

Abstract
The carboxyl terminus of the G protein α subunit plays a key role in interactions with G protein-coupled receptors. Previous studies that have incorporated covalently attached probes have demonstrated that the carboxyl terminus undergoes conformational changes upon G protein activation. To examine the conformational changes that occur at the carboxyl terminus of Gα subunits upon G protein activation in a more native system, we generated a semisynthetic Gα subunit, site-specifically labeled in its carboxyl terminus with 13C amino acids. Using expressed protein ligation, 9-mer peptides were ligated to recombinant Gαi1 subunits lacking the corresponding carboxyl-terminal residues. In a receptor-G protein reconstitution assay, the truncated Gαi1 subunit could not be activated by receptor; whereas the semisynthetic protein demonstrated functionality that was comparable with recombinant Gαi1. To study the conformation of the carboxyl terminus of the semisynthetic G protein, we applied high resolution solution NMR to Gα subunits containing 13C labels at the corresponding sites in Gαi1: Leu-348 (uniform), Gly-352 (α carbon), and Phe-354 (ring). In the GDP-bound state, the spectra of the ligated carboxyl terminus appeared similar to the spectra obtained for 13C-labeled free peptide. Upon titration with increasing concentrations of AlF4−, the 13C resonances demonstrated a marked loss of signal intensity in the semisynthetic Gα subunit but not in free peptide subjected to the same conditions. Because AlF4− complexes with GDP to stabilize an activated state of the Gα subunit, these results suggest that the Gα carboxyl terminus is highly mobile in its GDP-bound state but adopts an ordered conformation upon activation by AlF4−.
Heterotrimeric G proteins mediate signal transduction between G protein-coupled receptors (GPCRs)1 and a host of downstream intracellular effectors. The α subunit binds GDP and forms a tight complex with βγ subunits. Activated GPCRs can catalyze the exchange of GDP for GTP, which leads to dissociation of Gα from the complex. The structure of the Gα subunit has been solved bound to GDP (1), GTPγS (2), GDP-AlF4− (3), and in complex with Gβγ (4). Gα has two domains, a GTPase domain that is similar to the Ras-like GTPase proteins and an amino-terminal helical domain that is unique to the heterotrimeric G proteins. The GTPase domain consists of five helices surrounding a six-stranded β-sheet, whereas the helical domain has one long helix surrounded by five short helices. In the GTPase domain, GDP to GTP exchange results in conformational changes at three regions near the guanine nucleotide binding pocket, referred to as switch I (11 amino acids), switch II (21 amino acids), and switch III (10 amino acids) (1). The switch regions of Gα subunits either bind to βγ subunits or to downstream effectors, such as adenylyl cyclase and phospholipases, with the specificity of the Gα interactions dictated by GTP exchange and the conformation of the switch regions.
Although the crystal structures reveal the conformational changes in the switch regions and thus provide a structural basis for the specificity of interactions of the GDP-bound versus GTP-bound Gα subunits, the structures of Gα subunits and heterotrimeric G proteins provide little information regarding how receptors catalyze GTP exchange. The α subunit of transducin, Gαt, has been shown by mutational studies to contain three regions critical for rhodopsin interaction: 1) the amino-terminal 23 residues, 2) an internal sequence consisting of positions 311–329, and 3) the carboxyl-terminal eleven residues, 340–350 (reviewed in Ref. 5). When comparing the heterotrimeric structures versus the activated GTPγS-Gα structures, the internal sequence (311–329) does not change conformation significantly in the GDP-versus GTP-bound states, and the amino and carboxyl termini are not resolved in most of the structures. The structure of the carboxyl terminus of Gα is of particular interest, because this region confers receptor-specific interactions, and peptide analogs of the tail bind to rhodopsin and stabilize the MII activation state of the receptor. In most structures of G proteins, the carboxyl terminus is not resolved, suggesting that this region is relatively mobile. However, in three independent crystal structures, a structured carboxyl terminus is observed, although the conformations differ. In the structure of RGS 4 bound to AlF4−-activated Gαi1 (6), the carboxyl terminus adopts an ordered continuous helix terminated by a carboxyl-terminal capping motif. Of note, this structure is consistent with transferred nuclear Overhauser effect structures of carboxyl-terminal peptides of Gαt bound to photo-activated rhodopsin (7–9). In contrast, the crystal structure of Gαt complexed with GTPγS shows the carboxyl-terminal residues in an extended linear structure, and in one molecule of the asymmetric unit cell, residues 343–349 are in van der Waals contact with residues 212–215 of the α2/β4 loop, which is part of the switch II region of Gα (2). In the crystal structure of Gαi1 bound to GDP, the carboxyl tail forms an extended α-helical structure that is cradled between the amino terminus and the body of the GTPase domain (10). Although the crystal structures demonstrate disordered termini or different, and seemingly conflicting, structures of the carboxyl tail of Gα subunits, cross-linking and biophysical studies using fluorescent probes provide compelling evidence that the conformation of the carboxyl tail does change upon G protein activation (11, 12).
What happens to the Gα structures at the receptor/G protein interface upon receptor activation? Several studies have implicated the carboxyl terminus to be involved in regulating receptor interaction and affecting GDP affinity (13–16) and GDP-GTP exchange (17, 18). In addition, carboxyl-terminal peptides from several Gα subunits can both competitively inhibit G protein activation and stabilize the active conformation of G protein-coupled receptors (7, 8, 19–21). Taken together, these studies suggest that interactions between the Gα carboxyl terminus and GPCRs might play essential roles in communication between the receptor and the GDP binding pocket of the Gα subunit. Further understanding of these conformational changes should thus provide a better understanding of the mechanism of nucleotide exchange on α subunits of G proteins.
To date, most biophysical studies have focused either on peptide segments of the G protein or bulky fluorophores inserted by standard molecular techniques on the receptor or G proteins. Here, we use expressed protein ligation (EPL) to incorporate 13C isotope-labeled amino acids into the carboxyl-terminal tail of Gα. EPL takes advantage of the ability of intein domains to generate thioester intermediates in recombinant proteins, which can then be ligated to peptides containing an amino-terminal cysteine residue. Inteins, the protein equivalents of introns, are found in prokaryotes, archaebacteria, and simple eukaryotes and possess the unique ability to autocatalytically splice themselves from newly translated proteins and, in the process, regenerate a native amide bond at the splice junction (22). By modifying the intein, a thioester intermediate can be trapped and purified for use in native chemical ligations (23). For this study, we ligated a nine-amino-acid carboxyl-terminal peptide to a recombinant Gα (1–345) thioester lacking the carboxyl terminus to create the full-length Gα (1–354). We demonstrated the ability to generate a semisynthetic Gα subunit containing 13C-labeled amino acids in its carboxyl terminus that retains native Gα activity as assayed by its ability to be activated by receptors. Furthermore, we have studied the mobility of the carboxyl terminus of this semisynthetic Gα by high resolution solution NMR. The experiments show that the carboxyl terminus is highly mobile when in the inactive conformation and that binding of AlF4− restricts this motion.
MATERIALS AND METHODS
Peptide Synthesis
Peptides were manually synthesized by fluorenylmethoxycarbonyl (Fmoc) solid phase peptide synthesis. 13C-labeled amino acids were obtained from Cambridge Isotope Laboratories (Andover, MA) and were treated with Fmoc-(oxy)succinimide to afford the amino terminus Fmoc protection. After the final cleavage step, crude peptides were purified by preparative high pressure liquid chromatography using a C18 column and characterized by electrospray mass spectrometry.
Cloning, Expression, and Purification of Proteins
A plasmid encoding for His6Gαi1 (pQE-6 expression vector) was kindly provided by Dr. M. Linder, Washington University, St. Louis, MO. Plasmid pBN1018, which encodes His6Gαi1 containing a N342C mutation, was generated by site-directed mutagenesis with Pfu Turbo DNA polymerase (Stratagene). The plasmid pBN905, which expresses Gαi1ΔCT, truncated by nine residues at its carboxyl terminus, fused in-frame to intein-CBD cDNA from the pTXB3 expression vector (New England Biolabs) was constructed as follows. Initially, a His6Gαi1/αt chimera (Chi6, a gift from Dr. H. Bourne, UCLA; originally from Dr. H. Hamm, Vanderbilt University) was digested with PstI/BglII to generate a 3.4-kb fragment. Intein-CBD cDNA from pTXB3 was digested with PstI/SpeI and sub-cloned along with a BglII/SpeI oligonucleotide cassette into the pChi6 expression vector to generate pBN834. To generate pBN905, pHis6Gαi1 was digested with EcoRI/AatII and subcloned, along with an AatII/SpeI oligonucleotide cassette into pBN834, which had been digested with EcoRI/SpeI. This strategy resulted in an expression vector containing cDNA encoding Gαi1ΔCT (truncated by nine residues at the carboxyl terminus) fused in-frame with intein-CBD, without the introduction of extra amino acids. Plasmids were transformed into Escherichia coli BL21 cells. The cells were grown in 3 liters of T7 medium (20 g of tryptone, 10 g of yeast extract, 5 g of NaCl, 2 ml of glycerol, and 50 ml of 1 m KH2PO4, pH 7.2, per liter) in the presence of 50 μg/ml ampicillin at 25 °C up to an A600 of 0.5–0.8. The cells were then induced with 50 μ m isopropyl-1-thio-β-d-galactopyranoside at 25 °C for 4–6 h.
After recovery by centrifugation, the cell pellet was resuspended in 1:20 of cell culture volume with lysis buffer (25 mm Tris-HCl, pH 8.0, 250 mm NaCl, 0.1 mm EDTA, 5% glycerol, 5 mm MgCl2, 50 μm GDP, 0.1 mm phenylmethylsulfonyl fluoride). The lysis buffer for His6Gαi1 and His6Gαi1N342C also contained 5 mM β-mercaptoethanol, and lysis buffer for His6Gαi1ΔCT-intein-CBD contained 0.1% Triton X-100. The cell suspension was lysed by sonication, and crude cell lysate was cleared by low speed centrifugation followed by centrifugation at 100,000 × g for 60 min.
His6 purification of His6Gαi1 and His6Gαi1N342C was performed as described previously (24). Briefly, the supernatant was adjusted to 500 mm NaCl, 20 mm imidazole with the addition of 8× binding buffer (160 mm Tris HCl, pH 8.0, 4 m NaCl, and 160 mm imidazole). The resultant mixture was loaded onto two 5-ml metal chelating columns (Pharmacia Corporation) charged with 100 mm NiCl2 and prepared according to the manufacturer’s protocol, at a flow rate of 1 ml/min. The column was washed with 10 volumes of 1× binding buffer, and the bound protein was eluted with 5 column volumes of 1× binding buffer containing 80 mm imidazole (100 mm total). GDP, MgCl2, and β-mercaptoethanol were added to each fraction at a concentration of 25 μm, 2 mm, and 5 mm, respectively. Fractions were analyzed by SDS-PAGE for protein purity, quantitated by comparison to known bovine serum albumin standards, pooled, and dialyzed overnight in dialysis buffer (20 mm Tris-HCl, pH 8.0, 150 mm Nacl, 2 mm MgCl2, 50 μm GDP, and 20% glycerol). Samples were aliquoted and stored at −80 °C. The final yield of Gα subunits was 1–2 mg/liter.
Protein Ligation
Purification of His6Gαi1ΔCT-intein-CBD was as follows. After cell lysis, the clarified cell lysate was applied to an affinity column containing chitin matrix that had been equilibrated with 10 column volumes of column buffer (20 mm Tris-HCl, pH 8.0, 250 mm NaCl, 1 mm EDTA, and 0.1% Triton X-100). The column was then washed with 20 column volumes of column buffer. Treatment with cleavage buffer (20 mm Tris-Cl, pH 8.0, and 500 mm NaCl) containing 100 mm mercaptoethanesulfonic acid overnight at 4 °C resulted in self-cleavage of the intein, releasing the Gαi1ΔCT thioester from the chitin-bound intein. Fractions were eluted with 3 column volumes of column buffer (minus Triton X-100). Collected fractions were treated as above, and the Gαi1ΔCT protein was stored at −80 °C and used as a control in functional assays. Expressed protein ligation of the synthetic peptides to GαΔCT-intein-CBD was carried out as follows. Following loading of the Gα-intein-CBD fusion protein onto the chitin matrix, cleavage was initiated by quickly flushing the column with cleavage buffer containing 1% mercaptoethanesulfonic acid. Synthetic peptides, corresponding to the carboxyl-terminal tail of Gα, were dissolved into cleavage buffer and applied to the chitin column in the presence of 1% mercaptoethanesulfonic acid at an excess of 10–20-fold over protein concentration. The ligation reaction proceeded at 4 °C and was found to be complete within 24 h. The column was eluted with column buffer (minus Triton X-100). The fractions were treated as above, with the exception of an additional reduction step before dialysis. In this step, the pooled fractions were treated with 15 mm dithiothreitol for 30 min at 30 °C to remove peptide linked to the Gα through nonspecific disulfide interaction. The semi-synthetic proteins were analyzed by SDS-PAGE using antibodies corresponding to the carboxyl-terminal synthetic peptide and quantitated on Coomassie-stained gels using bovine serum albumin standards. Protein was concentrated to 5 mg/ml using a Centricon centrifugal device (molecular weight cut off 10,000; Amicon). Ligation was estimated to be 50–70%.
Quantification of Ligation Efficiency
The efficiency of ligation was estimated by quantitative immunoblotting with anti-Giα1, carboxyl-terminal (residues 345–354), and anti-Gαi-(40–54) antibodies (EMD Biosciences) and treatment with goat anti-rabbit IgG conjugated to horseradish peroxidase (Chemicon International), followed by incubation with Lumi-LightPLUS Western blotting substrate (Roche Applied Science). Images were acquired using the ChemiDoc Gel Documentation System (Bio-Rad) and analyzed with the program Quantity One (version 4). For each set of immunoblots, known amounts of purified GαiN346C subunit (0, 210, 420, and 630 ng) served as internal standards with which serial dilutions of the ligated sample were compared. To determine the yield of the ligation reaction, the amount of protein determined by blotting with the anti-Gαi antibody (ligated plus unligated protein) was compared with the amount of protein determined using the anti-Gα carboxyl-terminal antibody (ligated protein only). Yields generally ranged from 50 to 70%.
Membrane Preparation
Chinese hamster ovary membranes containing C5aR were prepared as described previously (25, 26). Briefly, Chinese hamster ovary cells stably expressing C5aRs were harvested, lysed with buffer containing 50 mm Tris, pH 7.5, 1 mm EDTA, 20 μg/ml aprotinin, and 0.5 mm phenylmethylsulfonyl fluoride, and homogenized by aspirating through a 27-gauge needle 10 times. The lysed solution was first centrifuged at 960 × g for 5 min twice, followed by an additional centrifugation at 217,000 × g for 30 min at 4 °C using a sucrose cushion (250 mm sucrose, 50 mm Tris, pH 7.5, 1 mm EDTA). The supernatant was aspirated, and the pellet was resuspended in 1 ml of 6 m urea (10 mm HEPES, pH 7.3, 2.5 mm EGTA), incubated at 4 °C for 30 min, and the membranes were pelleted at 217,000 × g for 30 min at 4 °C to strip the membranes of GTP-binding proteins. After a second urea wash and sedimentation, the membranes were resuspended in sucrose buffer, frozen, and stored at −80 °C.
Gβγ Purification
Gβγ was purified from Sf9 insect cells as described previously (27). The baculovirus encoding Gβ1His6-γ2 was obtained from M. Linder, Washington University, St. Louis, MO. Peak fractions from the MonoQ were concentrated and exchanged into a buffer containing 20 mm HEPES, pH 8.0, 0.1 mm EDTA, 50 mm NaCl, 2 mm β-mercaptoethanol, and 0.7% CHAPS. The final protein concentration was estimated against the known concentrations of bovine serum albumin.
Receptor-catalyzed GDP/GTPγS Exchange Assay
The receptor-catalyzed exchange of GDP for GTPγS was based on a modification of procedures by Fawzi et al. (28). Reactions were performed in a total assay volume of 20 μl. Urea-washed membranes containing C5aR were mixed with G protein subunits on ice in buffer to give a final concentration of 20 mm HEPES, pH 7.6, 100 mm NaCl, 2 mm MgCl2, 0.3 mM β-mercaptoethanol, and 1 μm GDP. The reactions were initiated by the addition of 3 μm [35S]GTPγS (5000 cycles/min/pmol) and 100 nm C5a ligand, reacted at 30 °C for 10 min, and terminated by the addition of ice-cold wash buffer (20 mm Tris-Cl, pH 8.0, 25 mm MgCl2, and 100 mM NaCl) followed by filtration over BA85 nitrocellulose filters. Filters were washed with the ice-cold wash buffer, dried, and radioactivity measured by liquid scintillation counting.
Sample Preparation
The concentration of the protein sample was 50 μm. A total of 1 mg of semisynthetic Gα protein (13C-labeled in the carboxyl terminus) was used. The concentrations of AlF4− were 100 and 200 μm (100 μm AlCl3, 10 mM NaF) corresponding to 2 and 4 molar equivalents of AlF4−.
NMR Spectroscopy
NMR spectra were collected with a Bruker Avance NMR spectrometer at a 1H frequency of 700.13 MHz using a 5-mm triple resonance HCN probe. The two-dimensional 1H-13C HSQC spectrum was obtained using 256 increments in the t1 dimension, and 1200 transients were collected at each increment and averaged. The recycle delay was 1.5 s. The temperature was maintained at 25 °C.
RESULTS
Peptide Design
The scheme for the generation of semisynthetic Gα subunits is shown in Fig. 1. The junction between the recombinant thioester lacking the carboxyl terminus (designated Gαi1ΔCT) and the synthetic peptide was selected based on the criteria that a cysteine substitution would likely be tolerated at this position, because the EPL mechanism requires a cysteine at the splice point. An alanine-scanning mutagenesis study of Gαt (13) examined the effects of substitutions throughout the entire protein and demonstrated that an alanine point mutation at Glu-342 within the carboxyl-terminal tail was well tolerated. The Glu-342 position in Gαt corresponds to an Asn-346 residue in Gαi1; thus the N346C mutation was made, and the peptide synthesized was CNLKDCGLF (residues 346–354 of Gαi1).
Fig. 1. Expressed protein ligation.
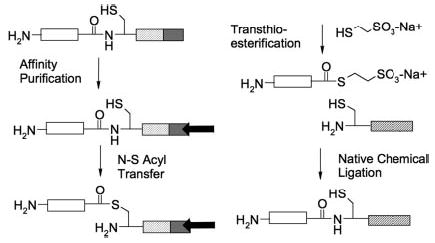
Gαi1ΔCT (white box) is expressed in E. coli as a fusion protein, Gαi1ΔCT-intein-CBD. The chitin-binding domain (CBD; gray box) allows affinity purification on the chitin matrix (black arrow). Intein domain (diagonal lines) catalyzes N→S acyl transfer. Incubation with mercaptoethanesulfonic acid results in cleavage of the target protein from the intein-CBD domains, generating recombinant Gαi1ΔCT thioester. The thioester protein thereafter undergoes native chemical ligation with CNLKDCGLF peptide (cross-hatched).
The remaining sequences of the carboxyl termini of Gαi1 and Gαt are identical after positions Glu-342 and Asn-346, respectively; therefore, we reasoned that previous results of mutational data of Gαt and biophysical studies of transducin peptides could be applied directly to Gαi1. For NMR studies, we selected which amino acids in the synthetic carboxyl-terminal peptide to 13C label based on the transferred nuclear Overhauser effect structure of the α-peptide (11 carboxyl-terminal residues of Gαt) bound to R* (8). In this structure, Gly-348 is critical for forming the C-cap turn observed upon photoactivation, and Leu-344 and Phe-350 are members of the hydrophobic cluster. The peptide synthesized for ligation was labeled at the corresponding sites in Gαi1: Leu-348 (U-13C), Gly-352 (2-13C), and Phe-354 (ring-13C). The 13C-LGF labels provide a total of 12 unique 13C sites to monitor structural changes in the carboxyl terminus via NMR.
Preparation of Semisynthetic Gαi1
Initial experiments to optimize peptide ligation and demonstrate functionality of the recombinant Gα subunit were performed using an unlabeled synthetic peptide (CNLKDCGLF) corresponding to the carboxyl terminus of Gαi1 and recombinant Gαi1ΔCT. For the EPL methodology, Gαi1 offers the advantage of much higher yields in bacterial expression systems when compared with Gαt. A recombinant protein consisting of residues 1–345 of Gαi1 was fused to an intein-CBD. Ligation of the recombinant protein (Gαi1-(1–345)) to the synthetic peptide was carried out as described under “Materials and Methods.” The following control proteins were also generated: 1) the unligated Gαi1ΔCT protein, obtained by eluting the thioester form of Gαi1 from the chitin column, and 2) full-length Gαi1N346C, because ligation of synthetic peptide to the recombinant protein introduces a cysteine at the ligation junction. The ligation products were analyzed using antibodies corresponding to the carboxyl-terminal synthetic peptide (anti-Gαi1; KNNLKDCGLF). Because there was no cross-reactivity with the recombinant thioester (Fig. 2, lane 1), Western blot analysis proved to be a convenient and sensitive method for detecting and monitoring the ligation reaction (Fig. 2). Analysis of the ligation product revealed that the reaction was complete after 24 h at 4 °C (Fig. 2, lanes 3 and 4) and that the ligation products remained stable even after 72 h (Fig. 2, lanes 7 and 8).
Fig. 2. SDS-PAGE/Western analysis of semisynthetic Gα.
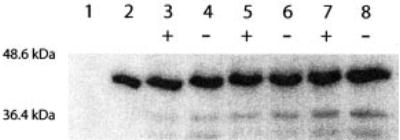
Ligated product was observed after 24 h. Lane 1, purified Gαi1ΔCT thioester (negative control); lane 2, purified Gαi1 (N346C). Time points were taken at 24 (lanes 3 and 4), 48 (lanes 5 and 6), and 72 h (lanes 7 and 8) and treated with (+) or without (−) 15 mm dithiothreitol before separation on 10% SDS-polyacrylamide gels. Products were transferred to membranes and blotted with anti-carboxyl-terminal Gαi1 antibody.
The yield of the ligation reaction was estimated by quantitative Western blot analysis. In this instance, two individual Gα antibodies were used, one to the carboxyl terminus (to monitor ligation efficiency) and the second to an internal site within Gα (to quantify the total amount of Gα subunit present). Direct comparison of the total amount of Gα, including both ligated and unligated components, to the amount of detected ligated product provided an estimated ligation efficiency of 50–70% (data not shown). In other EPL studies, higher yields of protein ligation (approaching >90%) can be achieved with the use of chaotropic agents (i.e. guanidinium-hydrochloride and urea), organic solvents (i.e. Me2SO), or higher temperatures (usually 25–30 °C) (29). However, such agents result in protein denaturation, thereby requiring a strategy for refolding the semisynthetic protein when it is to be used for biological assays. Here, all procedures were carried out at conditions optimal for maintaining stability and function of the Gα subunit, which might have lowered the efficiency of chemical ligation.
Semisynthetic Gαi1 Is Functional
Using membrane reconstitution assays, we characterized the ability of the semisynthetic Gαi1 subunits to be activated by GPCRs. Given the large number of studies implicating the carboxyl terminus as essential for the ability of receptors to activate G proteins, we reasoned that this would serve as the strictest test of functionality of ligated G proteins. Intact membranes were isolated from Chinese hamster ovary cells stably expressed with human C5a receptors, a chemoattractant receptor and member of the rhodopsin family of GPCRs. C5a receptors activate Gαi subunits and mediate neutrophil chemotaxis in response to gradients of the C5a ligand, a 74-amino-acid cleavage product of the complement cascade. The receptor-containing membranes were washed with 6 m urea to remove endogenous G proteins. Recombinant Gα proteins, expressed and purified from BL21 (DE3) E. coli, and Gβγ, purified from Sf9 insect cells, were incubated with whole membranes, C5a ligand, and 35S-labeled GTPγS. Fig. 3 demonstrates the receptor-catalyzed GDP-GTP exchange. The ligated Gαi bound 4.8 pmol of GTPγS in response to C5a ligand versus 3.0 pmol in the absence of ligand (1.6-fold stimulation). The amount of GTPγS binding observed for the native Gαi subunit was 7.2 pmol bound (4.3-fold stimulation). For a 10-min reaction, this amount of GTPγS binding corresponds to a turnover rate of ~50 G proteins/ligand-stimulated receptor. The majority of the basal GTPγS binding that occurs in the absence of ligand most likely represents the intrinsic GTPγS binding activity of the Gα subunit (a function of the GDP off rate), because each of the recombinant G proteins in the absence of membranes bound similar amounts of GTPγS (data not shown). The extent of receptor activation observed for the G proteins in these assays is comparable with those published for activation of purified Gα subunits by other GPCRs in similar membrane reconstitutions systems (30, 31).
Fig. 3. C5a receptor activation of ligated G proteins.
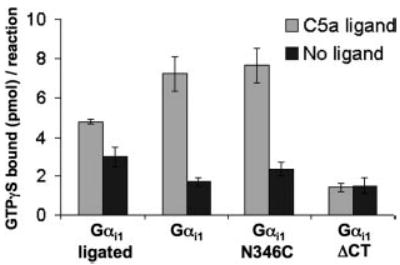
C5a receptors (1–5 nm) in urea-stripped membranes were incubated with C5a ligand (100 nm) and Gα proteins (1 μm) and Gβγ (0.5 μm) and GTPγS (3 μm) in 20-μl reactions for 10 min at 30 °C and then filtered and counted. Data shown are the average ± S.E. of six experiments.
To ensure that the introduction of a cysteine substitution at Asn-346 did not affect the function of the carboxyl terminus in receptor activation, we also assessed the ability of the recombinant GαiN346C mutant to bind GTPγS in response to ligand stimulation of the C5a receptor in reconstituted membranes. Confirming our prediction that this position should tolerate a cysteine mutation, we found that the C5a receptor activated the GαiN346C mutant to an extent equivalent to the wild-type Gαi1 subunit (Fig. 3). We also tested Gαi that lacked the terminal nine amino acids. The GαiΔCT protein demonstrated increased basal levels of GTPγS binding but did not respond to receptor stimulation. The fact that the truncated Gα subunit Gαi1ΔCT displayed increased basal GTPγS binding is consistent with previous work that demonstrates that truncation of the carboxyl-terminal 10 residues of Gαo resulted in decreased affinity of the G protein for GDP but with no change in affinity for GTPγS (14). The complete lack of receptor stimulation of the GαiΔCT mutant underscores the importance of the carboxyl terminus for activation of G proteins by receptors.
Truncated Gαi1 Acts as Competitive Inhibitor for Receptor Activation
Although the ligated Gαi1 demonstrated receptor-catalyzed nucleotide exchange (1.6-fold stimulation), the activity of the ligated G protein is less than that observed for full-length Gαi1 or Gαi1N346C (3–5-fold stimulation). One potential explanation for this decreased activity might be that the unligated component of G proteins present in the semisynthetic Gαi1 preparations (ranging from 30 to 50% of the total G protein) might compete for receptor sites on our membranes and thereby inhibit receptor activation of full-length ligated Gαi subunits. We tested this possibility by performing competition assays, adding increasing amounts of unligated G protein (GαiΔCT) to full-length Gαi1N346C and monitoring the receptor-catalyzed nucleotide exchange. We found that the addition of Gαi1ΔCT protein inhibited the receptor-catalyzed exchange of GDP for GTP for Gαi1N346C (Fig. 4). Because the efficiency of ligation of our semisynthetic Gα subunits is 50–70%, we can translate our finding to be consistent with what we are observing upon the addition of 125–250 nm unligated protein in Fig. 4. These data demonstrate that the addition of 125 nm unligated protein (33% of total) to 250 nm recombinant Gαi1N346C severely masks the full functional activity of the active subunit (8.4-fold stimulation versus 1.9-fold stimulation) and implicates the unligated protein as a competitive inhibitor. For the following solution NMR studies, we assumed that the presence of unligated Gα subunit would not affect the conformational changes that might occur during G protein activation. Because the unligated protein does not contain isotope labels, it should not contribute to the obtained spectra. The fact that our semi-synthetic proteins displayed receptor-catalyzed nucleotide exchange even in the presence of a competitive inhibitor was quite compelling. These findings further implicate the carboxyl terminus to be essential for receptor activation but not GTPγS binding.
Fig. 4. Competitive inhibition of C5a receptor activation by GαiΔCT.
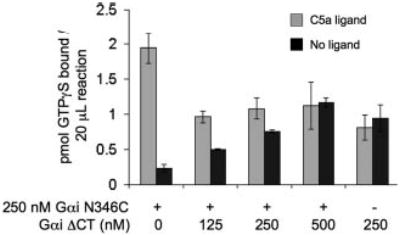
C5a receptors (1–5 nm) in urea-stripped membranes were incubated with C5a ligand (100 nm) and GαiN346C proteins (250 nM) and Gβγ (0.5 μm) and GTPγS (3 μm) in 20-μl reactions. Indicated amounts of GαiΔCT were added, and reactions proceeded for 10 min at 30 °C and then filtered and counted. Data shown are the average ± S.E. of three experiments.
HSQC NMR Spectrum of 13C-Gα Protein
Fig. 5 presents the 1H-13C HSQC NMR spectrum of the semisynthetic Gαi1 subunit that has been 13C-labeled at Leu-348, Gly-352, and Phe-54. The Cβ-H, Cδ-H, and Cγ-H resonances from Leu-48 are observed in Fig. 5A, the Cα-H resonances of Gly-352 and Leu-348 are observed in Fig. 5B, and the Cδ-H resonances of the Phe-354 aromatic ring are observed in Fig. 5C. Both the 1H and 13C chemical shifts are close to their positions in the free amino acids, indicating no dispersion due to protein interactions.
Fig. 5. 1H-13C HSQC spectrum of Gα protein labeled at Leu-348, Gly-352, and Phe-354.
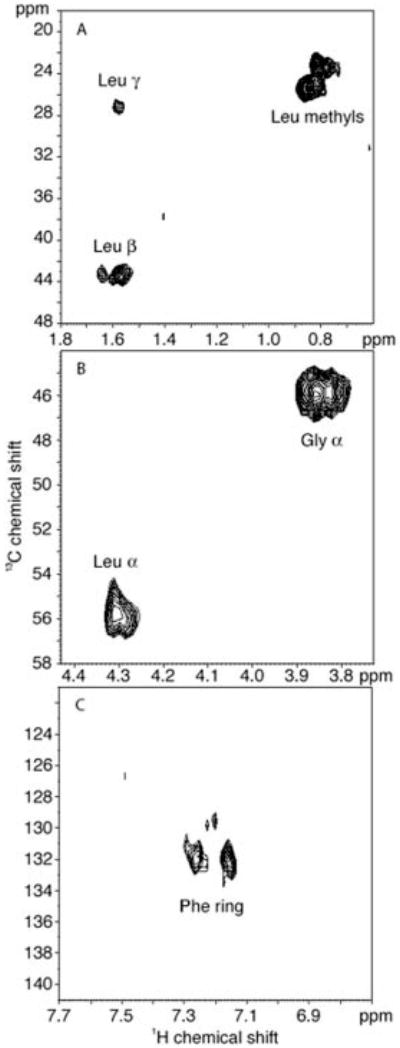
The panels display the regions containing the Leu-348 methyl Cβ-H and Cγ-H resonances. The Gly-352 (A) and Leu-348 (B) Cα-H resonances and the Phe354-13C-ring resonances (C).
The 1H line widths in the HSQC spectrum range from 27 Hz for Leu-348 methyl protons to 50 Hz for the Gly-352 Cα-H protons. For comparison, the methyl line widths in the HSQC spectrum of ubiquitin, a small 76-amino-acid protein, are on the order of 24 Hz. Based on the average line widths in ubiquitin and the Gα subunit, an analysis of the rotational correlation times indicates that the carboxyl terminus in the Gα subunit is much more mobile than expected for a 40-kDa protein.
Mobility of Carboxyl Terminus; Titration with AlF4−
Activation of Gα subunits results in characteristic changes in both their structure and function. Such activation can be induced using nonhydrolyzable analogs of GTP (such as GTPγS) or by AlF4−, an analog of the γ-phosphate of GTP that coordinates with GDP to mimic the active state of the G protein. To characterize the influence of AlF4− on the NMR spectrum of the Gα subunit, the semisynthetic 13C-labeled Gαi1 protein was titrated with 2 and 4 molar equivalents of AlF4−. The addition of AlF4− resulted in a loss of intensity in all of the resonances observed in the HSQC spectrum for the 13C-LGF protein. Examination of slices from the two-dimensional HSQC spectra at different concentrations of AlF4− showed that there is a loss of total intensity in the lines and not an increase in the broad component at the foot of the peak (Fig. 6). Although all of the resonances decreased, the intensity losses of the Gly-352 and Phe-354 resonances were approximately twice the losses in the Leu-348 resonances. As a control, the addition of AlF4− to a solution of the 13C-labeled nine-residue peptide corresponding to the carboxyl terminus of Gα did not influence the line width, consistent with the carboxyl-terminal peptide alone being free and mobile (data not shown). The loss of intensity can only be attributed to loss of mobility of the carboxyl-terminal tail upon the addition of AlF4−. The larger influence on Gly-352 and Phe-354 suggests that these carboxyl-terminal residues may become buried upon an AlF4−-induced conformational change.
Fig. 6. Intensity of the HSQC NMR resonances as a function of added AlF4−.
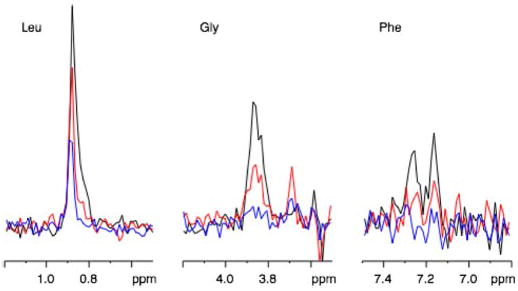
Slices from the two-dimensional HSQC spectra at different concentrations of AlF4−. Black line is spectra obtained with no additional AlF4−. Red indicates two molar equivalents of AlF4−. Blue represents four molar equivalents of AlF4−.
Fig. 7 presents the 1H spectrum of the Cα-H region of the ligated Gα protein before and after the addition of AlF4−. The absence of any significant change in the proton line widths indicates that the dramatic loss of intensity observed in the 13C resonances of the carboxyl terminus does not represent a global change in the motion of the protein due, for instance, to oligomerization or aggregation but is restricted to a large change in the mobility of the carboxyl-terminal tail.
Fig. 7. One-dimensional 1H spectra of the Gα protein before and after the addition of AlF4−.
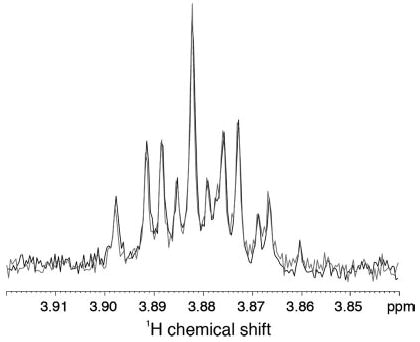
The spectra are virtually identical, indicating that there is no overall change in the mobility of the protein due to the binding of AlF4−. The changes in the carboxyl-terminal region of the protein are dramatic and localized.
DISCUSSION
Crystal structures and biochemical studies of G proteins have identified putative receptor interaction sites and regions that undergo conformational changes as a result of nucleotide exchange. The receptor binding regions on Gα are known to be located at some distance (25–30 Å) from the GDP binding pocket. Still, several questions regarding G protein activation remain. For instance, how does receptor binding trigger GDP release? How does GTP binding lead to dissociation of Gα from Gβγ and the receptor?
Here, we probed the conformational changes of the carboxyl terminus of Gα associated with G protein activation using solution NMR. We have developed a strategy, using expressed protein ligation, to site-specifically introduce 13C-labeled amino acids into the carboxyl terminus of a recombinant Gαi1 subunit. Solution NMR studies on the ligated 13C Gα subunit revealed a loss in intensity of all 13C resonances upon titration with AlF4−, which when bound to Gα serves to mimic the activated state of the G protein. This loss in intensity can be directly attributed to loss of mobility of the carboxyl-terminal tail upon the addition of AlF4−. The spectra show that there is a loss of total intensity in the relatively narrow NMR resonances (Fig. 6). This would be consistent with the carboxyl terminus being in slow exchange between a free (mobile) conformation and an ordered (bound) conformation. The resonance corresponding to the bound conformation was not observed, even as a broad component in the spectrum. The complete loss of intensity for the bound conformation suggests that the wild-type Gα subunit is oligomeric or aggregated in the absence of the Gβ and Gγ subunits. If the carboxyl terminus is in dynamic equilibrium (i.e. in the intermediate exchange regime) between the free and bound conformations, we would expect to see a change in the line width as a function of added AlF4−, which we did not observe. Lowering the temperature to 4 °C from 25 °C did not change the observed line widths, suggesting that the carboxyl terminus is not in fast exchange (data not shown). Also, the greater intensity loss observed at Gly-352 and Phe-354 compared with Leu-348 further suggests that the extreme carboxyl-terminal residues become buried upon an AlF4−-induced conformational change. One can speculate that the carboxyl-terminal aromatic group (Phe-354) finds a binding site in the GTP-stabilized form of Gα. However, we cannot determine this from our study, because the loss of signal intensity that occurs with the addition of AlF4− makes an assignment of a specific structure difficult.
Our finding that the carboxyl terminus of Gα subunit becomes ordered upon activation by AlF4− is supported by previous work by Hamm and co-workers (11). Using lucifer yellow, an environmentally sensitive fluorescent probe, to label Cys-347 on the carboxyl terminus of Gαt/Gαi chimera, Hamm and co-workers observed an increase in fluorescence upon binding of AlF4− to the G protein. The increased fluorescence can be interpreted as the fluorescent label moving from a relatively hydrophilic environment to one that is more hydrophobic. Moreover, the kinetics of the fluorescence change of the lucifer yellow probe correlated well with the rate of formation of the active state, as monitored by the intrinsic fluorescence change of Trp-207 in switch II. Consistent with the idea that Gα activation buries the carboxyl terminus, the ability to label Cys-347 with lucifer yellow decreased after AlF4− binding. It is difficult to know what effect, if any, the lucifer yellow probe (Mr 457.2) might introduce on the conformation of the tail. Modification of the Cys-347 by the lucifer yellow would be predicted to disrupt receptor activation, much like ADP-ribosylation of Cys-347 by pertussis toxin. Therefore, the findings of our solution NMR studies demonstrating AlF4−-dependent ordering of the carboxyl terminus provide strong validation of the results obtained with fluorescent labeling. Moreover, our studies introduce isotope labels that should not perturb functionality and therefore are more likely to reflect conformational changes that occur in the native Gα subunit.
How do the NMR results of the carboxyl terminus of Gαi compare with crystallography studies? In the GDP-bound state, the carboxyl terminus appeared highly mobile, which is consistent with the observation that the carboxyl terminus is not resolved in all but one of the crystal structures of the heterotrimeric G proteins and Gα subunits bound to GDP. In the one example where the carboxyl terminus is ordered in the GDP-bound state of the Gα subunit (Gαi1), the tertiary and quaternary structure revealed microdomains composed of regions from the amino terminus of one monomer and the carboxyl terminus of another monomer, suggesting the presence of head-to-tail Gα polymers (10). Based on these data and the observation that the amino and carboxyl termini of Gα subunits appear disordered in many structures of GTP-bound Gα subunits, the authors suggest that polymers of α subunits form as a result of GTP hydrolysis and that the polymers might provide a functional role in signal transduction. The data obtained in our solution NMR studies do not support this model, because the carboxyl terminus of Gαi1 bound to GDP appeared freely mobile, whereas the tail became ordered upon activation of G protein by the addition of AlF4−. In two crystals of activated Gα subunits determined subsequent to the Gαi1-GDP structure, the carboxyl terminus was ordered; the tail of Gαt bound to GTPγS formed an extended linear structure, and the carboxyl terminus of Gαi1 bound to regulator of G protein signaling 4 adopted the carboxyl-terminal capping motif (6) observed in the transferred nuclear Overhauser effect structures of carboxyl-terminal peptides bound to photo-activated rhodopsin. We did not observe any spin-spin interactions between Leu-348 and Phe-354, as would be predicted by the carboxyl-terminal capping motif. However, the loss of signal intensity associated with ordering of the carboxyl terminus also decreased the sensitivity of these studies to observe any potential spin-spin interactions or to make any specific assignments to confirm other potential conformations of the carboxyl terminus.
A role for the carboxyl terminus in nucleotide exchange was initially proposed by Denker et al. (14), in which a carboxyl-terminal deletion of Gαo resulted in enhanced activation of the subunit in the presence of high GTP concentrations. In their study, truncated Gαo subunits with deletions of 5, 10, or 14 amino acids from the carboxyl terminus of αo are monitored for their ability to bind GTP or GTPγS. Truncation of 5 or 10 amino acids have little effect on the affinity of the G protein for GDP, which is similar to what we observed for the Gα subunit that lacked the carboxyl-terminal nine amino acids (data not shown). However, deletion of 14 amino acids resulted in a diminished affinity for GDP, resulting in enhanced activation of the Gα subunit in the presence of high concentrations of GTP. The region 10–14 amino acids from the carboxyl terminus is located at the end of the α5 helix, which connects the carboxyl-terminal tail and a loop contacting the nucleotide-binding pocket. More recent studies by Marin et al. (18) further define a role for the α5 helix in nucleotide exchange. Point mutations at Thr-325, Val-328, or Phe-332 in the α subunit of transducin resulted in dramatic increases in both basal nucleotide exchange and receptor-catalyzed nucleotide exchange (18). These three residues (located 19–26 amino acids from the carboxyl terminus) face inward toward the body of the G protein and likely comprise a functional microdomain in G proteins that affects basal nucleotide release.
To promote nucleotide exchange, conformational changes likely must be transmitted from the carboxyl-terminal tail, an eight-amino-acid region known to be involved in receptor recognition through the α5 helix (18 amino acids) to the nucleotide-binding pocket located 25–30 Å from the membrane. To demonstrate this proposed mechanism, the Artemyev group (32) made recombinant Gα subunits modified between the carboxyl terminus and the α5 helix with either a flexible glycine linker or an extended α helix. They found that mutants containing the flexible linker could not be activated by the receptor, whereas those extending the helix by three turns were capable of activation by photo-excited rhodopsin. These results provide direct evidence for structural requirements of the carboxyl-terminal tail in G protein activation by GPCRs.
In our membrane reconstitution experiments, we found that the carboxyl terminus is essential for receptor activation, consistent with abundant evidence for essential roles of amino acids in the carboxyl terminus of Gα subunits. Notably, in our membrane reconstitution experiments, the truncated Gα subunit (Gαi1ΔCT) acted as a competitive inhibitor for receptor activation, thus demonstrating that the carboxyl terminus is not required for receptor binding, but is likely more important for the receptor-catalyzed exchange of GTP for GDP on the G protein (Fig. 4). The ability of the truncated Gα subunit to compete with the full-length Gα subunit seems paradoxical, because numerous biochemical studies demonstrate the carboxyl-terminal peptides can bind to activated receptors, although with micromolar affinities. Therefore, one would expect that the carboxyl terminus should provide additional binding energy for the full-length G protein and that the truncated Gα subunit would be a poor competitor. How can this be reconciled? One possibility is that the truncated Gα subunit does have reduced binding affinity for the receptor; however, the lack of the carboxyl terminus prevents G protein activation and subsequent release. In this way, the truncated Gα subunit, once bound to a receptor, might act as a dominant negative protein for other G protein activation. Alternately, the peptide binding studies might represent an intermediate in the activation process, such as stabilization of the MII activation state of rhodopsin. Thus, during the activation process, the carboxyl terminus might adopt a conformation that could increase the affinity of the receptor for G protein. Consistent with this notion, G proteins in the empty state have high affinity for receptors (33, 34), and it is tempting to speculate that the structures of the carboxyl termini might be functionally linked to the empty state of the G protein. Binding of the GTP by the Gα subunit would then be predicted to remove this interaction, presumably by altering the conformation of the tail.
The results obtained here suggest that activation of Gα subunits alters the conformation of the carboxyl tail. From a mechanistic standpoint, it could be considered that activation of the Gα subunit induces the carboxyl-terminal tail to adopt an ordered conformation in the GTP-bound state. In this conformation, the carboxyl terminus may sterically interfere with binding to the receptor interface, thus decreasing binding affinity for the activated Gα subunit to the receptor and thereby encouraging the release of the Gα subunit from the receptor. Additionally, this model also allows for competition by the truncated G protein for the receptor, because without the carboxyl terminus, the truncated version of the G protein cannot adopt the conformation of the full-length G protein and thus would not reduce receptor-binding affinity.
In addition, we demonstrate the utility of expressed protein ligation for the study of G proteins and signal transduction pathways. The ability to ligate structural probes site-specifically onto proteins provides many applications for this technology. This is particularly important and interesting for G proteins, because they undergo many conformational changes to function in signal transduction. The work presented here provides one example of how one region of a large protein can be monitored, in terms of structural conformation, without the requirement for modifying a large region of the protein. Future directions will focus on performing solid state NMR experiments on isotope-labeled rhodopsin in complex with segment-labeled G protein to allow determination of a structural picture of the docking of the G protein α subunit to the ligand-activated receptor.
Acknowledgments
We thank Vivek Mittal and Dr. Maurine Linder for help in the purification of G proteins and Jeffrey Klco for supplying C5a receptor-containing membranes.
Footnotes
This research was supported in part by grants from the Lucille P. Markey Predoctoral Fellowship (to L. L. A.), the Chemistry Biology Interface Pathway T32GM0878 (to L. L. A.), the Culpeper Award, Rockefeller Brothers Fund (to T. J. B.), and National Institutes of Health Grants EY12113 (to G. R. M.), and GM63720-01 (to T. J. B.).
The abbreviations used are: GPCR, G protein-coupled receptor; GTPγS, guanosine 5′-3-O-(thio)triphosphate; EPL, expressed protein ligation; HSQC, heteronuclear single quantum correlation; CBD, chitin-binding domain; Fmoc, fluorenylmethoxycarbonyl; CHAPS, 3-[(3-chol-amidopropyl)dimethylammonio]-1-propanesulfonic acid.
References
- 1.Lambright DG, Noel JP, Hamm HE, Sigler PB. Nature. 1994;369:621–628. doi: 10.1038/369621a0. [DOI] [PubMed] [Google Scholar]
- 2.Noel JP, Hamm HE, Sigler PB. Nature. 1993;366:654–663. doi: 10.1038/366654a0. [DOI] [PubMed] [Google Scholar]
- 3.Sondek J, Lambright DG, Noel JP, Hamm HE, Sigler PB. Nature. 1994;372:276–279. doi: 10.1038/372276a0. [DOI] [PubMed] [Google Scholar]
- 4.Lambright DG, Sondek J, Bohm A, Skiba NP, Hamm HE, Sigler PB. Nature. 1996;379:311–319. doi: 10.1038/379311a0. [DOI] [PubMed] [Google Scholar]
- 5.Hamm HE. Proc Natl Acad Sci U S A. 2001;98:4819–4821. doi: 10.1073/pnas.011099798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tesmer JJ, Berman DM, Gilman AG, Sprang SR. Cell. 1997;89:251–261. doi: 10.1016/s0092-8674(00)80204-4. [DOI] [PubMed] [Google Scholar]
- 7.Dratz EA, Furstenau JE, Lambert CG, Thireault DL, Rarick H, Schepers T, Pakhlevaniants S, Hamm HE. Nature. 1993;363:276–281. doi: 10.1038/363276a0. [DOI] [PubMed] [Google Scholar]
- 8.Kisselev OG, Kao J, Ponder JW, Fann YC, Gautam N, Marshall GR. Proc Natl Acad Sci U S A. 1998;95:4270–4275. doi: 10.1073/pnas.95.8.4270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Koenig BW, Kontaxis G, Mitchell DC, Louis JM, Litman BJ, Bax A. J Mol Biol. 2002;322:441–461. doi: 10.1016/s0022-2836(02)00745-3. [DOI] [PubMed] [Google Scholar]
- 10.Mixon MB, Lee E, Coleman DE, Berghuis AM, Gilman AG, Sprang SR. Science. 1995;270:954–960. doi: 10.1126/science.270.5238.954. [DOI] [PubMed] [Google Scholar]
- 11.Yang CS, Skiba NP, Mazzoni MR, Hamm HE. J Biol Chem. 1999;274:2379–2385. doi: 10.1074/jbc.274.4.2379. [DOI] [PubMed] [Google Scholar]
- 12.Dhanasekaran N, Wessling-Resnick M, Kelleher DJ, Johnson GL, Ruoho AE. J Biol Chem. 1988;263:17942–17950. [PubMed] [Google Scholar]
- 13.Onrust R, Herzmark P, Chi P, Garcia PD, Lichtarge O, Kingsley C, Bourne HR. Science. 1997;275:381–384. doi: 10.1126/science.275.5298.381. [DOI] [PubMed] [Google Scholar]
- 14.Denker BM, Schmidt CJ, Neer EJ. J Biol Chem. 1992;267:9998–10002. [PubMed] [Google Scholar]
- 15.Conklin BR, Herzmark P, Ishida S, Voyno-Yasenetskaya TA, Sun Y, Farfel Z, Bourne HR. Mol Pharmacol. 1996;50:885–890. [PubMed] [Google Scholar]
- 16.Conklin BR, Bourne HR. Cell. 1993;73:631–641. doi: 10.1016/0092-8674(93)90245-l. [DOI] [PubMed] [Google Scholar]
- 17.Marin EP, Krishna AG, Sakmar TP. Biochemistry. 2002;41:6988–6994. doi: 10.1021/bi025514k. [DOI] [PubMed] [Google Scholar]
- 18.Marin EP, Krishna AG, Sakmar TP. J Biol Chem. 2001;276:27400–27405. doi: 10.1074/jbc.C100198200. [DOI] [PubMed] [Google Scholar]
- 19.Hamm HE, Deretic D, Arendt A, Hargrave PA, Koenig B, Hofmann KP. Science. 1988;241:832–835. doi: 10.1126/science.3136547. [DOI] [PubMed] [Google Scholar]
- 20.Rasenick MM, Watanabe M, Lazarevic MB, Hatta S, Hamm HE. J Biol Chem. 1994;269:21519–21525. [PubMed] [Google Scholar]
- 21.Martin EL, Rens-Domiano S, Schatz PJ, Hamm HE. J Biol Chem. 1996;271:361–366. doi: 10.1074/jbc.271.1.361. [DOI] [PubMed] [Google Scholar]
- 22.Perler FB. Cell. 1998;92:1–4. doi: 10.1016/s0092-8674(00)80892-2. [DOI] [PubMed] [Google Scholar]
- 23.Dawson PE, Muir TW, Clark-Lewis I, Kent SB. Science. 1994;266:776–779. doi: 10.1126/science.7973629. [DOI] [PubMed] [Google Scholar]
- 24.Skiba NP, Bae H, Hamm HE. J Biol Chem. 1996;271:413–424. doi: 10.1074/jbc.271.1.413. [DOI] [PubMed] [Google Scholar]
- 25.Sheikh SP, Vilardarga JP, Baranski TJ, Lichtarge O, Iiri T, Meng EC, Nissenson RA, Bourne HR. J Biol Chem. 1999;274:17033–17041. doi: 10.1074/jbc.274.24.17033. [DOI] [PubMed] [Google Scholar]
- 26.Klco JM, Lassere TB, Baranski TJ. J Biol Chem. 2003;278:35345–35353. doi: 10.1074/jbc.M305606200. [DOI] [PubMed] [Google Scholar]
- 27.Kozasa T, Gilman AG. J Biol Chem. 1995;270:1734–1741. doi: 10.1074/jbc.270.4.1734. [DOI] [PubMed] [Google Scholar]
- 28.Fawzi AB, Fay DS, Murphy EA, Tamir H, Erdos JJ, Northup JK. J Biol Chem. 1991;266:12194–12200. [PubMed] [Google Scholar]
- 29.Ayers B, Blaschke UK, Camarero JA, Cotton GJ, Holford M, Muir TW. Biopolymers. 1999;51:343–354. doi: 10.1002/(SICI)1097-0282(1999)51:5<343::AID-BIP4>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 30.Hellmich MR, Battey JF, Northup JK. Proc Natl Acad Sci U S A. 1997;94:751–756. doi: 10.1073/pnas.94.2.751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hansen JL, Servant G, Baranski TJ, Fujita T, Iiri T, Soren PS. Circ Res. 2000;87:753–759. doi: 10.1161/01.res.87.9.753. [DOI] [PubMed] [Google Scholar]
- 32.Natochin M, Moussaif M, Artemyev NO. J Neurochem. 2001;77:202–210. doi: 10.1046/j.1471-4159.2001.t01-1-00221.x. [DOI] [PubMed] [Google Scholar]
- 33.Yu B, Simon MI. J Biol Chem. 1998;273:30183–30188. doi: 10.1074/jbc.273.46.30183. [DOI] [PubMed] [Google Scholar]
- 34.Haga K, Haga T, Ichiyama A. J Biol Chem. 1986;261:10133–10140. [PubMed] [Google Scholar]