

Abstract
Hutchinson-Gilford progeria syndrome (HGPS) is a childhood premature aging disease caused by a spontaneous point mutation in lamin A, one of the major architectural elements of the mammalian cell nucleus1–4. The HGPS mutation activates an aberrant cryptic splice site in the lamin A pre-mRNA, leading to synthesis of a truncated lamin A protein and concomitant reduction of wild type lamin A levels3,4. Fibroblasts from HGPS patients have severe morphological abnormalities in nuclear envelope structure. Here we show that the cellular disease phenotype is reversible in cells from HGPS patients. Introduction of wild type lamin A protein does not rescue the cellular disease symptoms. The mutant lamin A mRNA and protein can be efficiently eliminated by correction of the aberrant splicing event using a modified oligonucleotide targeted to the activated cryptic splice site. Upon splicing correction, HGPS fibroblasts assume normal nuclear morphology, the aberrant nuclear distribution and cellular levels of lamina-associated proteins are rescued, defects in heterochromatin-specific histone modifications are corrected, the dynamic properties of lamin A are restored, and proper expression of several misregulated genes is reestablished. Our results establish proof of principle for the application of small molecules to the correction of the premature aging phenotype in HGPS patients.
Hutchinson-Gilford Progeria Syndrome is most commonly caused by a de novo heterozygous silent substitution at codon 608 (G608G: GGC>GGT) of the lamin A/C (LMNA) gene3,4. The mutation resides in exon 11 of LMNA and activates an exonic cryptic donor splice site four nucleotides upstream. The pre-mRNA derived from the mutated allele is spliced using this aberrant donor splice site and the correct exon 12 acceptor splice site, giving rise to a truncated lamin A mRNA lacking the terminal 150 nucleotides of exon 113,4. As a consequence of the aberrant splicing event a mutant protein, Δ50 lamin A, containing a 50 aa internal deletion in its globular tail domain is generated3,4. As previously reported3–6, we observed that fibroblasts from HGPS patients are characterized by the presence of dysmorphic nuclei with altered size and shape, exhibiting lobules, wrinkles and herniations as evident by staining for lamin A (Fig. 1a,b). We found aberrant morphology (as assessed by a nuclear envelope contour ratio of < 0.7; see Methods for definition) in typically ~ 70% of cells in multiple HGPS cell lines (Supplementary Table 1 online). In addition to these well-established morphological changes, we found several novel defects in HGPS fibroblasts. In ~ 70% of the cells, lamin B, the major lamin A-interacting partner, and multiple members of the family of lamina-associated polypeptide (LAP2s) were depleted from their typical localization at the nuclear envelope and the global cellular amount of these proteins was reduced up to 6-fold compared to the control cells when assessed by total cellular fluorescence intensity measurements (See Methods) (Fig 1a,b; Supplementary Table 1 online). Furthermore, ~ 60% of HGPS cells showed an at least 2-fold reduction in the cellular level of the heterochromatin protein HP1α, one of the adaptors between the nuclear lamina and chromatin7 (Fig. 1a,b; Supplementary Table 1 online). The reduced level of HP1α coincided with aberrant histone modification status of chromatin, with reduction or complete loss of the heterochromatin marker tri-methyl lysine 9 of core histone H3 (Tri-Me-K9)8 in ~ 60% of the cells (Fig. 1a,b; Supplementary Table 1 online). Reduced levels of lamina-associated proteins, HP1 and Tri-Me-K9 in the whole cell population were confirmed by Western blotting (data not shown).
Fig. 1. Wild type GFP-lamin A is insufficient for phenotypic rescue of HGPS cells.
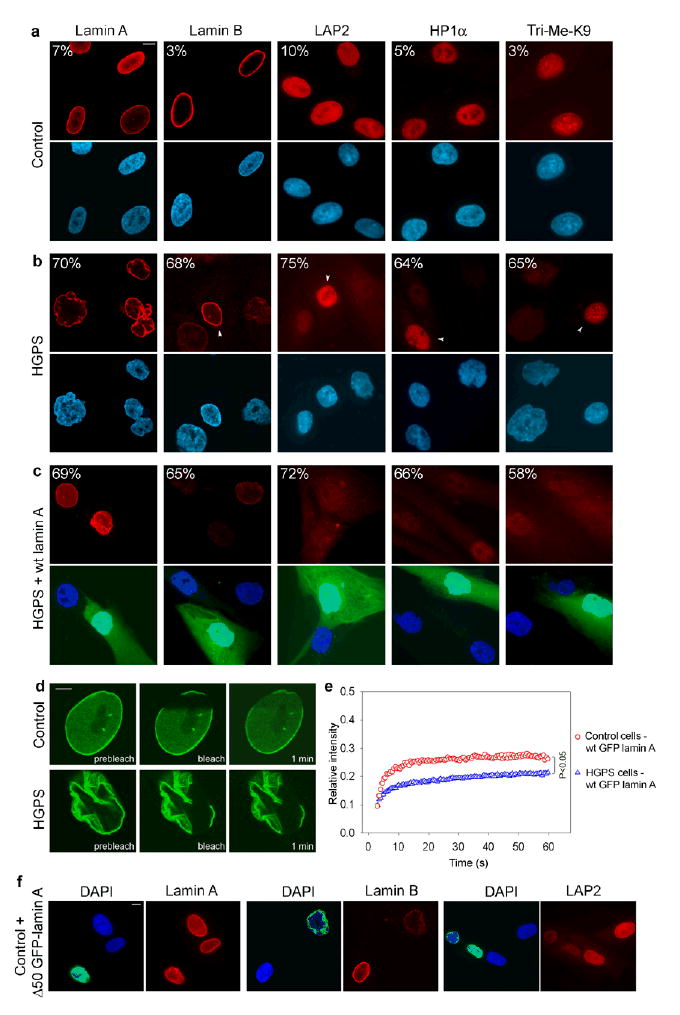
(a–c) Immunofluorescence microscopy on primary dermal fibroblasts from a healthy control individual (AG08469; population doublings: 25–30) (a) and a HGPS patient (AG01972; population doublings: 25–30), untreated (b) or transfected with wild type lamin A and GFP as a transfection marker (c). Cells were stained with DAPI (blue) and antibodies (red) against the indicated proteins. Four cells (indicated by arrowheads) with normal staining respectively for lamin B, LAP2, HP1α and Tri-Me-K9 are shown in panel b to directly compare the cellular level of the proteins in unaffected and affected cells. The percentage of cells showing aberrant phenotype is indicated. Scale bar: 10 μm. (d) FRAP analysis of wild type GFP-lamin A in living control and HGPS cells. Nuclei were imaged before and during recovery after the bleach pulse. Scale bar: 5 μm. (e) Kinetics of recovery of the fluorescence signal in the whole bleached area. The statistical significance of the difference between the two recovery curves is indicated. (f) Immunofluorescence microscopy on primary dermal fibroblasts from a healthy control individual (AG08469; population doublings: 25–30) transfected with GFP-Δ50 lamin A. Cells were stained with DAPI (blue) and antibodies (red) against lamin A/C, lamin B, LAP2 proteins. The green fluorescent signal from GFP-Δ50 lamin A is overlaid with the DAPI signal. Scale bar: 10 μm.
Using these characteristics of HGPS patient fibroblasts, we set out to ask whether the morphological and cellular changes associated with progeria are permanent or can be reversed. Since in HGPS cells the presence of the mutant lamin A is accompanied by a reduction in the amount of the wild type protein, the cellular defects might either be due to a decrease in the level of fully functional wild type lamin A or due to a dominant-negative effect of the mutant Δ50 lamin A3,4. Transfection of HGPS patient cells with a plasmid simultaneously expressing untagged lamin A and GFP to identify transfected cells failed to lower the percentage of cells with aberrant overall morphology compared to untransfected cells (Fig. 1c; Supplementary Table 1 online) and was insufficient to restore the normal cellular levels of the lamina-associated proteins and HP1α (Fig. 1c; Supplementary Table 1 online). Furthermore, it did not reestablish proper histone modifications in heterochromatin in HGPS cells (Fig. 1c; Supplementary Table 1 online). Even high levels and prolonged expression of wild type lamin A up to ten days were not effective in correcting the cellular HGPS defects (data not shown). Identical results were obtained using a FLAG-tagged and GFP-tagged lamin A, previously shown to complement lamin A-deficient cells9–11 (data not shown). Analysis of the dynamic properties of the introduced wild type GFP-lamin A suggested that the failure to rescue the progeria phenotype was likely due to the inability of wild type lamin A to function properly in HGPS cells. In Fluorescence Recovery after Photobleaching (FRAP) experiments recovery kinetics of wild type GFP-lamin A were reduced in HGPS cells compared to control cells (P < 0.05), indicating that wild type lamin A becomes more tightly associated with its nuclear binding sites in the presence of the mutant protein (Fig. 1d,e). The observation that the cellular behavior of wild type lamin A is altered in the presence of the mutant lamin A suggests a dominant negative effect of Δ50 lamin A in HGPS cells. In support, expression of GFP-Δ50 lamin A in control fibroblasts induced nuclear morphological defects similar to those observed in HGPS cells, and led to a reduction in the cellular levels of lamin B and LAP2 (Fig. 1f).
The dominant negative nature of the HGPS-causing mutant lamin A protein means that reversal of the cellular phenotype in HGPS cells requires the elimination of the mutant protein. To do so, we sought to correct the aberrant splicing of the mutant lamin A pre-mRNA. We designed a 25-mer morpholino oligonucleotide (exo11) complementary to the region containing the HGPS mutation in exon 11 to sterically block the activated cryptic splice site, thus preventing the access of the splicing machinery to the aberrant splice site12 (Fig. 2a). In contrast to unmodified oligonucleotides, morpholinos are stable and they do not induce RNAse H degradation of the heteroduplex nor do they interfere with translation13,14. The exo11 oligonucleotide was able to correct aberrant splicing of a lamin A minigene in HeLa cells (Supplementary Fig. 1 online). To test whether aberrant splicing of the endogenous LMNA transcript could be corrected, exo11 oligonucleotide was introduced into HGPS fibroblasts by two sequential electroporations. The splicing pattern of endogenous lamin A was analyzed after four days by RT-PCR using primers in exon 9 and exon 12. Oligonucleotide treatment effectively blocked aberrant splicing of endogenous lamin A mRNA with a half maximal effect at ~ 7 μM and resulted in the removal up to 90% of the truncated lamin A mRNA (Fig. 2b,c). Similar results were obtained in three dermal fibroblast and two B-lymphocyte cell lines derived from HGPS patients, indicating that correction of lamin A aberrant splicing can be achieved with similar efficiency in multiple cell types (Fig. 2b,c). No change in the relative amount of splice products was observed after treatment with a control scrambled oligonucleotide, indicating that correction of aberrant splicing occurs in an oligonucleotide sequence-specific manner (Fig. 2b,c). The exo11 oligonucleotide was specific for correction of the aberrant splicing event and had no effect on other splicing events in lamin A pre-mRNA as demonstrated by similar RNA levels and the absence of aberrant splice products when probed along the entire mRNA (Fig. 2d–f). Concentrations of 40 μM and above resulted in a slight increase of lamin C production. This is most likely due to a partial block of the normal exon 11-intron 11 junction which shares 16 out of 25 nucleotides with the activated splice site, resulting in the activation of the lamin C alternative splice site in exon 10 (ref. 15) (Fig. 2e,f). In agreement with previous reports demonstrating high sequence-specificity of morpholino oligonucleotides14,16, no effect was seen on splicing of several cellular RNAs (Supplementary Fig. 2 online).
Fig. 2. Correction of aberrant splicing in the endogenous lamin A transcript in HGPS cells.
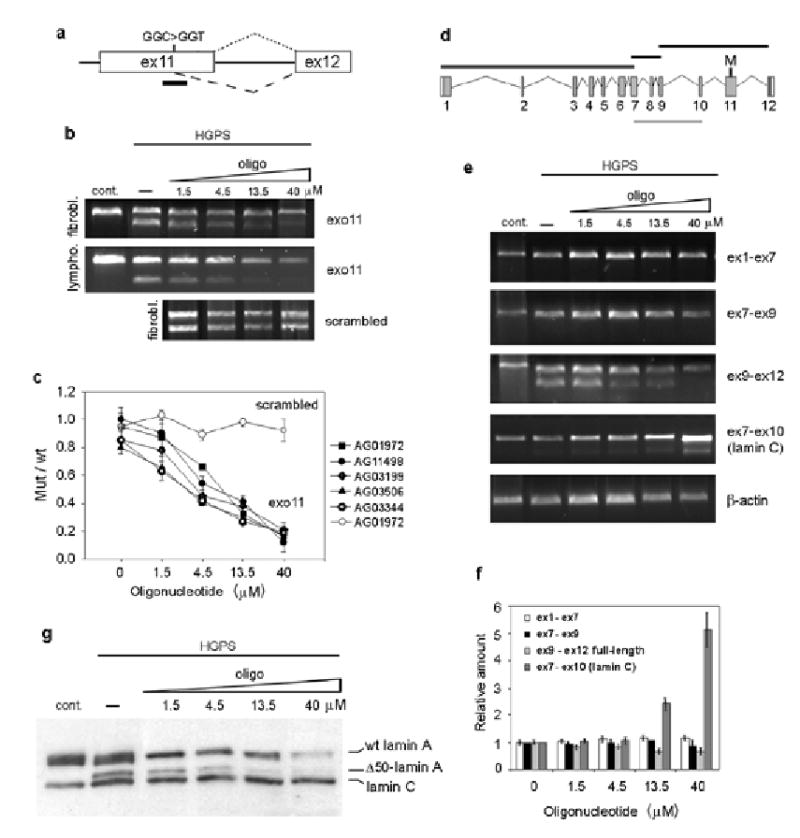
(a) Schematic representation of the region of lamin A pre-mRNA targeted by exo11 antisense oilgonucleotide. The predicted splicing events (dotted line for the normal splicing, dashed line for the aberrant splicing) and the position of exo11 oligonucleotide (black bar) are indicated. (b) RT PCR analysis of control fibroblasts and lymphocytes and HGPS fibroblasts and lymphocytes subjected to 2 sequential electroporations with oligonucleotide or mock treated. (c) Quantitation of exo 11 (black symbols) and scrambled (with symbols) oligonucleotides titration in different cell lines. All values represent averages from 5 independent experiments ±S.D. (d) Schematic representation of the different fragments of lamin A (black bars) and lamin C (gray bar) cDNAs amplified by RT-PCR. (e) RT-PCR analysis and (f) quantification of the indicated fragments after oligonucleotide titration. The intensity of each band was normalized to the intensity of the corresponding β-actin band. (g) Western blot analysis of control cells and HGPS fibroblasts subjected to 3 sequential electroporations with oligonucleotide or mock treated, analyzed 6 days after the first delivery.
To confirm that correction of aberrant splicing of the LMNA pre-mRNA also resulted in a reduction of the mutant lamin A protein, Western blot analysis was performed on treated HGPS patient fibroblasts. Titration of exo11 oligonucleotide induced progressive reduction of mutant lamin A, leaving less than 5% of the protein at concentrations above 13.5 μM (Fig. 2g). Consistent with the effects observed at the RNA level, wild type lamin A did not increase upon treatment, and at high concentrations of oligonucleotide lamin C was moderately increased relative to lamin A (Fig. 2g).
We used several criteria to test whether correction of the splicing event restored the normal cellular phenotype in HGPS patient fibroblasts. Treatment of HGPS cells with 13.5 or 40 μM exo11 oligonucleotide corrected the morphological abnormalities of cell nuclei in more than 90% of cells (Fig. 3a–c; Supplementary Table 1 online). Nuclei lost the severe wrinkles, herniations and lobes typically observed in HGPS cells and acquired a normal ellipsoid shape, indistinguishable from that of control cells (contour ratio = 0.89 ± 0.1) (Fig. 3a–c; Supplementary Table 1 online). The normal cellular level of lamin B, LAP2 proteins and HP1α was restored in ~90% of treated cells (Fig. 3a–d; Supplementary Table 1 online). Upon treatment normal levels of Tri-Me-K9 were observed in HGPS cells (Fig. 3a–c; Supplementary Table 1 online). Finally, normal cellular behavior of the wild type lamin A protein was restored after correction of aberrant splicing. In the FRAP assay, GFP-lamin A was dynamically exchanged at similar rates in control cells and in HGPS cells treated with oligonucleotide, suggesting that correction of splicing restores proper dynamic interactions of the lamin A protein in vivo (Fig. 3e,f).
Fig. 3. Phenotypic rescue of HGPS cells by treatment with morpholino oligonucleotide.
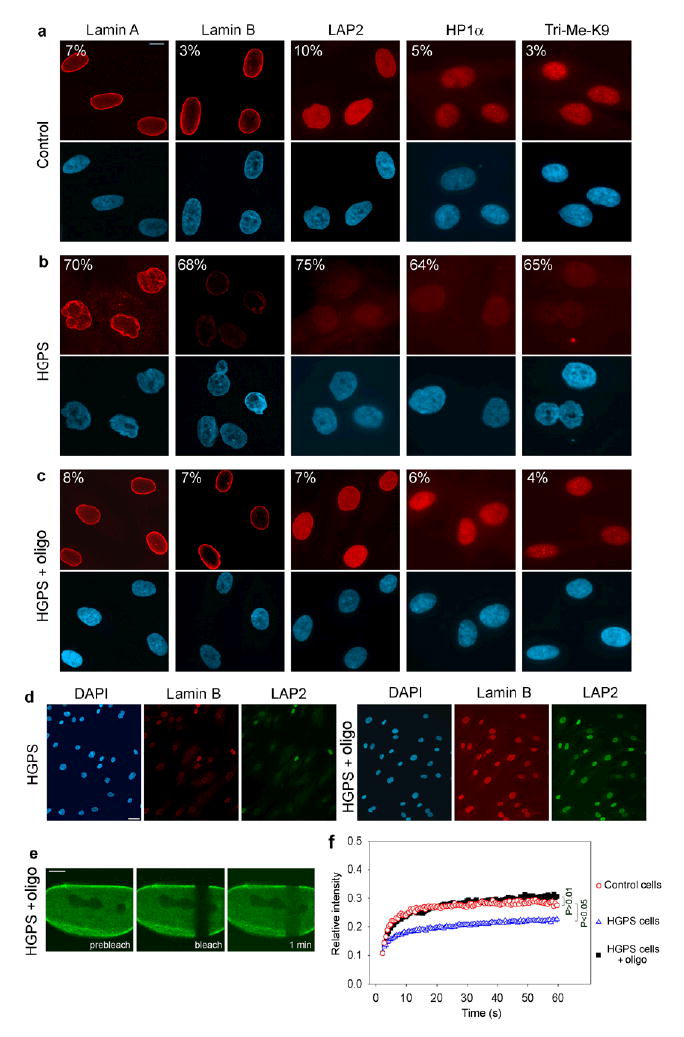
(a–d) Immunofluorescence microscopy on primary dermal fibroblasts from a healthy control individual (a) and a HGPS patient (AG01972) untreated (b) or treated with oligonucleotide (c). Cells were stained with DAPI (blue) and antibodies (red) against the indicated proteins. The percentage of cells showing aberrant phenotype is indicated. Scale bar: 10 μm. (d) Low magnification images of untreated and treated HGPS cells. Scale bar: 35 μm. (e) FRAP analysis of wt GFP-lamin A in HGPS cells upon splicing correction. A nucleus was imaged before and during recovery after the bleach pulse. Scale bar: 5 μm. (f) Kinetics of recovery of the fluorescence signal in the whole bleached area compared to control and untreated HGPS cells. The statistical significance of the differences between the curves is indicated.
Reversal of the cellular phenotype in HGPS fibroblasts did not require mitosis or nuclear lamina disassembly. Analysis of single living cells expressing GFP-lamin A by in vivo imaging showed restoration of normal nuclear morphology without mitosis (Supplementary Fig. 3 online). In addition, when cell division was prevented during oligonucleotide treatment by either contact inhibition or arrest of cells in G0, the normal cellular phenotype as assessed by lamin A, LAP2 or HP1 staining was restored in most cells (Supplementary Fig. 3 online).
As a final criterion for restoration of normal cellular function of HGPS cells we analyzed the expression levels of genes that are misregulated in progeria patient fibroblasts. Several genes have been shown to be differentially expressed in HGPS cells by microarray experiments17,18. Consistent with those results we found by RNase protection (see Supplementary Methods online) a 2-fold reduction of matrix metalloproteinase 3 (MMP3), hyaluron synthase III (HASIII) and tissue inhibitor of metalloproteinase 3 (TIMP3) and a 2-fold increase in matrix metalloproteinase 14 (MMP14) and chemokine (C-C motif) ligand 8 (CCL8) mRNA in HGPS cells compared to age matched control cells (Fig. 4a,b). Upon treatment of HGPS cells with oligonucleotide, the expression of all five genes returned to the levels observed in age matched healthy control cells (Fig. 4a,b). CCL8 and MMP14 levels were reduced and TIMP3, MMP3 and HASIII were increased to levels similar to control cells. As a control signal transducer and activator of transcription 3 (STAT3), which is unchanged by the lamin A mutation, was not affected by the presence of the oligonucleotide. These results demonstrate that correction of the aberrant splicing defect of lamin A pre-mRNA restores normal expression of several genes misregulated in HGPS.
Fig. 4. Restoration of normal gene activity in HGPS cells by treatment with oligonucleotide.
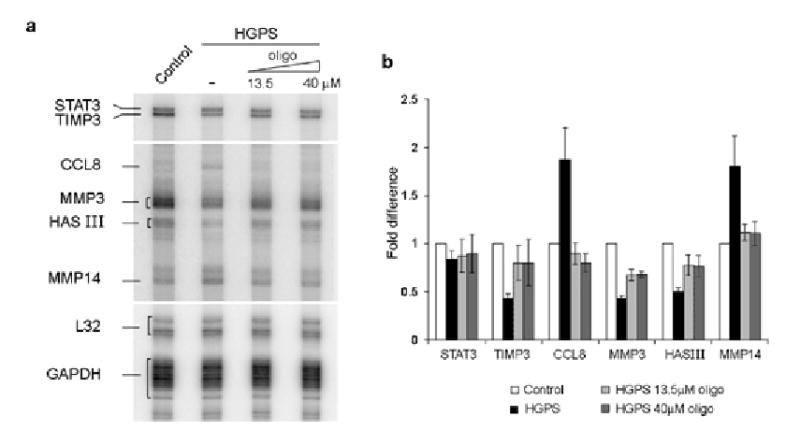
(a) RNAse protection assay on control cells (CRL-1474) and HGPS cells (AG11498) subjected to 3 electroporations with oligonucleotide or mock treated. A longer exposure of the gel is shown for CCL8, MMP3, HAS III and MMP14 due to the low expression level of those genes compared to STAT3, TIMP3, L32 and GAPDH. (b) Quantitation of gene expression levels. Values represent fold differences of the indicated mRNAs levels between untreated and treated HGPS cells and control cells. Values are averages from at least 3 independent experiments ±S.D.
Our findings demonstrate that elimination of the mutant HGPS lamin A protein is necessary and sufficient to rescue the progeria cellular phenotype in HGPS patient fibroblasts. Together with the inability to restore the normal cellular phenotype by overexpressing wild type lamin A in HGPS cells, these data indicate that the cellular defects in HGPS patient cells are due to a dominant-negative mode of action of the mutant lamin A protein. Furthermore, overexpression of Δ50 lamin A in wild type cells induces abnormalities in nuclear morphology. The observation that in HGPS cells the presence of Δ50 lamin A severely compromises the cellular behavior of wild type lamina A and the composition of the nuclear rim is further direct evidence for the dominant negative role of mutant lamin A.
The reversibility of the cellular disease phenotype by all tested criteria via correction of the aberrant splicing event may be exploited as a strategy for therapeutic purposes. The observation that cells can be rescued independently of mitosis suggests that reversal of the cellular phenotype might also be feasible in non-dividing tissues. A mouse model displaying several progeria symptoms19 is available, however this model it is not suitable for further testing of our approach since the molecular basis of this animal model is a L530P mutation rather than the G608G mutation found in humans and does not result in simple activation of a cryptic splice site19. Although morpholinos and oligonucleotides of other chemistries have successfully been used for sustained delivery in animals and humans12,20,21, splicing correction may also be achieved by use of trans-splicing expression systems as applied to other splicing diseases or by use of small molecule inhibitors22. Regardless of the method, our results establish proof of principle for the reversal of the cellular effects of premature aging in progeria.
Materials and Methods
Cell culture and transfection
Primary dermal fibroblast cell lines and lymphoblastoid cell lines from patients and healthy donors were obtained from the Aging Repository of the Coriell Cell Repository (CCR) and from the American Type of Culture Collection (ATCC). HGPS fibroblast cell lines were AG01972 (patient age 14 years, CCR, PDs 22 at purchase), AG11498 (14 years, CCR, PDs 10), AG03199 (10 Years, CCR, PDs 10). HGPS lymphoblatoid cell lines were AG03506 (13 years, CCR), AG03344 (10 years, CCR). Control fibroblasts cell lines were AG08469 (38 years old healthy male, CCR, PDs 4) and CRL-1474 (7 years, ATCC, PDs 12). Control lymphoblatoid cell line was AG03504 (41 years old healthy female, CCR). Cells were grown in medium (Minimum Essential Medium (Gibco) for fibroblasts and RPMI (Gibco) for lymphocytes) supplemented with 15% FCS, 2 mM L-glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin at 37 °C in 5% CO2. Fibroblasts were analyzed over up to 30 population doublings while the cells maintained a constant growth rate with a typical double time of 48 h. Electroporations were carried out with a BTX electroporator ECM 830 (175 V, 1 ms pulse, 5 pulses, and 0.5 s interval between pulses). Cells were electroporated in serum-free medium at ~ 2 × 107 cells/ml (fibroblasts from one 75 cm2 flask at 80–90% confluence in 100 μl) in a 2 mm gap cuvette using either 5 μg plasmid DNA and 15 μg sheared salmon sperm carrier DNA, or the indicated amount of morpholino oligonucleotide without carrier DNA. Oligonucleotides were delivered in two or three rounds at 48 h intervals for fibroblasts, and at 24 h intervals for lymphocytes. To prevent cell division, 24 h before treatment fibroblasts were either plated at 100% confluence or serum starved in 0.5% FBS. Contact inhibition and serum starvation were maintained throughout the treatment (2 deliveries over 4 days). HeLa cells were grown in DMEM supplemented with 10% FCS, 2 mM L-glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin at 37 °C in 5% CO2.
Phenotypic evaluation and statistical analysis
To quantify changes in the cellular phenotype upon treatment 200 cells were analyzed in each experiment for each parameter. Morphological abnormalities in the nucleus of control and HGPS cells were quantify based on contour ratio (4Π × area/perimeter2) as described6. The contour ratio for a circle is 1. As the nucleus becomes more lobulated, this ratio approaches 0. Nuclei with contour ratio < 0.7 were considered abnormal. The cellular levels of LAP2, lamin B, HP1α and Tri-Me-k9 in the nuclei of HGPS and control cells were quantify by imaging cells with identical settings and measuring the average intensity of the fluorescent signal in the whole nucleus using Metamorph software. The intensity from 20 control cells was averaged and used as reference to determine the fold reduction in every cell. Cells showing at least a 2-fold reduction were considered abnormal. Statistical significance of the differences in the percentage of abnormal cells was determined by contingency table analysis and χ2 test. To assess the phenotype of HGPS fibroblasts upon expression of wild type lamin A, cells were fixed 24, 72 h or 10 days after transfection. To assess the phenotype of HGPS fibroblasts upon treatment with morpholino oligonucleotide, cells were fixed after three rounds of electroporation 7 days after the first delivery.
Modified antisense oligonucleotide
Morpholino oligonucleotide were purchased from Gene Tools. Exo11: 5′-GGGTCCACCCACCTGGGCTCCTGAG-3′. Control scrambled: 5′-GCTGCCACGTGACGTGGCAGCCTCC-3′
Acknowledgments
We thank J. Boers, T. Jenuwein, K. Wilson and G. Almouzni for reagents and K. Wilson, C. Stewart, B. Burke and J. Caceres for comments on the manuscript. T.M. is Fellow of the Keith R. Porter Endowment for Cell Biology.
References
- 1.DeBusk FL. The Hutchinson-Gilford progeria syndrome. Report of 4 cases and review of the literature. J Pediatr. 1972;80:697–724. doi: 10.1016/s0022-3476(72)80229-4. [DOI] [PubMed] [Google Scholar]
- 2.Uitto J. Searching for clues to premature aging. . Trends Endocrinol Metab. 2002;13:140–141. doi: 10.1016/s1043-2760(02)00595-7. [DOI] [PubMed] [Google Scholar]
- 3.Eriksson M, et al. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature. 2003;423:293–298. doi: 10.1038/nature01629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.De Sandre-Giovannoli A, et al. Lamin A truncation in Hutchinson-Gilford progeria. Science. 2003;300:2055. doi: 10.1126/science.1084125. [DOI] [PubMed] [Google Scholar]
- 5.Csoka AB, et al. Novel lamin A/C gene (LMNA) mutations in atypical progeroid syndromes. J Med Genet. 2004;41:304–308. doi: 10.1136/jmg.2003.015651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goldman RD, et al. Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci USA. 2004;101:8963–8968. doi: 10.1073/pnas.0402943101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ye Q, Callebaut I, Pezhman A, Courvalin JC, Worman HJ. Domain-specific interactions of human HP1-type chromodomain proteins and inner nuclear membrane protein LBR. J Biol Chem. 1997;272:14983–14989. doi: 10.1074/jbc.272.23.14983. [DOI] [PubMed] [Google Scholar]
- 8.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 9.Holt I, et al. Effect of pathogenic mis-sense mutations in lamin A on its interaction with emerin in vivo. J Cell Sci. 2003;116:3027–3035. doi: 10.1242/jcs.00599. [DOI] [PubMed] [Google Scholar]
- 10.Vaughan A, et al. Both emerin and lamin C depend on lamin A for localization at the nuclear envelope. J Cell Sci. 2001;114:2577–2590. doi: 10.1242/jcs.114.14.2577. [DOI] [PubMed] [Google Scholar]
- 11.Johnson BR, et al. A-type lamins regulate retinoblastoma protein function by promoting subnuclear localization and preventing proteasomal degradation. Proc Natl Acad Sci U S A. 2004;101:9677–9682. doi: 10.1073/pnas.0403250101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sazani P, Kole R. Therapeutic potential of antisense oligonucleotides as modulators of alternative splicing. J Clin Invest. 2003;112:481–486. doi: 10.1172/JCI19547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Giles RV, Spiller DG, Clark RE, Tidd DM. Antisense morpholino oligonucleotide analog induces missplicing of C-myc mRNA. Antisense Nucleic Acid Drug Dev. 1999;9:213–220. doi: 10.1089/oli.1.1999.9.213. [DOI] [PubMed] [Google Scholar]
- 14.Summerton J. Morpholino antisense oligomers: the case for an RNase H-independent structural type. Biochim Biophys Acta. 1999;1489:141–158. doi: 10.1016/s0167-4781(99)00150-5. [DOI] [PubMed] [Google Scholar]
- 15.Lin F, Worman HJ. Structural organization of the human gene encoding nuclear lamin A and nuclear lamin C. J Biol Chem. 1993;268:16321–16326. [PubMed] [Google Scholar]
- 16.Summerton J, et al. Morpholino and phosphorothioate antisense oligomers compared in cell-free and in-cell systems. Antisense Nucleic Acid Drug Dev. 1997;7:63–70. doi: 10.1089/oli.1.1997.7.63. [DOI] [PubMed] [Google Scholar]
- 17.Ly DH, Lockhart DJ, Lerner RA, Schultz PG. Mitotic misregulation and human aging. Science. 2000;287:2486–2492. doi: 10.1126/science.287.5462.2486. [DOI] [PubMed] [Google Scholar]
- 18.Csoka AB, et al. Genome-scale expression profiling of Hutchinson-Gilford progeria syndrome reveals widespread transcriptional misregulation leading to mesodermal/mesenchymal defects and accelerated atherosclerosis. Aging Cell. 2004;3:235–243. doi: 10.1111/j.1474-9728.2004.00105.x. [DOI] [PubMed] [Google Scholar]
- 19.Mounkes LC, Kozlov S, Hernandez L, Sullivan T, Stewart CL. A progeroid syndrome in mice is caused by defects in A-type lamins. Nature. 2003;423:298–301. doi: 10.1038/nature01631. [DOI] [PubMed] [Google Scholar]
- 20.Moulton HM, Nelson MH, Hatlevig SA, Reddy MT, Iversen PL. Cellular uptake of antisense morpholino oligomers conjugated to arginine-rich peptides. Bioconjug Chem. 2004;15:290–299. doi: 10.1021/bc034221g. [DOI] [PubMed] [Google Scholar]
- 21.Gebski BL, Mann CJ, Fletcher S, Wilton SD. Morpholino antisense oligonucleotide induced dystrophin exon 23 skipping in mdx mouse muscle. Hum Mol Genet. 2003;12:1801–1811. doi: 10.1093/hmg/ddg196. [DOI] [PubMed] [Google Scholar]
- 22.Garcia-Blanco MA, Baraniak AP, Lasda EL. Alternative splicing in disease and therapy. Nat Biotechnol. 2004;22:535–546. doi: 10.1038/nbt964. [DOI] [PubMed] [Google Scholar]