the rather flat β-sheet surface does not provide a complementary shape for binding to CD58, and hence does not have well-defined epitopes for the design of small molecular inhibitors. The structure of CD2 is similar to those of CD4 and other IgSF molecules.19,34,36 In the D1 domain of CD4, the β-turn near the CC′ loop appears to be important for binding to its receptor.34 β-Turn peptides based on CD4 have been shown to be effective in inhibiting CD4 interactions.34 Analysis of the crystal structure of CD2 revealed that on either side of the binding surface of CD2, there are β-turns which stabilize the β-strands. Thus, these β-turns may serve as good surface epitopes for the design of peptides to inhibit CD2–CD58 interactions. Meanwhile, the crystal structure of the human CD2–CD58 complex became available.18 Examination of the CD2–CD58 crystal structure indicated that the interface of the CD2–CD58 complex has poor shape complementarity in the center region of interaction (Figure 2A). Most of the interaction is via salt bridges with charge neutralization and hydrogen bonds. Furthermore, the β-strand surface of CD2 that interacts with CD58 is stabilized by β-turns on either side. These β-turn regions seem to be important in holding the CD2–CD58 interface intact with β-sheet and salt bridges. The residues in the β-turn in the human CD2 sequence are Thr38, Ser39, Asp40, and Lys41; Arg48, Lys49, Glu50, and Lys51; and Asp87, Thr88, Lys89, and Gly90. Lys41 of the β-turn of the Thr38-Ser39-Asp40-Lys41 sequence is involved in the hydrogen bonding interaction with CD58. Arg48 and Lys51 of the β-turn of the Arg48-Lys49-Glu50-Lys51 fragment are involved in hydrogen bonding with CD58. Mutagenesis studies carried out by alanine scanning indicated the complete loss of adhesion to CD58 upon replacement of Arg48 and Lys51 in the CD2 protein.3 Similarly, in the β-turn at the Asp87-Thr88-Lys89-Gly90 sequence, the Gly90 backbone carbonyl carbon is involved in hydrogen bonding interaction with CD58. The flanking residue Tyr86 of the β-turn in the Asp87-Thr88-Lys89-Gly90 fragment has been shown to be important in CD2–CD58 interaction.3 Therefore, we hypothesized that stable β-turn conformations mimicking the native CD2 surface-binding region with CD58 may be important for the inhibitory activity of the peptide. In the β-turn of the Thr38-Ser39-Asp40-Lys41 sea-quence, Thr38, Ser39, and Asp40 did not seem to be involved in any interaction with CD58; hence, that peptide was not designed for the study of β-turn peptides.
The adhesive domains of both proteins, CD2 and CD58, have a V-set topology composed of nine β-strands distributed in two sheets.3 The contact area involves mainly the C, C′, and C′′ β-strands and the FG, CC′, and C′C′′ loops of these domains. Ten salt bridges and five hydrogen bonds were identified in the CD2–CD58 interaction. The charged residues at the adhesive interface form the following interprotein salt bridges (first residue from CD2, second residue from CD58): Asp32Lys34, Asp31Arg44, Lys34Glu78, Lys41Asp84, Lys43Glu25, Arg48Glu37, Arg48Glu39, Lys51Glu42, Lys51Glu39, and Glu95Lys32.19 The only hydrophobic contact, from the CD2 side, involves the packing of the aromatic ring of CD2 Tyr86 with the aliphatic portion of hCD58 Lys34, Phe46 Cδ2 of CD58 with Gly90 Cα of hCD2, and Pro80 Cβ of CD58 with Lys42 Cδ of hCD2. The point mutations of the CD2 and CD58 proteins on cell-to-cell adhesion showed that mutation of residues Lys41, Lys51, Asn92, and Glu95 (G and F strands or CC′ and C′C′′ loops) to alanine affects CD58 binding (with an approximately ≤10-fold reduction in binding affinity); mutation of residues Asp31, Asp32, Lys34, Lys43, and Arg48 (map to the central C and C′ strands) alters binding by more than 1 order of magnitude (manifesting a 47–127-fold decrease in binding affinity).3 In addition, the Tyr86 to Ala mutation (F strand) resulted in a significant loss of binding of CD2 to CD58 (more than 1000-fold), while the Tyr86 to Phe mutation in the same region did not affect the binding. Of the six point mutations, the strongest effect observed is among Asp32Ala, Arg48Ala, and Lys91Ala of CD2 and Asp33Ala, Lys34Ala, and Glu37Ala of CD58. The Asp32Lys34 salt bridge is directly involved in a region identified as the energetic hot spot of the CD2–CD58 interaction.3 Thus, the residues in the β-strand regions were important for adhesion of CD2 to CD58, and the peptides from these regions are potential targets for the design of peptides.
On the basis of the structures of the CD2 N-terminal adhesion domain and the bimolecular complex between the N-terminal adhesion domains of CD2 and CD58,18–24 rational design of the peptides has been carried out according to the following considerations: (1) The conformational feature of the β-turn region and the residues important in binding to CD58 should be conserved. (2) The conformational features of the segment encompassing C, C′, and C′′ β-strands and FG and C′C′′ loops of these domains, which are critical for CD58 receptor binding, should be conserved. (3) The backbone flexibility of the peptides should be minimized to stabilize the bioactive conformation. To identify the “minimal“ structural parts of CD2-derived peptides responsible for CD58 binding, 12-residue peptides DS, SE, IK, lIL, and lAQ were designed (Figures 1 and 2 and Table 1). The peptides generated from the β-turn regions were cyclized with a disulfide bond. Penicillamine is used in position 1 to induce conformational rigidity of the cyclic peptide.27 We chose to cyclize the peptides by disulfide bonds with the introduction of amino acids penicillamine and cysteine at the two ends of the peptide sequence. Cyclization using a disulfide bond is relatively easy, and a good yield is obtained after purification. Penicillamine with two bulky methyl groups is known to stabilize disulfide bonds.27 Pen is used in position 1 because in previous work we have been successful in improving the conformational stability of the cyclic peptides by using Pen at position 1.37 Initially, 12-mer peptides were designed (Table 1). After a preliminary examination of the biological activity of 12-mer linear and cyclic peptides, the peptides were truncated to six amino acid residues in an effort to elucidate the minimum number of critical amino acids necessary for biological activity. These hexapeptides were cyclized by amide bonds to stabilize the structure. In the hexapeptides, cyclization by a disulfide bond was not examined since addition of penicillamine and cysteine to form a disulfide bond will increase the number of amino acids in the peptide. The information obtained from truncating the peptides will be useful in the design of future pharmacophores. A control peptide (Table 1) was designed to compare the importance of primary and secondary structures in the designed peptides. The control peptide sequence was chosen from the hot-spot3 region of the hCD2–hCD58 interface on the CD2 protein. The sequence was then reversed, and the important amino acid Tyr86 as well as Tyr81 was replaced with Ala to generate the control peptide sequence shown in Table 1.
Structure and Biological Activity.
The biological activity of the peptides was evaluated by two cell adhesion inhibitory assays. Statistical analysis of the results showed that in the heterotypic adhesion assay, almost all the peptides at all three concentrations that were tested displayed a significant difference with respect to the control peptide. These data suggest that the designed peptides were effective in inhibiting cell adhesion. On the basis of these results, cyclic 12-residue peptide cAQ from the C′C′′ loop and cIL from the FG loop, encompassing the hot-spot Tyr86, were identified as more potent inhibitory peptides than their linear counterparts, peptides lAQ and lIL, in the series of peptides derived from loop regions. The CD study showed that both linear peptides displayed random structures in solution, while the cyclic peptides demonstrated β-turn conformations. NMR and molecular modeling study of the cAQ and cIL peptides indicated that they had β-turn conformations consistent with our design. Comparison of the cAQ and cIL peptides with the crystal structure of hCD2 and rat CD2 protein indicated that these β-turn peptides mimic the conformation of the protein fragment, which is consistent with their biological activity.
However, truncation of the larger cyclic peptides into six-residue cyclic peptides (peptides cFE and cYK) in the same series decreased the CD58 receptor antagonist potency. This suggests that directly linking the first flanking residues of the β-turn structure could not stabilize the conformation or mimic the CD2 conformation as expected. Detailed structural studies of cYK by NMR showed that the hexapeptide does not form a β-turn structure in solution; instead, it forms a rectangle-shaped structure. Comparison of the cYK peptide structure with the crystal structure of hCD2 protein did not show good overlap between the two fragments. Also, the side chain of Tyr1 deviates significantly in the cYK peptide from that of Tyr86 in human CD2. Thus, it is possible that the change in the conformation of peptide cYK (compared to the protein crystal structure) results in the loss of activity of inhibition of the peptide as observed in E-rosetting and heterotypic adhesion assays.
For the other hexapeptide cFE, the NMR-derived structure (Figure 9B) exhibits two possible β-turns. The superimposition of peptide cFE on the respective fragment in human CD2, Phe47-Arg48-Lys49-Glu50-Lys51-Glu52, showed that the side chains of Arg2 and Glu4 do not overlap very well with the crystal structure. These two residues correspond to two important residues, Arg48 and Lys51 in CD2, which are in the group of salt bridges providing the specificity for the CD58 binding. Interestingly, when we overlayed the cFE structure with the Tyr86-Asp87-Thr88-Lys89-Gly90-Lys91 fragment of the hCD2 crystal structure, overlapping of the backbone atoms was observed. Tyr86, Asp87, and Lys91 of the CD2 fragment overlapped with Phe1, Glu6, and Lys3 of the cFE peptide (Figure 9C). The proper positioning of these residues may result in the 25% inhibition activity of the cFE peptide compared to hexapeptide cYK which showed <20% inhibition activity (Figure 4). Mutagenesis study has indicated that the Tyr86 Phe mutation did not have any significant functional consequence, in contrast to the “fatal” Tyr86 Ala mutation.3 Actually, the π-cation interaction between the positively charged ε-amino group of CD58 Lys34 and the two aromatic rings of CD58 Phe46 and CD2 Tyr86 plays the key role in CD2–CD58 binding. The distribution of the electrostatic potential in peptide cFE (Phe1-Arg2-Lys3-Glu4-Lys5-Glu6) contrasts greatly with the Tyr86-Asp-Thr-Lys-Gly-Lys segment in human CD2 (Arg2 vs Asp87, and two additional positively charged residues, Lys3 and Lys5). These may affect the specificity of binding of peptide cFE to the interface of the CD58 protein and the binding energy as well. Therefore, to improve the binding energy and specificity of peptide cFE, oppositely charged residues should be strategically replaced.
The linear peptides derived from the β-strand region of CD2 demonstrated low inhibitory activity. The CD study showed that all the linear peptides displayed random structures in solution. Thus, for peptides derived from the β-strand region, conformational constraint might be necessary for activity. Compared with the two peptides SE and IK which are generated from C′ and F strands, peptide DS displayed a relatively higher inhibitory activity. Moreover, there seems to be a correlation between the number of important residues and the inhibitory activity (DS > SE > IK).
To conclude, we have designed linear and cyclic peptides from the β-turn regions and hot-spot region of hCD2 (hCD2–hCD58 interaction). The designed β-turn peptides exhibit T-cell adhesion in the E-rosetting assay, and in the heterotypic cell adhesion assay. Cyclic peptides exhibited better inhibitory activity and seem to mimic the protein epitopes. Thus, the introduction of conformational constraints on the peptide backbone through disulfide bridges in the two termini of the peptides from C′C′′ and FG loop regions imparts large changes in the potency of inhibition of CD2–CD58 interaction. Furthermore, reduction in the ring size in cyclic peptides by hexapeptides can change the β-turn conformation which may affect biological activity. The NMR studies suggest a deviation of the conformations of peptides cFE and cYK with the topologies in the CD2 protein, not only in the side chain orientation of aromatic residues Phe and Tyr but also in mimicking the β-turn conformation. In this regard, further study should be aimed at stabilizing the β-turn conformation by introducing β-turn mimetics, D-isomers, or novel χ-constrained amino acids38 to modify the orientation of the aromatic side chains.
Materials and Methods
Abbreviations.
AET, 2-aminoethylisothiouronium hydro-bromide; BCECF-AM, bis-carboxyethyl-carboxyfluorescein ac-etoxymethyl; CD, cluster of differentiation; DMSO, dimethyl sulfoxide; FBS, fetal bovine serum; FITC, fluorescein isothio-cyanate; hCD2, human CD2; hCD58, human CD58; MHC, major histocompatibility complex; MEM-α, minimum essential medium-α; MTT assay, mitochondrial dehydrogenase activity assay; PAL resin, 5-(4-aminomethyl-3,5-dimethoxyphenoxy)-valeryl resin; TCR, T-cell receptor; TFA, trifluoroacetic acid.
Peptides.
The linear and cyclic peptides (Table 1) were designed and synthesized, and peptides cAQ, cFE, and cYK were purchased from Multiple Peptide Systems (San Diego, CA). The pure products were analyzed by HPLC and fast atom bombardment mass spectroscopy (FAB-MS). The HPLC chromatogram showed that the purities of peptides were >90%, and FABMS showed the correct molecular ion for the peptides. Linear and cyclic peptides lAQ, lIL, DS, SE, and IK and the control peptide were synthesized using an automatic solid-phase peptide synthesizer (Pioneer, Perspective Biosystem) using Fmoc chemistry. The Fmoc-protected amino acids were obtained from Novabiochem. All the solvents used in the Pioneer Protein Synthesizer were obtained from Applied Biosystems (Foster City, CA).
Synthesis of Linear Peptides.
PAL resin [5-(4-N-Fmoc-aminomethyl-3,5-dimethoxyphenoxy)valeryl] was used as a solid support for the linear peptides. First, the Fmoc protecting group on the resin was removed by treatment with a 20% piperidine/DMF mixture. The Nα-Fmoc-amino acids were preactivated by mixing them with the coupling reagent HATU/ DIPEA (1:1); the activated amino acid was then added to the resin and mixed at room temperature. Cycles of deprotection of Fmoc and coupling with the subsequent amino acids were repeated until the desired peptide-bound resin was completed. The resin was washed manually with DMF, DCM, and methanol successively to remove the excess solvents and dried in vacuo over KOH overnight before cleavage and deprotection. The dried peptidyl resin was mixed with the cleavage cocktail (90% TFA, 5% thioanisole, and 5% ethanedithiol, 5 mL/g of resin) and precipitated with cold ether.
Synthesis of Acm-Protected Linear Peptide lAQ and Disulfide-Bridged Cyclic Peptide cIL.
Disulfide-bridged cyclic peptide cIL and Acm-protected linear peptide lAQ were synthesized on Fmoc-Cys(Acm)-2-chlorotrityl chloride resin utilizing a standard Fmoc solid-phase synthesis protocol mentioned above.39 Linear peptide lAQ was cleaved and deblocked from the resin with a 95:5:5 trifluoroacetic acid (TFA)/anisole/water mixture. The Acm protection group is not removed by TFA, and thus was kept during cleavage of the resin to prevent the formation of interchain or intrachain disulfide bridges in solution. Although the 2-chlorotrityl chloride resin has been reported to be partially cleaved by treatment with a 1:1 TFA/DCM mixture to give fully protected peptides,40 cyclic peptide cIL was completely cleaved and deblocked (except S-Acm) after being treated with a 1:1 TFA/ DCM mixture with a higher yield and purity than with treatment of neat TFA (data not shown). Cyclized peptide cIL was simultaneously created by the simple and safe procedure of iodine oxidation of the crude peptide without purification. To avoid the overoxidation of the thiol functionality to the corresponding sulfonic acid, as well as to prevent or minimize modification of other sensitive amino acid side chains (Tyr), the reaction was monitored by MS, and it was found that 30 min was enough for complete oxidation.
Peptides were purified by preparative HPLC (Waters 600 HPLC system), on a reversed-phase C18 column (Inertsil, 10 mm × 250 mm, 5 μm, 300 Å) with a linear gradient of solvent A (0.1% TFA/H2O) and solvent B (0.1% TFA/acetonitrile). The peptides were detected by UV at 215 and 280 nm (λ). The purity of each peptide was verified with an analytical HPLC column (Shimadzu LC-10AT VP) using a reversed-phase C18 column (Lichrosorb RP18, 4.6 mm × 200 mm, 10 μm) with the same solvent system as in the preparative HPLC system. The molecular weight of the peptides was confirmed by using electrospray ionization mass spectrometry (ESI-MS, Finnigon MAT). The HPLC chromatogram showed that the purity of the peptides was >95%, while ESI-MS showed the correct molecular ion for the peptide.
Cell Lines.
The Jurkat T-leukemia and human colon adenocarcinoma cell Caco-2 lines were obtained from American Type Culture Collection (Rockville, MD). Jurkat cells were maintained in suspension in RPMI-1640 medium supplemented with 10% heat-inactivated fetal bovine serum (FBS) and 100 mg/L penicillin/streptomycin. Caco-2 cells were maintained in minimal essential medium-α containing 10% FBS, 1% nonessential amino acids, 1 mM sodium pyruvate, 1% l-glutamine, and 100 mg/L penicillin/streptomycin. Caco-2 cells were used between passages 50 and 60. Sheep blood in Alsever’s solution was purchased from TCS Biosciences Ltd. (Singapore).
E-Rosetting.
Sheep red blood cells (SRBCs) were isolated by centrifuging sheep blood in Alsever’s solution at 200g for 5 min. SRBCs were washed three times with PBS and incubated with 4 volumes of an AET solution at 37 °C for 15 min. The cells were washed three times in PBS and resuspended in RPMI-1640 medium containing 20% FBS to give a 10% suspension. For use, the cell suspension was diluted 1:20 (0.5%) with medium. Serial dilutions of peptides in PBS were added to 0.2 mL of 0.5% AET-treated SRBCs, and incubated at 37 °C for 30 min. After that, 0.2 mL of a Jurkat cell suspension (2 × 106 cells/mL) was added to the mixture, and incubated for an additional 15 min. The cells were pelleted by centrifugation (200g for 5 min at 4 °C) and then incubated at 4 °C for 1 h. The cell pellet was gently resuspended, and the E-rosettes were counted with a hemocytometer.16 Cells with five or more SRBCs bound were counted as rosettes. At least 200 cells were counted to determine the percentage of E-rosette cells. The inhibitory activity was calculated.
Lymphocyte–Epithelial Adhesion Assay.
Caco-2 cells were used between passages 50 and 60 and were plated onto a 48-well plate at a density of approximately 2 × 104 cells/ well. When the cells reached confluency, the monolayers were washed once with MEM-α. Jurkat cells were labeled the same day as the adhesion assay by loading with 2 μM fluorescent dye BCECF at 37 °C for 1 h. Peptide dissolved in MEM-α was added at various concentrations to Caco-2 cell monolayers. After incubation at 37 °C for 30 min, the labeled Jurkat cells (1 × 106 cells/well) were added onto the monolayers. After incubation at 37 °C for 45 min, nonadherent Jurkat cells were removed by washing three times with PBS, and the monolayer-associated Jurkat cells were lysed with 2% Triton X-100 in 0.2 N NaOH. Soluble lysates are transferred to 96-well plates for reading with a microplate fluorescence analyzer.28 Data are presented as the relative fluorescence or percent inhibition. Relative fluorescence (FL) was found by reading the values of fluorescence intensity corrected for the reading of background (cell monolayers only).
Statistical analyses of the results from the cell adhesion assay were carried out using the Mann Whitney U test (n) 4) using SPSS software. Peptides were grouped into three categories for the analysis based on their sequence origin: group 1, lAQ, cAQ, and cFE; group 2, lIL, cIL, and cYK; and group 3, SE, IK, and DS. Within each group, the peptides are linear, cyclic, and truncated form or from β-strand.
Cell Viability Assay.
Peptides which exhibited effects on Jurkat–Caco-2 cell adherence were tested by the MTT assay29 to determine if their effects were due to frank toxicity. A final peptide concentration of 180 μM was added to Jurkat or Caco-2 cells for 1 or 2 h, which is the maximum time of exposure of Caco-2–Jurkat cells during the adherence assay. The cell viabilities were validated by incubating with 5 mg/mL MTT at 37 °C for 3 h. The MTT-labeled cells were lysed by DMSO, and the absorbance was measured with a microplate reader at a wavelength of 570 nm.
Circular Dichroism Measurement.
Circular dichroism experiments were carried out at room temperature on a Jasco J-715 spectropolarimeter flushed with nitrogen. Spectra were collected from 240 to 190 nm using a 1 mm path length cylindrical quartz cell. Each spectrum was the average of three scans taken at a scan rate of 50 nm/min with a spectral bandwidth of 1 nm. The concentration of peptides was varied from 0.15 to 2.4 mM. For the final representation, the baseline was subtracted from the spectrum.
NMR Spectroscopy.
The samples for the NMR spectra of the peptide were prepared by dissolving 1–3 mg of the peptides in 0.6 mL of a 90% H2O/10% D2O mixture at the pH with the best dispersion. For pH titration experiments, the pH of the solution was varied by the addition of DCl or NaOD (pH was not corrected for isotopic effects). The temperature dependence of the amide proton chemical shift was measured by collecting data from 283 to 303 K in steps of 5 K using a variable-temperature probe. The one- and two-dimensional NMR experiments were performed and processed on 300, 500, and 600 MHz Bruker DRX spectrometers equipped with a 5 mm probe, at proton frequencies of 300.3414, 500.134, and 600.1299 MHz, respectively, using XWINNMR, version 1.0. Spectra were acquired at 298 and 293 K unless otherwise specified. TOC-SY,41 DQF-COSY,42 rotating frame nuclear Overhauser spectroscopy (ROESY),43 and NOESY44 experiments were performed by presaturation of water during the relaxation delay in the 500 MHz NMR instrument. In the 600 MHz instrument, a Watergate sequence with gradient suppression was used for water suppression.45 Data were collected by the TPPI method46 with a sweep width of 5000 Hz. Analysis of the data and assignments of the resonance were carried out using Sparky.47 ROE cross-peak volumes were measured using ROESY spectra with 200–300 ms spin–lock times, and NOESY cross-peak volumes for hexapeptides were measured with a mixing time of 200 ms. Coupling constants (3JHNα) were measured from the one- or two-dimensional DQF-COSY spectrum with a curve fitting program (MRQfit, N. Jacobson, University of Arizona). Intensities were assigned as strong, medium, and weak with upper and lower boundaries of distances for dNα(i,i), dαN(i,i+1), and dNN(i,i+1) of 1.9–3.0, 2.2–3.6, and 3.0–5.0 Å, respectively.35 Side chain protons were not stereospecifically assigned; hence, ROE and NOE restraints for the side chain protons were calculated by considering pseudoatoms.35
Determination of Peptide Structures.
Conformational space was searched for the peptides using CYANA48,49 and discover program version 2000 (Accelrys Inc., San Diego, CA) to identify conformations consistent with the experimental ROE and NOE and coupling constant data.50 Briefly, structures were generated using distance geometry-based algorithms with NMR constraints using CYANA. Disulfide bond constraints were also included in the calculations. A total of 100 structures were generated, and these were subjected to the simulated annealing procedure. Twenty structures that have a low target function48,49 were selected for further calculations. The target function takes into account ROE and NOE violations, peptide bond lengths, and bond angle restraints. Each of these structures was then soaked with water molecules, followed by MD simulations at 300 K with all the ROE and NOE constraints using the discover program (Accelrys Inc.). These structures were further energy-minimized with solvent molecules using the steepest descent method and conjugate gradient method until the rms derivative was 0.3 kcal mol−1 Å−1. The structures were checked for quality using the Ramachandran map and InsightII (bond length, bond angle, and peptide bond torsion angle). The final structures which satisfy all of the NMR distance constraints were clustered together on the basis of the rms deviation of the backbone atoms, and the structures which had similar NOE and ROE violations were clustered together as one family. Each family or cluster had 10–12 structures. An average structure was chosen from this family as a representative structure.
Acknowledgments
We gratefully acknowledge the Super Computer and Visualization Unit, National University of Singapore, for the use of computational facilities and the NMR facilities at the National University of Singapore and University of Arizona. We also thank Dr. Neil Jacobson (University of Arizona) for his suggestions. This research was funded by an academic research grant (R-148-000-026-112) from the National University of Singapore. NMR studies for cyclic peptides cAQ and cIL were carried out in the Department of Chemistry, University of Arizona, during S.D.S.’s summer study leave at the University of Arizona. The summer study leave and the research during that period were supported by U.S. Public Health Service Grants DK 17420 and 334781 (V.J.H.).
Footnotes
Supporting Information Available: Analytical data for peptides studied in this work (Table 1), NMR chemical shift tables for cAQ, cIL, cYK, and cFE peptides (Tables 2–5, respectively), and 600 MHz TOCSY and NOESY/ROESY (Figures 1–6) data for peptides cAQ and cIL and different families of structures for peptide cYK (Figure 7). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Bierer BE, Burakoff SJ. T cells adhesion molecules. FASEB J. 1988;2:2584–2590. doi: 10.1096/fasebj.2.10.2838364. [DOI] [PubMed] [Google Scholar]
- 2.Wang JH, Reinherz E. Structure basis of cell–cell interactions in the immune system. Curr Opin Struct Biol. 2000;10:656–661. doi: 10.1016/s0959-440x(00)00150-0. [DOI] [PubMed] [Google Scholar]
- 3.Kim M, Sun ZYJ, Byron O, Campbell G, Wagner G, Wang JH, Reinherz EL. Molecular dissection of the CD2-CD58 counter-receptor interface identifies CD2 Tyr86 and CD58 Lys34 residues as the functional “hot spot”. J Mol Biol. 2001;312:711–720. doi: 10.1006/jmbi.2001.4980. [DOI] [PubMed] [Google Scholar]
- 4.Davis SJ, van der Merwe PA. The structure and ligand interactions of CD2: Implications for T cell function. Immunol Today. 1996;17:177–187. doi: 10.1016/0167-5699(96)80617-7. [DOI] [PubMed] [Google Scholar]
- 5.Bachmann MF, Barner M, Kopf M. CD2 sets quantitative thresholds in T cell activation. J Exp Med. 1999;190:1383–1391. doi: 10.1084/jem.190.10.1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mojcik CF, Shevach E. Adhesion molecules: A rheumatologic perspective. Arthritis Rheum. 1997;6:991–1004. doi: 10.1002/art.1780400602. [DOI] [PubMed] [Google Scholar]
- 7.Kaplon RJ, Hochman PS, Michler RE, Kwiatkowski PA, Edwards NM, Berger CL, Xu H, Meier W, Wallner BP, Chisholm P, Marboe CC. Short course single agent therapy with an LFA-3-IgG1 fusion protein prolongs primate cardias allograft survival. Transplantation. 1995;61:356–363. doi: 10.1097/00007890-199602150-00004. [DOI] [PubMed] [Google Scholar]
- 8.Sultan P, Schechner JS, McNiff JM, Hochman PS, Hughes CCW, Lorber MI, Askenase PW, Pober JS. Blockade of CD2-LFA-3 interactions protects human skin al-lografts in immunodeficient mouse/human chimeras. Nat Bio-technol. 1997;15:759–762. doi: 10.1038/nbt0897-759. [DOI] [PubMed] [Google Scholar]
- 9.Qin LH, Chavin KD, Lin JX, Yagita H, Bromberg JS. Anti-CD2 receptor and anti-CD2 ligand (CD48) antibodies synergize to prolong allograft survival. J Exp Med. 1994;179:341–346. doi: 10.1084/jem.179.1.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hirahara H, Tsuchida M, Wanatabe T, Haga M, Matsumoto Y, Abo T, Eguchi S. Long-term survival of cardiac allografts in rats treated before and after surgery with monoclonal antibody to CD2. Transplantation. 1995;59:85–90. doi: 10.1097/00007890-199501150-00015. [DOI] [PubMed] [Google Scholar]
- 11.Sido B, Otto G, Zimmermann R, Müller P, Meuer S, Dengler TJ. Prolonged allograft survival by the inhibition of costimulatory CD2 signals but not by modulation CD48 (CD2 ligand) in the rat. Transplant Int. 1996;9:S323–S327. doi: 10.1007/978-3-662-00818-8_80. [DOI] [PubMed] [Google Scholar]
- 12.Sido B, Otto G, Zimmermann R, Müller P, Meuer S, Dengler TJ. Modulation of the CD2 receptor and not disruption of the CD2/CD48 interaction is the principal action of CD2-mediated immunosuppression in the rat. Cell Immunol. 1997;182:57–67. doi: 10.1006/cimm.1997.1204. [DOI] [PubMed] [Google Scholar]
- 13.Aruffo A, Hollenbaugh D. Therapeutic intervention with inhibitors of co-stimulatory pathways in autoimmune disease. Curr Opin Immunol. 2001;13:683–686. doi: 10.1016/s0952-7915(01)00279-5. [DOI] [PubMed] [Google Scholar]
- 14.Przepiorka D, Phillips GL, Ratanatharathorn V, Michele CF, Sehn LH, Antin JH, Lebherz D, Awwad M, Hope J, McClain JB. A phase II study of BTI-322, a monoclonal anti-CD2 antibody, for treatment of steroid-resistant acute graft-versus-host disease. Blood. 1998;92:4066–4071. [PubMed] [Google Scholar]
- 15.Branco L, Barren P, Mao SY, Pfarr D, Kaplon R, Psotema C, Langermann S, Koenig S, Johnson S. Selective deletion of antigen-specific, activated T cells by a humanized mAb to CD2 (MEDI-507) is mediated by NK cells. Transplantation. 1999;68:1588–1596. doi: 10.1097/00007890-199911270-00026. [DOI] [PubMed] [Google Scholar]
- 16.Albert-Wolf M, Meuer SC, Wallich R. Dual function of recombinant human CD58: Inhibition of T-cell adhesion and activation via the CD2 pathway. Int Immunol. 1991;3:1335–1347. doi: 10.1093/intimm/3.12.1335. [DOI] [PubMed] [Google Scholar]
- 17.Jining L, Makagiansar I, Makagiansar HY, Chow VTK, Satyanarayanajois DS. Design, structure and biological activity of β-turn peptides of CD2 protein for inhibition of T-cell adhesion. Eur J Biochem. 2004;271:2873–2886. doi: 10.1111/j.1432-1033.2004.04198.x. [DOI] [PubMed] [Google Scholar]
- 18.Wang JH, Smolyar A, Tan K, Liu JH, Kim M, Sun ZYJ, Wagner G, Reinherz EL. Structure of a heterophilic adhesion complex between human CD2 and CD58 (LFA-3) counter-receptors. Cell. 1999;97:791–803. doi: 10.1016/s0092-8674(00)80790-4. [DOI] [PubMed] [Google Scholar]
- 19.Ikemizu S, Sparks LM, van der Merwe A, Stuart DI, Jones EY, Davis SJ. Crystal structure of the CD2-binding domain of CD58 (lymphocyte function-associated antigen 3) at 1.8 Å resolution. Proc Natl Acad Sci USA. 1999;96:4289–4294. doi: 10.1073/pnas.96.8.4289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bodian DL, Jones EY, Harlos K, Stuart DI, Davis SJ. Crystal structure of the extracellular region of the human cell adhesion molecule CD2 at 2.5 Å resolution. Structure. 1994;2:755–766. doi: 10.1016/s0969-2126(94)00076-x. [DOI] [PubMed] [Google Scholar]
- 21.Withka JM, Wyss DF, Wagner G, Arulanandam ARN, Reinherz EL, Recny MA. Structure of the glycosylated adhesion domain of human T lymphocye glycoprotein CD2. Structure. 1993;1:69–81. doi: 10.1016/0969-2126(93)90009-6. [DOI] [PubMed] [Google Scholar]
- 22.Jones EY, Davis SJ, Williams AF, Harlos K, Stuart DI. Crystal structure at 2.8 Å resolution of soluble form of the cell adhesion molecule CD2. Nature. 1992;360:232–239. doi: 10.1038/360232a0. [DOI] [PubMed] [Google Scholar]
- 23.Sun ZYJ, Dõtsch V, Kim M, Li J, Reinherz EL, Wagner G. Functional glycan-free adhesion domain of human cell surface receptor CD58: Design, production and NMR studies. EMBO J. 1999;18:2941–2949. doi: 10.1093/emboj/18.11.2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Davis SJ, van der Merwe PA. The structure and ligand interactions of CD2: Implications for T-cell function. Immunol Today. 1996;17:177–187. doi: 10.1016/0167-5699(96)80617-7. [DOI] [PubMed] [Google Scholar]
- 25.Bachmann MF, Barner M, Kopf M. CD2 sets quantitative thresholds in T cell activation. J Exp Med. 1999;190:1383–1391. doi: 10.1084/jem.190.10.1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Van der Merwe AP, Davis SD. Molecular interactions mediating T cell antigen recognition. Annu Rev Immunol. 2003;21:659–684. doi: 10.1146/annurev.immunol.21.120601.141036. [DOI] [PubMed] [Google Scholar]
- 27.Hruby VJ. Conformational and topographical considerations in the design of biologically active peptides. Biopolymers. 1993;33:1073–1082. doi: 10.1002/bip.360330709. [DOI] [PubMed] [Google Scholar]
- 28.Jining L, Chow VTK, Satyanarayanajois DS. A novel, rapid and sensitive heterotypic cell adhesion assay for CD2-CD58 interaction, and its application for testing inhibitory peptides. J Immunol Methods. 2004;291:39–49. doi: 10.1016/j.jim.2004.04.026. [DOI] [PubMed] [Google Scholar]
- 29.Mosmann T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;5:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 30.Perczel A, Fasman GD. Analysis of the circular dichroism spectrum of proteins using the convex constraint algorithm: A practical guide. Anal Biochem. 1992;203:83–93. doi: 10.1016/0003-2697(92)90046-a. [DOI] [PubMed] [Google Scholar]
- 31.Perczel A, Fasman GD. Quantitative analysis of cyclic β-turn models. Protein Sci. 1992;1:378–395. doi: 10.1002/pro.5560010310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Anderson NH, Neidigh WJ, Harris NS, Lee GM, Liu Z, Tong H. Extracting Information from the Temperature Gradients of Polypeptide NH Chemical Shifts. 1. The Importance of Conformational Averaging. J Am Chem Soc. 1997;119:8547–8561. [Google Scholar]
- 33.Rose GD, Gierasch LM, Smith JA. Turns in peptides and proteins. Adv Protein Chem. 1985;37:1–97. doi: 10.1016/s0065-3233(08)60063-7. [DOI] [PubMed] [Google Scholar]
- 34.Huang Z, Li S, Korngold R. Immunoglobulin superfamily proteins: Structure, mechanisms and drug discovery. Biopolymers. 1997;43:367–382. doi: 10.1002/(SICI)1097-0282(1997)43:5<367::AID-BIP3>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 35.Wûthrich, K. NMR of Proteins and Nucleic Acids; John Wiley & Sons: New York, 1986.
- 36.Wang JH, Meijers R, Xiong Y, Liu JH, Sakihama T, Zhang R, Joachimiak A, Reinherz EL. Crystal structure of the human CD4 N-terminal two-domain fragment complexed to a class II MHC molecule. Proc Natl Acad Sci USA. 2001;98:10799–10804. doi: 10.1073/pnas.191124098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Satyanarayanajois DS, Siahaan TJ, Tibbetts SA, Chan MA. A Ca2+ binding cyclic peptide derived from the α-subunit of LFA-1: Inhibitor of ICAM-1/LFA-1 mediated T-cell adhesion. J Pept Res. 1999;53:18–29. doi: 10.1111/j.1399-3011.1999.tb01613.x. [DOI] [PubMed] [Google Scholar]
- 38.Hruby VJ, Li G, Luevarno HC, Shendlrovich MD. Design of peptides, proteins, and peptidomimetics in χ space. Biopolymers. 1997;43:219–266. doi: 10.1002/(SICI)1097-0282(1997)43:3<219::AID-BIP3>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 39.Peptide synthesis. Advanced Chemtech Handbook of Combinatorial and Solid-Phase Organic Chemistry: A Guide to Principles, Products and Protocols; 1998; pp 342.
- 40.Lloyd-Williams, P.; Albericio, F.; Giralt, E. Chemical Approaches to the Synthesis of Peptides and Proteins; CRC: Boca Raton, FL, 1997.
- 41.Bax A, Davis DG. MLEV-17-based two-dimensional homo-nuclear magnetization transfer spectroscopy. J Magn Reson. 1985;65:355–360. [Google Scholar]
- 42.Rance M, Sorensen OW, Bodenhausen G, Wagner G, Ernst RR, Wüthrich K. Improved spectral resolution in COSY 1H NMR spectra of protein via double quantum filtering. Biochem Biophys Res Commun. 1983;117:479–485. doi: 10.1016/0006-291x(83)91225-1. [DOI] [PubMed] [Google Scholar]
- 43.Bax A, Davis DG. Practical aspects of two-dimensional transverse NOE spectroscopy. J Magn Reson. 1985;63:207–213. [Google Scholar]
- 44.Kumar A, Wagner G, Ernst RR, Wuthrich K. Buildup rates of the nuclear Overhauser effect measured by two-dimensional proton magnetic resonance spectroscopy: Implications for studies of protein conformation. J Am Chem Soc. 1981;103:3654–3658. [Google Scholar]
- 45.Sklenar V. Suppression of Radiation Damping in Multidimensional NMR Experiments Using Magnetic Field Gradients. J Magn Reson, Ser A. 1995;114:132–135. [Google Scholar]
- 46.Marion D, Wüthrich K. Application of phase sensitive two-dimensional correlated spectroscopy (COSY) for measurements of 1H–1H spin–spin coupling constants in proteins. Biochem Biophys Res Commun. 1983;113:967–974. doi: 10.1016/0006-291x(83)91093-8. [DOI] [PubMed] [Google Scholar]
- 47.Goddard, T. D.; Kneller, D. G. SPARKY3; University of Califor-nia: San Francisco, 2003.
- 48.Guntert P, Mumenthaler C, Wuthrich K. Torsion angle dynamics for NMR structure calculation with the new program DYANA. J Mol Biol. 1997;273:283–298. doi: 10.1006/jmbi.1997.1284. [DOI] [PubMed] [Google Scholar]
- 49.Herrmann T, Guntert P, Wuthrich K. Protein NMR Structure Determination with Automated NOE Assignment Using the New Software CANDID and the Torsion Angle Dynamics Algorithm DYANA. J Mol Biol. 2003;319:209–227. doi: 10.1016/s0022-2836(02)00241-3. [DOI] [PubMed] [Google Scholar]
- 50.Sutcliffe, M. J. Structure determination from NMR data II. Computational Approaches. In NMR of Macromolecules: A Practical Approach; Roberts, G. C. K., Ed.; Oxford University Press: New York, 1993; pp 359–390.