

Summary
A panel of human glioma cell explants was screened for integrin expression by flow cytometry using ανβ-specific antibodies. A lower percentage of the glioma cells were positive for the ανβ3 (mean % positive = 20.8%) integrin, whereas higher percentages were positive for the ανβ5 (mean % positive = 72.7%), VLA5α (mean % positive = 87%) and VLAβ1 (mean % positive = 41.7%) integrins. A series of RGD peptides was designed, synthesized and tested for binding to integrin receptors. Based on the results of the binding to the isolated integrin receptors and the expression of integrins on glioma cell lines, a peptide that binds potently to the ανβ3, ανβ5 and α5β1 was selected for further investigations with regards to its effect on glioma cells. The peptide, Ac-c[(Pen)-Tyr(Me)-Ala-Arg-Gly-Asp-Asn-Tic-Cys]NH2 (RGD peptide), exhibited high potential for use in clinical intracranial administration since it had good stability in rat brain cell homogenates placed into artificial cerebrospinal fluid. Using an HPLC method for quantification of peptides in rat brain cell homogenates, we could demonstrate the half-life of the RGD peptide approximated 20 hr. Relative to a scrambled peptide control (non-RGD sequence, same amino acids), the experimental RGD peptide significantly decreased glioma cell proliferation of the entire panel of rat and human glioma cells tested. Adhesion of recently passaged glioma cells to glioma-derived extracellular matrix protein-coated plates was inhibited significantly by the RGD peptide. The peptide also reversed attachment of plated glioma cells. The RGD peptide caused some, but not substantial, glioma cell injury, as evidenced by a quantitative in vitro nuclear DNA morphologic assay and by a flow cytometric assay employing 7-amino actinomycin D (7AAD). We histologically monitored for toxicity caused by various doses of the RGD peptide infused repeatedly into normal cannulated rat brain. At safe doses, the experimental RGD peptide-treated brains did not show significant differences from those infused with scrambled peptide or buffer-treated controls. In tumor-bearing brains, slightly smaller tumor areas were measured with a higher necrotic-to-tumor index in the RGD peptide treated relative to the scrambled peptide-treated controls. This was obtained with intracranial peptide administrations or combined intracranial and intraperitoneal injections. From this in vitro work, we conclude that the anti-glioma effects of the RGD peptide tested resulted from lowered glioma proliferation and adhesion/mobility, rather than from significant glioma cell injury in the timeframe analyzed. Although other mechanisms not discerned from our limited histopathological observations may be operational, from our in vivo work, we conclude that repeated administration of RGD peptide into brain is safe but that better delivery of the peptides to infiltrating tumor cells is necessary.
Keywords: RGD peptides, brain tumors, apoptosis, adhesion, motility, proliferation
I. Introduction
Glial neoplasms are the most common primary tumors of the central nervous system. The prognosis for patients with nervous system tumors is discouraging (CBTRUS 2002). They remain a significant cause of death in young adults and in children (Prados et al, 1998). The dismal outlook for malignant brain tumor patients is due to the inability of conventional therapies (surgery, radiation and chemotherapy) to completely eliminate gliomas. Several factors contribute to the inefficacy of these treatments including the precarious locations of the tumors within the brain and the infiltrative nature of malignant gliomas. As such, there is a necessity to explore alternative experimental therapies.
Concentrated efforts in the area of angiogenesis research are leading to the discovery of a number of anti-angiogenic substances for treatment of angiogenic disorders and cancer. The involvement of integrins as regulators of angiogenic and apoptotic processes and in brain tumor cell and astrocyte recognition, adhesion and migration on extracellular matrix (ECM) are documented (Ruoslahti and Reed 1999; MacDonald et al, 2001; Ding et al, 2002; Hynes 2002; Milner and Campbell 2002). Many integrin receptors on cells are known to bind to the tripeptide Arg-Gly-Asp (RGD) sequences present in various ECM components such as fibronectin, vitronectin, collagen and fibrinogen. Due to the lack of existing effective treatments for gliomas and other brain cancers, integrin antagonists were explored as an alternative treatment for gliomas. RGD-containing peptides were found to be efficacious in in vitro studies with glioma cells and in animal brain tumor models (Chatterjee et al, 2000; MacDonald and Ladisch 2001; MacDonald et al, 2001; Taga et al, 2002). The studies by Chatterjee and colleagues focused on the cyclic RGDfV integrin antagonist and its linear homolog, the cyclic form of which was known to bind to the αν– integrins (Chatterjee et al, 2000). The accumulation of preclinical data has led to several clinical trials testing systemic administration of integrin antagonists for patients with gliomas or with advanced solid tumors (Eskens et al, 2003; Phuphanich et al, 2004).
In this investigation, a broader screen of the integrin expression by a panel of gliomas was conducted. After synthesizing a variety of RGD-containing peptides, the binding affinities to integrin receptors were screened. Stability of one RGD peptide displaying broad specificity was obtained upon its in vitro exposure to rat brain cell homogenates in artificial cerebrospinal fluid. We then noted effects of the RGD peptide on glioma cells by a number of in vitro assays, including proliferation, adhesion and apoptosis induction. Finally, in pilot studies we explored their toxicity when introduced intracranially into normal cannulated rat brain and into tumor-bearing rat brain.
II. Materials and Methods
A. Cells and cell culture
Early passage (≤P10) human glioma cell explants used in these experiments, 13-06-MG, 04-11-MG, 10-08-MG and 14-07-MG, were obtained according to Institutional Regulatory Body guidelines and practices. The similarities of cell explants to those of primary tissues have been validated by gene array patterns (Zhang et al, 1997). Minced and enzymatically digested tissue cells were placed into culture as described (Gomez and Kruse 2003). Some of the cell explants at higher passage number received partial characterization (Kruse et al, 1998; Kleinschmidt-DeMasters et al, 1999; Read et al, 2003). Other human glioma cell lines, U-373MG, U-87MG and U-251MG, were graciously supplied by Drs. D. Bigner (Duke University, Durham, NC) or M. Jadus (Veterans Medical Center, Long Beach, CA). The cells were maintained in F12/DMEM medium (1:1 v/v, InVitrogen Life Technologies, Carlsbad, CA) or RPMI 1640 medium supplemented with 10% heat inactivated fetal bovine serum (FBS, Gibco, Grand Island, NY), all at a pH of 7.2. The cells were incubated at 37°C in a humidified 5% CO2 atmosphere. The cells that attached to the plastic were passaged when confluent with 0.025% trypsin in phosphate buffered saline (PBS) containing 1 mM EDTA and placed into culture medium containing 20% conditioned medium from their previous passage. Fischer rat 9L gliosarcoma cells and Lewis rat CNS-1 glioma cells were maintained in RPMI-1640 and Dulbecco's Modified Eagle's Medium (2:1 v/v) supplemented with 10% FBS (Gibco), 2 mM L-glutamine, 100 U/ml penicillin and 0.1 mg/ml streptomycin.
B. Integrin expression by glioma cells
Cell cultures at ∼80% confluency were disaggregated with 1 mM EDTA and 1% bovine serum albumin in PBS. Cell clumps were further dissociated by tituration. Five × 105 cells were pelleted at 200 × g for 5 min and the supernatants decanted. After washing, cells were then incubated on ice for 45 min with monoclonal antibodies to identify integrin expression. FITC-conjugated antibodies were as follows: mouse anti-human integrin αvβ5 (MAB 1961F, Chemicon International, Temecula, CA), mouse anti-human integrin αvβ3 (MAB 1976F, Chemicon International), mouse anti-human antibody was used for VLAβ1 chain expression or CD29 (CBL 481F, Cymbus Biotechnology LTD), along with their isotype control, mouse IgG1 (CBL 600F, Cymbus Biotechnology LTD, Hants, UK). Additionally, a FITC-conjugated mouse anti-human VLA5α chain or CD49e (CBL 497F Cymbus Biotechnology LTD, Hants, UK) was used along with its isotype control, a mouse IgG2b (PharMingen, San Diego, CA). The cells were washed with PBS containing 1% FBS and pelleted at 200 × g for 5 min. The stained cells were then fixed with 1% paraformaldehyde (Sigma, St. Louis, MO) in PBS and kept refrigerated until analysis on an EPICS XL-MCL flow cytometer. The percentage of positive cells as well as the mean fluorescence intensities (MFI) were obtained, the latter of which is an expression of the relative antigen density.
C. Synthesis of peptides
The peptides were synthesized by stepwise coupling of 9-fluorenylmethoxycarbonyl- (Fmoc)-amino acid derivatives on solid-phase Rink amide methylbenzhydrylamine (MBHA) resin (Novabiochem, San Diego, CA) using standard coupling procedures. Usually, diisopropylcarbodiimide (DIC) and hydroxybenzotriazole in N-methylpyrrolidinone were used as activation reagents for peptide bond formation. The reactions were carried out in the presence of 2 equivalents of N,N-diisopropylethylamine. For the more hindered coupling to the secondary amino group in tetrahydroisoquinoline carboxylic acid (Tic), 2-(7-Aza-1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HATU) was utilized. The sensitive amino acid cysteine was coupled using a combination of DIC and 0.5 M 1-hydroxybenzotriazole (HOBt)-solution without any base to minimize the risk of racemization. The side chain protecting groups were the trityl group for cysteine, the tert-butyl group for aspartic acid and threonine and 2,2,4,6,7-pentamethyldihydrobenzofuran-5-sulfonyl (Pbf) for arginine. Cleavage of the peptides from resin and simultaneous deprotection of all side chain protecting groups were accomplished by treatment with a trifluoroacetic acid cocktail, which contains triisopropylsilane, phenol and water as scavengers. Disulfide bond formation was completed by air oxidation in aqueous solution. Peptide purification was performed by preparative reverse phase-high performance liquid chromatography (RP-HPLC) on a Vydac C18 column (WR Grace & Co.-Conn, Columbia, MD) using a gradient of acetonitrile in water with 0.1% trifluoroacetic acid.
D. Binding affinity assay
Each well of a microtiter plate (Nunc MaxiSorp, Naperville, IL) was coated with 120 μl of purified receptor (0.5 μg/ml in assay buffer composed of 2 mM CaCl2, 1 mM MgCl2, 50 mM TRIS, 150 mM NaCl at pH 7.4) with 4 mM octyl glucoside overnight at room temperature with shaking. The receptor solution was removed and each well was washed with 200 μl of 0.5% bovine serum albumin in assay buffer for ten minutes. This step was repeated for a total of three washes. Fifty microliters of ten-fold dilutions (from 0.0002 to 200 μg/ml) of the inhibitory compounds in assay buffer was added to the wells. Fifty μl of biotinylated ligand (fibrinogen for αIIbβ3, fibronectin for α5β1 and vitronectin for αvβ3 and αvβ5) in assay buffer was added to the wells. The plates were sealed and incubated overnight at room temperature with shaking. The ligand/competitor solution was removed, then each well was washed with 250 μl wash buffer (0.05 % Tween 20, 50 mM TRIS, 150 mM NaCl2, pH 7.4) for 5 min. This step was repeated for a total of three washes. One hundred microliters of an avidin biotin peroxidase complex (Pierce Chemical ABC kit 32050, Urbana, IL) in wash buffer was added to each well. The plates were incubated for 30 min at room temperature with shaking. The ABC solution was removed, then each well was washed with 250 μl wash buffer for 5 min. This step was repeated for a total of three washes. One hundred microliters of a peroxidase substrate (3, 3′, 5, 5′ tetramethylbenzidine, Pierce Chemical TMB substrate kit 34021) was added to each well. The conversion of the substrate was monitored kinetically in a microtiter plate reader (Molecular Devices, Sunnyvale C A) at 650 nm. Optical density readings were made of each well at 12 sec intervals for 10 min. The software for the plate reader was used to calculate the concentration at which 50% of the binding of the ligand to the receptor was inhibited (IC50). The maximal velocity of the enzymatic conversion (Vmax) was calculated for each well and expressed in milli-optical density units per min (mOD/min). The Vmax values were plotted as a function of inhibitor concentration and a four parameter logistic curve was fitted to the data. The inflection point of this curve was the IC50.
E. Stability of RGD peptide in rat brain cell lysates suspended in artificial cerebrospinal fluid (aCSF)
The peptides were tested for stability in rat brain homogenate suspended in aCSF (Grosshans et al, 2002). The rat brains were removed from the skull of sacrificed animals and kept frozen. The aCSF was prepared by dissolving 8.6 g of sodium chloride, 0.224 g of potassium chloride, 0.206 g of calcium chloride dihyrate and 0.163 g of magnesium chloride hexahydrate in 500 ml of sterile water and combining that solution at a ratio of 1:1 with a solution of 0.214 g of sodium hydrogen phosphate heptahydrate and 0.027 g sodium phosphate monohydrate in 500 ml of sterile water. The pH was adjusted to 7.45. A 350 mg portion of rat brain was homogenized in 3.1 ml of aCSF in a glass tissue homogenizer. Then 400 μl of RGD peptide was dissolved in aCSF to a final concentration of 3.5 mg/ml and that solution added to the homogenate. Samples were agitated gently at 37°C and aliquots were taken after 5, 15, 30, 45, 60, 90 and 1200 min, filtered and analyzed by RP-HPLC. The RP-HPLC method involved a determination of the RGD peptide peak area (in millions) present at various times.
F. Effects of RGD peptide on glioma cell proliferation
A quantitative in vitro colorimetric cell proliferation assay was performed with glioma cells seeded into triplicate wells (3 × 103 cells/well) of a 96-well tissue culture plate. The cells were allowed to attach overnight. The medium was gently aspirated, rinsed once with PBS and replaced with 300 μl of fresh complete medium or that containing 0.001 M of a scrambled non-RGD peptide analog (Ac-[(Pen)RY(Me)AGND(Tic)C]-NH2) or experimental RGD peptide. Cells were incubated at 37°C in a humidified atmosphere at 5% CO2 and the colorimetric assay was performed at 1, 3, 6 or 8 days. On the day of assay, the relative metabolic activity was estimated from triplicate wells using a CellTiter 96 AQ kit (Promega, Madison, WI) according to the manufacturer's instructions. Absorbance at 562 nm was plotted with SigmaPlot (SPSS Inc., Chicago, IL) and was a reflection of the relative metabolic activity as measured by viable cell dehydrogenase activity.
G. Effects of RGD peptide on glioma cell adhesion to extracellular matrix (ECM) proteins
Culture plates coated with glioma-derived ECM proteins were produced by plating 5 × 103 glioma cells/well in 96 well tissue culture plates and grown in complete medium until confluent (Berens and Giese 1996). Medium was carefully removed and the monolayers were rinsed once with PBS. Cells were then lysed with 100 μl of 0.5% Triton X-100 and placing the plates at 120 rpm on a horizontal rotator for 30 min. This was followed by incubation with 0.1 M NH4OH for 3 min. Plates then were rinsed three times with PBS. The wells were covered with 200 μl PBS and stored at 4°C until use in the adhesion assays. Relevant ECM proteins such as collagen type IV, fibronectin, tenascin, laminin, and vitronectin are produced by glioma cells, although the specific types and ratios may vary from one to another (Berens and Giese, 1996).
The scrambled non-RGD and experimental RGD peptides were tested for their ability to reverse the adhesion of attached glioma cells plated onto ECM-coated plates or to interfere with adhesion of recently passaged glioma cells (Berens and Giese 1996). In the first set of experiments, glioma cells were harvested from monolayer cultures and placed into 96-well flat bottom plates coated with autologous ECM (i.e., the ECM used for analysis of adhesion of the individual glioma cells were derived from the same cells to be tested) at a concentration of 5 × 105 cells/ml (50,000 cells/well) and incubated overnight. Medium was removed and replaced with 200 μl of fresh medium, or that containing scrambled peptide or the experimental RGD peptide (0.001 M). After 4 hr incubation at 37°C, the plates were subjected to 350 rpm agitation on a horizontal rotator for 6 min. The medium containing nonattached cells was removed and the wells were rinsed with PBS. Attached cells were fixed in 1% glutaraldehyde for 10 min before staining with crystal violet (0.1% in H2O). Spectrophotometric absorbance of the stained nuclei was quantified at 595 nm. In the second set of experiments, similar assays were performed with nonattached glioma cells. 5 × 104 cells/well were seeded in 96 well ECM-coated plates with medium or that containing the scrambled or RGD peptides. Plates were incubated 30 min on ice and then at 37°C for 60 min to allow adhesion. The wells were rinsed, fixed, stained and adhesion quantified as previously described (Berens and Giese 1996).
H. Effects of RGD peptide on glioma cell injury by a quantitative morphologic assay and by a 7AAD flow cytometric assay
1. Apoptosis assessment by a quantitative in vitro morphologic assay
Cell morphology studies were performed to discern apoptotic figures in hematoxylin and eosin (H&E) stained glioma cells (10-08-MG, 04-11-MG, U-251MG, U-373MG) after a 18 hr coincubation with either scrambled or RGD peptide. The glioma cells (4 × 104) were plated onto sterile Lab-Tek 4 well glass chamber slides (Nalge Nunc International, Naperville, IL) for 48 hr. Then medium was gently aspirated from the wells and replaced with fresh medium, or that containing scrambled or RGD peptides (0.001 M). The mixtures were incubated at 5% CO2 in a 37°C humidified chamber for 18 hr. The medium was removed, the chambers gently rinsed with HBSS (Life Technologies) and fresh medium added to the wells. The adherent cells were incubated for another 24 hr before fixation in 10% phosphate buffered formalin for 30 min. The cells were stained with H&E. Glioma cell nuclear morphology was examined by light microscopy using a 60X objective (Olympus BX40, Melville, NY). Results are expressed as the percentage of adherent cells identified as live or apoptotic, obtained from an approximate 250-300 total cell count (Gomez et al, 2004).
2. Cell injury assessment by a flow cytometric 7AAD assay
Glioma cells (13-06-MG, 04-11-MG, 10-08-MG and 14-07-MG) were plated into sterile 6 well plates for 24 hr. RGD peptide or scrambled peptide (0.001 M), or fresh culture medium alone were then added to the wells. Coincubation for 4 or 18 hr at 37°C in a humidified 5% CO2 chamber was followed by collection of both the cells in suspension and the adherent cells, which were harvested with 1 mM EDTA and 1% bovine serum albumin in PBS. The combined adherent and non-adherent cells were centrifuged and the supernatants decanted. A 50 μl volume of 7AAD (20 μg/ml in PBS) was added to the resuspended pellet. After staining for 20 min at 4°C, cells were rinsed once and resuspended in 400 μl of PBS. Samples were analyzed by flow cytometry within 30 min at the University of Colorado Flow Cytometry Core facility. Scattergrams were generated by 7AAD fluorescence of the labeled glioma population. Regions were drawn around clear-cut populations having negative (live cells), bright (late apoptotic/dead cells) and dim (apoptotic) fluorescence (Schmid et al, 1994; Philpott et al, 1996). The percentages of glioma cells within the segregated live, apoptotic and late apoptotic/necrotic populations were determined (Gomez et al, 2004).
I. Instillation of cannulas and infusion of peptides into normal and tumor-bearing rat brain
1. Surgical implantation of cannulas
Anesthetized F344 rats (190-210 g, Harlan, Indianapolis, IN) underwent one surgical procedure for the permanent implantation of a stainless steel cannula into the right frontal brain (3 mm lateral to midline, 2 mm anterior to bregma, 4 mm depth) as described (Fleshner et al, 1992; Kruse et al, 1994b). A sterile stylet inserted into the cannula maintained patency. The rats were allowed to recover from surgery for one week before being further manipulated. Intracranial infusions (RGD peptide, scrambled peptide, saline, CNS-1 or 9L tumor cells) never exceeded a 10 μl volume and were given to rats in the awake state. Tumor was allowed to establish for one week before starting treatment. Intraperitoneal injections (RGD peptide or scrambled peptide at 0.01 M) never exceeded a 100 μl volume.
2. In vivo toxicity assessments
To establish the highest tolerable dose, in vivo dosing assays were conducted. Cannulated F344 rats were randomized into groups (n = 4 rats/group) that received either three or six 10 μl intracranial infusions of either 0.0, 0.1, 0.5, 0.75, 1.0, 2.0, or 10 mg/ml of RGD peptide every other day. Animals were euthanized at 1, 2, 7, or 14 days after the last infusion and the brains were removed for histopathologic examination. Animal group weights were monitored and plotted every 2-3 days during and after treatment, as we previously established that weight loss (>20%) is evidence of toxicity in non-tumor bearing rats (Kruse et al, 1993). As well, animals were observed daily for gross neurological abnormalities (tremors and ataxic gait) and general reflex behavior (placing/stepping and righting reflexes) as described (Heimberger et al, 2000). In a second toxicity screen, cannulated rats received three 10 μl (0.001 M or 0.114 mg/ml) intracranial infusions every other day. Some animals also received scrambled peptide at the same dose, or saline. They were sacrificed at 1, 7 and 14 days after the last infusion and brains collected for histopathologic evaluation.
3. In vivo efficacy assessments
To obtain preliminary efficacy testing in Fischer or Lewis tumor-bearing rats (n = 4-7 rats/group), 9L (5 × 103/10 μl PBS) or CNS-1 (1 × 104/10 μl PBS) cells, respectively, were infused one week post-surgery into conscious cannulated rats over a 5 min period (Fleshner et al, 1992). These doses were previously determined to give an approximate 1 month survival for sham treated animals (Kruse et al, 1994a). The tumor was allowed to establish for 1 week after which scrambled peptide or RGD peptide (10 μl at 0.001 M) were intracranially infused six times through the cannula every other day. Some groups of rats received daily intraperitoneal injections (0.1 ml at 0.01 M) of scrambled and RGD peptides for 12 days. Systemic administration of RGD peptides had been tested in rodents and ten-fold higher concentrations were safe (data not shown). Other groups of rats received 6 intracranial infusions every other day along with 12 daily intraperitoneal injections. Rats were sacrificed 2 days following the last infusion and the brains were collected for histopathologic analyses and calculation of tumor and necrotic areas at the instillation site. For statistics, an unpaired t test with Welch correction was performed because of the variance in the SD. The p values were considered significant if ≤0.05.
4. Histopathological assessments
Brain tissue specimens were fixed in 10% buffered formalin. Brains were placed into a Jacobowitz rat brain slicer (Zivic Miller, Allison Park, PA) and a coronal slice made at the instillation site and at 4 mm posterior and anterior to that site. The two brain sections were placed face down in a tissue cassette and paraffin-embedded. Five μm coronal brain sections were taken at the instillation site and at 50 μm intervals out to 250 μm to assure that we appropriately analyzed the instillation site. Paraffin sections were dewaxed by placing the slides in a 60°C incubator overnight. After rinsing the slides with saline solution, they were stained with Harris H&E for histopathologic analysis and photomicroscopy. Stained sections were digitally imaged using a Sprint Scan 35 digital scanner (Polaroid, Cambridge, MA) adapted with a Path Scan Enabler (Meyer Instruments, Houston, TX) adapted to image microscope slides (Kruse et al, 2000). Digital images were analyzed using SigmaScan software (SPSS Inc., Chicago, IL). Tumor area was traced and expressed as a percentage of total tissue area using the tissue section at or nearest the cannulation site (Owens et al, 1998). Necrotic area was traced and expressed as a percentage of total tumor area.
III. Results
A. Expression of integrins by human glioma cells
The expression of integrins on the surfaces of human glioma cell explants was analyzed using 4 separate monoclonal antibodies to integrins αvβ3, αvβ5, the VLAβ1 chain and the VLA5α chain of the α5β1 receptor (Table 1). Low passage cell explants were used since they were considered to be more reflective of the integrin expression found on tissues in situ (Zhang et al, 1997). Previously, Chatterjee and colleagues used the U-373MG and U-251MG glioma cell lines as positive and negative controls for αvβ3 expression, respectively (Chatterjee et al, 2000). We included these two cell lines in our analyses and our data confirm their findings with 73% of the U-373MG cells being positive compared to only 0.9% of the U-251MG cells. A low percentage of the glioma cells displayed a fair to moderate expression of the vitronectin receptor, αvβ3 (mean % positive= 20.8, MFI=4.3), whereas the αvβ5 integrin was expressed by a majority of the glioma cells (mean 72.7%) more intensely (mean MFI 6.2). The VLAβ1 (CD29) and VLA5α (CD49e) chains complex to form α5β1 (the fibronectin receptor). The VLAβ1 was even more intensely expressed (mean MFI 17.8) by higher percentages (mean 87%) of the glioma cells, whereas, they exhibited slightly lower positivity at moderate levels for the VLA5α chain (mean 41.7% with mean MFI 4.7). Overall, the findings indicate that the ανβ5 and α5β1 integrins are expressed at consistently higher levels than ανβ3 by human glioma cells. Additionally, we screened the rat 9L and CNS-1 cells with the anti-human antibodies to ανβ3 and ανβ5. The 9L and CNS-1 cells were negative for ανβ3, however, 9L was 88.5% positive and CNS-1 was 28.5% positive for ανβ5. A limitation of the latter analyses is the unknown cross reactivities of the two antibodies; as such, the negative results obtained for ανβ3 can not be reliably interpreted.
Table 1.
Integrin expression by human glioma cell explants*
| ανβ3 |
ανβ5 |
VLAβ1 |
VLA5 |
|||||
|---|---|---|---|---|---|---|---|---|
| Cell explant | % Positive (MFI) | % Positive (MFI) | % Positive (MFI) | % Positive (MFI) | ||||
| 13-06-MG | 28.2 | (4.1) | 75.7 | (6.2) | 90.4 | (24.1) | 46.3 | (4.8) |
| 10-08-MG | 14.6 | (4.2) | 72.2 | (6.2) | 89.2 | (15.6) | 36.7 | (4.3) |
| 04-11-MG | 20.8 | (4.5) | 70.9 | (5.9) | 81.2 | (14.2) | 52.4 | (4.9) |
| 14-07-MG | 19.6 |
(4.5) |
71.8 |
(6.6) |
87.1 |
(17.4) |
31.5 |
(4.8) |
| Mean % + & MFI | 20.8 | (4.3) | 72.7 | (6.2) | 87.0 | (17.8) | 41.7 | (4.7) |
| Cell line | ||||||||
| U-373MG** | 73.0 | (5.5) | 68.7 | (4.6) | 98.7 | (13.4) | 25.3 | (3.0) |
| U-251MG*** | 0.9 | (4.0) | 45.9 | (4.3) | 97.9 | (10.5) | 3.4 | (3.6) |
Flow cytometry was used to determine the percentage of positive cells and the relative antigen density, which was expressed as the mean fluorescence intensity (MFI) of the integrin.
The U-373MG glioma cell line was used as a positive control for ανβ3 (Chatterjee et al, J. Neurooncol. 46:135-144, 2000).
The U-251MG glioma cell line was used as a negative control for ανβ3 (Chatterjee et al, ibid).
B. Binding affinities of RGD-containing peptides to integrin receptors
Six different cyclic RGD-containing peptides were synthesized and characterized for their ability to bind to purified integrins. The sequences as well as the binding affinities of the synthesized peptides to isolated integrin receptors are given (Table 2). The data shown are the IC50 (nM) concentration at which 50% inhibition of binding by the peptide to the receptor occurred. The specificities (bolded numbers) were indicated if lower concentrations provided the inhibition. Compounds 1 and 2 have individual specificity for ανβ3, whereas compound 3 has specificity for ανβ5 receptors. Compound 4 is a potent and selective antagonist for α5β1. Compound 6 displayed a broader selectivity to ανβ3 and ανβ5 receptors. However, peptide 5, Ac-c[(Pen)-Tyr(Me)-Ala-Arg-Gly-Asp-Asn-Tic-Cys]NH2, which we will subsequently refer to as RGD peptide, was selected for further study based on the fact that 1) it exhibited not only potent binding to the ανβ3 and ανβ5 receptors but also to the α5β1 fibronectin receptor and 2) our screen for integrin expression demonstrated that all three receptors, especially the latter two, were commonly displayed by our human glioma cell explants. Thus, we would be able to test whether an RGD peptide with broader specificity may have more potent effects on glioma cell growth, adhesion, migration and induction of apoptosis. The structures of the RGD peptide and a control scrambled peptide, synthesized with the same amino acids in a non-RGD sequence, are shown in the footnote of Table 2.
Table 2.
Binding affinities of RGD-containing peptides to integrin receptors determined from the IC50*
| RGD -containing peptide/sequence** | ανβ3 | ανβ5 | α5β1 | αIIbβ3 |
|---|---|---|---|---|
| 1 c[RGDD(t-B uG)(M amb)] | 3 | 2 0 | 4 2 | 2 40 |
| 2 c[(Mpa)RGDD(t-B uG)C]-N H2 | 2 0 | 2 10 | 3 90 | 7 0 |
| 3 G-c[(Pen)FRG DSFC]-A | 2 70 0 | 4 6 | 1 80 0 | 3 40 0 |
| 4 G-c[(Pe n)R AR GD N PC]-A | 5 2 | 3 30 | 2 | 3 0 |
| 5 Ac -c[(Pen)Y(Me)AR GD N(Tic)C]-N H2*** | 2 | 6 | 5 | 1 90 |
| 6 A cF-c[CR GD TFC]-N H2 | 1 4 | 8 | 2 60 | 1 25 0 |
IC50 is the concentration (nM) at which 50% of the binding of ligand to integrin receptor is inhibited.
Abbreviations: Ac, Acetyl; t-BUG, tert-Butylglycine; Mamb, m-aminomethylbenzoic acid; Mpa, 3-Mercaptopropionic acid; Pen, Penicillamine; Tic, Tetrahydroisoquinoline-3-carboxylic acid; Y(Me), O-Methyltyrosine; AcF, acetylphenylalanine
Molecular structures of Ac-c[(Pen)Y(Me)ARGDN(Tic)C]-NH2 (left) and its scrambled analog Ac-[(pen)RY(Me)AGND(Tic)C]-NH2 (right) are: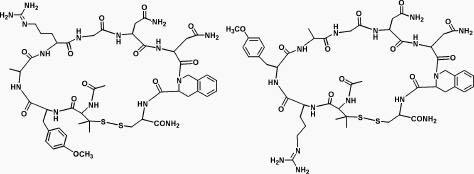
C. In vitro stability of the RGD peptide
To analyze whether the RGD peptide might have appropriate pharmacokinetic properties for in vivo use, an HPLC method was selected for testing the stability of the RGD peptide at 37°C in rat brain homogenate suspended in artificial cerebrospinal fluid. The testing was performed at multiple earlier time points between 5 and 90 min (a range likely to be physiologically-relevant in vivo) and at one later time point at 20 hr (data not shown). At 90 min or less the RGD peptide exhibited good stability and no significant differences existed between the peak areas. At 20 hr, the peptide concentration in the sample was approximately 50% of the initial concentration, i.e., one half-life. These in vitro data suggest that the half life of the RGD peptide will be quite good if placed into the microenvironment of the brain.
D. RGD peptide effects on human and rat glioma cell proliferation
The relative metabolic activity, a measure of cell proliferation, was monitored over time with a panel of human and rat glioma cells when they were untreated, or treated with scrambled non-RGD peptide or RGD peptide (Figure 1). Although not apparent on days 1 and 3, all of the gliomas treated with RGD peptide exhibited a decrease in growth rate by days 6 and 8 when compared to the growth rates of glioma cells treated with scrambled peptide or when placed into medium alone. Thus, the RGD peptide significantly inhibited the proliferation of the entire panel of human glioma cells and the rat CNS-1 glioma cells.
Figure 1.
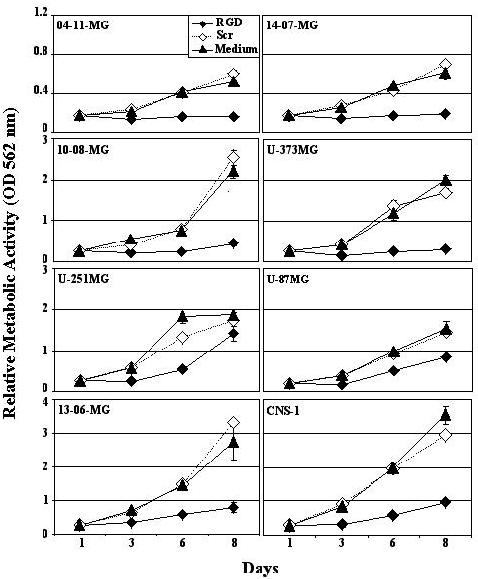
RGD peptide effects on glioma cell proliferation. A panel of glioma cells were assayed when incubated with fresh medium, or that containing scrambled non-RGD peptide (Scr) or RGD peptide. The relative metabolic activity was determined by an in vitro colorimetric method. The mean of the absorbances obtained at 562 nm from supernates harvested from triplicate wells ± SE is shown at 1, 3, 6 and 8 days.
E. RGD peptide effects on glioma cell adhesion to glioma ECM protein-coated plates.
To determine the effect of RGD peptide on glioma cell adhesion to glioma cell-derived ECM, two assays were performed. In the first assay, scrambled peptide or RGD peptide was added to cells pre-plated on ECM protein-coated plates. The spectrophotometric absorbances obtained from the wells with crystal violet stained cells revealed that the RGD peptide successfully competed for and reversed the adhesion of attached glioma cells bound to ECM-coated plates (Figure 2).
Figure 2.
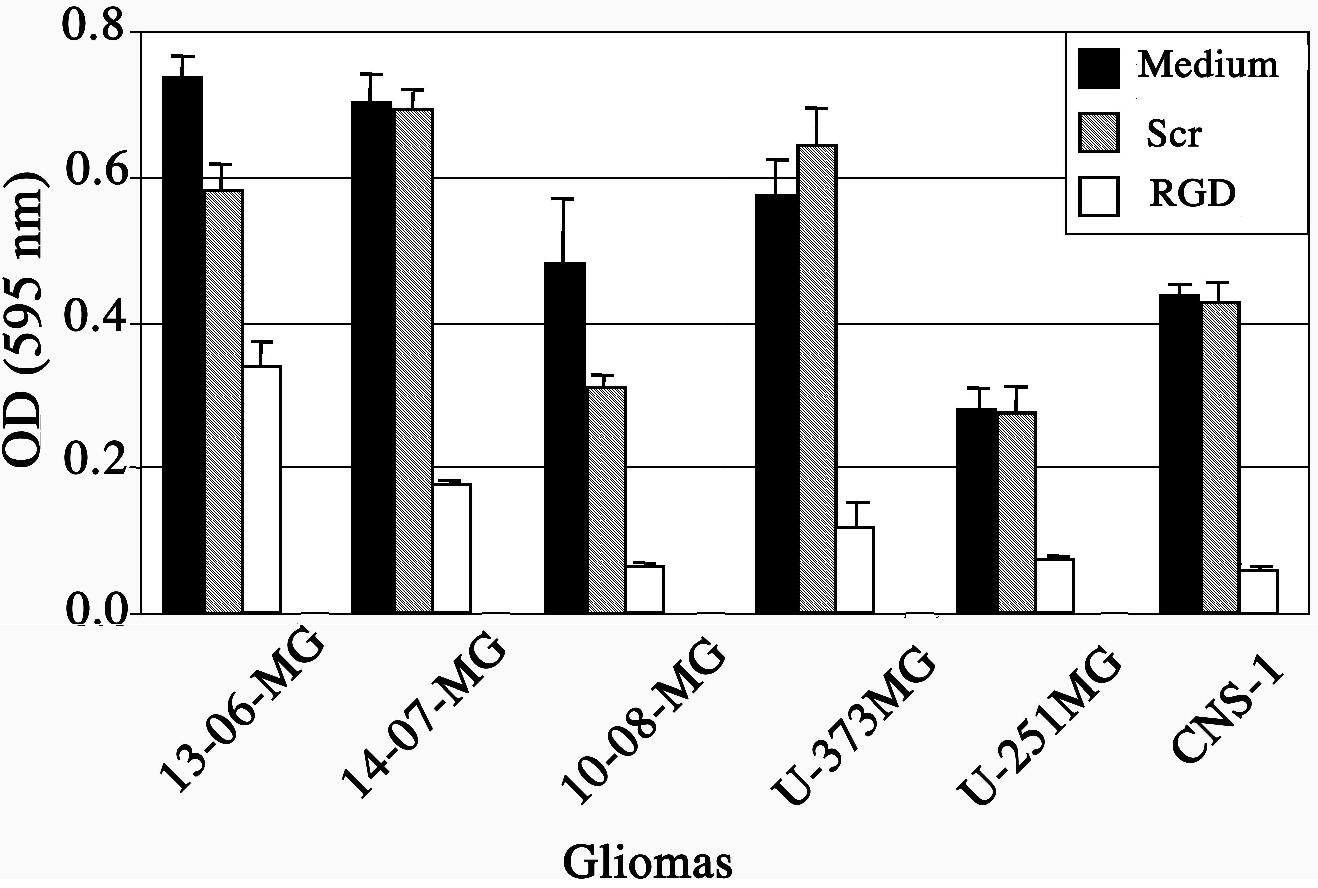
RGD peptide effects on adhesion of glioma cells to autologous glioma cell extracts of extracellular matrix proteins. The mean absorbance values ± SE obtained at 595 nm were from triplicate wells containing crystal violet stained cells. Cells were incubated for 4 hr with fresh medium (■) or that containing scrambled peptide () or RGD peptide (□).
The scrambled peptide did not significantly alter the adhesion of glioma cells compared to the medium control. As examples, representative wells from 14-07-MG and U-373MG glioma cells show few crystal-violet stained cells left on the plate after exposure to the RGD peptide compared to the glioma cells incubated with scrambled peptide or in medium alone (Figure 3). The second assay entailed a determination of whether RGD peptide, when added to glioma cells in suspension, would interfere with their subsequent attachment to ECM protein-coated plates. Similar findings were obtained (data not shown). The RGD peptide interfered with the adhesion of recently passaged glioma cells in suspension to ECM-coated plates. Thus, RGD peptide can interfere with glioma cell adhesion or reverse the adhesion of plated glioma cells.
Figure 3.
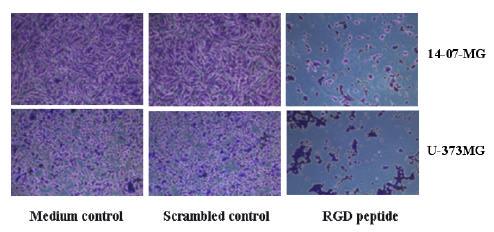
Crystal violet stained 14-07-MG and U-373MG glioma cells adhered to ECM protein-coated wells after 4 hr incubation in fresh medium, in medium containing scrambled peptide, or RGD peptide.
F. Influence of RGD peptide on apoptosis induction in glioma cells
A quantitative morphologic assay was conducted to determine the apoptotic effects of RGD peptide on human and rat glioma cell populations. The numbers of apoptotic and live glioma cells were obtained, as determined by counting high power fields of H&E-stained adherent cells by light microscopy (Figure 4). Representative light microscopic photographs of human 10-08-MG glioma cells and rat 9L gliosarcoma cells exposed to RGD peptide for 18 hr (Figures 4a and 4c, respectively) show cells with nuclear changes consistent with apoptosis, as opposed to the control cell monolayers (Figures 4b and 4d, respectively). With a panel of four human glioma cells, quantitative data from the morphologic assay were collected, which showed a 4-6 fold increase in apoptotic cell percentages in two of the four glioma cell populations tested (U-251MG and U-373MG), although the degree of cell injury was not exceptionally high, i.e., only 11-12%. In the other two cases, 10-08-MG and 04-11-MG, few cells remained attached to the slides for counting, thus we were unable to accurately determine the degree of cell injury from a high number of total cell counts. Since we were unable by this method to determine whether the cells that had lifted were viable, or were permanently versus reversibly injured in the 24 hr following 18 hr RGD exposure, we used an alternative flow cytometric technique with 7AAD to examine glioma cell injury in combined adherent/nonadherent cells (Table 3). Early (4 hr) and later (18 hr) time points after glioma cell exposure to RGD peptide were examined. In the assay, another set of four human glioma cell populations were incubated with medium, or with medium containing scrambled or RGD peptide. Again, the percentages of apoptotic cells only reached 18% for two of the four human glioma cell populations by 18 hr. The cell damage observed was not significantly enhanced relative to the cells exposed to the scrambled peptide controls.
Figure 4.
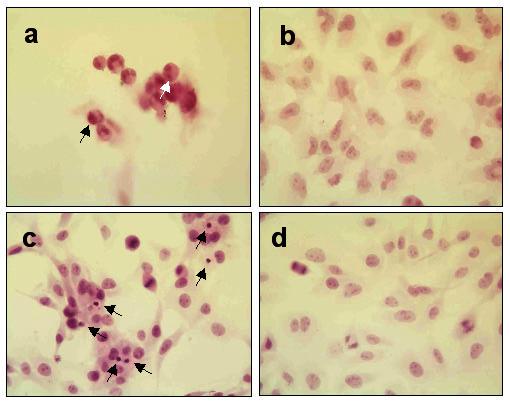
Quantitative morphologic assay showing H&E stained brain tumor cell monolayers that were or were not exposed to RGD peptide (a) human 10-08-MG glioma cells after 18 hr coincubation with RGD peptide demonstrate fewer attached cells and rounded shapes. Some of the cells exhibit typical apoptotic morphological changes such as those with condensed nuclei (black arrow) and fragmented DNA (white arrow) (b) 10-08-MG glioma cells not exposed to RGD peptide exhibited fewer apoptotic cells. The cells were larger with oval nuclei and abundant cytoplasm (c) rat 9L gliosarcoma cells after 4 hr coincubation with RGD peptide. Apoptotic cells with condensed nuclei (black arrows) are visible in the monolayer (d) 9L gliosarcoma cells in monolayer and not exposed to peptide. The healthy cells are well attached to the surface and mitotic figures are readily apparent.
Table 3.
RGD effects on glioma cell apoptosis as assessed by a quantitative in vitro morphologic assay on adherent cells (Experiment 1) or by a 7AAD assay where nonadherent and adherent cells were analyzed (Experiment 2).
| Tumor |
10-08-MG |
04-11-MG |
13-06-MG |
14-07-MG |
|
|---|---|---|---|---|---|
| Additive to Medium | Incubation time (hr) | Apoptotic % | Apoptotic % | Apoptotic % | Apoptotic % |
| none | 4 | 1.1 | 1.6 | 2.8 | 3.6 |
| 18 | 7.5 | 0.8 | 3.3 | 0.4 | |
| Scrambled | 4 | 4.7 | 0.8 | 4.9 | 8.3 |
| 18 | 13.6 | 0.6 | 4.8 | 12.0 | |
| RGD peptide | 4 | 8.4 | 2.1 | 4.2 | 15.8 |
| 18 | 18.7 | 3.0 | 7.2 | 17.8 |
** Glioma cells were plated into 6 well plates for 24 hr. RGD peptide or scrambled peptide (0.001 M), or fresh culture medium were then added to the wells. Coincubation for 4 or 18 hr at 37°C in a humidified 5% CO2 chamber was followed by collection of the adherent and nonadherent cells. Cells were incubated with 7AAD (20 μg/ml in PBS) for 20 min at 4 °C, rinsed once and resuspended in 400 μlof PBS. Samples were analyzed by flow cytometry. The percentages of glioma cells within the segregated live, apoptotic, and late apoptotic/necrotic populations were determined.
Scattergrams from a 7AAD flow cytometric assay, which show live, apoptotic and dead (necrotic/late apoptotic) cell percentages after a 3 hr treatment of rat 9L gliosarcoma cells with scrambled peptide and RGD peptide is shown in Figure 5. An approximate 2-fold increase in injured 9L gliosarcoma cells was observed when they were incubated with RGD peptide (20.9%) compared to the scrambled counterpart (10.1%). The damaged cell population in the medium control was 6.0% (data not shown). Thus, at this early time point apoptosis was slightly induced upon incubation of the 9L rat gliosarcoma cells with RGD peptide, as it was with the human glioma cells.
Figure 5.
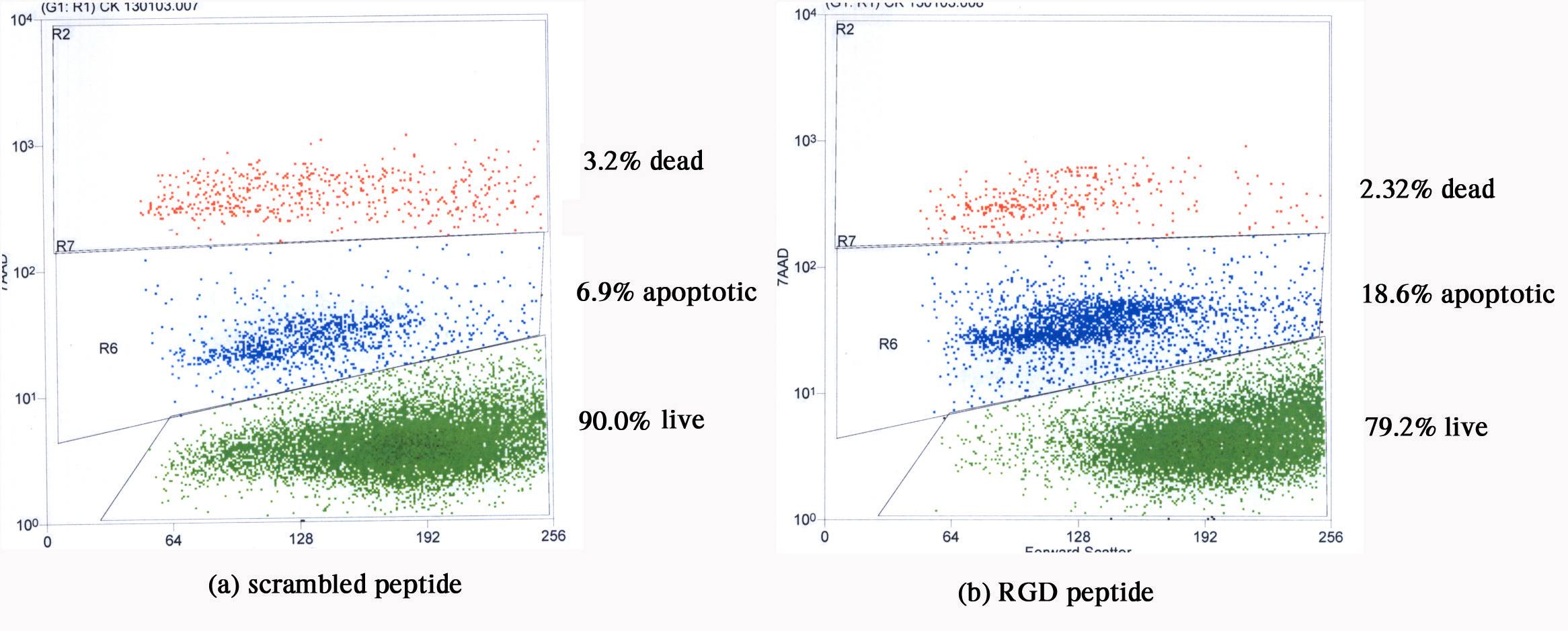
Scattergrams from a 7AAD flow cytometric assay, which give live, apoptotic and dead (necrotic/late apoptotic) cell percentages after a short 3 hr treatment of rat 9L gliosarcoma cells with (a) scrambled non-RGD peptide and (b) RGD peptide. An approximate 2-fold increase in injured 9L gliosarcoma cells is seen when incubated with RGD peptide compared to the scrambled counterpart. Forward scatter (abscissa) is plotted versus 7AAD intensity (ordinate).
G. In vivo toxicology and efficacy of the RGD peptide
Based upon in vitro testing that showed the high levels of ανβ5 integrin expression by the rat 9L and CNS-1 cells and the decreased proliferation of CNS-1 cells and apoptosis induction in 9L cells upon their exposure to the RGD peptide, we proceeded with in vivo toxicity and pilot efficacy assessments with RGD peptide in the rat brain tumor models. For such testing, RGD peptide was repeatedly introduced intracranially into normal cannulated rat brain and into tumor-bearing cannulated rat brain.
To establish appropriate doses for repeated intracranial administrations of RGD peptide, seven different doses (0.1, 0.5, 0.75, 1.0, 1.5, 2.0 and 10 mg/ml), as well as saline controls, were evaluated in cannulated F344 rat brains. Six infusions (10 μl) were given every other day. Groups of rats at the different dose levels (n=4) were evaluated for side effects that included weight loss and symptoms for neurotoxicity. Half or more animals in the groups at the five highest peptide doses exhibited an immediate onset of abnormal motor function, including ataxic gait and some tremor. The animals given the highest intracranial dose displayed the most severe and long-lasting (>3 hr) symptoms. We discontinued testing of the 10 mg/ml dose after the first two doses. No disturbances in behavior were monitored at the lowest dose (0.1 mg/ml) and transient but tolerable effects were seen in several animals at the 0.5 mg/ml dose. As a gross estimate of long term toxicity due to repeated administration of treatment agent, mean group weights were monitored during and after treatment. The mean group weights of the animals were maintained or increased over time, indicating their ability to thrive upon exposure to multiple intracranial infusions. The histopathological evaluation of normal brain that received higher doses of the RGD peptide (≥ 1.0 mg/ml) showed a degree of stress to the brain as evidenced by the presence of congested blood vessels (black arrows) and capillaries (white arrows) (Figure 6a). Some neurons stained pink with eosin, possibly an indication of apoptosis induction and where neuronal dropout would eventually occur. In the brains treated with more dilute doses of RGD peptide (≤0.5 mg/ml), at 24 hr following the last infusion, some brains showed an influx of polymorphonuclear leukocytes (black arrows) near the instillation site (Figure 6b), suggestive of an acute inflammatory response. Scattered mononuclear cells also were evident in the brains treated with the RGD peptide (Figure 6b). Although not shown in this particular photomicrograph, some of the cells presented with morphology characteristic of plasma cells. At 7 or 14 days following the last infusion, some brains revealed formation of small focal sterile granulomas (Figure 6c) at the instillation site. A necrotic center (Nc) was surrounded by a fibrotic wall (Fb) composed of fibroblasts, histiocytes and neutrophils; that was immediately adjacent to normal brain (NB). Granulomas may have formed as a result of the cannulation procedure itself, as some were also seen in animals administered saline through the cannulas. Overall, the areas of damage were small and well contained. The histological differences between the brains given low doses of RGD and scrambled peptides or saline treated were relatively insignificant.
Figure 6.
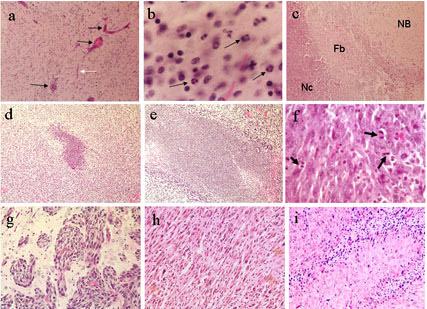
Photomicrographs of RGD or scrambled peptide repeatedly introduced intracranially into cannulated normal rat brain (a-c) or 9L tumor-bearing rat brain (d-i). At 24 hr following the last infusion: (a) Rat brains administered RGD peptide at high doses show signs of stress including congested vessels (black arrows) and capillaries (white arrow). (b) At higher power, brains given RGD peptide at higher doses also showed infiltration of mononuclear cells and polymorphonuclear cells (black arrows), indicative of an acute inflammatory reaction. At 7 or 14 days following the last infusion: (c) Small focal sterile granulomas often formed in cannulated rat brain, whether treated with saline or peptide, as visualized by a necrotic center (Nc) surrounded by a fibrotic wall (Fb). Normal brain (NB) was immediately adjacent to the granuloma. (d) At low power, a representative sized area of necrosis characteristic of scrambled peptide treated brains is shown next to (e) a representative larger sized area of necrosis characteristic of RGD peptide treated brains. The (f) saline-treated tumor has viable cells showing a number of mitotic figures (black arrows) and (g) tumor cells at the periphery of the solid tumor mass are infiltrating into perivascular spaces within normal brain. The (h) RGD peptide treated brain sections show many cells with pyknotic nuclei interspersed within the solid tumor mass adjacent to the instillation site (i) often the palisading areas of growth better show the pyknotic cells that are assumed undergoing apoptosis. Healthy tumor cells are also shown growing in perivascular spaces of the RGD peptide treated brains, however, similar to that shown in g. Magnifications are: d,e = 40X, a,c = 100X, h,i = 200X, f,g = 400X, b = 600X
In the efficacy studies, 9L tumors were visually discernible in the right frontal quadrants upon gross examination of the brains regardless of whether they were treated with saline, the scrambled non-RGD peptide, or the experimental RGD peptide. Microscopically, at low power, the 9L-tumor bearing brains treated with scrambled peptide had smaller areas of necrosis at the instillation site (Figure 6d), as opposed to those treated with RGD peptide (Figure 6e). Shown at higher power and immediately adjacent to the instillation site, the scrambled peptide or saline treated 9L tumor cells were highly viable and presented in pseudopalisade formation; denoting rapid growth, there were many mitotic figures per microscopic field (black arrows, Figure 6f). Analysis at the periphery of the solid tumor growth revealed perivascular extension of the tumor cells into normal brain (Figure 6g). Characteristic of the RGD peptide treated brains and within the solid tumor growth adjacent to the instillation site, large numbers of cells appeared with pyknotic nuclei, a characteristic of cells undergoing apoptosis (Figure 6h). Damaged cells were perhaps more readily apparent in areas of pseudopalisading tumor growth (Figure 6i). In the RGD peptide treated brains, however, healthy and viable tumor cells appeared in perivascular spaces as they did in control treated groups.
Since tumor cells visible at the periphery of the areas of solid tumor growth would have eventually resulted in the demise of the animals in all treatment groups, including those given the experimental RGD peptide, we performed several pilot experiments to compare delivery of the RGD peptide by intracranial, intraperitoneal, or both administration routes in animals bearing 9L or CNS-1 tumors. Rats with 1 week established CNS-1 tumors were either treated with six infusions, given every other day, of intracranially administered RGD or scrambled peptide (10 μl of 0.001 M), or twelve daily intraperitoneal injections of both types of peptide (100 μl of 0.01 M), or the treatments were combined. At 48 hr following the last treatment, brains were collected and multiple H&E-stained sections were examined; sections were chosen for area analysis that corresponded to at or near the instillation site, where tumor growth would be expected to be the largest. We also assessed the necrotic areas within the tumor mass. As examples, Figure 7a shows a gross section of brain where the majority of the hemisphere contains a tumor with centralized necrosis, whereas Figure 7b shows a tumor without necrosis. Figure 8a shows the mean tumor area for each treated group as a percentage ± SD of the total brain area on the sections. Figure 8b shows the mean necrotic area as a percentage ± SD of the total tumor area on the sections. The trend was that smaller tumor areas with higher necrotic indices were present in the RGD treated brain slices compared to the scrambled peptide treated controls when peptides were administered by intracranial or intracranial/intraperitoneal combined routes. There were no differences in tumor areas of groups given RGD or scrambled peptide by the intraperitoneal route. Combining intracranial with intraperitoneal administrations did not result in greater anti-tumor effect than the intracranial administrations. Interestingly, none of the animal tumors given intraperitoneally-administered peptides had necrotic areas (data not shown), perhaps implying that the RGD peptide was not delivered across the blood-brain-barrier.
Figure 7.

H&E stained sections macroscopically showing tumor filling the majority of the upper part of the hemispheres. Panel (a) shows a tumor containing a substantial degree of necrosis derived from an animal that was treated with IC, IP RGD peptide. Panel (b) shows a tumor not displaying necrosis that was derived from an animal that was treated with IC, IP scrambled peptide. Magnifications are 4X
Figure 8.
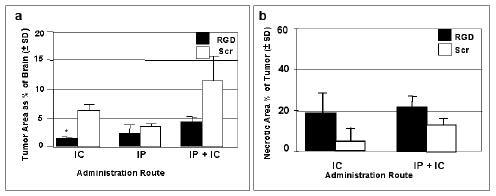
Tumor areas as percentages of total brain areas at the instillation site and necrotic areas as percentages of the total tumor areas, evaluated in groups of RGD and scrambled (Scr) peptide treated rat brains. The peptides were infused 6 times over a two week period. Sacrifice was at 48 hr following the last infusion. (a) The CNS-1 tumor areas are given as percentages of total brain area when administration of RGD or scrambled peptides was by intracranial (IC) or intraperitoneal (IP) routes, or both (IP + IC). (b) Necrotic areas as percentages of the tumor areas at the instillation site when administered by IC or IP + IC routes. The tumors given peptides by the IP route were not necrotic (data not shown).* By unpaired t test with Welch correction, comparing RGD vs Scr given IC the p value was significant at 0.0263. Also, comparing RGD given IP vs IP+IC was not considered quite significant with p=0.0547.
IV. Discussion
Previous studies have shown that anti-integrin antagonists (i.e. cyclic RGD peptides) block glioma tumor growth, however, analysis of them in various tumor models, integrin knockout mouse models and established glioma cell lines indicates that further characterization of the role of αv integrins in glioma biology is necessary. In this study we have focused on the characterization of integrin expression by a panel of low passage glioma cells and identified significant differences in the ratio of integrin αvβ3 and αvβ5 expression in these cells. We propose that the analysis of integrin expression in these low passage and infiltrative glioma cells will be an important consideration in the design of specific integrin antagonists. In these studies we show that although integrin αvβ3 is expressed in these low passage glioma cells, consistent with previous observations (Gladson and Cheresh 1991), we also observed a significantly higher level of integrins αvβ5 receptor and β1-containing integrins. Using various cyclic RGD integrin antagonists we suggest that targeting integrins αvβ5 and α5β1– may be more important to glioma cell biology than those inhibitors that are primarily restricted to αvβ3.
We synthesized a number of RGD-containing peptides and continued study with one that could potently bind to αvβ3, αvβ5 and α5β1 integrins. It suppressed glioma cell growth, inhibited adhesion of glioma cells to glioma-derived ECM, and induced a small degree of apoptosis in glioma cells in vitro. The findings that the RGD peptide induced reductions of both cell growth and adhesion may be surprising given the direct association of adhesion with migration, and extrapolation of cell migration occurring at the expense of proliferation as put forth in the “go or grow concept” (Berens and Giese, 1996). Also, it was surprising that the degree of apoptosis induction was relatively low at the few time points we assayed in vitro. The only reason the apoptosis percentages might be higher than assayed is if the cells had already lysed (i.e., not able to adhere in the in vitro morphologic assay or unrecoverable by centrifugation in the 7AAD flow cytometric assay). Of course, the percentages of injured cells might vary if other times after glioma cell exposure to RGD peptide are examined in vitro, or if the assay is performed with ECM substrate. Indeed, Taga and colleagues showed that the αv integrin antagonist EMD12197, which is a cyclic RGD-pentapeptide, induced apoptosis in human and pediatric brain tumor cells by detaching them from vitronectin and tenascin (Taga et al, 2002). One group reported that synthetic peptides containing the RGD sequence directly induced apoptosis by entering the cells and inducing autoprocessing and enzymatic activity of pre-caspase 3 (Buckley et al, 1999). Chatterjee and collaborators studied the effects of linear and cyclic RGD peptides on gliomas (Chatterjee et al, 2000). They found that conformation of the peptide was important, as the linear peptide had little killing effect. Furthermore, their cyclic RGD peptide was capable of killing glioma cell lines expressing αvβ3 receptors on their cellular membranes, like U-373MG or U-87MG, whereas it had relatively little effect on the U-251MG cell line that did not express the αvβ3 receptors. We assume the reason that the RGD peptide we tested was able to induce a similar level of apoptosis in U-373MG and U-251MG cells was because of its broader specificity. All of the published data are in correlation with the findings by MacDonald and Ladisch, where antisense oligonucleotides to the αv integrin inhibited growth and induced apoptosis of medulloblastoma cells (MacDonald and Ladisch 2001). Thus, our experimental findings conform with those from a number of studies with RGD peptide integrin antagonists that demonstrate similar anti-tumor actions on brain tumor cells.
The in vitro stability of the RGD peptide was very good in rat brain homogenates suspended in aCSF. We believe these data should be indicative of the stability of peptide if administered intracranially. Our data also showed that the RGD peptides displayed a consistent, high stability in plasma upon subcutaneous bolus injection (half life between 3-4 hours, data not shown). Therefore, the pharmacokinetic data demonstrate that they have useful half-lives when administered by these routes. In animal studies, Taga and colleagues showed that the daily systemic administration of the RGD peptide inhibited the growth of orthotopically implanted brain tumors in athymic mice (Taga et al, 2002). The Chatterjee group also intratumorally treated brain tumors with RGD peptide and found anti-tumor effects (Chatterjee et al, 2000). Our animal studies incorporated testing of intracranial or systemic administration of RGD peptide, or a combination of these two administration routes. The tumor areas traced in the groups of animals given RGD peptide intracranially or by combined intracranial and intraperitoneal administration were distinguishably smaller than the groups given scrambled peptide. The RGD peptide treated groups also had a higher necrotic index. The histopathological findings were from animal brains collected at 48 hr following the last administration of RGD peptide. Based upon the number of cells with pyknotic nuclei seen dispersed through the tumor, the necrotic index analyzed at later times might have been even higher. Interestingly, none of the animal tumors given intraperitoneally-administered peptides had necrotic areas, indicating delivery of the peptide to the tumor may have been compromised by the blood-brain-barrier. Since the animal study findings are a reflection of one early time point following the last RGD administration, a time course would be warranted before firm conclusions could be reached regarding the toxicity/efficacy of RGD peptide for brain tumor treatment. Nonetheless, the experimental peptide did have some in vitro and in vivo anti-glioma effects that cumulatively appeared to provide limited benefit in vivo. The treatment would need to be experimentally optimized, as healthy tumor cells existing in perivascular spaces would eventually cause the demise of the animals. Delivery of the RGD peptide appeared to be part of the problem. RGD peptide deposited at the instillation site was capable of reducing the tumor burden centrally. However, infiltrating glioma cells escaped the local intracranial delivery and systemic administration appeared to have little effect at the tumor periphery when combined with the intracranial administration. For a significant improvement in survival to occur, it is likely an improvement to delivery of the peptide, such as by convection enhanced delivery and/or transient opening of the blood-brain-barrier would be necessary (Neuwelt and Rapoport 1984; Bartus et al, 1996; Kroll and Neuwelt 1998; Chen et al, 1999). Further testing is warranted.

Carol A. Kruse
Acknowledgements
We thank Dr. Ron Ingram for helpful discussions regarding the peptide specificities. University of Colorado Cancer Summer Student Fellows, Ms. Esperanza Salazar and Stacy Muffly provided technical assistance associated with the animal studies. CAK was a member of the University of Colorado Cancer Center at the initiation of this project. We gratefully acknowledge financial support from the National Institutes of Health (DK51938-03 and NS046463). This work also was partially supported by Integra Neurosciences, the R. Herbert and Alma S. Manweiler Memorial Fund and the La Jolla Foundation for Molecular Medicine.
Abbreviations
- (7AAD)
7-amino actinomycin D
- (Fmoc)
9-fluorenylmethoxycarbonyl-
- (RGD peptide)
Ac-c[(Pen)-Tyr(Me)-Ala-Arg-Gly-Asp-Asn-Tic-Cys]NH2
- (aCSF)
artificial cerebrospinal fluid
- (DIC)
diisopropylcarbodiimide
- (ECM)
extracellular matrix
- (FBS)
fetal bovine serum
- (Fb)
fibrotic wall
- (H&E)
hematoxylin and eosin
- (HATU)
hexafluorophosphate
- (HOBt)
hydroxybenzotriazole
- (MFI)
mean fluorescence intensities
- (MBHA)
methylbenzhydrylamine
- (mOD/min)
milli-optical density units per min
- (Nc)
necrotic center
- (NB)
normal brain
- (PBS)
phosphate buffered saline
- (RP-HPLC)
reverse phase-high performance liquid chromatography
- (Scr)
scrambled
- (Tic)
tetrahydroisoquinoline carboxylic acid
References
- Bartus RT, Elliott PJ, Dean RL, Hayward NJ, Nagle TL, Huff MR, Snodgrass PA, Blunt DG. Controlled modulation of BBB permeability using the bradykinin agonist, RMP-7. Exp Neurol. 1996;142:14–28. doi: 10.1006/exnr.1996.0175. [DOI] [PubMed] [Google Scholar]
- Berens ME, Giese A. What's malignant about astrocytomas? Studies of brain tumor proliferation and migration. Barrows Neurol Inst Quarterly. 1996;12:15–22. [Google Scholar]
- Buckley CD, Pilling D, Henriquez NV, Parsonage G, Threlfall K, Scheel-Toellner D, Simmons DL, Akbar AN, Lord JM, Salmon M. RGD peptides induce apoptosis by direct caspase-3 activation. Nature. 1999;397:534–9. doi: 10.1038/17409. [DOI] [PubMed] [Google Scholar]
- CBTRUS . Central Brain Tumor Registry of the United States. 2002. CBTRUS: Statistical Report: Primary Brain Tumors in the United States, 1995-1999. [Google Scholar]
- Chatterjee S, Matsumura A, Schradermeier J, Gillespie GY. Human malignant glioma therapy using anti-α(v)β3 integrin agents. J Neurooncol. 2000;46:135–44. doi: 10.1023/a:1006444300504. [DOI] [PubMed] [Google Scholar]
- Chen MY, Lonser RR, Morrison PF, Governale LS, Oldfield EH. Variables affecting convection-enhanced delivery to the striatum: a systematic examination of rate of infusion, cannula size, infusate concentration and tissue-cannula sealing time. J Neurosurg. 1999;90:315–20. doi: 10.3171/jns.1999.90.2.0315. [DOI] [PubMed] [Google Scholar]
- Ding Q, Stewart J, Jr., Prince CW, Chang PL, Trikha M, Han X, Grammer JR, Gladson CL. Promotion of malignant astrocytoma cell migration by osteopontin expressed in the normal brain: differences in integrin signaling during cell adhesion to osteopontin versus vitronectin. Cancer Res. 2002;62:5336–43. doi: 10.1100/tsw.2002.247. [DOI] [PubMed] [Google Scholar]
- Eskens FA, Dumez H, Hoekstra R, Perschl A, Brindley C, Bottcher S, Wynendaele W, Drevs J, Verweij J, van Oosterom AT. Phase I and pharmacokinetic study of continuous twice weekly intravenous administration of Cilengitide (EMD 121974), a novel inhibitor of the integrins αvβ3 and αvβ5 in patients with advanced solid tumours. Eur J Cancer. 2003;39:917–26. doi: 10.1016/s0959-8049(03)00057-1. [DOI] [PubMed] [Google Scholar]
- Fleshner M, Watkins LR, Redd JM, Kruse CA, Bellgrau D. A 9L gliosarcoma transplantation model for studying adoptive immunotherapy into the brains of conscious rats. Cell Transplant. 1992;1:307–12. doi: 10.1177/096368979200100408. [DOI] [PubMed] [Google Scholar]
- Gladson CL, Cheresh DA. Glioblastoma expression of vitronectin and the avB3 integrin: adhesion mechanism for transformed glial cells. J Clin Invest. 1991;88:1924–32. doi: 10.1172/JCI115516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez GG, Kruse CA. The isolation and culture of human brain tumor cells. In: Langdon S, editor. Methods in Molecular Medicine, Cancer Cell Culture: Methods and Protocols. Vol. 88. Humana; Totowa, NJ: 2003. pp. 101–109. [DOI] [PubMed] [Google Scholar]
- Gomez GG, Read SB, Gerschenson LE, Santoli D, Zweifach A, Kruse CA. Interactions of the allogeneic effector leukemic T cell line, TALL-104, with human malignant brain tumors. Neurooncol. 2004;6:83–95. doi: 10.1215/S1152851703000140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosshans DR, Clayton DA, Coultrap SJ, Browning MD. Science's STKE. 2002. Analysis of glutamate receptor surface expression in acute hippocampal slices. [DOI] [PubMed] [Google Scholar]
- Heimberger AB, Archer GE, McLendon RE, Hulette C, Friedman AH, Friedman HS, Bigner DD, Sampson JH. Temozolomide delivered by intracerebral microinfusion is safe and efficacious against malignant gliomas in rats. Clin Cancer Res. 2000;6:4148–53. [PubMed] [Google Scholar]
- Hynes RO. A reevaluation of integrins as regulators of angiogenesis. Nat Med. 2002;8:918–21. doi: 10.1038/nm0902-918. [DOI] [PubMed] [Google Scholar]
- Kleinschmidt-DeMasters BK, Orr EA, Savelieva E, Owens GC, Kruse CA. Paucity of retinoic acid receptor α (RAR α) nuclear immunostaining in gliomas and inability of retinoic acid to influence neural cell adhesion molecule (NCAM) expression. J Neurooncol. 1999;41:31–42. doi: 10.1023/a:1006162211296. [DOI] [PubMed] [Google Scholar]
- Kroll RA, Neuwelt EA.Outwitting the blood-brain barrier for therapeutic purposes: osmotic opening and other means Neurosurg 1998421083–99.discussion 99-100 [DOI] [PubMed] [Google Scholar]
- Kruse CA, Lamb C, Hogan S, Smiley WR, Kleinschmidt-Demasters BK, Burrows FJ. Purified herpes simplex thymidine kinase retroviral particles. II. Influence of clinical parameters and bystander killing mechanisms. Cancer Gene Ther. 2000;7:118–27. doi: 10.1038/sj.cgt.7700097. [DOI] [PubMed] [Google Scholar]
- Kruse CA, Mitchell DH, Kleinschmidt-DeMasters BK, Bellgrau D, Eule JM, Parra JR, Kong Q, Lillehei KO. Systemic chemotherapy combined with local adoptive immunotherapy cures rats bearing 9L gliosarcoma. J Neurooncol. 1993;15:97–112. doi: 10.1007/BF01053931. [DOI] [PubMed] [Google Scholar]
- Kruse CA, Molleston MC, Parks EP, Schiltz PM, Kleinschmidt-DeMasters BK, Hickey WF. A rat glioma model, CNS-1, with invasive characteristics similar to those of human gliomas: a comparison to 9L gliosarcoma. J Neurooncol. 1994a;22:191–200. doi: 10.1007/BF01052919. [DOI] [PubMed] [Google Scholar]
- Kruse CA, Schiltz PM, Bellgrau D, Kong Q, Kleinschmidt-DeMasters BK. Intracranial administrations of single or multiple source allogeneic cytotoxic T lymphocytes: chronic therapy for primary brain tumors. J Neurooncol. 1994b;19:161–8. doi: 10.1007/BF01306458. [DOI] [PubMed] [Google Scholar]
- Kruse CA, Varella-Garcia M, Kleinschmidt-Demasters BK, Owens GC, Spector EB, Fakhrai H, Savelieva E, Liang BC. Receptor expression, cytogenetic and molecular analysis of six continuous human glioma cell lines. In Vitro Cell Dev Biol Anim. 1998;34:455–62. doi: 10.1007/s11626-998-0078-x. [DOI] [PubMed] [Google Scholar]
- MacDonald TJ, Ladisch S. Antisense to integrin αv inhibits growth and induces apoptosis in medulloblastoma cells. Anticancer Res. 2001;21:3785–91. [PubMed] [Google Scholar]
- MacDonald TJ, Taga T, Shimada H, Tabrizi P, Zlokovic BV, Cheresh DA, Laug WE. Preferential susceptibility of brain tumors to the antiangiogenic effects of an α(v) integrin antagonist. Neurosurg. 2001;48:151–7. doi: 10.1097/00006123-200101000-00026. [DOI] [PubMed] [Google Scholar]
- Milner R, Campbell IL. The integrin family of cell adhesion molecules has multiple functions with the CNS. J Neurosci Res. 2002;69:286–91. doi: 10.1002/jnr.10321. [DOI] [PubMed] [Google Scholar]
- Neuwelt EA, Rapoport SI. Modification of the blood-brain barrier in the chemotherapy of malignant brain tumors. Fed Proc. 1984;43:214–9. [PubMed] [Google Scholar]
- Owens GC, Orr EA, DeMasters BK, Muschel RJ, Berens ME, Kruse CA. Overexpression of a transmembrane isoform of neural cell adhesion molecule alters the invasiveness of rat CNS-1 glioma. Cancer Res. 1998;58:2020–8. [PubMed] [Google Scholar]
- Philpott NJ, Turner AJ, Scopes J, Westby M, Marsh JC, Gordon-Smith EC, Dalgleish AG, Gibson FM. The use of 7-amino actinomycin D in identifying apoptosis: simplicity of use and broad spectrum of application compared with other techniques. Blood. 1996;87:2244–51. [PubMed] [Google Scholar]
- Phuphanich S, Brat DJ, Olson JJ. Delivery systems and molecular targets of mechanism-based therapies for GBM. Expert Rev Neurotherapeutics. 2004;4:649–63. doi: 10.1586/14737175.4.4.649. [DOI] [PubMed] [Google Scholar]
- Prados MD, Berger MS, Wilson CB.Primary central nervous system tumors: advances in knowledge and treatment CA: Cancer J Clin 199848331–60.21 [DOI] [PubMed] [Google Scholar]
- Read SB, Kulprathipanja NV, Gomez GG, Paul DB, Winston KR, Robbins JM, Kruse CA. Human alloreactive CTL interactions with gliomas and with those having upregulated HLA expression from exogenous IFN-γ or IFN-γ gene modification. J Int Cyt Res. 2003;23:379–93. doi: 10.1089/107999003322226032. [DOI] [PubMed] [Google Scholar]
- Ruoslahti E, Reed J. New way to activate caspases. Nature. 1999;397:479–80. doi: 10.1038/17229. [DOI] [PubMed] [Google Scholar]
- Schmid I, Uittenbogaart CH, Keld B, Giorgi JV. A rapid method for measuring apoptosis and dual-color immunofluorescence by single laser flow cytometry. J Immunol Methods. 1994;170:145–57. doi: 10.1016/0022-1759(94)90390-5. [DOI] [PubMed] [Google Scholar]
- Taga T, Suzuki A, Gonzalez-Gomez I, Gilles FH, Stins M, Shimada H, Barsky L, Weinberg KI, Laug WE. αv-Integrin antagonist EMD 121974 induces apoptosis in brain tumor cells growing on vitronectin and tenascin. Int J Cancer. 2002;98:690–7. doi: 10.1002/ijc.10265. [DOI] [PubMed] [Google Scholar]
- Zhang L, Zhou W, Velculescu VE, Kern SE, Hruban RH, Hamilton SR, Vogelstein B, Kinzler KW. Gene expression profiles in normal and cancer cells. Science. 1997;276:1268–72. doi: 10.1126/science.276.5316.1268. [DOI] [PubMed] [Google Scholar]