

Abstract
Proteasome inhibitors have emerged as promising anticancer therapeutic agents. Bortezomib (PS-341), a specific proteasome inhibitor, exhibits antitumor activity against a wide range of malignancies and has been approved by the U.S. Food and Drug Administration for the treatment of relapsed or refractory multiple myeloma. However, the molecular mechanisms of bortezomib-mediated apoptosis remain unclear. To characterize the mechanisms of apoptosis induction by proteasome inhibitors, we examined levels of Bcl-2 protein family members (Bik/NBK, Bax, Bak, Bcl-2, and Bcl-XL), release of cytochrome C, and activation of caspase-9 and caspase-3 in human colon cancer cell lines DLD-1, LOVO, SW620, and HCT116; human lung cancer cell line H1299; and human ovarian cancer cell line SKOV3 after they were treated with bortezomib. The result showed that bortezomib induced rapid accumulation of Bik/NBK but not other Bcl-2 family members in all six cell lines. Bortezomib-mediated Bik/NBK accumulation and apoptosis were also observed in human embryonic kidney cells 293 and normal human bronchial epithelial cells. Moreover, dramatic Bik/NBK accumulation and apoptosis induction were observed when cells were treated with proteasome inhibitor MG132 and calpain inhibitor I (ALLN). Furthermore, no detectable changes in IκBα levels or in NFκB functionality were found after treatment with bortezomib. Finally, Bik/NBK accumulation was caused by stabilization of the protein from degradation and was associated with bortezomib cytotoxicity and apoptosis induction. Pretreatment of DLD-1 cells with Bik/NBK siRNA reduced bortezomib-mediated Bik/NBK accumulation and cell death. Our results suggested that Bik/NBK is one of mediators of proteasome inhibitor-induced apoptosis.
Keywords: Proteasome Inhibitor, Apoptosis, Bik, PS-341, Bortezomib
The abbrevations used are: ALLN, Calpain inhibitor I; DMSO, dimethyl sulfoxide; PBS, phosphate-buffered saline; PMSF, Phenylmethylsulphonyl fluoride; XTT, 3-bis-(2-methoxy-4-nitro-5-sulphenyl)-(2H)-tetrazolium-5-carboxanilide
INTRODUCTION
The ubiquitin-proteasome pathway is critical for maintaining the homeostasis of most intracellular proteins in eukaryotic cells. In particular, the 26S proteasome plays a central role in eliminating damaged or misfolded proteins and is responsible for more than 80% of intracellular protein degradation (Palombella et al., 1994). Cell cycle progression, transcription factor activation, apoptosis, and other cellular events may be directly or indirectly controlled by the ubiquitin-proteasome pathway (Palombella et al., 1994; King et al., 1996). Several important regulators, such as cyclin-dependent inhibitors (p21 and p27), p53, and Bax, are degraded via this pathway (Palombella et al., 1994; Pagano et al., 1995; Li et al., 2000), and inhibiting proteasome activity leads to the accumulation of these proteins and results in cell cycle arrest and apoptosis. Furthermore, evidence has shown that transformed cells are more susceptible than nontransformed cells to proteasome inhibitor-induced apoptosis (Adams et al., 2002b). Targeting the proteasome pathway has thus emerged as a novel approach to cancer therapy.
Bortezomib (PS-341, Velcade [Millenium, Boston, MA]), a dipeptidyl boronic acid, is a specific and selective inhibitor of 26S proteasome (Adams et al., 1999; Adams et al., 2002b). Studies of this drug have established its antitumor activity against a wide range of malignancies, including myeloma, prostate cancer, breast cancer, colon cancer, and lung cancer (Adams et al., 2002b; Lenz et al., 2003). Recently, bortezomib became the first proteasome inhibitor approved by the U.S. Food and Drug Administration for the treatment of relapsed or refractory multiple myeloma. Ongoing clinical trials of bortezomib for prostate cancer and lung cancer have revealed promising results (Lara, Jr. et al., 2004). Like other proteasome inhibitors, bortezomib can stabilize several protein-including p21, p27, p53, various transcriptional factors (e.g., c-myc, c-fos, and c-jun), IκBα, cyclins, and some Bcl-2 family members (e.g., Bak and Bax) by inhibiting their degradation (Lara, Jr. et al., 2004; Pei et al., 2003). However, the mechanisms of apoptosis induction by bortezomib are not well delineated. To elucidate the mechanisms of bortezomib-mediated apoptosis we analyzed changes of Bcl2 family members after bortezomib treatment and found that bortezomib induced rapid accumulation of Bik/NBK by preventing its degradation and that Bik/NBK accumulation correlated with bortezomib cytotoxicity and apoptosis induction.
Results
Rapid accumulation of Bik/NBK by bortezomib in various cancer cells
Several members of the Bcl-2 family, including Bax, Bak, Bcl-2, and Bcl-XL, are known targets of bortezomib (Lara, Jr. et al., 2004; Pei et al., 2003). To evaluate which Bcl-2 family members are critically affected by bortezomib treatment, we determined their protein levels in the colon cancer cell lines DLD-1, LOVO, SW620 and HCT116 after treatment with 0.1 to 5.0 μM bortezomib for 6 hours. Western blot analysis showed that Bik/NBK expression was rapidly and very noticeably up-regulated by bortezomib in all four cell lines (Fig. 1A), even at 0.1 μM. Furthermore, in these cells, bortezomib-induced Bik/NBK accumulation was time dependent: accumulation started within 3 hours after treatment and became much stronger over time (Fig. 1B). The levels of other Bcl-2 family members (Bax, Bak, Bcl-2, and Bcl-XL) did not obviously change at any bortezomib concentration or time point.
Fig. 1.

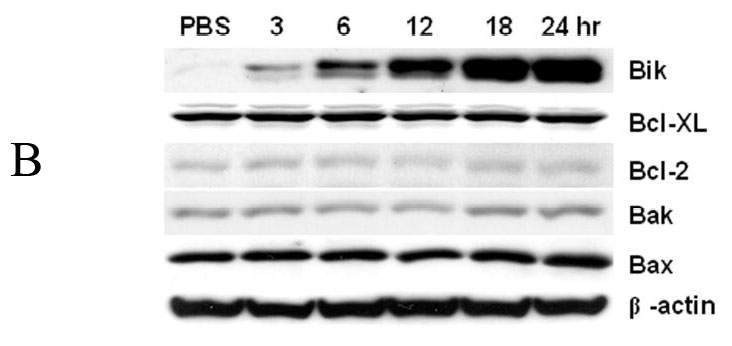
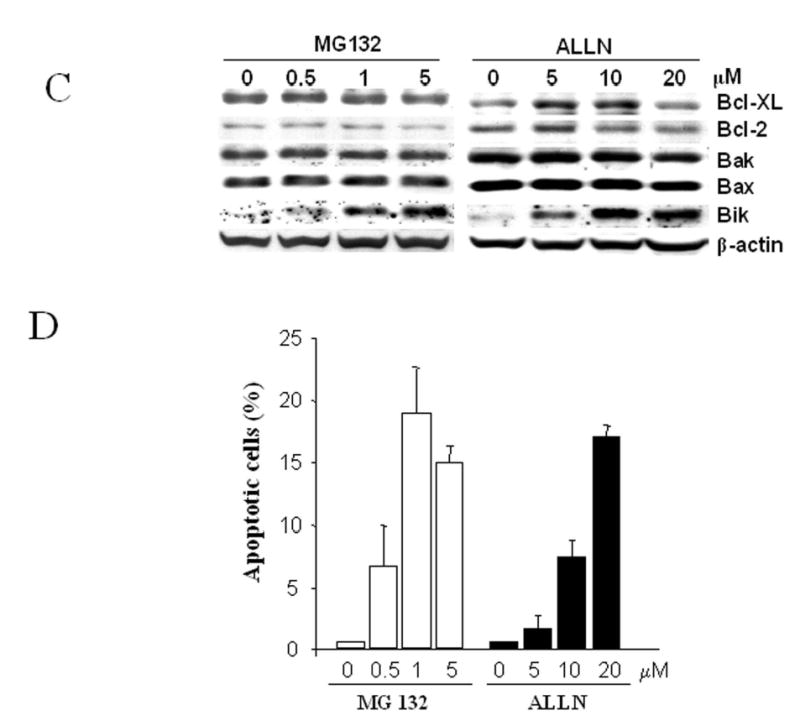
Western blot Profiles of Bcl-2 family members after treatment with bortezomib. (A) Human colon carcinoma cell lines (DLD-1, LOVO, SW620 and HCT116), human lung cancer cell H1299 and human ovarian cancer cell SKOV3 were treated with 0.1 to 5- μM bortezomib for 6 hours. (B) DLD1 cells were treated with 1 μM bortezomib for 3–24 hours. (C) Western blot analysis of DLD1 cells treated with MG132 (0.5 to 5 μM) or ALLN (5 to 20 μM) for 24 h. Data represent one of two independent experiments with similar results. (D) Apoptotic cells determined by SubG1 assay in cell samples treated as in (C). The values represent mean + SD of three assays.
To evaluate whether bortezomib-induced Bik/NBK accumulation was cell- or tissue-type specific, we performed the same experiment with human lung cancer cell line H1299, and ovarian cancer cell line SKOV3. Bik/NBK accumulation was observed in these two cell lines, although the amount of accumulation varied and depended on endogenous Bik/NBK levels (Figure 1A). We observed similar results when we used two other proteasome inhibitors, MG132 and ALLN, to treat all six cell lines. For example, Bik/NBK accumulation and apoptosis induction were observed in DLD1 cells treated with 0.5 to 5 μM of MG132 or 5 to 20 μM of ALLN in a dose-dependent manner (Figure 1C, D).
Bortezomib induced Bik/NBK accumulation is NFκB-independent
To further characterize the effect of bortezomib on Bik/NBK accumulation, we evaluated levels of Bik/NBK or Bax in DLD1, 293 and normal human bronchial epithelial cells (HBE) after treatment with low dose of bortezomib. Bik/NBK accumulation was obvious at 24 h after treatment with 50 nM of bortezomib in all three cells (Figure 2A). The apoptotic cells detected by SubG1 analysis in those cell samples were 44% (DLD1), 17% (293) and 22% (HBE), respectively. However, at this time point, Bik/NBK accumulation was not detectable in these cells at the dose of 10 nM bortezomib. The apoptotic cells in these cell samples were also low (less than 5%).
Fig. 2.


Western blot analysis and NFκB function assay. (A) DLD1, 293 and normal human bronchial epithelial cells (HBE) treated with 10 or 50 nM bortezomib for 24 h. (B) DLD1 cells treated with 100 nM paclitaxel or 50 nM bortezomib for various times as indicated. (C) DLD1 cells treated with 0.1 to 5 μM bortezomib for 6 hours as described in Figure 1A. (D) NFκB function detected by EMSA. DLD1 cells treated with 100 nM bortezomib for 0 to 24h or with 10 to 500 nM bortezomib for 24h. DLD1 cells treated with PBS or 20 nM TNFα were used as mock (−) or positive (+) controls. Data represent one of two independent experiments with similar results.
We also tested whether Bik/NBK is accumulated after treatment with other chemotherapeutic agents. For this purpose, we compared Bik/NBK levels in DLD1 cells treated with 100 nM of paclitaxel or 50 nM of bortezomib. Western blot analysis showed that Bik/NBK was only barely detectable after treatment with paclitaxel. However, a time-dependent Bik/NBK accumulation was obvious after treatment bortezomib (Figure 2B). Of note, IC50 of paclitaxel in DLD1 cells was about 2 nM. 100 nM paclitaxel is sufficient to induce apoptosis in DLD1 cells at 24 h. This result suggested that bortezomib-mediated Bik/NBK accumulation is not caused non-specifically by apoptosis.
Proteasome inhibitors were reported to inhibit NFκB activation by blocking proteolysis of the inhibitor protein IκBα (Lin et al., 1995). To test whether bortezomib-mediated Bik/NBK accumulation was related to functional changes of NFκB, we evaluated IκBα levels in DLD1 after treatment with 0.1 μM to 5 μM of bortezomib for 6h. Unlike Bik/NBK whose accumulation was easily detected at those conditions (Figure 1A), no obvious change in IκBα levels was detected in all the doses tested (Figure 2C). Moreover, nuclear extracts and electrophoretic mobility shift assay (EMSA) (Schmidt et al., 2003) showed no detectable changes in NFκB functionality in DLD1 cells after treatment with 100 nM of bortezomib for 3 to 24 h or with 10 to 500 nM of bortezomib for 24 h (Figure 2D). Thus, treatment with bortezomib did not induce detectable changes in NFκB function and bortezomib-mediated Bik/NBK accumulation is NFκB-independent.
Bortezomib Enhancement of Bik/NBK Protein Stability
Because bortezomib inhibits proteasome-mediated protein degradation, bortezomib-mediated Bik/NBK accumulation is likely caused by stabilization of the protein. To test this hypothesis, we treated DLD1 cells with DMSO, 1 μM bortezomib, or 5-μM MG132 for 6 hours and then add cycloheximide to block protein synthesis in DLD1 cells. Cells were then harvested over time and Bik/NBK levels assessed by Western blot analysis. Bik/NBK protein was rapidly degraded in cells treated with DMSO (Fig. 3A) and had a mean half-life of about 1 hour (Fig. 3B). In contrast, in cells treated with bortezomib or MG132, the Bik/NBK protein level and mean half-life were stable even after 6 hours of cycloheximide treatment, indicating that Bik/NBK degradation was blocked by treatment of bortezomib.
Fig. 3.
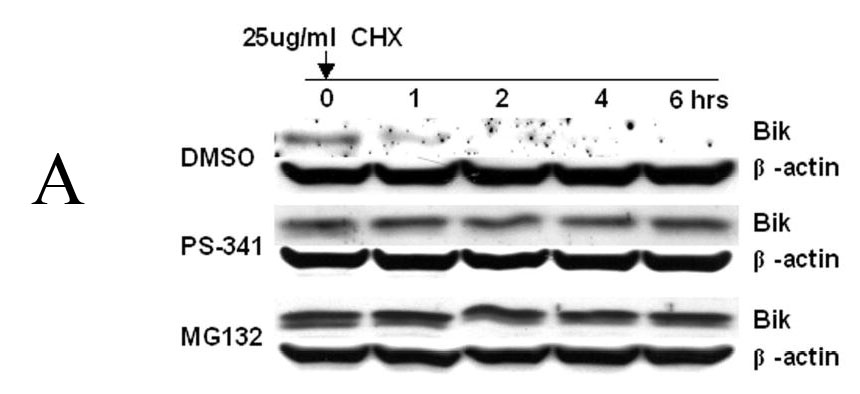

Effect of proteasome inhibitors on Bik/NBK degradation. (A) DLD1 cells were treated with DMSO, 1 μM bortezomib, or 5 μM MG132 for 6 hours, and then with 25 μg/ml of cycloheximide to block protein synthesis. Western blot analysis was performed. Data represent one of three independent experiments with similar results. (B) Quantitative analysis of the Western blot results shown in (A) using Optimas software. Data represent means ± SD of three assays.
Association between Bortezomib-Induced Apoptosis and Bik/NBK Accumulation
Bortezomib induced obvious accumulation of Bik/NBK, and over-expression of Bik/NBK can induce apoptosis in various cancer cells (Boyd et al., 1995; Shimizu et al., 2000). Thus, it is conceivable that Bik/NBK accumulation contributes to bortezomib-induced apoptotosis. To test whether Bik/NBK accumulation was associated with apoptosis induction, we analyzed the release of cytochrome C from DLD1 cells after treatment with 1 μM bortezomib for various times. Release of cytochrome C started within 3 hours of treatment and became strong after 18 hours (Fig. 4A). This change was consistent with the time-dependent Bik/NBK accumulation we noted. Similarly, cleavage of caspase-9 and caspase-3 started within 3 hours of treatment and became stronger after 18 hours (Fig. 4B). Together, these data suggested that Bik/NBK accumulation was associated with bortezomib-initated apoptosis in cancer cells.
Fig. 4.
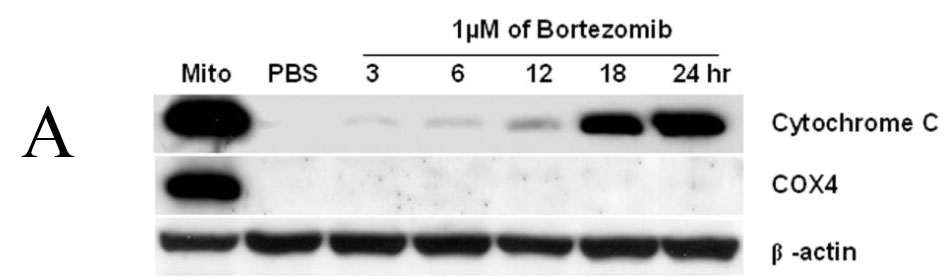

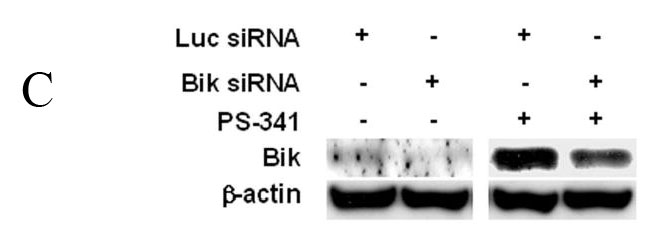

Bortezomib-induced apoptosis in DLD1 cells. (A) and (B) Apoptosis induction. Cells were treated with 1 μM bortezomib for 3–24 hours. Cell lysates were then analyzed by Western blotting. Mito, mitochondria control. Data represent one of two independent experiments with similar results. (C) Bik/NBK levels after treatment with siRNA ± bortezomib. (D) Bortezomib-mediated cell killing. Cells were pretreated with PBS, luciferase siRNA or Bik/NBK siRNA for 24 h and then treated with 1 μM bortezomib for another 24 h. Cell viability was determined in quadruplet and was normalized with cells treated with PBS alone, which was arbitrarily set as 1. Each value represents the mean +SD. * indicates p <0.01.
To further test the correlation between Bik/NBK accumulation and bortezomib-mediated cytotoxicity, we evaluated the effect of Bik/NBK knockdown by siRNA. For this purpose, DLD1 cells were treated with 200 nM siRNA specific for Bik/NBK or luciferase (control) as reported (Hur et al., 2004). 24 h later, the cells were treated with 1-μM bortezomib. Levels of Bik/NBK protein was determined by western blot analysis 24 h later. In comparison with control siRNA, pretreatment with Bik/NBK siRNA can dramatically reduce Bik/NBK protein level (about 50% when determined by band intensity and normalized with β-actin). Nevertheless, treatment with bortezomib resulted in obvious Bik/NBK accumulation in cells pretreated with Bik/NBK siRNA, although the level of this accumulation was dramatically lower (also about 50%) than that pretreated by control siRNA (Fig. 4C), suggesting that bortezomib-mediated Bik/NBK accumulation was very efficient and can occur when Bik/NBK level was very low. Cell viability analysis showed that treatment with Bik/NBK or control siRNA alone did not lead to cell viability loss. No detectable viability loss was observed at 12 h after addition of bortezomib in all groups (data not shown). Treatment with bortezomib for 24 h led to significant viability loss in all groups. However, in comparison with cells pretreated with PBS or control siRNA, pretreatment with Bik/NBK siRNA significantly reduced bortezomib-mediated cell killing (Fig. 4D) (p<0.01). Thus, reduced level of Bik/NBK accumulation correlates with reduced cell killing by bortezomib.
Association between Cancer Cell Sensitivity to Bortezomib and Bik/NBK Accumulation
Although bortezomib induced accumulation of Bik/NBK in various cancer cells, the amount of Bik/NBK accumulation in these cell lines varied dramatically (Fig. 5A). To test whether this amount affected the sensitivity of cancer cells to bortezomib, we determined susceptibility of these cells to the treatment of bortezomib by analyzing the concentration required to inhibit 50% of cell growth after exposure to this agent for 4 days. The result showed that the cell lines with higher Bik/NBK accumulation (DLD-1, LOVO, SW620 and HCT116) had low IC50, or were more sensitive to bortezomib treatment than was the cell line with lower Bik/NBK accumulation (SKOV3) (Fig. 5B). Analysis of apoptosis by Annexin V staining revealed a similar result. After treatment with the same concentration of bortezomib, percentage of apoptotic cells were detected in Lovo and DLD1 cells were dramatically high than that in SKOV3 cells (data not shown). The H1299 cell line was an exception: although it had a low level of Bik/NBK accumulation, it was sensitive to bortezomib.
Fig. 5.
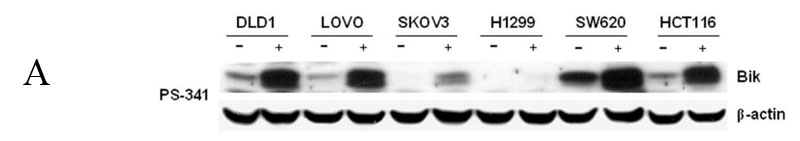
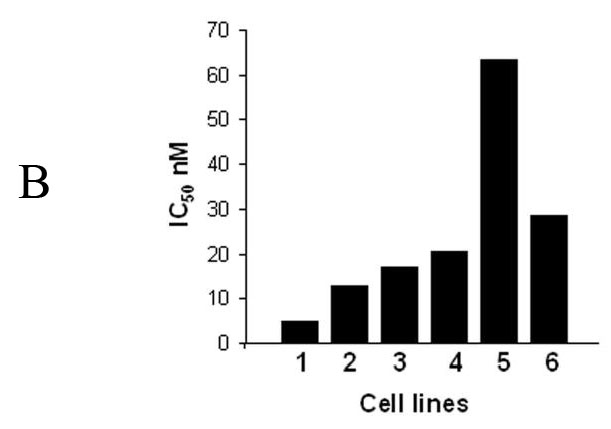
Bortezomib-induced Bik/NBK accumulation and apoptosis different cancer cell lines. (A) Bortezomib-induced Bik/NBk accumulation. Cells were treated with 1 μM bortezomib (+) for 6 hours and harvested for Western blotting. Control cells (−) received no bortezomib. Data represent one of two independent experiments with similar results. (B) IC50 determined by XTT assay at 4 days for the six cell lines listed in (A). (1) Lovo; (2) SW620; (3) HCT116; (4) DLD1; (5) SKVO3; (6) H1299.
Discussion
We demonstrated that bortezomib induced rapid and dramatic accumulation of Bik/NBK protein in several cancer cell lines, particularly human colon cancer cell lines, and that this accumulation was due mainly to stabilization of Bik/NBK protein by bortezomib. Bortezomib did not alter the expression of other Bcl-2 family members reportedly degraded by proteasome. Our results also showed that the occurrence of bortezomib-mediated apoptosis was associated with Bik/NBk accumulation in cancer cells. Moreover, similar to bortezomib, other two proteasome inhibitors, MG132 and ALLN, also induced dramatic Bik/NBK accumulation in these cells, suggesting that bortezomib-induced Bik/NBK accumulation is not related to the other compounds presented in the clinical formulation of bortezomib. Taken together, these findings suggest that Bik/NBK accumulation after treatment with a proteasome inhibitor might be a general phenomenon in cancer cells. This finding may help to delineate mechanisms of apoptosis-inductions by proteasome inhibitors.
Bortezomib has been reported to up-regulate Bax and Bak protein expression through the accumulation of p53 (Yu et al., 2003). However, we tested several cell lines with different p53 status (LOVO and HCT116, wild type, and DLD1 and SW620, mutant) and found no obvious changes in the expression of these two proteins. This result is not consist with Bax/Bak accumulation in p53 wide type HCT116 cells that was reported by others (Yu et al., 2003). It is possible that biological property of the same cell line used in different laboratory may vary. However, because Bax/Bak accumulation upon bortezomib treatment was not observed in all the cell lines tested in this study, such an accumulation may be not a generalized phenomenon. Bortezomib could also inhibit activation of NFκB by preventing degradation of IκBα, leading to down-regulation of Bcl-2 and Bcl-XL protein expression (Lara, Jr. et al., 2004; Pei et al., 2003). However, we did not find any changes in the expression of Bcl-2 or Bcl-XL in cell lines tested here and observed no substantial accumulation of IκBα after 6 h treatment with 0.1 to 5 μM bortezomib. NFκB functional assay also did not reveal detectable changes after treatment with bortezomib at various doses and time points. Together, these data indicated that Bik/NBK accumulation after exposure to bortezomib might a major contribution to apoptosis induction and that p53 and NFκB may not be critical in Bik/NBK accumulation. It remains unclear, however, why only Bik/NBK accumulation was detected after treatment with bortezomib and other proteasome inhibitors. A possible explanation could be that E3 ubiquitin ligase specific for Bik/NBK is more sensitive to these proteasome inhibitors. Further investigation on this issue may provide insight on selective apoptosis-induction in transformed cells by proteasome inhibitors (Adams et al., 2002a).
Bik/NBK is reported to interact with Bcl-xL and Bcl-2, and triggers Bax-dependent apoptosis that coincides with cytochrome c release (Gillissen et al., 2003). Using GFP-tagged cytochrome c to monitor cytochrome c release during apoptosis, Goldstein et al showed that release of cytochrome c during apoptosis is rapid, complete and kinetically invariant (Goldstein et al., 2000). In this study, we found cytochrome c release was detectable as early as 3 h after treatment with bortezomib, coinciding with Bik/NBK accumulation (Fig. 1B and 3A). However, dramatic cytochrome c was detected at 18 h after bortezomib treatment. Whereas the cytochrome c release reported by Goldstein et al (Goldstein et al., 2000) represents a process occurring at a single cell level, the cytochrome c release detected in this study represents a process for whole population of cells in culture. It is likely that response to bortezomib in cultured cells is not a synchronized process. Apoptosis may occur at early time points in a few cells but increase dramatically over time.
Results from our six different cell lines showed that the amount of Bik/NBK accumulation is generally associated with the cells susceptibility to bortezomib. The exception was H1299 cells, which expressed a low level of endogenous Bik/NBK but were sensitive to bortezomib. In H1299 cells, Bik/NBK accumulation after bortezomib treatment was detectable but not as dramatic as that in the other cell lines. Bax and Bak were detected in this cell line, however, not accumulated after bortezomib treatment. One explanation is that low accumulation of Bik/NBK is sufficient to kill H1299 cells or suppress their growth. In fact, using a GFP/Bik fusion construct, we have previously reported that detectable expression of exogenous Bik (green color) was sufficient to induce cell death in all cells we have tested (Kagawa et al., 2001), suggesting that apoptosis induction by Bik is potent and effective. Alternatively, proteins other than Bik/NBK may contribute to the cell killing activities of proteasome inhibitors in these cells. Interestingly, although H1299 is sensitive to bortezomib by cell viability assay, apoptotic cells detectable by SubG1 assay was only less than 5% at 24 h after treatment with 0.1 to 1 μM of bortezomib. Treatment with Bik/NBK siRNA has no significant impact on bortezomib-mediated cell killing in H1299 cells (data not shown). Thus, it is possible that Bik/NBK accumulation and apoptosis-induction is one but not the sole mechanism of proteasome inhibitor-mediated antitumor activity. Because a large number of cellular proteins are degraded by ubiquitin/proteasome pathway, it is not unexpected that inhibition of proteasome will alter the levels of a variety of proteins, including many molecules that may induce cell death or suppress cell proliferation but are not analyzed in this manuscript. Nevertheless, the generally dramatic accumulation of Bik/NBK in all cancer cell lines after treatment with proteasome inhibitors and its association with apoptosis induction may provide useful information for evaluating of bortezomib-based cancer therapy.
Materials and Methods
Cells and Cell Culture
Human colon cancer cell lines DLD-1, LOVO, SW620, and HCT116; human lung cancer cell lines H1299; human ovarian cancer cell line SKOV3; and human embryonic kidney 293 cells were maintained in RPMI 1640 or Dulbecco’s modified Eagle’s medium supplemented with 10% (v/v) heat-inactivated fetal bovine serum, 1% glutamine and 1 × antibiotics-antimycotics mixture (Invitrogen, Carlsbad, CA). Human bronchial epithelial cells were purchased from Clonetics (San Diego, CA) and cultured in media recommended by the manufacturer. All cells were cultured at 37°C in a humidified incubator containing 5% CO2.
Chemicals
Bortezomib was obtained from the Pharmacy of The University of Texas M. D. Anderson Cancer Center and dissolved in phosphate-buffered saline (PBS) at 5 mM as a stock solution. Proteasome inhibitor MG132 and calpain inhibitor I (ALLN) were purchased from Calbiochem (La Jolla, CA) and diluted in dimethyl sulfoxide (DMSO) at stock concentrations of 10 mM and 20 mM, respectively. Cycloheximide and DMSO were bought from Sigma (St. Louis, MO).
Western Blot Analysis
Cells were lyzed in Laemmli’s lysis buffer. Equal amounts of lysate were separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and subjected to Western bloting, as described previously (Wu et al., 2003). Rabbit anti-human Bik/NBK, Bcl-XL, Bcl-2, Bax, caspase-3, and caspase-9 antibodies and goat anti-human antibody Bak were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Mouse anti-human cytochrome C and COX4 antibodies were bought from BD Pharmingen (San Diego, CA). Mouse anti-human β-actin antibody was obtained from Sigma and was used as a loading control.
Extraction of cytosol
Cells (1×106) were plated onto 10-cm dishes, incubated overnight, and then treated with 1 μM bortezomib for various times. The medium was removed and cells were trypsinized and collected into Eppendorf tubes. After being washed with PBS once, cells were resuspended with 100 μl of digitonin buffer (0.02% digitonin, 5-mM EDTA, 1 mM PMSF, 75-mM sucrose, 25-mM NaCl, 2.5-mM PIPES, 0.75-mM MgCl2·6H2O), incubated on ice for 10 minutes and then spun at 10,000 rpm for 25 minutes. The supernatants were collected as cytosol components. Mitochondria were extracted as previously reported (Wu et al., 2003) and were used as a control.
Nuclear Extracts and Electrophoretic Mobility Shift Assay (EMSA)
EMSAs were performed as described (Schmidt et al., 2003). (32P)-labeled double-stranded oligonucleotides containing the κB site (underlined) found in the HIV-1 LTR (5′-CTCAACAGAGGGGACTTTCCGAGAGGCCAT) were incubated as probes. Unlabeled double-stranded oligonucleotides (50-fold molar excess) containing the mutant HIV-κB (5′CTCAACAGAGTTGACTTTTCGAGAGGCCAT-3′) and wild-type HIV-κB were used for competition studies. For antibody supershifts, 2 μl of the polyclonal antibodies against p65 and p50 (Santa Cruz Biotechnology) were preincubated for 30 min at room temperature before the probe was added. The probe was allowed to bind for 20 min on ice. Reaction mixtures were analyzed on 4% polyacrylamide gels containing 0.25 × TBE (22.5 mM Tris, 22.5 mM borate, 0.5 mM EDTA, pH 8.0) buffer.
Cell Viability Assay
Cell viability was determined by seeding 4×103 cells/well in 96-well plates, incubating them for 1 day, treating them with bortezomib (0.01–100 μM) continuously for 4 days. Cell viability was then determined by XTT assay (Cell Proliferation Kit; II, Roche Molecular Biochemicals, Indianapolis, IN), as described previously (Wu et al., 2003).
siRNA Transfection
The sequences for oligodubplex Bik siRNA was the same as reported (Hur et al., 2004). Both Bik siRAN and control luciferase siRNA were obtained from Dharmacon (Lafayette, CO). Transfection of siRNA was performed using Oligofectamine (Invitrogen) according to the manufacturer’s instruction.
Protein stability Assay
To determine protein stability, we treated cells with DMSO, MG132, or bortezomib for up to 6 hours and then added 25-μg/ml cycloheximide to block protein synthesis (Kim et al., 2002). Collected protein samples were subjected to Western blot analysis using anti-Bik/NBK antibody. Band densities were qualified using Optimas software (Media Cybernetics, Silver Spring, MD), and the mean half-life of BiK/NBK was calculated.
Acknowledgments
We thank Elizabeth L. Hess for editorial review and Alma J. Vega for assistance in preparing the manuscript. This article represents partial fulfillment of the requirements for a Ph.D. degree for J. J. Davis.
References
- Adams J. Oncologist. 2002a;7:9–16. doi: 10.1634/theoncologist.7-1-9. [DOI] [PubMed] [Google Scholar]
- Adams J. Cur Opinion Oncol. 2002b;14:628–634. doi: 10.1097/00001622-200211000-00007. [DOI] [PubMed] [Google Scholar]
- Adams J, Palombella VJ, Sausville EA, Johnson J, Destree A, Lazarus DD, Maas J, Pien CS, Prakash S, Elliott PJ. Cancer Res. 1999;59:2615–2622. [PubMed] [Google Scholar]
- Boyd JM, Gallo GJ, Elangovan B, Houghton AB, Malstrom S, Avery BJ, Ebb RG, Subramanian T, Chittenden T, Lutz RJ, et al. Oncogene. 1995;11:1921–8. [PubMed] [Google Scholar]
- Gillissen B, Essmann F, Graupner V, Starck L, Radetzki S, Dorken B, Schulze-Osthoff K, Daniel PT. EMBO J. 2003;22:3580–3590. doi: 10.1093/emboj/cdg343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein JC, Waterhouse NJ, Juin P, Evan GI, Green DR. Nat Cell Biol. 2000;2:156–162. doi: 10.1038/35004029. [DOI] [PubMed] [Google Scholar]
- Hur J, Chesnes J, Coser KR, Lee RS, Geck P, Isselbacher KJ, Shioda T. Proc Natl Acad Sci U S A. 2004;101:2351–2356. doi: 10.1073/pnas.0307337101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagawa S, He C, Gu J, Koch P, Rha SJ, Roth JA, Curley SA, Stephens LC, Fang B. Cancer Res. 2001;61:3330–3338. [PubMed] [Google Scholar]
- Kim GY, Mercer SE, Ewton DZ, Yan Z, Jin K, Friedman E. J Biol Chem. 2002;277:29792–29802. doi: 10.1074/jbc.M201299200. [DOI] [PubMed] [Google Scholar]
- King RW, Deshaies RJ, Peters JM, Kirschner MW. Science. 1996;274:1652–1659. doi: 10.1126/science.274.5293.1652. [DOI] [PubMed] [Google Scholar]
- Lara PN, Jr, Davies AM, Mack PC, Mortenson MM, Bold RJ, Gumerlock PH, Gandara DR. Sem Oncol. 2004;31:40–46. doi: 10.1053/j.seminoncol.2003.12.013. [DOI] [PubMed] [Google Scholar]
- Lenz HJ. Cancer Treat Rev. 2003;29(Suppl 1):41–48. doi: 10.1016/s0305-7372(03)00082-3. [DOI] [PubMed] [Google Scholar]
- Li B, Dou QP. Proc Natl Acad Sci U S A. 2000;97:3850–3855. doi: 10.1073/pnas.070047997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YC, Brown K, Siebenlist U. Proc Natl Acad Sci U S A. 1995;92:552–556. doi: 10.1073/pnas.92.2.552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagano M, Tam SW, Theodoras AM, Beer-Romero P, Del Sal G, Chau V, Yew PR, Draetta GF, Rolfe M. Science. 1995;269:682–685. doi: 10.1126/science.7624798. [DOI] [PubMed] [Google Scholar]
- Palombella VJ, Rando OJ, Goldberg AL, Maniatis T. Cell. 1994;78:773–785. doi: 10.1016/s0092-8674(94)90482-0. [DOI] [PubMed] [Google Scholar]
- Pei XY, Dai Y, Grant S. Leukemia. 2003;17:2036–2045. doi: 10.1038/sj.leu.2403109. [DOI] [PubMed] [Google Scholar]
- Schmidt C, Peng B, Li Z, Sclabas GM, Fujioka S, Niu J, Schmidt-Supprian M, Evans DB, Abbruzzese JL, Chiao PJ. Mol Cell. 2003;12:1287–1300. doi: 10.1016/s1097-2765(03)00390-3. [DOI] [PubMed] [Google Scholar]
- Shimizu S, Tsujimoto Y. Proc Natl Acad Sci U S A. 2000;97:577–582. doi: 10.1073/pnas.97.2.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S, Zhu H, Gu J, Zhang L, Teraishi F, Davis JJ, Jacob DA, Fang B. Cancer Res. 2003;64:1110–1113. doi: 10.1158/0008-5472.can-03-2790. [DOI] [PubMed] [Google Scholar]
- Yu J, Tiwari S, Steiner P, Zhang L. Cancer Biol Ther. 2003;2:694–699. [PubMed] [Google Scholar]