

Abstract
Gene expression profiling provides a quantitative molecular framework for the study of human lymphomas. This genomic technology has revealed that existing diagnostic categories are comprised of multiple molecularly and clinically distinct diseases. Diffuse large B cell lymphoma (DLBCL), for example, consists of three gene expression subgroups, termed germinal center B cell-like (GCB) DLBCL, activated B cell-like (ABC) DLBCL, and primary mediastinal be cell lymphoma (PMBL). These DLBCL subgroups arise from different stages of normal B cell differentiation, utilize distinct oncogenic mechanisms, and differ in their ability to be cured by chemotherapy. Key regulatory factors and their target genes are differentially expressed among these subgroups, including BCL-6, Blimp-1, and XBP1. ABC DLBCL and PMBL depend upon constitutive activation of the NF-κB pathway for their survival but GCB DLBCL does not, demonstrating that this pathway is a potential therapeutic target for certain DLBCL subgroups. In DLBCL, mantle cell lymphoma, and follicular lymphoma, gene expression profiling has also been used to create gene expression-based models of survival, which have identified the biological characteristics of the tumors that influence their clinical behavior. In mantle cell lymphoma, the length of survival following diagnosis is primarily influenced by the tumor proliferation rate, which can be quantitatively measured by a proliferation gene expression “signature”. Based on this accurate measure, the proliferation rate can now be viewed as an integration of several oncogenic lesions that each increase progression from G1 to S phase of the cell cycle. In DLBCL and follicular lymphoma, gene expression profiling has revealed that the molecular characteristics of non-malignant tumor-infiltrating immune cells have a major influence on the length of survival. The implications of these insights for the diagnosis and treatment of non-Hodgkin lymphomas are discussed.
INTRODUCTION
The study of human diseases presents a unique challenge to the experimental biologist. Traditional modes of biological inquiry often require the manipulation of a biological system, which may be difficult or impossible to achieve when studying a human disease. Therefore, initial insights into human diseases are often observational in nature, and the depth of these insights relies on the precision and breadth of the observational tools at hand.
A traditional and successful approach to the study of human cancer begins with the identification of genes that are mutated and/or misexpressed in one or more cancer specimen. These potential oncogenes and tumor suppressor genes are then assessed for their ability to create or modify a transformed phenotype in cultured cells or in mice. By assessing such genes one by one, an edifice of knowledge has been created that provides a framework for understanding cancer biology (Hanahan and Weinberg, 2000).
An orthogonal approach to human cancer has been provided by two key methodological advances: the completion of the human genome sequence and the development of DNA microarrays to measure the activity of the genome. A genomewide measurement of mRNA levels in a cancer specimen, known as its gene expression profile, can reveal aspects of the cancer phenotype that are difficult to perceive by studying individual oncogenes and tumor suppressor genes. Even more powerful insights can be derived when gene expression profiles are obtained from a large number of cancer specimens for which clinical data are available. Clinical data such as response to treatment and length of survival can be used to tease biological insights out of the large data sets that DNA microarrays generate. The beauty of gene expression profiling data is that it is quantitative and highly reproducible. Because of this, these data can be used to generate multivariate statistical models of the clinical behavior of cancer that have great predictive power.
From this new perspective, human cancer is found to be mathematically tractable. Various biological phenotypes of a tumor, such as the proliferation rate, the activity of survival pathways, and the nature of non-malignant tumor-infiltrating cells, can now be accurately measured because they are reflected in the gene expression profile. Each biological phenotype can be associated with the expression of a characteristic set of genes, known as its gene expression signature, and the expression of these signature genes serve as a quantitative measure of the phenotype (Shaffer et al., 2001). The length of survival of cancer patients and their response to therapy can be traced to biological differences in their tumors that are reflected quantitatively in gene expression signatures. Often, several gene expression signatures are needed to adequately model the complex clinical behavior of cancer, thus revealing cancer as the integration of multiple abnormal biological mechanisms. By combining gene expression profiling with traditional analysis of oncogenes and tumor suppressors, it becomes clear that the multiple oncogenic abnormalities in a single tumor act synergistically and quantitatively to alter discrete tumor phenotypes. This mathematical view of the cancer process will ultimately change our approach to cancer therapy, particularly as agents that target discrete biological pathways become available
This review will provide examples from studies of lymphoid malignancies that illustrate the ability of gene expression profiling to illuminate cancer biology. The first section demonstrates how gene expression profiling can reveal that current diagnostic categories are comprised of several molecularly and clinically distinct diseases. The second section demonstrates how mathematical models of survival in cancer can be created by gene expression profiling, thereby revealing biological attributes of the tumors that influence their clinical behavior.
MOLECULAR DIAGNOSIS OF LYMPHOID MALIGNANCIES
Molecularly and clinically distinct diseases within diffuse large B cell lymphoma
Diffuse large B cell lymphoma (DLBCL) is the largest diagnostic category among the non-Hodgkin lymphomas, accounting for roughly 40 percent of all cases. Within the United States, this translates into more than 23,000 new cases of DLBCL diagnosed each year. Multi-agent chemotherapy can cure approximately 40% of these patients, and this represents one of the successes of modern cancer therapy. Unfortunately, DLBCL still accounts for approximately 10,000 deaths per year in the United States. Why some patients with DLBCL can be cured and others not has been a longstanding and frustrating puzzle.
The first clue to this puzzle came when gene expression profiling studies revealed that DLBCL is comprised of at least two molecularly and clinically distinct diseases (Alizadeh et al., 2000;Rosenwald et al., 2002;Wright et al., 2003). One subgroup of DLBCL, termed germinal center B cell-like (GCB) DLBCL, expresses genes that are hallmarks of normal germinal center B cells (Fig. 1A). By contrast, another DLBCL subgroup, termed activated B cell-like (ABC) DLBCL, lacks expression of germinal center B cell-restricted genes and instead expresses genes that are induced during mitogenic stimulation of blood B cells. These two subgroups of DLBCL differ in the expression of thousands of genes, and in this respect they are as different as acute myelogenous leukemia and acute lymphoblastic leukemia. More recently, a third subgroup of DLBCL has been defined by gene expression profiling, termed primary mediastinal B cell lymphoma (PMBL) (Rosenwald et al., 2003a;Savage et al., 2003). These three DLBCL subgroups should be considered separate disease entities since they arise from B cells at different stages of differentiation, utilize different oncogenic pathways, and have distinct clinical behaviors.
Figure 1.

A. Genes characteristically expressed by three subgroups diffuse large B-cell lymphoma (DLBCL): Primary mediastinal B-cell lymphoma (PMBL), germinal center B-cell-like (GCB) DLBCL, and the activated B cell-like (ABC) DLBCL (Rosenwald et al.,2002;Rosenwald et al., 2003a;Wright et al., 2003). Each of the 201 columns represents gene expression data from a single DLBCL biopsy sample and each row represents expression of a single gene. Relative gene expression is indicated according to the color scale shown. B. Kaplan-Meier plot of overall survival for the different DLBCL subgroups. C. Distinct oncogenic mechanisms in the DLBCL subgroups. D. Selective toxicity of a small molecule IκB kinase inhibitor for ABC DLBCL and PMBL cell lines (Lam et al., 2004).
DLBCL subgroups and the regulatory biology of B cell differentiation
Insight into the normal cellular counterparts of the DLBCL subgroups has been provided by an analysis of regulatory factors that control the differentiation of germinal center B cells to plasma cells (Fig. 2). BCL-6 is a transcriptional repressor that is required for mature B cells to differentiate into germinal center B cells during an immune response (Dent et al., 1997;Ye et al., 1997). Normal germinal center B cells express BCL-6 at high levels but BCL-6 expression is silenced during plasmacytic differentiation (Allman et al., 1996;Cattoretti et al., 1995;Onizuka et al., 1995). DLBCLs belonging to the GCB subgroup express BCL-6mRNA at significantly higher levels than DLBCLs belonging to the ABC subgroup (Alizadeh et al., 2000;Rosenwald et al., 2002;Wright et al., 2003). BCL-6 is deregulated by chromosomal translocations in roughly 20% of DLBCLs (Pasqualucci et al., 2003), but the high expression of BCL-6 in GCB DLBCLs is not accounted for by these translocations. Rather,BCL-6 is expressed in GCB DLBCLs along with a host of other germinal center B cell restricted-genes because these DLBCLs are derived from normal germinal center B cells and retain much of their biology. In keeping with this notion, GCB DLBCLs have ongoing somatic hypermutation of their immunoglobulin genes, a characteristic feature of normal germinal center B cells (Lossos et al., 2000).
Figure 2.

Schematic of the regulatory network governing differentiation of a germinal center B cell to a plasma cell.
To elucidate the mechanisms by which BCL-6 regulates germinal center B cell differentiation, gene expression profiling was used to identify the target genes of BCL-6 repression (Shaffer et al., 2000). DNA microarrays were used to identify genes that were downregulated when BCL-6 was introduced into cells lacking endogenous BCL-6 expression and that were upregulated when a dominant negative form of BCL-6 was introduced into cells that have endogenous BCL-6 expression. The BCL-6 target genes identified in this fashion were found to be expressed at much lower levels in germinal center B cells than in resting or activated blood B cells, in keeping with the germinal center B cell-restricted expression of BCL-6 (Shaffer et al., 2000). One group of BCL-6 target genes are genes that are induced when B cells are activated through the antigen receptor, including cyclin D2, CD69, CD44, and MIP-1 α. By blocking expression of B cell activation genes, BCL-6 may guide an antigen-stimulated B cell towards germinal center differentiation and away from the alternate fate of rapid plasmacytic differentiation (Shaffer et al., 2000). Another important BCL-6 target gene is p27kip1, which encodes a negative regulator of cell cycle progression. By repressing p27kip1, BCL-6 may contribute to the extraordinarily high proliferation rate of germinal center B cells.
One particularly illuminating BCL-6 target gene is Blimp-1 (Shaffer et al., 2000), which encodes a transcriptional repressor that is required for plasma cell differentiation (Shapiro-Shelef et al., 2003). BCL-6 binds to a motif in intron 5 of the Blimp-1 gene that is conserved between the human and mouse genes (Tunyaplin et al., 2004). Since BCL-6 is bound to this site in vivo, as judged by chromatin immunoprecipitation, Blimp-1 appears to be a direct target of BCL-6 repression. Dominant negative inhibition of BCL-6 in a Burkitt lymphoma cell line (which is of germinal center origin) caused the endogenous Blimp-1 gene to be derepressed, and this was followed by partial plasmacytic differentiation: multiple mature B cell markers were silenced while plasma cell markers such as CD38 were induced (Shaffer et al., 2000). In addition, c-mycand a host of other proliferation associated genes were turned off (Shaffer et al., 2000), which is consistent with the fact that c-myc is a direct target of Blimp-1 repression (Lin, Wong and Calame, 1997).
These results led to the hypothesis that one role for BCL-6 in normal germinal center B cells and in non-Hodgkin lymphomas is to block differentiation into plasma cells (Shaffer et al., 2000). In keeping with this notion, mice in which the BCL-6 gene is disrupted generate plasma cells more readily in response to antigenic stimulation (Tunyaplin et al., 2004). In non-Hodgkin lymphomas with BCL-6 translocations, the promoter of BCL-6is substituted with the promoter from the translocation partner gene, which may prevent the physiological downregulation of BCL-6 mRNA expression that occurs during plasmacytic differentiation. This deregulation of BCL-6 may contribute to malignant transformation by keeping Blimp-1 repressed, thereby preventing plasmacytic differentiation and the attendant cell cycle arrest.
Gene expression profiling has also been used to elucidate the mechanisms by which Blimp-1 promotes plasmacytic differentiation (Shaffer et al., 2002;Shaffer et al., 2004). Ectopic expression of Blimp-1 in B cells silences the expression of virtually all mature B cell-restricted genes (Shaffer et al., 2002). Blimp-1 exerts its powerful effect by directly repressing genes encoding key B cell transcription factors, including Pax5, Spi-B, Id3, and CIITA, thereby indirectly blocking the expression of the genes that they transactivate (Lin et al., 2002;Piskurich et al., 2000;Shaffer et al., 2002).
One of the genes silenced by Blimp-1, directly or indirectly, is BCL-6 (Shaffer et al., 2002). Blimp-1 and BCL-6 therefore create a double negative regulatory loop (Shaffer et al., 2002), a form of regulation first described by Monod and Jacob four decades ago (Monod and Jacob, 1961). An important feature of this regulatory design is its plasticity: a change in the expression or activity of either repressor, even transiently, can swing the system towards one of two cellular states. In germinal center B cells, strong stimulation by antigen through the immunoglobulin receptor or by activated T cells through CD40 downregulates BCL-6 mRNA and/or protein expression (Allman et al., 1996;Niu, Ye and Dalla-Favera, 1998), which would tip the regulatory balance in favor of Blimp-1 and plasmacytic differentiation.
Strong support for this double negative model has recently been provided by the analysis of MTA3, a germinal center B cell-restricted subunit of the Mi-2/NuRD corepressor complex (Fujita et al., 2003). MTA3 tethers the Mi-2/NuRD complex to BCL-6 by binding to a central region of BCL-6 that is required for full transcriptional repression activity (Albagli et al., 1996;Chang et al., 1996;Seyfert et al., 1996). The Mi-2/NuRD complex has histone deacetylase activity, which BCL-6 uses to create a repressive chromatin structure at its target genes. Knockdown of MTA3 by RNA interference in germinal center B cell-derived lymphoma cell lines caused Blimp-1 to be derepressed and partial plasmacytic differentiation to ensue (Fujita et al., 2003). Remarkably, coexpression of BCL-6 and MTA3 in a plasmacytic cell line derived from a patient with multiple myeloma caused the cells to “dedifferentiate” to a mature B cell phenotype: the plasma cell genes Blimp-1, XBP1, and syndecan-1 were silenced while the B cell genes CD19, CD20, BLNK, syk, Igβ and MHC class II were expressed. Although this dedifferentiation is surprising, it is an inherent feature of the plasticity built into it double negative regulatory circuit (Monod and Jacob, 1961).
In addition to Blimp-1, plasmacytic differentiation requires the transcriptional activator XBP1 (Reimold et al., 2001). XBP1 lies downstream of Blimp-1 in the regulatory hierarchy: XBP1 mRNA expression is induced during plasmacytic differentiation, in part, because Blimp-1 represses Pax5, which encodes a repressor of XBP1 (Lin et al., 2002;Reimold et al., 1996;Shaffer et al., 2002). Recent experiments demonstrated that XBP1 is the master regulator of the secretory phenotype of plasma cells (Shaffer et al., 2004). Gene expression profiling revealed that XBP1 induces the expression of a large set of genes encoding components of the endoplasmic reticulum and golgi, leading to a dramatic expansion of the secretory apparatus (Shaffer et al., 2004). Unexpectedly, XBP1 also increases cell size, mitochondrial and lysosomal biogenesis, mitochondrial respiration, total cellular protein content, and the overall rate of protein synthesis (Shaffer et al., 2004). XBP1 is a mammalian homologue of the yeast transcription factor Hac1, which controls the unfolded protein response that is triggered by cellular events that disturb the processing of proteins in the endoplasmic reticulum. In mammalian cells, XBP1 coordinates a “physiological” unfolded protein response that allows permanent differentiation to a secretory state characterized by a continuous, high burden of proteins entering the secretory pathway (Shaffer et al., 2004). The pleiotropic effects of XBP1 on cellular structure, function and energetics are all likely to contribute to the high-level secretion of immunoglobulin that characterizes plasma cells.
Based on the regulatory biology of plasmacytic differentiation, it is possible to speculate that the cell of origin of ABC DLBCL may be a plasmablastic B cell that is poised to exit the germinal center. In comparison to GCB DLBCLs, ABC DLBCLs express higher levels of many plasma cell genes (Wright et al., 2003). Among these is XBP1 and a variety of its target genes, which encode endoplasmic reticulum and golgi. Further, ABC DLBCLs express Blimp-1mRNA at levels comparable to those in multiple myeloma cells (unpublished). This phenotype is similar to that of a rare subpopulation of plasmablasts in the germinal center, which are thought to be in the process of migrating to the bone marrow where they differentiate fully into plasma cells (Angelin-Duclos et al., 2000;Falini et al., 2000). ABC DLBCLs do not express a variety of other genes that characterize normal plasma cells and multiple myeloma, suggesting that they are derived from a cell that is intermediate between a germinal center B cell and a plasma cell. In support of this notion, ABC DLBCLs have somatically mutated immunoglobulin genes, and therefore are derived from a B cell that has likely traversed the germinal center (Lossos et al., 2000). However, in contrast to GCB DLBCLs, ABC DLBCLs have a fixed complement of immunoglobulin gene mutations, suggesting that the somatic hypermutation machinery has been inactivated as occurs normally during plasmacytic differentiation.
The increased understanding of the regulatory biology of DLBCLs may provide avenues for therapy. BCL-6 itself could be a therapeutic target given that inhibition of BCL-6 function in a Burkitt lymphoma cell line caused cell cycle arrest by derepressing the cell cycle inhibitor p27kip1 and by derepressing Blimp-1, thereby downregulating c-myc expression (Shaffer et al., 2000). The activity of BCL-6 as a transcriptional repressor could be inhibited by blocking its interaction with corepressor complexes containing MTA3 or those containing SMRT, NCoR, or BCoR that bind to its aminoterminal POZ/BTB domain (Dhordain et al., 1997;Huynh et al., 2000;Melnick et al., 2002;Muscat, Burke and Downes, 1998;Wong and Privalsky, 1998). The function of BCL-6 is also inhibited by acetylation, and therefore drugs that inhibit deacetylation of BCL-6 might prove useful (Bereshchenko, Gu and Dalla-Favera, 2002). Further work is needed to understand which DLBCLs might be susceptible to BCL-6 inhibition. ABC LBCLs may be less susceptible than GCB DLBCLs since that have lower BCL-6 expression. Curiously, ABC DLBCLs have high Blimp-1 mRNA expression and yet are highly proliferative and express c-myc at high levels. It is therefore conceivable that ABC DLBCLs may have a means to inactivate Blimp-1 function, and understanding this mechanism might provide additional molecular targets for this DLBCL subgroup.
Primary mediastinal B cell lymphoma: a DLBCL subgroup related to Hodgkin lymphoma
Primary mediastinal B cell lymphoma (PMBL) was initially defined as a subgroup of DLBCL based primarily on its unusual clinical presentation: these patients are often young and have an aggressive lymphoma originating in the mediastinum. Since this lymphoma subgroup was solely defined by its clinical characteristics, it was likely to be heterogeneous and include other lymphoma types that happened by chance to present in the mediastinum. Gene expression profiling was able to clarify this diagnostic category by identifying a clear gene expression signature that distinguishes this type of DLBCL from the other DLBCL subgroups (Fig. 1A) (Rosenwald et al., 2003a;Savage et al., 2003). Importantly, one quarter of the samples submitted with the clinical diagnosis of PMBL were found to lack the PMBL gene expression signature, and are therefore likely to be cases of GCB or ABC DLBCL with predominant mediastinal involvement (Rosenwald et al., 2003a).
Patients with a molecular diagnosis of PMBL were found to be young (median age 33), as compared to patients with GCB and ABC DLBCL (median age over 60), and the majority of the young patients were women (Rosenwald et al., 2003a). PMBL tumors often involved other thoracic sites, such as the pleura, pericardium, lung, and breast, and these sites were infrequently involved in cases of GCB or ABC DLBCL (Rosenwald et al., 2003a). Conversely, sites of frequent extranodal involvement in GCB and ABC DLBCL are the gastrointestinal tract, bone marrow, liver, and muscle, and PMBL tumors never involved these sites. Thus, PMBL tumors appear to spread by local extension from the mediastinum whereas GCB and ABC DLBCL may spread through the bloodstream.
Gene expression profiling uncovered a striking and unexpected relationship between PMBL and Hodgkin lymphoma (Rosenwald et al., 2003a;Savage et al., 2003). Over one-third of the genes that distinguish PMBL from the other DLBCL subgroups ere also highly expressed in Hodgkin lymphoma cell lines as compared with GCB DLBCL cell lines. Several of the PMBL signature genes (e.g. MAL, IL4I) were found to be expressed in primary Hodgkin Reed-Sternberg cells microdissected from cases of nodular-sclerosing Hodgkin lymphoma (Rosenwald et al., 2003a). Likewise, the PMBL signature includes several genes that are known to be characteristically expressed in Hodgkin Reed-Sternberg cells, such as CD30, IL-13 receptor α chain, and TARC (Peh, Kim and Poppema, 2001;Schwab et al., 1982;Skinnider et al., 2001). However, the gene expression programs of PMBL and Hodgkin lymphoma are not identical. A subset of the PMBL signature genes is not expressed in Hodgkin lymphoma cells (Rosenwald et al., 2003a). Moreover, Hodgkin lymphoma cells characteristically lack expression of many mature B cell genes, but these are expressed by PMBLs.
Once realized, it is evident that PMBL and Hodgkin lymphoma have many clinical and pathological features in common (Jaffe and Muller-Hermelink, 1999). Both lymphoma types are common in young individuals, especially women. Furthermore, Hodgkin lymphoma often originates in the mediastinum, like PMBL, and upon pathological examination both lymphoma types can be associated with thymic remnants. PMBL is characterized frequently by prominent sclerotic reactions histologically as is also the case in nodular-sclerosing Hodgkin lymphoma. Interestingly, case reports have documented that patients with PMBL can relapse after treatment with Hodgkin lymphoma (Gonzalez, Medeiros and Jaffe, 1991;Zarate-Osorno et al., 1992).
This spectrum of molecular, pathological, and clinical similarities suggests that PMBL and Hodgkin lymphoma are pathogenetically related. One explanation for these similarities could be that both lymphoma types may originate from a rare B cell population in the thymus (Addis and Isaacson, 1986). In support of this notion, a gene that is characteristically expressed in PMBL and Hodgkin lymphoma, MAL, is also expressed in a subset of thymic medullary B cells (Copie-Bergman et al., 2002). Thus, it is possible that PMBL and some cases of Hodgkin lymphoma inherit a gene expression program that is characteristic of these thymic B cells. That being said, the origin and function of thymic B cells is obscure at present. Thymic B cells have a high load of somatic mutation in their immunoglobulin genes without evidence of ongoing somatic hypermutation, as do PMBLs (Csernus et al., 2004;Flores, Li and Hale, 2001;Kuppers, Rajewsky and Hansmann, 1997;Leithauser et al., 2001;Pileri et al., 2003;Tonnelle et al., 1997). These data suggest that thymic B cells are post-germinal center in origin, but it is unclear whether these B cells diversify in the thymus or diversify elsewhere and home to this organ.
The gene expression similarities between PMBL and Hodgkin lymphoma are also due to the fact that they share several oncogenic mechanisms. The NF-κB signaling pathway is constitutively active in both the lymphoma types, leading to the shared expression of many NF-κB target genes (see below) (Rosenwald et al., 2003a;Savage et al., 2003). In addition, both PMBL and Hodgkin lymphoma have recurrent gains and amplifications of a chromosomal region in cytoband 9p24. These abnormalities are present in 40-50% of PMBL tumors, and 10% have a high-level amplification (Rosenwald et al., 2003a). This region is also amplified in Hodgkin lymphoma cell lines and in primary Hodgkin Reed-Sternberg cells (Bentz et al., 2001;Copie-Bergman et al., 2002;Joos et al., 2000;Joos et al., 1996;Rosenwald et al., 2003a). The amplicon contains the genes encoding the tyrosine kinase JAK2, the B7 family members PDL1 and PDL2, and the putative chromatin regulator SMARCA2 (Rosenwald et al., 2003a). Of these, JAK2 has received the most attention due to its involvement in stimulating cytokine signaling pathways. Several PMBL signature genes are known to be induced by IL-4, including IL4I/Fig1 (Chu and Paul, 1997), and MAL(Kovanen et al., 2003). STAT6, which mediates signaling by IL-4 and IL-13, is phosphorylated and present in the nucleus of PMBL cell lines and most primary PMBL tumors (Guiter et al., 2004). Indeed, JAK2 is phosphorylated and potentially active in PMBL cell lines and primary tumors (Guiter et al., 2004). The mechanisms underlying the activity of JAK2 remain to be elucidated, as neither IL-4 nor IL-13 are expressed in PMBL.
Genomic copy number changes in cancer may be selected because they simultaneously alter the expression or activity of several genes in the affected region. It is therefore possible that the 9p24 amplicon is important in PMBL and Hodgkin lymphoma because it causes overexpression of PDL1 and/or PDL2, in addition to JAK2. PDL2 was found to be the gene that distinguished PMBL from other DLBCLs most significantly (Rosenwald et al., 2003a). PDL2 was found to be highly expressed in PMBL tumors even in the absence of a 9p24 gain or amplification and was further upregulated in those tumors in which the gene was amplified. PDL1was likewise overexpressed in PMBL tumors but to a lesser degree than PDL2. Both genes are also expressed in Hodgkin lymphoma cells (Rosenwald et al., 2003a). PDL1 and PDL2 belong to the B7 family of surface proteins that affect T cell responses (Curiel et al., 2003;Dong et al., 2002;Dong et al., 1999;Freeman et al., 2000;Latchman et al., 2001). Both proteins bind PD-1, a cell surface receptor on T cells that bears sequence homology with CD28 and CTLA4 (Dong et al., 1999;Freeman et al., 2000;Latchman et al., 2001). Engagement of PD-1 by PDL1 or PDL2 delivers a negative signal that inhibits signaling through the T cell receptor (Freeman et al., 2000;Latchman et al., 2001). It has been suggested that tumors that express PDL1 or PDL2 may inhibit tumor-specific T cell responses (Dong and Chen, 2003;Dong et al., 2002). In addition, however, PDL2 can costimulate T cells under certain conditions, which is mediated by another receptor that has not yet been identified (Liu et al., 2003). Given the probable origin of PMBL from a thymic B cell, the cell surface expression of PDL2 or PDL1 may allow these lymphoma cells to coexist with the T cells in the thymus. On one hand, PDL1 and PDL2 may inhibit T cell responses to the lymphoma. On the other hand, the ability of PDL2 to costimulate thymic T cells may lead to the production of cytokines that promote proliferation and/or survival of the PMBL cells.
Oncogenic pathways in DLBCL subgroups
One of the most compelling arguments supporting the view that the DLBCL subgroups represent distinct diseases is that they utilize distinct oncogenic mechanisms (Fig. 1C). The t(14;18) translocation deregulates the BCL-2 gene by placing it near the enhancer elements of the immunoglobulin heavy chain locus. This oncogenic event was found to be common in GCB DLBCL, occurring in 45% (29/65) of cases analyzed, and also was detected in 18% (2/11) of PMBL cases (Huang et al., 2002;Rosenwald et al., 2002). By contrast, out of 42 cases of ABC DLBCL analyzed, none had a BCL-2 translocation. Nonetheless, the majority of ABC DLBCLs express BCL-2 mRNA at high levels, presumably due to transcriptional deregulation of BCL-2 (Rosenwald et al., 2002;Wright et al., 2003). Furthermore, the anti-apoptotic BCL-2 family member A1 is expressed in ABC DLBCLs due to their constitutive NF-κB activity (see below). In the ace of the high expression of BCL-2 and/or A1 in ABC DLBCL, there would be no selective pressure for a t(14;18) translocation. On the other hand, normal germinal center entroblasts express little if any BCL-2, which favors apoptosis as the default pathway in these cells (Martinez-Valdez et al., 1996;Tuscano et al., 1996). Therefore, the t(14;18) translocation would provide a strong selective advantage in a germinal center B cell-derived lymphomas such as GCB DLBCL.
Another recurrent oncogenic abnormality in DLBCL is amplification of the c-rellocus on chromosome arm 2p. This oncogenic event occurs in 16% of GCB DLBCLs and in 25% of PMBLs, but has never been detected in ABC DLBCLs (Rosenwald et al., 2002;Bea et al., 2005). c-rel encodes a member of the anti-apoptotic NF-κB family of transcription factors. However, it is currently unclear what selective advantage is conferred by c-rel amplification in GCB DLBCLs since cases with this abnormality do not express NF-κB target genes at higher levels than those with a wild type c-rel copy number (unpublished). This conundrum might be explained by the fact that NF-κB transcription factors require the activity of IκB kinase to become localized to the nucleus. During a germinal center reaction, stimulation through the antigen receptor or through CD40 can activate IκB kinase, and a germinal center B cell with a c-rel amplification might receive a stronger anti-apoptotic signal and be positively selected. In this scenario, c-rel amplifications may confer a selective advantage early in the genesis of a GCB DLBCL, but may not be functionally important in the tumor at diagnosis.
Comparative genomic hybridization analysis has revealed multiple differences in chromosomal copy number alterations between the DLBCL subgroups (unpublished). In ABC DLBCLs, trisomy 3 or gain of the chromosome 3q arm was detected in 18% (11/62) and 24% (15/62) of cases, respectively, but these abnormalities were never detected in GCB DLBCLs (0/63). High-level amplification of chromosome band 18q21, which contains the BCL-2 gene, was detected in one sixth of ABC DLBCLs but in only 3% of GCB DLBCLs. This abnormality may occur more commonly in ABC DLBCL because BCL-2 is transcriptionally active in the majority of these tumors. Interestingly, among the few GCB DLBCLs with this abnormality, 75% (3/4) had a t(14;18) translocation and thus expressed BCL-2. Finally, as discuss in detail above, 43% of PMBLs have a gain or amplification of cytoband 9p24 whereas this abnormality has not been detected in GCB DLBCLs (0/63) and only rarely in ABC DLBCLs (2/62). The uneven distribution of these chromosomal abnormalities among the DLBCL subgroups suggests that the subgroups utilize distinct oncogenic pathways.
One of the most important differences among the DLBCL subgroups is the constitutive activity of the NF-κB pathway in ABC DLBCL and PMBL but not GCB DLBCL. By gene expression profiling, ABC DLBCLs were found to have high expression of known NF-κB target genes when compared with GCB DLBCLs (Davis et al., 2001). Cell line models of ABC DLBCL have constitutive nuclear NF-κB due to constitutive activity of IκB kinase, and these were not features of GCB DLBCL cell lines. Inhibition of the NF-κB pathway using a dominant active form of IκBα or a dominant negative version of the IκB kinase β subunit was toxic to ABC DLBCL cell lines but not to GCB DLBCL cell lines. More recently, PMBL cells were found to express NF-κB target genes and have nuclear NF-κB, demonstrating the activity of the NF-κB pathway in this DLBCL subgroup as well (Rosenwald et al., 2003a;Savage et al., 2003). In this respect, PMBLs again resemble Hodgkin lymphoma, which is also characterized by constitutive NF-κB activity (Bargou et al., 1997;Bargou et al., 1996;Cabannes et al., 1999;Emmerich et al., 1999;Krappmann et al., 1999;Wood, Roff and Hay, 1998).
These findings suggest that the NF-κB pathway is a potential therapeutic target for ABC DLBCL and PMBL, but not GCB DLBCL. In support of this idea, small molecule inhibitors of IκB kinase were found to be selectively toxic for ABC DLBCL and PMBL cell lines, but had no effect on GCB DLBCL cell lines (Fig. 1D) (Lam et al., 2004). These findings support the further development of IκB kinase inhibitors for the treatment of lymphomas with NF-κB activity. In addition, an understanding of the mechanism(s) underlying the constitutive IκB kinase activity in ABC DLBCL and PMBL may lead to the identification of further molecular targets.
Clinical differences between DLBCL subgroups
Given that the DLBCL subgroups appear to arise from different stages of B cell differentiation, utilize distinct oncogenic pathways, and differ in the expression of thousands of genes, it is perhaps not surprising that they differ in their care rate following anthracycline-based multiagent chemotherapy (Alizadeh et al., 2000;Rosenwald et al., 2002;Wright et al., 2003). Patients with ABC DLBCL, GCB DLBCL, and PMBL have five-year survival rates of 31%, 59% and 64%, respectively (Fig. 1B). Relatively few relapses occur beyond five years, so these survival rates roughly reflect the probability that patients in each DLBCL subgroup will be cured by chemotherapy.
To determine if these survival differences are reproducible in different DLBCL cohorts, it is necessary to establish a reliable method for the molecular diagnosis of the DLBCL subgroups. To this end, a method based on Bayesian statistics was developed that is able to assign a probability that a given DLBCL tumor sample belongs to the GCB DLBCL, ABC DLBCL or PMBL subgroups (Rosenwald et al., 2003a;Wright et al., 2003). An important feature of this method is that in allows a patient sample to be declared “unclassified” if its gene expression profile does not closely matched that of any of the subgroups. Based on this method, GCB DLBCL, ABC DLBCL, and PMBL account for roughly 40%, 34%, and 8% of DLBCLs. Thus, 18% of DLBCL samples remain unclassified, and may represent additional DLBCL subgroups. Alternatively, some samples may fail to yield a diagnosis for technical reasons such as inadequate content of malignant cells within the biopsy sample, which may happen in cases with an abundant infiltration of non-malignant immune cells.
An attractive feature of this molecular diagnosis method is that the statistical algorithm is independent of the type of DNA microarray used to generate the gene expression data (Wright et al., 2003). When this Bayesian predictor was applied to data from 58 DLBCL samples profiled using Affymetrix oligonucleotide microarrays, GCB DLBCL and ABC DLBCL subgroups were identified, and their 5-year survival rates were 62% and 26%, respectively (Wright et al., 2003). More recently, many groups have used immunohistochemical stains for a few proteins to approximate the distinction between GCB DLBCL and ABC DLBCL and have consistently found that GCB DLBCL is the more favorable prognostic group (Chang et al., 2004;de Leval et al., 2003;Hans et al., 2004;Tzankov et al., 2003).
GENE EXPRESSION-BASED SURVIVAL PREDICTORS
The distinction between the DLBCL subgroups detailed above was discovered using an “unsupervised” approach that relies on methods that detect prominent patterns in the gene expression data. A complementary analytical approach is termed “supervised” because it uses statistical methods to find associations between gene expression data and external clinical, pathological, or molecular data (Staudt, 2003). Supervised methods typically begin by determining the statistical correlation between the expression pattern of each gene on a microarray and an external parameter such as the length of survival following diagnosis. Supervised methods face a mammoth multiple comparisons problem: thousands of statistical correlations can be made using the expression profiles of each gene on the microarray and a large number of these correlations will occur by chance alone. Because of this, the indiscriminant choice of the genes with the best correlations can lead to the development of statistical models that are overfit to the particular data set on which they were based (Ransohoff, 2004).
This problem can be circumvented by dividing samples into a “training” set that is used to generate a statistical model and a “test” set that is used to validate the model. Nevertheless, it is likely that most models generated in the training set will fail to validate in the test set unless the model is formed from genes whose expression patterns reflect meaningful biological variation that is related to the external parameter. A useful method to derive biological meaning from gene expression data is to classify genes into gene expression “signatures” (Shaffer et al., 2001). This method relies on the fact that genes that encode proteins with similar biological function are often transcriptionally coregulated. A gene expression signature may reflect a particular cellular lineage or stage of differentiation, the activity of an intracellular signaling pathway, or an aspect of cellular physiology such as the proliferation rate (Shaffer et al., 2001). A successful approach has been to use supervised methods to identify genes with expression patterns that are correlated with an external parameter, and then to classify these genes into gene expression signatures (Rosenwald et al., 2002). Next, a statistical model of the external parameter is created using the gene expression signatures instead of the individual genes. By creating statistical models from a limited number of gene expression signatures, the multiple comparisons problem is mitigated. The following sections demonstrate that this analytical approach has been used successfully to create survival predictors in DLBCL, mantle cell lymphoma, and follicular lymphoma.
A gene expression-based survival predictor for diffuse large B cell lymphoma
The subdivision of diffuse large B cell lymphoma into subgroups explains some, but not all, of the variation in survival of these patients. Although patients with GCB LBCL have a relatively favorable prognosis, roughly 30% die within the first two years of diagnosis. Patients with ABC DLBCL have a relatively poor prognosis yet roughly 20% are long term survivors. Some of this clinical heterogeneity is explained by the fact that certain clinical variables have prognostic significance in DLBCL, and their influence may not be reflected in tumor gene expression. Clinical parameters with prognostic significance in DLBCL include age, tumor stage, serum lactate dehydrogenase level (a measure of tumor bulk), performance status, and the number of extranodal sites (Shipp, 1993). These five clinical parameters form the International Prognostic Index (IPI), which is a strong predictor of survival in DLBCL (Shipp, 1993). The DLBCL subgroup distinction is statistically independent of the IPI in predicting survival, demonstrating that the subgroup distinction is not acting as a surrogate for clinical prognostic variables (Alizadeh et al., 2000;Rosenwald et al., 2002).
Further scrutiny of DLBCL gene expression profiling data, using a supervised method revealed that the DLBCL subgroup distinction captures only part of the biological variation among DLBCL tumors that influences survival (Rosenwald et al., 2002). This study began by identifying “survival predictor” genes with expression patterns in DLBCL tumor biopsies that were statistically associated with the length of survival of the patients (Rosenwald et al., 2002). Separately, gene expression signatures were defined as sets of genes that were coordinately expressed across the DLBCL tumor samples. Notably, more than half of the survival predictor genes could be classified into one of four gene expression signatures, termed germinal center B cell, lymph node, MHC class II, and proliferation (Fig. 3). A multivariate model of survival was formed from these four signatures and one additional predictive gene, BMP6, which was not part of a gene expression signature. This model was used to assign a “survival predictor score” to each patient based on tumor gene expression. Patients were ranked according to these survival predictor scores and divided into four quartile groups that had 5-year survival rates of 73%, 71%, 34%, and 15% (Rosenwald et al., 2002). This model had a stronger statistical association with survival than the DLBCL subgroup distinction, demonstrating that it accounted for a greater degree of the clinical heterogeneity among these patients. This model may prove useful clinically: it places one half of the patients in a favorable prognostic group for whom anthracycline-based chemotherapy has a reasonable chance of producing a cure, and one quarter of the patients in a poor prognosis group for whom alternative therapies should be considered.
Figure 3.

A gene expression-based multivariate model of survival following chemotherapy for DLBCL. The left panel shows the expression of the four gene expression signatures used to create the survival model in 201 DLBCL biopsy samples. Expression of the germinal center, lymph node, and MHC class II signatures is associated favorable prognosis while expression of the proliferation signature is associated with poor prognosis (Rosenwald et al., 2002). Representative genes from each signature that were used to create the survival model are shown. For each signature, patients were divided into four equal quartiles based on the expression of the signature in their biopsy samples. The four Kaplan Meier plots in the center depict the survival of patients in each signature quartile. These four signatures were combined into a multivariate model of survival and patients were divided into four quartiles groups based on this model (Rosenwald et al., 2002). The Kaplan Meier plot at the right depicts the overall survival of each quartile group of the multivariate model.
Germinal center B cell signature
The components of this multivariate model reveal biological features of DLBCL tumors that appear to influence the probability of being cured by chemotherapy. The germinal center B cell signature includes those genes that are characteristically expressed at the germinal center B cell stage of differentiation. This signature mirrors the DLBCL subgroup distinction in that both GCB DLBCLs and PMBLs express germinal center B cell signature genes, but ABC DLBCLs do not. The three germinal center B cell signature genes that were found to be most strongly associated with survival were BCL6, SERPINA9, and GCET2 (Rosenwald et al., 2002). Both SERPINA9 and GCET2 were identified as germinal center B cell signature genes because the Lymphochip DNA microarrays used for this study included a large number of DNA clones selected from a germinal center B cell cDNA library (Alizadeh et al., 1999). SERPINA9 (GCET1) encodes a serine protease inhibitor whereas GCET2 (HGAL) is of unknown function (Lossos et al., 2003;Pan et al., 2003). Interestingly, GCET2 is the human homologue of the mouse M17 gene, which is restricted in expression to germinal center B cells (Christoph, Rickert and Rajewsky, 1994).
It is important to emphasize, however, that these three germinal center B cell signature genes may not themselves influence survival in DLBCL, but merely represent the fact that those tumors of germinal center B cell origin have a more favorable prognosis. A second study also used supervised methods to discover survival predictor genes in DLBCL (Shipp et al., 2002). Two of the survival predictor genes highlighted in the study were protein kinase C β(PRKCB1) and phosphodiesterase 4B (PDE4B), both of which are more highly expressed in ABC DLBCLs than in GCB DLBCLs (Davis and Staudt, 2002;Rosenwald et al., 2002). Likewise, another study generated a model of survival in DLBCL using six genes, two of which are germinal center B cell signature genes (BCL6, LMO2) and two others are more highly expressed in ABC DLBCLs (CCND2, BCL2)(Lossos et al., 2004). Thus, not unexpectedly, each supervised model of DLBCL survival that has been created incorporates the DLBCL subgroup distinction since this is a predominant biological distinction among DLBCLs that is associated with survival.
Lymph node signature
The “lymph node” signature includes many genes whose expression patterns were found to be associated with favorable survival in DLBCL (Rosenwald et al., 2002). Despite its name, the genes in this signature do not merely reflect normal lymph node cells, but rather reflect a host response to malignant DLBCL cells in the lymph node. In keeping with this notion, the lymph node signature genes are not highly expressed in most lymph node biopsies from mantle cell lymphoma, small lymphocytic lymphoma, or follicular lymphoma. The survival predictor genes within the lymph node signature include genes that are expressed in macrophages (e.g. α actinin 1/ACTN1, urokinase plasminogen activator/PLAU) or fibroblasts (e.g.connective tissue growth factor/CTGF), and genes that encode extracellular matrix components (Fibronectin/FN1, collagen III α1/COL3A1)(Rosenwald et al., 2002). Involved lymph nodes from some DLBCL patients are heavily infiltrated with macrophages and some have a prominent sclerotic eaction; expression of the lymph node signature genes appears to reflect these host responses. Indeed, gene expression profiling analysis of separated malignant and nonmalignant cells from DLBCL biopsy specimens confirms that the large majority of lymph node signature genes are expressed in the non-malignant cells (unpublished). The DLBCLs subgroups have somewhat different expression of lymph node signature genes, with PMBLs having universally high expression, GCB DLBCLs having intermediate expression, and most ABC DLBCLs having a relatively low expression. Nonetheless, among ABC DLBCLs, lymph node signature expression is associated with a more favorable prognosis, even though this DLBCL subgroup as a whole has a relatively poor prognosis.
The mechanisms accounting for the association of lymph node signature gene expression and favorable outcome in DLBCL have not been elucidated. One possibility is that the infiltrating cells that generate the lymph node signature are mounting an immune response to the tumor. The immune response might be ineffective on its own, but might help contribute to curative responses following chemotherapy. A second possibility is that some DLBCLs rely on signals derived from host cells for survival and/or proliferation. In this “extracellular signal addiction” hypothesis, the dependence of the malignant cells on microenvironmental signals in the lymph node may prevent their spread to other anatomical sites in which these signals are absent. In this regard, it is interesting that high expression of the lymph node signature is associated with low tumor stage (unpublished). Tumor stage is a measure of how many lymph node groups and extranodal sites are involved in a patient, and thus could reflect how well a malignant DLBCL cell can survive and proliferate in new anatomical locations. In DLBCLs with high lymph node signature expression, the malignant cells may receive signals by direct cell-cell contact with tumor-infiltrating host cells. Alternatively, since many lymph node signature genes encode extracellular matrix components, it is conceivable that cytokine or chemokine signaling could be augmented by their deposition on extracellular matrix in DLBCLs with high lymph node signature expression.
Comparative genomic hybridization analysis has revealed that some chromosomal copy number changes in DLBCLs are associated with high or low lymph node signature expression (unpublished). This observation suggests that particular oncogenic events create a DLBCL that is dependent upon lymph node microenvironmental signals while other oncogenic events create a DLBCL that is independent of such signals. Whereas most oncogenes render a cell independent of extracellular signals, it has been postulated that certain oncogenes may create an interdependence between the malignant cell and its microenvironment (Hanahan and Weinberg, 2000). One example is the oncogene c-maf, which is frequently overexpressed in multiple myeloma (Hurt et al., 2004). c-maf encodes a B-ZIP transcription factor, and gene expression profiling was used to discover the genes that it transactivates (Hurt et al., 2004). One c-maf target gene is integrin β7, which encodes an adhesion molecule that, together with integrin αE, binds to E-cadherin. E-cadherin is expressed on the surface of bone marrow stromal cells, and the upregulation of integrin β7 by c-maf causes myeloma cells to adhere more strongly to these stromal cells. As a result, the bone marrow stromal cells secrete more vascular endothelial growth factor (VEGF), which is a proliferation factor for myeloma cells as well as an angiogenic factor. This example suggests that an understanding of mechanisms that may cause certain DLBCLs to be “addicted” to the lymph node microenvironment could provide new targets for therapeutic intervention.
MHC class II signature
Expression of the MHC class II signature predicts favorable survival in DLBCL (Rosenwald et al., 2002). This signature is comprised of the genes encoding the alpha and beta chains of MHC class II molecules as well as the gene encoding invariant chain, which plays a role in the MHC class II antigen presentation pathway. The variation in MHC class II signature expression among DLBCLs is due to differences in expression within the malignant cells, and not to differences in MHC class II-expressing nonmalignant cells (Rimsza et al., 2004). In fact, some DLBCLs are virtually devoid of MHC class II molecules on their surface. Since the MHC class II molecules and invariant chain are encoded on different chromosomes, a single structural alteration in the genome cannot account for differences in MHC class II signature expression. Therefore, these differences are likely due to variation in a regulatory factor that coordinates the expression of all of the MHC class II signature genes. Notably, the prognostic significance of the MHC class II signature is independent of the DLBCLs subgroup distinction. The mechanisms accounting for the favorable prognosis associated with MHC class II signature expression are not clear. One possibility is that loss of MHC class II molecules on the surface of DLBCLs prevents an immune response from developing against the malignant cells. In this regard, it is interesting that DLBCLs lacking MHC class II expression have significantly fewer CD8+ T cells infiltrating the tumor (List et al., 1993;Rimsza et al., 2004). MHC class II is associated with antigen presentation to CD4+ T cells, which are known to be required for the generation of optimal CD8+ T cell responses, but further work is needed to evaluate whether such immunological mechanisms account for the prognostic significance of MHC class II signature expression.
Proliferation signature
Many of the genes with expression patterns that are correlated poor outcome in DLBCL belong to the proliferation signature (Rosenwald et al., 2002). The proliferation signature includes genes that are expressed highly in proliferating cells and at low levels in quiescent cells (Shaffer et al., 2001). Typically, one eighth of all the genes represented on a DNA microarray belong to the proliferation signature. These genes encode a functionally diverse set of proteins that play roles in cell cycle progression, DNA replication and repair, protein translation, cell growth, and cellular metabolism. To some degree, the proliferation signature genes that encode proteins involved in the same aspect of cellular proliferation (e.g. mitosis regulators, glycolysis enzymes, ribosomal proteins) are more tightly coregulated than are proliferation signature genes as a whole (Shaffer et al., 2001).
Given the breadth of the proliferation signature, it is important to understand which subset of these genes is associated with survival in DLBCL. Interestingly, proliferation signature genes that encode cell cycle progression proteins are not associated with survival in DLBCL. Instead, the oncogene c-myc is one of the genes in the proliferation signature that is most strongly associated with the outcome (Rosenwald et al., 2002). c-myc is a transcription factor that controls many aspects of cellular metabolism, but is particularly associated with cell growth (i.e. an increase in cell size) (Levens, 2002). c-myc is well known to play a role in the pathogenesis of non-Hodgkin lymphomas in that it is deregulated by translocation to the immunoglobulin locus in all cases of Burkitt lymphoma and in a small subset of DLBCLs. Notably, normal germinal center B cells have very low expression of c-myc and some of its known target genes such as those that encode the ribosomal proteins (Klein et al., 2003;Shaffer et al., 2001). Teleologically, germinal center centroblasts may have low c-myc so as to avoid expending energy on cell growth, thereby favoring maximal proliferation. This bias in favor of proliferation allows centroblasts to undergo the multiple rounds of somatic hypermutation that are required for optimal positive selection of B cells with high affinity for antigen.
Among the DLBCL subgroups, ABC DLBCL has the highest expression of cmyc, which is consistent with its origin from a post-germinal center cell that has engaged signaling pathways that transcriptionally activate c-myc. Stimulation of normal B cells through a variety of cell surface receptors increases c-myc expression and, in particular, the NF-κB signaling pathway can increase c-myc transcription (Schauer et al., 1996). Therefore, the constitutive activity of the NF-κB pathway in ABC DLBCL may contribute to its high c-myc expression. The transcriptional regulation of c-myc is complex, however, and this as unlikely to be the entire explanation for elevated c-myc expression in ABC DLBCLs.
Interestingly, another proliferation signature gene associated with poor outcome in DLBCL is nucleostemin (Rosenwald et al., 2002), which encodes a nucleolar protein that is a direct transcriptional target of c-myc (O'Connell et al., 2003;Schlosser et al., 2003;Tsai and McKay, 2002;Zeller et al., 2003). One function of nucleostemin may be to bind p53 and sequester it in the nucleolus (Tsai and McKay, 2002). However, nucleostemin is likely to have other functions since RNA interference-mediated knockdown of nucleostemin decreases the number of cells in S-phase of the cell cycle (Tsai and McKay, 2002). c-myc transactivates a large number of genes that encode nucleolar proteins and also enhances processing of ribosomal RNA in the nucleolus (Schlosser et al., 2003). A third proliferation signature gene in the DLBCL survival predictor is nucleophosmin-3/NPM3, which is structurally related to the nucleolar protein nucleophosmin-1/NPM1(Rosenwald et al., 2002). Together, these observations suggest hat the particular subset of proliferation signature genes that is associated with poor outcome in DLBCL are those that contribute to nucleolar function, many of which are c-myc targets. c-myc thus appears to play an important role in lymphoma biology that extends beyond those malignancies in which c-myc is translocated.
A gene expression-based survival predictor for mantle cell lymphoma
Mantle cell lymphoma accounts for roughly 6% of non-Hodgkin lymphomas but it contributes disproportionately to the deaths due to lymphoma because there is no curative treatment. This lymphoma type is derived in the vast majority of cases from a pre-germinal center B cell since its rearranged immunoglobulin genes are usually unmutated. As a consequence, the biology of mantle cell lymphoma is substantially different from that of other non-Hodgkin lymphomas that originate from B cells that have traversed the germinal center. The characteristic t(11;14) translocation of mantle cell lymphoma deregulates the cyclin D1 gene by placing it in proximity to the immunoglobulin heavy chain locus. D-type cyclins form heterodimers with the cyclin-dependent kinases cdk4 and cdk6, thereby forming active kinase complexes that are key regulators of the G1/S phase transition of the cell cycle (Sherr and McCormick, 2002). Of the three D-type cyclins, cyclin D1 is not normally expressed by B lymphocytes, whereas cyclin D2 is expressed by mitogenically activated B cells and cyclin D3 is expressed by a germinal center B cells. The t(11;14) translocation points to the central role of cell cycle dysregulation in the pathophysiology of mantle cell lymphoma.
Mantle cell lymphoma has a characteristic gene expression signature that can be used to distinguish it from other non-Hodgkin lymphomas (Rosenwald et al., 2003b). The vast majority (>95%) of cases that are histologically compatible with a diagnosis of mantle cell lymphoma have cyclin D1 overexpression (Rosenwald et al., 2003b). However, gene expression profiling revealed a rare subtype of mantle cell lymphoma in which cyclin D1 is not expressed, but which are nevertheless histologically and molecularly indistinguishable from cyclin D1-positive mantle cell lymphoma (Rosenwald et al., 2003b). Not surprisingly, cyclin D2 or cyclin D3 is expressed in some of these cases by mechanisms that are as yet unknown.
The survival of patients with mantle cell lymphoma ranges from under one year to more than five years. Using the gene expression signature of mantle cell lymphoma, it was possible to identify some patients with an extremely indolent form of this lymphoma who survived more than 10 years following diagnosis (Rosenwald et al., 2003b). Although mantle cell lymphoma patients receive a variety of chemotherapy regimens, none has been shown to alter the length of survival.
A supervised analysis of gene expression in biopsy specimens from patients with cyclin D1-positive mantle cell lymphoma was used to uncover the molecular basis for the varying lengths of survival of these patients (Rosenwald et al., 2003b). All of the genes whose expression was associated with short survival belonged to the proliferation gene expression signature (Fig. 4A). In contrast to DLBCL, the proliferation signature genes that were associated with survival in mantle cell lymphoma included genes involved in cell cycle progression and DNA synthesis, but did not include c-myc. This suggests that the length of survival in mantle cell lymphoma is predicted by proliferation signature genes that quantitatively reflect the tumor proliferation rate.
Figure 4.
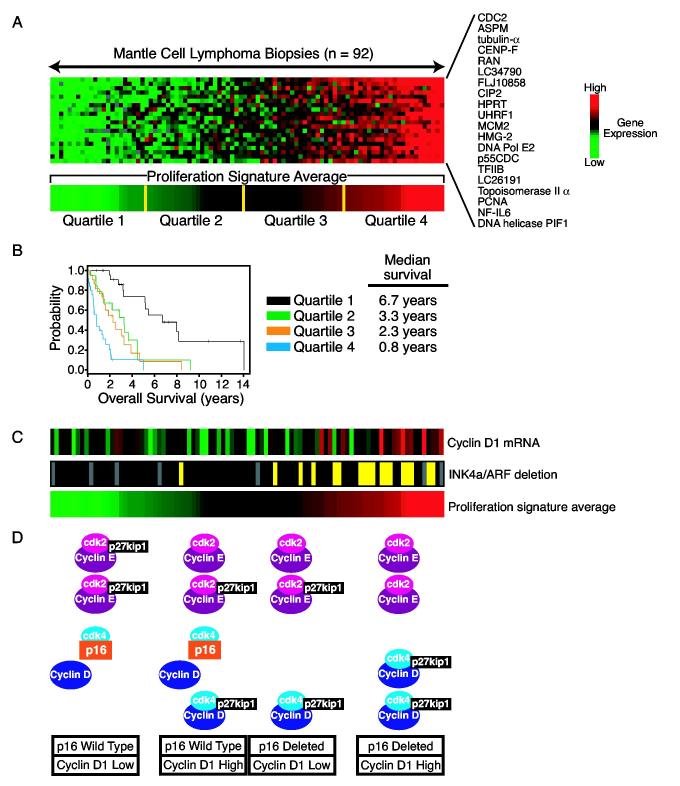
A. Differential expression of proliferation signature genes in biopsy samples from mantle cell lymphoma. The expression patterns of 20 genes from the proliferation signature are shown across 92 mantle cell lymphoma biopsy samples. The expression of each of these genes was found to being associated with short survival (Rosenwald et al., 2003b). The expression levels of these 20 genes were averaged to create the proliferation signature average, which was divided into four quartile groups as shown. B. Kaplan Meier plot of overall survival of patients with mantle cell lymphoma,divided into four quartile groups according to the expression of the proliferation signature in their tumors. C. The proliferation signature integrates distinct oncogenic events in mantle cell lymphoma. The expression of the cyclin D1 mRNA was determined by a quantitative RT-PCR assay for the coding region. Deletion of the INK4a/ARF locus was determined by a quantitative PCR using genomic DNA. Yellow indicates heterozygous or homozygous deletion; black indicates wild type copy number. Tumors with higher expression of the proliferation signature tend to have higher cyclin D1 mRNA expression and or deletion of the INK4a/ARF locus. D. Model depicting how increased cyclin D1 expression and deletion of the INK4a/ARF locus may contribute to a higher proliferation rate in mantle cell lymphoma. See text for details.
The average expression of 20 proliferation signature genes was used to calculate a survival predictor score for each mantle cell lymphoma patient (Rosenwald et al., 2003b). The patients were ranked according to these scores and divided into four equal quartile groups with median survival times of 0.8 years, 2.3 years, 3.3 years, and 6.7 years (Fig. 4B). Other studies have used immunohistochemical or cytological techniques to estimate the proliferation rate in mantle cell lymphoma but at best, the prognostic groups that were identified differed by 2.7 years in median survival (Argatoff et al., 1997;Bosch et al., 1998;Raty et al., 2002;Velders et al., 1996). The proliferation signature average provides a more quantitative measure of tumor proliferation rate than these other methods, which most likely explains why it is able to identify risk groups of patients that differ by 5.9 years in median survival. From a clinical standpoint, this molecular predictor of survival could provide valuable prognostic information to these patients. For those patients with indolent forms of this lymphoma, a watchful waiting approach is appropriate. Patients with aggressive forms of the disease should be considered for newer therapeutic approaches, some of which show early promise (see below).
From a biological standpoint, the most remarkable feature of the proliferation signature average is that it quantitatively integrates the effects of multiple oncogenic events that affect cell cycle progression. Unexpectedly, a quantitative RT-PCR assay for the cyclin D1 coding region revealed higher expression in many of the mantle cell lymphomas with the highest proliferation signature average (Fig. 4C) (Rosenwald et al., 2003b). Consequently, higher expression of cyclin D1 coding region mRNA was associated with shorter survival. Several molecular mechanisms account for the varying expression of cyclin D1 coding region transcripts. Various forms of cyclin D1 mRNA have been observed in mantle cell lymphoma: a full-length 4.7 kb isoform that contains a ∼3 kb 3′ untranslated region and shorter isoforms that essentially consist of the coding region. The cyclin D1 3′ untranslated region contains an mRNA destabilizing element and therefore, with an equivalent amount of transcription, the long mRNA isoform will accumulate to lower levels than the short mRNA isoforms (Lebwohl et al., 1994;Lin et al., 2000;Rimokh et al., 1994). Some mantle cell lymphomas were found to lack expression of the long mRNA isoform and exclusively expressed shorter, more stable, mRNA isoforms, and these were the cases with higher levels of cyclin D1 coding region transcripts (Rosenwald et al., 2003b). In some mantle cell lymphoma cases, the genomic region encoding the cyclin D1 3′ untranslated region is deleted, resulting in expression of a short cyclin D1 mRNA (de Boer et al., 1995;Rimokh et al., 1994;Seto et al., 1992;Withers et al., 1991). Another molecular mechanism leading to exclusive expression of a short cyclin D1 mRNA is the acquisition of a somatic mutation immediately downstream of the stop codon that generates a new polyadenylation signal, thereby truncating the mRNA (unpublished).
Deletion of the INK4a/ARF locus is a frequent oncogenic event in mantle cell lymphoma, occuring in roughly one fifth of cases (Dreyling et al., 1997;Pinyol et al., 1998;Pinyol et al., 1997;Pinyol et al., 2000;Rosenwald et al., 2003b). INK4a/ARF deletions are more frequent in mantle cell lymphomas with high expression of the proliferation signature and are consequently associated with short survival (Fig. 4C) Rosenwald et al., 2003b). The INK4a/ARF locus encodes two key tumor suppressor proteins, p16INK4a and p14ARF (Sherr and McCormick, 2002). p16INK4a prevents the assembly of cdk4 and cdk6 with D-types cyclins and thus is an important negative regulator of the G1/S phase transition of the cell cycle. p14ARF blocks to the ability of MDM2 to target p53 for degradation, and thus potentiates p53-dependent apoptosis and cell cycle arrest. In addition, loss of p14ARF retards proliferation of a mouse embryonic fibroblasts and pre-B cells (Kamijo et al., 1998;Randle et al., 2001). Unlike some cancers, mantle cell lymphomas do not sustain inactivating mutations in the p16INK4a coding region (Pinyol et al., 1997;Pinyol et al., 2000), suggesting that the selective advantage of INK4a/ARF deletions in mantle cell lymphoma is due to loss of both p16INK4a and p14ARF.
Interestingly, INK4a/ARF deletions and elevated cyclin D1 expression were statistically independent in their associations with high proliferation signature expression and short survival (Rosenwald et al., 2003b). In fact, many mantle cell lymphomas have deletions of both the cyclin D1 3′ untranslated region and the INK4a/ARF locus. This observation suggests a model in which these two oncogenic events act synergistically to accelerate cell cycle progression. One way this could occur is by increasing the frequency (or probability) that a lymphoma cell moves from G1 to S phase. A current view of this cell cycle transition is that cyclin D/cdk4(6) complexes bind the cyclin-dependent kinase inhibitors p21 and p27kip1, but are not inhibited by them. In so doing, they titrate these inhibitors away from cyclin E/cdk2 complexes, whose activity is critical for entry into S phase (Fig. 4D). In keeping with this model, most p27kip1 in mantle cell lymphomas is physically associated with cyclin D1/cdk4(6) (Quintanilla-Martinez et al., 2003).
This model could have therapeutic implications. p27kip1 is ubiquitinylated by Skp2, which targets it for proteosomal degradation. Intriguingly, the proteasome inhibitor Velcade/PS-341 has shown activity against mantle cell lymphoma in early clinical trials. If p27kip1 levels are quantitatively titrated by cyclin D1/cdk4(6) complexes, proteosomal inhibition might allow more p27kip1 to accumulate and tip this regulatory balance in favor of a block in G1 phase of the cell cycle. A theoretically ideal inhibitor of the G1/S phase transition, yet to be developed, would be a small molecule mimetic of p16INK4a that would inhibit assembly of cdk4 and cdk6 with cyclin D1.
As mentioned above, loss of p14ARF in some mantle cell lymphomas may also contribute to cell growth and a higher proliferation rate. Many of the mantle cell lymphomas with INK4a/ARF deletions are histologically “blastic”, meaning that they have a large cell size compared with other mantle cell lymphomas (Pinyol et al., 1997;Rosenwald et al., 2003b). This phenotype could be caused by increased c-myc activity, due to the absence of p14ARF (Datta et al., 2004;Qi et al., 2004). In this regard, it is notable that the BMI-1gene is amplified in some mantle cell lymphoma cases that lack INK4a/ARF deletions (Bea et al., 2001;Rosenwald et al., 2003b). BMI-1 is a transcriptional repressor that blocks expression p16INK4a and p14ARF and cooperates with c-myc in malignant transformation (Sherr and McCormick, 2002). Thus, enhanced cmyc function may be another way in which INK4a/ARF deletions synergize with cyclin D1 upregulation to enhance the proliferation rate of mantle cell lymphomas.
This analysis leads to the view that tumor proliferation rate is a rheostat that can vary continuously over a broad range. The setpoint of this rheostat results from the cellular integration of multiple oncogenic changes that affect the rate or probability that the cancer cell transits the G1/S phase boundary of the cell cycle. The fact that the proliferation rate in mantle cell lymphoma (as measured by the proliferation signature expression) can be modeled using cyclin D1 expression and INK4a/ARF deletion illustrates that complex cancer phenotypes may become mathematically tractable as more precise measurements become available. Mathematical models can have heuristic value in that they can highlight areas where further biological research is needed. For example, the multivariate model of survival based on cyclin D1 expression and INK4a/ARF deletion was not as statistically significant as the model based on proliferation signature expression (Rosenwald et al., 2003b). This observation suggests that additional oncogenic mechanisms remain to be discovered that affect the proliferation rate and, consequently, the length of survival in mantle cell lymphoma.
An important implication of this rheostat model of proliferation is that therapies that can dial down the proliferation setpoint in mantle cell lymphoma might be of great clinical benefit, even if they are not curative. In other words, it is possible to imagine a therapy that could decrease the proliferation rate so significantly that it might be able to move a patient from the least favorable prognostic group to the most favorable prognostic group, thereby prolonging the patient's life by more than five years. Although such therapies might affect normal cells as well, studies in mice have shown that the D-type cyclins have cell type-restricted functions. For example, mice deficient in cyclin D1 have discrete defects in embryonic retinal development and in pregnancy-associated mammary development but are normal in other respects (Sicinski et al., 1995). Indeed, in mice lacking all D-type cyclins, many cell types proliferate normally although hematopoietic stem cell proliferation is compromised (Kozar et al., 2004). It therefore seems conceivable that a therapeutic window may exist to allow prolonged treatment of mantle cell lymphoma patients with agents targeted at cyclin D/cdk4(6) complexes.
A gene expression-based survival predictor for follicular lymphoma
Follicular lymphoma, the second most common form of non-Hodgkin lymphoma, has a highly variable clinical course. Although the median survival is approximately 10 years following diagnosis, some patients have an aggressive illness that is fatal in under one year while others may live more than 20 years. Patients with follicular lymphoma are treated with a variety of chemotherapeutic and immunotherapeutic regimens (Bendandi et al., 1999;Colombat et al., 2001;Czuczman et al., 1999;Kwak et al., 1992;Maloney et al., 1997;Timmerman et al., 2002;Witzig et al., 2002), but it has not been demonstrated that these approaches change the length of survival of these patients (Horning, 2000).
Follicular lymphomas are derived from germinal center B cells and maintain the gene expression program of this stage of differentiation (Dave et al., 2004;Flenghi et al., 1995;Shaffer, Rosenwald and Staudt, 2002). In addition, most follicular lymphomas grow as pseudo-germinal centers that include T cells and follicular dendritic cells intermingled with the malignant cells. Unlike normal germinal center B cells, roughly 90% of follicular lymphomas express BCL-2 as a result of the characteristic t(14;18) translocation. In some cases, follicular lymphoma “transforms” into an aggressive lymphoma resembling DLBCL, and this transformation can be associated with a variety of oncogenic changes (Lossos and Levy, 2003). However, there is no evidence that these stochastic events affect the overall survival rate.
Gene expression profiling has revealed that the length of survival following diagnosis of follicular lymphoma can be predicted by biological differences among the tumors at the time of diagnosis (Dave et al., 2004). A new form of supervised analysis, termed survival signature analysis, was used to develop a gene expression based-survival predictor for follicular lymphoma (Dave et al., 2004). Like other supervised methods, survival signature analysis begins with the subdivision of the cases into a training set and a test set, followed by the identification of genes with expression patterns associated with long survival (good prognosis genes) or short survival (bad prognosis genes) within the training set. Hierarchical clustering is then used to organize the good prognosis genes and the bad prognosis genes according to their expression patterns within the training set of cases. Clusters of genes with highly correlated expression patterns are grouped together into survival-associated signatures. Within each survival associated signature, the expression levels of the component genes are averaged, creating a signature average for each patient. Multivariate statistical models are created using signature averages from the training set of cases, and these models are then tested for their reproducibility in the test set of cases. This algorithm is based on the assumption that within a particular survival-associated signature, the expression profile of each component gene reflects the same biological process that influences survival. By grouping coordinately-expressed genes together and creating a limited set of survival associated signatures, the multiple comparisons problem associated with the supervised analysis of DNA microarray data is avoided (Ransohoff, 2004).
Using this method, an optimal survival model in follicular lymphoma was created using two survival-associated signatures, one from the good prognosis gene set and one from the bad prognosis gene set (Fig. 5A). These two signatures were termed immune response-1 in immune response-2 because their component genes included genes known to be expressed in normal immune cells. Although the immune response-1 and immune response-2 signatures each predicted survival as single variables, they were strongly synergistic in a multivariate model. This suggests that the relative level of these two signatures is more important in predicting survival than the absolute level of either one alone. The model generated a “survival predictor score” for each patient, with a high score associated with long survival and a low score associated with short survival. The patients were ranked according to their survival predictor scores and divided into four equal quartiles groups that had strikingly different median survival times of 3.9 years, 10.8 years, 11.1 years, and 13.6 years, respectively (Fig. 5B). This finding demonstrates that although the follicular lymphoma genome may continue to be altred following diagnosis, such stochastic changes do not have in large effect on the length of survival. Rather, the biological heterogeneity already present in follicular lymphoma at that time of diagnosis dictates, in large measure, the clinical aggressiveness of the disease.
Figure 5.
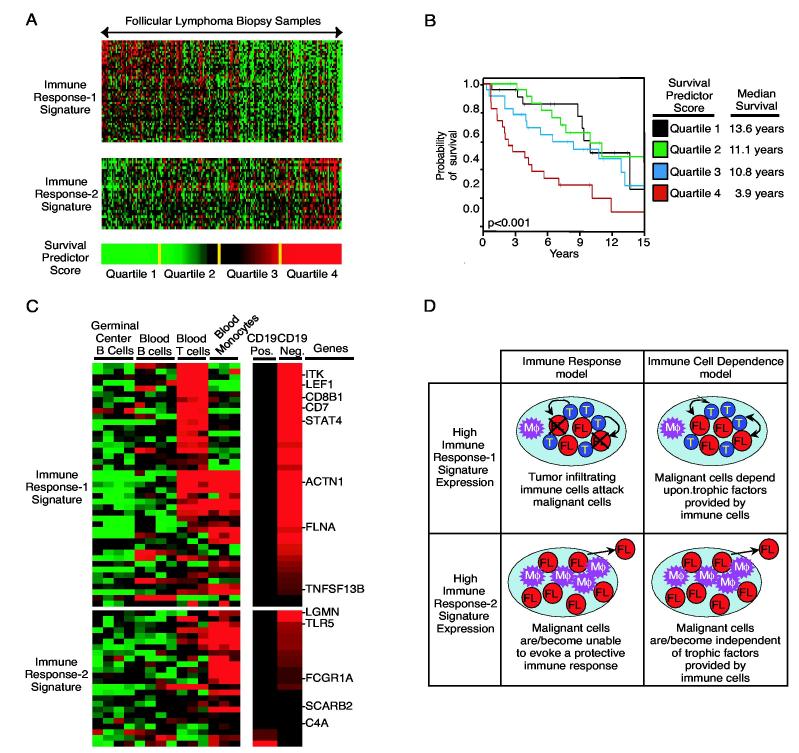
A. Two sets of coordinately expressed genes, termed the immune response-1 and immune response-2 signatures, are associated with survival in follicular lymphoma. The expression pattern of each gene in these two signatures in shown for 191 follicular lymphoma biopsy samples. Expression of the immune response-1 signature is associated with long survival following diagnosis and expression of the immune response-2 signature is associated with short survival (Dave et al., 2004). These signatures are combined into a multivariate model of survival that generates a survival predictor score for each patient. Patients are ranked according to this survival predictor and divided into four equal quartiles as shown. B. Kaplan-Meier plot of overall survival of patients in the four quartiles of the survival predictor. C. Expression of the genes that constitute the immune response-1 and immune response-2 signatures in normal immune cells. Tonsillar germinal center B cells, peripheral blood B cells, peripheral blood T cells and peripheral blood monocytes from healthy donors were profiled for gene expression. C. Relative expression of the immune response-1 and immune response-2 signature genes in the malignant (CD19-positive) and the non-malignant (CD19-negative) cells isolated from follicular lymphoma biopsy samples is shown. D. A schematic of how the immune response hypothesis and immune cell dependence hypothesis might explain the clinical behavior of tumors with high expression of the immune response-1 signature or the immune response-2 signature (see text for details).
The immune response-1 and immune response-2 signatures were found to be preferentially expressed in the CD19-negative non-malignant cells isolated from follicular lymphoma biopsies by flow sorting (Fig. 5C) (Dave et al., 2004). Furthermore, the genes that make up these two signatures are not highly expressed in normal germinal center B cells, the cell of origin of follicular lymphoma, but rather in normal blood T cells or monocytes. These results point to an interplay between the malignant cells and high the host immune system that influences the clinical behavior of follicular lymphoma.
The immune response-1 signature includes a number of genes that encode T cellrestricted proteins, such as CD7, CD8β1, ITK and LEF1 (Fig. 5C)(Dave et al., 2004). However, this signature is not merely a measure of the number of T cells in the follicular lymphoma because the expression patterns of a number of genes encoding T cell markers (e.g.CD2, CD4, LAT, TRIM, SH2D1A) were not found to be associated with survival in follicular lymphoma. This suggests that the expression of the T cell genes in the immune response-1 signature reflects the presence of a particular T cell subset(s) within the tumor. In this regard, it is notable that one of the immune response-1 signature genes is CD8β1,which could indicate the presence of cytotoxic T cells. However, this signature also includes several genes that are highly expressed in macrophages, such as ACTN1 (Allen and Aderem, 1996) and TNFSF13B(BLYS/BAFF) (Craxton et al., 2003). Therefore, the immune response-1 signature appears to reflect a complex mixture of immune cells, including T cells, that is associated with long survival in follicular lymphoma.
The immune response-2 signature, by contrast, does not include genes encoding T cell markers, but rather includes genes expressed in macrophages and/or dendritic cells Fig. 5C) (Dave et al., 2004). For instance, FCGR1A(CD64), TLR5, and C3AR encode receptors for immunoglobulin Fc regions, flagellin and complement component C3A, respectively, that are typically expressed in monocytic cells (Ames et al., 1996;Hayashi et al., 2001;Muzio et al., 2000;Roglic et al., 1996). Other immune response-2 signature genes are expressed in mature dendritic cells, including SEPT10, which encodes a septin and LGMN, which encodes a lysosomal protease (Li et al., 2003;Sui et al., 2003). Also included are the genes encoding the complement components C1qA, C1qB, and C4A, which are synthesized by phagocytes in the myeloid lineage (McPhaden and Whaley, 1993;Tsukamoto et al., 1992). The immune response-2 signature therefore reflects an immune infiltrate that is dominated by macrophages and/or dendritic cells that is associated with short survival in follicular lymphomas.
It is important to emphasize the strong statistical synergism of the immune response-1 and the immune response-2 signatures in predicting the length of survival (Dave et al., 2004). Biologically, this means that many follicular lymphoma tumors express both signatures, but their relative ratio is what is most predictive of the length of survival. This observation highlights the ability of gene expression profiling to reveal quantitative differences in immune responses that are clinically meaningful.
Why do patients with follicular lymphoma differ with respect to the type of immune infiltrate in their tumors? One possibility is that these differences reflect quantitative traits of the immune system that influence the way in which it responds to the malignant follicular lymphoma cells. Genetic differences in key regulators of the immune response are well known to influence susceptibility to autoimmune and inflammatory disorders, and may therefore play a role in anti-tumor responses as well. A second possibility is that the character of the immune infiltrate is dictated by the molecular features of the malignant cells in follicular lymphoma. To address this possibility comprehensively, the gene expression profiles and genetic abnormalities of sorted malignant cells from a large number of cases will need compared with the gene expression profiles of the corresponding non-malignant cells
Why is the immune response-1 signature associated with good prognosis and the immune response-2 signature associated with bad prognosis? Two hypotheses, which are not mutually exclusive, can be entertained: an “immune response” hypothesis and an “immune cell dependence” hypothesis (Fig. 5D). In the immune response hypothesis, the immune response-1 signature could reflect an active anti-tumor immune response. There is abundant clinical evidence suggesting that anti-tumor immune responses can be mounted in some patients with follicular lymphoma. Follicular lymphoma is the only lymphoma in which spontaneous regressions are observed, albeit rarely (Horning and Rosenberg, 1984). When this occurs in other cancers, such as melanoma and renal cell carcinoma, it is taken as an indication of an effective anti-tumor immune response. In addition, anti-idiotype vaccines can elicit long-term remissions in some patients with follicular lymphoma, demonstrating that this is one of the few cancers, so far, in which an immune response is clinically effective (Bendandi et al., 1999;Kwak et al., 1992;Timmerman et al., 2002). The immune response-1 signature may therefore reflect an anti-tumor immune response that is able to prolong the survival of these patients, but not eradicate the tumor. The inclusion of T cell genes in this signature, especially the cytotoxic T cell gene CD8β1, is consistent with this possibility. The immune response-2 signature lacks T cell-restricted genes, and instead includes genes expressed in innate immune cells. It is conceivable that the follicular lymphomas with high expression of the immune response-2 signature have evolved mechanisms to evade an adaptive immune response, and the short survival of these patients reflects this immune evasion.
If this “immune response” hypothesis is correct, the success of immune-based therapies in follicular lymphoma may depend upon the molecular profile of the tumor-infiltrating immune cells. Therapies that rely on T cell responses, such as anti-idiotype vaccination, may be more successful in tumors that express the immune response-1 signature highly since this signature reflects a type of T cell infiltrate that is associated with long survival. Furthermore, it is conceivable that non-specific augmentation of T cell responses, as can be achieved with anti-CTLA-4 therapy (Egen, Kuhns and Allison, 2002), could be of benefit to patients whose tumors express the immune response-1 signature highly.
The “immune cell dependence” hypothesis suggests that the tumor-infiltrating immune cells in some follicular lymphomas provide trophic signals that promote survival and/or proliferation of the malignant cells (Fig. 5D). The immune response-1 signature could reflect a pseudo-germinal center reaction in which the malignant cells receive signals from T cells and/or follicular dendritic cells. The malignant cells may be dependent upon these microenvironmental signals, making it difficult for them to leave the lymph node. This microenvironmental “addiction” could account for the relatively favorable prognosis associated with the immune response-1 signature. Follicular lymphomas with high expression of the immune response-2 signature may have lost their dependence on microenvironmental signals and may therefore be able to survive in other anatomical sites, possibly accounting for the relatively short survival of these patients.
Precedent for the immune cell dependence hypothesis is provided by certain mouse B cell lymphomas that require T cells for their growth (Ponzio and Thorbecke, 2000). These mouse lymphomas express an MMTV-encoded superantigen that stimulates CD4-positive T cells, which in turn secrete cytokines such as IL-4 that may provide survival signals to the lymphomas. Some intriguing data suggest that such mechanisms should be considered in human lymphomas as well. In follicular lymphoma, the pattern of immunoglobulin mutations is consistent with a role for antigen selection at some point in the genesis of the tumor (Bahler and Levy, 1992). In addition, the immunoglobulin variable regions in follicular lymphoma acquire N-linked glycosylation sites during the hypermutation process in 79% of cases, which is far in excess of the 9% frequency observed in normal B cells (Zhu et al., 2002). This surprising finding raises the possibility that carbohydrates attached to the immunoglobulin receptor of a follicular lymphoma could interact with lectin-like proteins present within the germinal center microenvironment. Further investigations will be needed to understand whether such interactions are of functional significance at the time of clinical presentation or play a role in the early development of these cancers. If malignant cells and immune cells communicate in follicular lymphoma, a molecular understanding of this communication could provide new targets for therapy.
CONCLUDING REMARKS
The comprehensive and exploratory nature of gene expression profiling has provided fresh insights into the pathobiology of human lymphomas, many of which are applicable to a wider range of cancers. Principles of cancer biology revealed by gene expression profiling include the following:
The clinical and biological behavior of cancers can be understood and modeled with mathematical precision. Complex clinical phenotypes such as the length of survival and the response to therapy can now be understood as the consequence of quantitative differences in tumor biology. The proliferation rate in cancer can be modeled as a continuous variable that integrates multiple oncogenic events that influence cell cycle progression. It is likely that other cancer phenotypes such as susceptibility to apoptosis or response to DNA damage can also be modeled, leading ultimately to a quantitative, systems biology view of cancer.
Existing diagnostic categories of cancer consist of multiple molecularly distinct diseases that differ in their cell of origin, oncogenic mechanisms and clinical outcome. The example of diffuse large B cell lymphoma illustrates this principle well, but gene expression profiling has also uncovered distinct disease entities in other cancers also (Sorlie et al., 2003).
The clinical heterogeneity of cancer can be ascribed to heterogeneity in the tumor at the time of diagnosis. This observation holds for each lymphoma type studied thus far, as well as for other cancers (Bernards and Weinberg, 2002). Although the stepwise acquisition of oncogenic changes certainly contributes to the cancer phenotype, stochastic changes in the tumor genome after diagnosis do not appear to strongly influence subsequent clinical behavior.
Multiple independently varying biological features of tumors contribute to their clinical behavior. Gene expression signatures can be used to measure different biological attributes in tumors samples. Optimal models of survival usually incorporate several gene expression signatures, reflecting tumor cell intrinsic properties as well as the influence of the tumor microenvironment.
Interactions between the tumor and its microenvironment are biologically and clinically crucial. For DLBCL and follicular lymphoma, survival was found to be strongly associated with the nature of the tumor-infiltrating immune cells. An understanding of the interaction between the malignant cell and its microenvironment should provide new therapeutic options.
Molecular diagnosis leads to new molecular targets. Of the three gene expression subgroups of DLBCL, two respond to inhibitors of the NF-κB pathway whereas the third is impervious to these agents. The importance of molecular diagnosis will continue to increase as drugs become available the target specific signaling pathways.
Several research challenges must be surmounted in order to build upon the recent progress in understanding human lymphomas. We must develop a comprehensive view of the abnormal signaling networks inside lymphoma cells, and functional genomics techniques such as RNA interference will be critical in this effort. Coupling RNA interference with gene expression profiling will lead to an understanding of the wiring diagrams of lymphoma cells, which will ultimately enable rational manipulation of signaling pathways. By necessity, much of this work must be carried out in lymphoma cell lines, and we must take care to use cell lines that are faithful mimics of particular lymphoma types. Concerted efforts should be made to generate additional cell lines that are representative of each lymphoma type. Finally, given the importance of the tumorinfiltrating immune cells in lymphomas, methods need to be developed to maintain patient tumor samples in tissue culture or in immunodeficient mice in a fashion that preserves the interactions between the malignant cells and their microenvironment. In all of these efforts, gene expression profiling will continue to provide a comprehensive and biologically informative framework.
ACKNOWLEDGEMENTS
This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. We are indebted to our colleagues in the Lymphoma/Leukemia Molecular Profiling Project for their many valuable insights.
REFERENCES
- Addis BJ, Isaacson PG. Histopathology. 1986;10:379. doi: 10.1111/j.1365-2559.1986.tb02491.x. [DOI] [PubMed] [Google Scholar]
- Albagli O, Dhordain P, Bernardin F, Quief S, Kerkaert JP, Leprince D. Biochem Biophys Res Commun. 1996;220:911. doi: 10.1006/bbrc.1996.0505. [DOI] [PubMed] [Google Scholar]
- Alizadeh A, Eisen M, Davis RE, Ma C, Sabet H, Tran T, Powell J, Yang L, Marti G, Moore T, Hudson J, Chan WC, Greiner T, Weisenburger D, Armitage JO, Lossos I, Levy R, Botstein D, Brown PO, Staudt LM. Cold Spring Harbor Symp Quant Biol. 1999;64:71. doi: 10.1101/sqb.1999.64.71. [DOI] [PubMed] [Google Scholar]
- Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, Boldrick JC, Sabet H, Tran T, Yu X, Powell JI, Yang L, Marti GE, Moore T, Hudson J, Lu L, Lewis DB, Tibshirani R, Sherlock G, Chan WC, Greiner TC, Weisenburger DD, Armitage JO, Warnke R, Levy R, Wilson W, Grever MR, Byrd JC, Botstein D, Brown PO, Staudt LM. Nature. 2000;403:503. doi: 10.1038/35000501. [DOI] [PubMed] [Google Scholar]
- Allen LA, Aderem A. J Exp Med. 1996;184:627. doi: 10.1084/jem.184.2.627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allman D, Jain A, Dent A, Maile RR, Selvaggi T, Kehry MR, Staudt LM. Blood. 1996;87:5257. [PubMed] [Google Scholar]
- Ames RS, Li Y, Sarau HM, Nuthulaganti P, Foley JJ, Ellis C, Zeng Z, Su K, Jurewicz AJ, Hertzberg RP, Bergsma DJ, Kumar C. J Biol Chem. 1996;271:20231. doi: 10.1074/jbc.271.34.20231. [DOI] [PubMed] [Google Scholar]
- Angelin-Duclos C, Cattoretti G, Lin KI, Calame K. J Immunol. 2000;165:5462. doi: 10.4049/jimmunol.165.10.5462. [DOI] [PubMed] [Google Scholar]
- Argatoff LH, Connors JM, Klasa RJ, Horsman DE, Gascoyne RD. Blood. 1997;89:2067. [PubMed] [Google Scholar]
- Bahler DW, Levy R. Proc Natl Acad Sci U S A. 1992;89:6770. doi: 10.1073/pnas.89.15.6770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bargou RC, Emmerich F, Krappmann D, Bommert K, Mapara MY, Arnold W, Royer HD, Grinstein E, Greiner A, Scheidereit C, Dorken B. J Clin Invest. 1997;100:2961. doi: 10.1172/JCI119849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bargou RC, Leng C, Krappmann D, Emmerich F, Mapara MY, Bommert K, Royer HD, Scheidereit C, Dorken B. Blood. 1996;87:4340. [PubMed] [Google Scholar]
- Bea S, Tort F, Pinyol M, Puig X, Hernandez L, Hernandez S, Fernandez PL, van Lohuizen M, Colomer D, Campo E. Cancer Res. 2001;61:2409. [PubMed] [Google Scholar]
- Bea S, Zettl A, Wright G, Salaverria I, Jehn J, Moreno V, Burek C, Ott G, Puig X, Yang L, Lopez-Guillermo A, Chan WC, Greiner TC, Weisenburger DD, Armitage JO, Gascoyne RD, Connors JM, Grogan TM, Braziel R, Fisher RI, Smeland EB, Kvaloy S, Delabie J, Simon R, Powell J, Wilson WH, Jaffe ES, Montserrat E, Muller-Hermelink HK, Staudt LM, Campo E, Rosenwald A. Blood. 2005. Diffuse large B cell lymphoma subtypes have distinct genetic profiles that influence tumor biology and improve gene expression-based survival prediction. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendandi M, Gocke CD, Kobrin CB, Benko FA, Sternas LA, Pennington R, Watson TM, Reynolds CW, Gause BL, Duffey PL, Jaffe ES, Creekmore SP, Longo DL, Kwak LW. Nat Med. 1999;5:1171. doi: 10.1038/13928. [DOI] [PubMed] [Google Scholar]
- Bentz M, Barth TF, Bruderlein S, Bock D, Schwerer MJ, Baudis M, Joos S, Viardot A, Feller AC, Muller-Hermelink HK, Lichter P, Dohner H, Moller P. Genes Chromosomes Cancer. 2001;30:393. doi: 10.1002/1098-2264(2001)9999:9999<::aid-gcc1105>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- Bereshchenko OR, Gu W, Dalla-Favera R. Nat Genet. 2002;32:606. doi: 10.1038/ng1018. [DOI] [PubMed] [Google Scholar]
- Bernards R, Weinberg RA. Nature. 2002;418:823. doi: 10.1038/418823a. [DOI] [PubMed] [Google Scholar]
- Bosch F, Lopez-Guillermo A, Campo E, Ribera JM, Conde E, Piris MA, Vallespi T, Woessner S, Montserrat E. Cancer. 1998;82:567. doi: 10.1002/(sici)1097-0142(19980201)82:3<567::aid-cncr20>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- Cabannes E, Khan G, Aillet F, Jarrett RF, Hay RT. Oncogene. 1999;18:3063. doi: 10.1038/sj.onc.1202893. [DOI] [PubMed] [Google Scholar]
- Cattoretti G, Chang C-C, Cechova K, Zhang J, Ye BH, Falini B, Louie DC, Offit K, Chaganti RSK, Dalla-Favera R. Blood. 1995;86:45. [PubMed] [Google Scholar]
- Chang CC, McClintock S, Cleveland RP, Trzpuc T, Vesole DH, Logan B, Kajdacsy-Balla A, Perkins SL. Am J Surg Pathol. 2004;28:464. doi: 10.1097/00000478-200404000-00005. [DOI] [PubMed] [Google Scholar]
- Chang CC, Ye BH, Chaganti RS, Dalla-Favera R. Proc Natl Acad Sci U S A. 1996;93:6947. doi: 10.1073/pnas.93.14.6947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christoph T, Rickert R, Rajewsky K. Int Immunol. 1994;6:1203. doi: 10.1093/intimm/6.8.1203. [DOI] [PubMed] [Google Scholar]
- Chu CC, Paul WE. Proc Natl Acad Sci U S A. 1997;94:2507. doi: 10.1073/pnas.94.6.2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombat P, Salles G, Brousse N, Eftekhari P, Soubeyran P, Delwail V, Deconinck E, Haioun C, Foussard C, Sebban C, Stamatoullas A, Milpied N, Boue F, Taillan B, Lederlin P, Najman A, Thieblemont C, Montestruc F, Mathieu-Boue A, Benzohra A, Solal-Celigny P. Blood. 2001;97:101. doi: 10.1182/blood.v97.1.101. [DOI] [PubMed] [Google Scholar]
- Copie-Bergman C, Plonquet A, Alonso MA, Boulland ML, Marquet J, Divine M, Moller P, Leroy K, Gaulard P. Mod Pathol. 2002;15:1172. doi: 10.1097/01.MP.0000032534.81894.B3. [DOI] [PubMed] [Google Scholar]
- Craxton A, Magaletti D, Ryan EJ, Clark EA. Blood. 2003;101:4464. doi: 10.1182/blood-2002-10-3123. [DOI] [PubMed] [Google Scholar]
- Csernus B, Timar B, Z FU, Bognar A, Szepesi A, Laszlo T, Jakso P, Warnke R, Kopper L, Matolcsy A. Leuk Lymphoma. 2004;45:2105. doi: 10.1080/1042819042000219467. [DOI] [PubMed] [Google Scholar]
- Curiel TJ, Wei S, Dong H, Alvarez X, Cheng P, Mottram P, Krzysiek R, Knutson KL, Daniel B, Zimmermann MC, David O, Burow M, Gordon A, Dhurandhar N, Myers L, Berggren R, Hemminki A, Alvarez RD, Emilie D, Curiel DT, Chen L, Zou W. Nat Med. 2003;9:562. doi: 10.1038/nm863. [DOI] [PubMed] [Google Scholar]
- Czuczman MS, Grillo-Lopez AJ, White CA, Saleh M, Gordon L, LoBuglio AF, Jonas C, Klippenstein D, Dallaire B, Varns C. J Clin Oncol. 1999;17:268. doi: 10.1200/JCO.1999.17.1.268. [DOI] [PubMed] [Google Scholar]
- Datta A, Nag A, Pan W, Hay N, Gartel AL, Colamonici O, Mori Y, Raychaudhuri P. J Biol Chem. 2004;279:36698. doi: 10.1074/jbc.M312305200. [DOI] [PubMed] [Google Scholar]
- Dave SS, Wright G, Tan B, Rosenwald A, Gascoyne RD, Chan WC, Fisher RI, Braziel RM, Rimsza LM, Grogan TM, Miller TP, LeBlanc M, Greiner TC, Weisenburger DD, Lynch JC, Vose JM, Armitage JO, Smeland EB, Kvaloy S, Holte H, Delabie J, Connors JM, Lansdorp P, Ouyang Q, Lister TA, Davies AJ, Norton AJ, Muller-Hermelink HK, Ott G, Campo E, Montserrat E, Wilson WH, Jaffe ES, Simon R, Yang L, Powell J, Zhao H, Goldschmidt N, Chiorazzi M, Staudt LM. N Engl J Med 2004. 2004;351:2159–2169. doi: 10.1056/NEJMoa041869. [DOI] [PubMed] [Google Scholar]
- Davis RE, Brown KD, Siebenlist U, Staudt LM. J Exp Med. 2001;194:1861. doi: 10.1084/jem.194.12.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis RE, Staudt LM. Curr Opin Hematol. 2002;9:333. doi: 10.1097/00062752-200207000-00011. [DOI] [PubMed] [Google Scholar]
- de Boer CJ, van Krieken JH, Kluin-Nelemans HC, Kluin PM, Schuuring E. Oncogene. 1995;10:1833. [PubMed] [Google Scholar]
- de Leval L, Braaten KM, Ancukiewicz M, Kiggundu E, Delaney T, Mankin HJ, Harris NL. Am J Surg Pathol. 2003;27:1269. doi: 10.1097/00000478-200309000-00011. [DOI] [PubMed] [Google Scholar]
- Dent AL, Shaffer AL, Yu X, Allman D, Staudt LM. Science. 1997;276:589. doi: 10.1126/science.276.5312.589. [DOI] [PubMed] [Google Scholar]
- Dhordain P, Albagli O, Lin RJ, Ansieau S, Quief S, Leutz A, Kerckaert JP, Evans RM, Leprince D. Proc Natl Acad Sci U S A. 1997;94:10762. doi: 10.1073/pnas.94.20.10762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong H, Chen L. J Mol Med. 2003;81:281. doi: 10.1007/s00109-003-0430-2. [DOI] [PubMed] [Google Scholar]
- Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, Roche PC, Lu J, Zhu G, Tamada K, Lennon VA, Celis E, Chen L. Nat Med. 2002;8:793. doi: 10.1038/nm730. [DOI] [PubMed] [Google Scholar]
- Dong H, Zhu G, Tamada K, Chen L. Nat Med. 1999;5:1365. doi: 10.1038/70932. [DOI] [PubMed] [Google Scholar]
- Dreyling MH, Bullinger L, Ott G, Stilgenbauer S, Muller-Hermelink HK, Bentz M, Hiddemann W, Dohner H. Cancer Res. 1997;57:4608. [PubMed] [Google Scholar]
- Egen JG, Kuhns MS, Allison JP. Nat Immunol. 2002;3:611. doi: 10.1038/ni0702-611. [DOI] [PubMed] [Google Scholar]
- Emmerich F, Meiser M, Hummel M, Demel G, Foss HD, Jundt F, Mathas S, Krappmann D, Scheidereit C, Stein H, Dorken B. Blood. 1999;94:3129. [PubMed] [Google Scholar]
- Falini B, Fizzotti M, Pucciarini A, Bigerna B, Marafioti T, Gambacorta M, Pacini R, Alunni C, Natali-Tanci L, Ugolini B, Sebastiani C, Cattoretti G, Pileri S, Dalla-Favera R, Stein H. Blood. 2000;95:2084. [PubMed] [Google Scholar]
- Flenghi L, Ye BH, Fizzotti M, Bigerna B, Cattoretti G, Venturi S, Pacini R, Pileri S, Lo Coco F, Pescarmona E, et al. Am J Pathol. 1995;147:405. [PMC free article] [PubMed] [Google Scholar]
- Flores KG, Li J, Hale LP. Hum Pathol. 2001;32:926. doi: 10.1053/hupa.2001.27106. [DOI] [PubMed] [Google Scholar]
- Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, Fitz LJ, Malenkovich N, Okazaki T, Byrne MC, Horton HF, Fouser L, Carter L, Ling V, Bowman MR, Carreno BM, Collins M, Wood CR, Honjo T. J Exp Med. 2000;192:1027. doi: 10.1084/jem.192.7.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita N, Jaye DL, Kajita M, Geigerman C, Moreno CS, Wade PA. Cell. 2003;113:207. doi: 10.1016/s0092-8674(03)00234-4. [DOI] [PubMed] [Google Scholar]
- Gonzalez CL, Medeiros LJ, Jaffe ES. Am J Clin Pathol. 1991;96:81. doi: 10.1093/ajcp/96.1.81. [DOI] [PubMed] [Google Scholar]
- Guiter C, Dusanter-Fourt I, Copie-Bergman C, Boulland ML, Le Gouvello S, Gaulard P, Leroy K, Castellano F. Blood. 2004;104:543. doi: 10.1182/blood-2003-10-3545. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Cell. 2000;100:57. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- Hans CP, Weisenburger DD, Greiner TC, Gascoyne RD, Delabie J, Ott G, Muller-Hermelink HK, Campo E, Braziel RM, Jaffe ES, Pan Z, Farinha P, Smith LM, Falini B, Banham AH, Rosenwald A, Staudt LM, Connors JM, Armitage JO, Chan WC. Blood. 2004;103:275. doi: 10.1182/blood-2003-05-1545. [DOI] [PubMed] [Google Scholar]
- Hayashi F, Smith KD, Ozinsky A, Hawn TR, Yi EC, Goodlett DR, Eng JK, Akira S, Underhill DM, Aderem A. Nature. 2001;410:1099. doi: 10.1038/35074106. [DOI] [PubMed] [Google Scholar]
- Horning SJ. Ann Oncol. 2000;11(Suppl 1):23. [PubMed] [Google Scholar]
- Horning SJ, Rosenberg SA. N Engl J Med. 1984;311:1471. doi: 10.1056/NEJM198412063112303. [DOI] [PubMed] [Google Scholar]
- Huang JZ, Sanger WG, Greiner TC, Staudt LM, Weisenburger DD, Pickering DL, Lynch JC, Armitage JO, Warnke RA, Alizadeh AA, Lossos IS, Levy R, Chan WC. Blood. 2002;99:2285. doi: 10.1182/blood.v99.7.2285. [DOI] [PubMed] [Google Scholar]
- Hurt EM, Wiestner A, Rosenwald A, Shaffer AL, Campo E, Grogan T, Bergsagel PL, Kuehl WM, Staudt LM. Cancer Cell. 2004;5:191. doi: 10.1016/s1535-6108(04)00019-4. [DOI] [PubMed] [Google Scholar]
- Huynh KD, Fischle W, Verdin E, Bardwell VJ. Genes Dev. 2000;14:1810. [PMC free article] [PubMed] [Google Scholar]
- Jaffe ES, Muller-Hermelink K. In: “Hodgkin's Disease”. Mauch PM, Armitage JO, Diehl V, Hoppe RT, Weiss LM, editors. Lippincott Williams & Wilkens; Philadelphia: 1999. p. 181. [Google Scholar]
- Joos S, Kupper M, Ohl S, von Bonin F, Mechtersheimer G, Bentz M, Marynen P, Moller P, Pfreundschuh M, Trumper L, Lichter P. Cancer Res. 2000;60:549. [PubMed] [Google Scholar]
- Joos S, Otano-Joos MI, Ziegler S, Bruderlein S, du Manoir S, Bentz M, Moller P, Lichter P. Blood. 1996;87:1571. [PubMed] [Google Scholar]
- Kamijo T, Weber JD, Zambetti G, Zindy F, Roussel MF, Sherr CJ. Proc Natl Acad Sci U S A. 1998;95:8292. doi: 10.1073/pnas.95.14.8292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein U, Tu Y, Stolovitzky GA, Keller JL, Haddad J, Jr., Miljkovic V, Cattoretti G, Califano A, Dalla-Favera R. Proc Natl Acad Sci U S A. 2003;100:2639. doi: 10.1073/pnas.0437996100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovanen PE, Rosenwald A, Fu J, Hurt EM, Lam LT, Giltnane JM, Wright G, Staudt LM, Leonard WJ. J Biol Chem. 2003;278:5205. doi: 10.1074/jbc.M209015200. [DOI] [PubMed] [Google Scholar]
- Kozar K, Ciemerych MA, Rebel VI, Shigematsu H, Zagozdzon A, Sicinska E, Geng Y, Yu Q, Bhattacharya S, Bronson RT, Akashi K, Sicinski P. Cell. 2004;118:477. doi: 10.1016/j.cell.2004.07.025. [DOI] [PubMed] [Google Scholar]
- Krappmann D, Emmerich F, Kordes U, Scharschmidt E, Dorken B, Scheidereit C. Oncogene. 1999;18:943. doi: 10.1038/sj.onc.1202351. [DOI] [PubMed] [Google Scholar]
- Kuppers R, Rajewsky K, Hansmann ML. Eur J Immunol. 1997;27:1398. doi: 10.1002/eji.1830270616. [DOI] [PubMed] [Google Scholar]
- Kwak LW, Campbell MJ, Czerwinski DK, Hart S, Miller RA, Levy R. N Engl J Med. 1992;327:1209. doi: 10.1056/NEJM199210223271705. [DOI] [PubMed] [Google Scholar]
- Lam LT, Davis RE, Wright G, Rosenwald A, Hurt EM,X,Y, Pierce J, Hepperle M, Xu Y, Adams J, Dang L, Staudt LM. Clin Cancer Res. 2005;11:1–13. [PubMed] [Google Scholar]
- Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, Chernova I, Iwai Y, Long AJ, Brown JA, Nunes R, Greenfield EA, Bourque K, Boussiotis VA, Carter LL, Carreno BM, Malenkovich N, Nishimura H, Okazaki T, Honjo T, Sharpe AH, Freeman GJ. Nat Immunol. 2001;2:261. doi: 10.1038/85330. [DOI] [PubMed] [Google Scholar]
- Lebwohl DE, Muise-Helmericks R, Sepp-Lorenzino L, Serve S, Timaul M, Bol R, Borgen P, Rosen N. Oncogene. 1994;9:1925. [PubMed] [Google Scholar]
- Leithauser F, Bauerle M, Huynh MQ, Moller P. Blood. 2001;98:2762. doi: 10.1182/blood.v98.9.2762. [DOI] [PubMed] [Google Scholar]
- Levens D. Proc Natl Acad Sci U S A. 2002;99:5757. doi: 10.1073/pnas.102173199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li DN, Matthews SP, Antoniou AN, Mazzeo D, Watts C. J Biol Chem. 2003;278:38980. doi: 10.1074/jbc.M305930200. [DOI] [PubMed] [Google Scholar]
- Lin KI, Angelin-Duclos C, Kuo TC, Calame K. Mol Cell Biol. 2002;22 doi: 10.1128/MCB.22.13.4771-4780.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S, Wang W, Wilson GM, Yang X, Brewer G, Holbrook NJ, Gorospe M. Mol Cell Biol. 2000;20:7903. doi: 10.1128/mcb.20.21.7903-7913.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Wong K, Calame K. Science. 1997;276:596. doi: 10.1126/science.276.5312.596. [DOI] [PubMed] [Google Scholar]
- List AF, Spier CM, Miller TP, Grogan TM. Leukemia. 1993;7:398. [PubMed] [Google Scholar]
- Liu X, Gao JX, Wen J, Yin L, Li O, Zuo T, Gajewski TF, Fu YX, Zheng P, Liu Y. J Exp Med. 2003;197:1721. doi: 10.1084/jem.20022089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lossos IS, Alizadeh AA, Eisen MB, Chan WC, Brown PO, Botstein D, Staudt LM, Levy R. Proc Natl Acad Sci U S A. 2000;97:10209. doi: 10.1073/pnas.180316097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lossos IS, Alizadeh AA, Rajapaksa R, Tibshirani R, Levy R. Blood. 2003;101:433. doi: 10.1182/blood-2002-06-1931. [DOI] [PubMed] [Google Scholar]
- Lossos IS, Czerwinski DK, Alizadeh AA, Wechser MA, Tibshirani R, Botstein D, Levy R. N Engl J Med. 2004;350:1828. doi: 10.1056/NEJMoa032520. [DOI] [PubMed] [Google Scholar]
- Lossos IS, Levy R. Semin Cancer Biol. 2003;13:191. doi: 10.1016/s1044-579x(03)00015-4. [DOI] [PubMed] [Google Scholar]
- Maloney DG, Grillo-Lopez AJ, White CA, Bodkin D, Schilder RJ, Neidhart JA, Janakiraman N, Foon KA, Liles TM, Dallaire BK, Wey K, Royston I, Davis T, Levy R. Blood. 1997;90:2188. [PubMed] [Google Scholar]
- Martinez-Valdez H, Guret C, de Bouteiller O, Fugier I, Banchereau J, Liu YJ. J Exp Med. 1996;183:971. doi: 10.1084/jem.183.3.971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPhaden AR, Whaley K. Immunol Res. 1993;12:213. doi: 10.1007/BF02918254. [DOI] [PubMed] [Google Scholar]
- Melnick A, Carlile G, Ahmad KF, Kiang CL, Corcoran C, Bardwell V, Prive GG, Licht JD. Mol Cell Biol. 2002;22:1804. doi: 10.1128/MCB.22.6.1804-1818.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monod J, Jacob F. Cold Spring Harb. Symp. Quant. Biol. 1961;26:389. doi: 10.1101/sqb.1961.026.01.048. [DOI] [PubMed] [Google Scholar]
- Muscat GE, Burke LJ, Downes M. Nucleic Acids Res. 1998;26:2899. doi: 10.1093/nar/26.12.2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muzio M, Bosisio D, Polentarutti N, D'Amico G, Stoppacciaro A, Mancinelli R, van't Veer C, Penton-Rol G, Ruco LP, Allavena P, Mantovani A. J Immunol. 2000;164:5998. doi: 10.4049/jimmunol.164.11.5998. [DOI] [PubMed] [Google Scholar]
- Niu H, Ye BH, Dalla-Favera R. Genes Dev. 1998;12:1953. doi: 10.1101/gad.12.13.1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connell BC, Cheung AF, Simkevich CP, Tam W, Ren X, Mateyak MK, Sedivy JM. J Biol Chem. 2003;278:12563. doi: 10.1074/jbc.M210462200. [DOI] [PubMed] [Google Scholar]
- Onizuka T, Moriyama M, Yamochi T, Kuroda T, Kazama A, Kanazawa N, Sato K, Kato T, Ota H, Mori S. Blood. 1995;86:28. [PubMed] [Google Scholar]
- Pan Z, Shen Y, Du C, Zhou G, Rosenwald A, Staudt LM, Greiner TC, McKeithan TW, Chan WC. Am J Pathol. 2003;163:135. doi: 10.1016/S0002-9440(10)63637-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasqualucci L, Bereschenko O, Niu H, Klein U, Basso K, Guglielmino R, Cattoretti G, Dalla-Favera R. Leuk Lymphoma. 2003;44(Suppl 3):S5. doi: 10.1080/10428190310001621588. [DOI] [PubMed] [Google Scholar]
- Peh SC, Kim LH, Poppema S. Am J Surg Pathol. 2001;25:925. doi: 10.1097/00000478-200107000-00011. [DOI] [PubMed] [Google Scholar]
- Pileri SA, Gaidano G, Zinzani PL, Falini B, Gaulard P, Zucca E, Pieri F, Berra E, Sabattini E, Ascani S, Piccioli M, Johnson PW, Giardini R, Pescarmona E, Novero D, Piccaluga PP, Marafioti T, Alonso MA, Cavalli F. Am J Pathol. 2003;162:243. doi: 10.1016/s0002-9440(10)63815-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinyol M, Cobo F, Bea S, Jares P, Nayach I, Fernandez PL, Montserrat E, Cardesa A, Campo E. Blood. 1998;91:2977. [PubMed] [Google Scholar]
- Pinyol M, Hernandez L, Cazorla M, Balbin M, Jares P, Fernandez PL, Montserrat E, Cardesa A, Lopez-Otin C, Campo E. Blood. 1997;89:272. [PubMed] [Google Scholar]
- Pinyol M, Hernandez L, Martinez A, Cobo F, Hernandez S, Bea S, Lopez-Guillermo A, Nayach I, Palacin A, Nadal A, Fernandez PL, Montserrat E, Cardesa A, Campo E. Am J Pathol. 2000;156:1987. doi: 10.1016/S0002-9440(10)65071-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piskurich JF, Lin KI, Lin Y, Wang Y, Ting JPY, Calame K. Nature Immunology. 2000;1:526. doi: 10.1038/82788. [DOI] [PubMed] [Google Scholar]
- Ponzio NM, Thorbecke GJ. Semin Cancer Biol. 2000;10:331. doi: 10.1006/scbi.2000.0330. [DOI] [PubMed] [Google Scholar]
- Qi Y, Gregory MA, Li Z, Brousal JP, West K, Hann SR. Nature. 2004;431:712. doi: 10.1038/nature02958. [DOI] [PubMed] [Google Scholar]
- Quintanilla-Martinez L, Davies-Hill T, Fend F, Calzada-Wack J, Sorbara L, Campo E, Jaffe ES, Raffeld M. Blood. 2003;101:3181. doi: 10.1182/blood-2002-01-0263. [DOI] [PubMed] [Google Scholar]
- Randle DH, Zindy F, Sherr CJ, Roussel MF. Proc Natl Acad Sci U S A. 2001;98:9654. doi: 10.1073/pnas.171217498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransohoff DF. Nat Rev Cancer. 2004;4:309. doi: 10.1038/nrc1322. [DOI] [PubMed] [Google Scholar]
- Raty R, Franssila K, Joensuu H, Teerenhovi L, Elonen E. Eur J Haematol. 2002;69:11. doi: 10.1034/j.1600-0609.2002.01677.x. [DOI] [PubMed] [Google Scholar]
- Reimold AM, Iwakoshi NN, Manis J, Vallabhajosyula P, Szomolanyi-Tsuda E, Gravallese EM, Friend D, Grusby MJ, Alt F, Glimcher LH. Nature. 2001;412:300. doi: 10.1038/35085509. [DOI] [PubMed] [Google Scholar]
- Reimold AM, Ponath PD, Li YS, Hardy RR, David CS, Strominger JL, Glimcher LH. J Exp Med. 1996;183:393. doi: 10.1084/jem.183.2.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rimokh R, Berger F, Bastard C, Klein B, French M, Archimbaud E, Rouault JP, Santa Lucia B., Duret L, Vuillaume M, et al. Blood. 1994;83:3689. [PubMed] [Google Scholar]
- Rimsza LM, Roberts RA, Miller TP, Unger JM, LeBlanc M, Braziel RM, Weisenberger DD, Chan WC, Muller-Hermelink HK, Jaffe ES, Gascoyne RD, Campo E, Fuchs DA, Spier CM, Fisher RI, Delabie J, Rosenwald A, Staudt LM, Grogan TM. Blood. 2004;103:4251. doi: 10.1182/blood-2003-07-2365. [DOI] [PubMed] [Google Scholar]
- Roglic A, Prossnitz ER, Cavanagh SL, Pan Z, Zou A, Ye RD. Biochim Biophys Acta. 1996;1305:39. doi: 10.1016/0167-4781(95)00209-x. [DOI] [PubMed] [Google Scholar]
- Rosenwald A, Wright G, Chan WC, Connors JM, Campo E, Fisher RI, Gascoyne RD, Muller-Hermelink HK, Smeland EB, Giltnane JM, Hurt EM, Zhao H, Averett L, Yang L, Wilson WH, Jaffe ES, Simon R, Klausner RD, Powell J, Duffey PL, Longo DL, Greiner TC, Weisenburger DD, Sanger WG, Dave BJ, Lynch JC, Vose J, Armitage JO, Montserrat E, Lopez-Guillermo A, Grogan TM, Miller TP, LeBlanc M, Ott G, Kvaloy S, Delabie J, Holte H, Krajci P, Stokke T, Staudt LM. N Engl J Med. 2002;346:1937. doi: 10.1056/NEJMoa012914. [DOI] [PubMed] [Google Scholar]
- Rosenwald A, Wright G, Leroy K, Yu X, Gaulard P, Gascoyne RD, Chan WC, Zhao T, Haioun C, Greiner TC, Weisenburger DD, Lynch JC, Vose J, Armitage JO, Smeland EB, Kvaloy S, Holte H, Delabie J, Campo E, Montserrat E, Lopez-Guillermo A, Ott G, Muller-Hermelink HK, Connors JM, Braziel R, Grogan TM, Fisher RI, Miller TP, LeBlanc M, Chiorazzi M, Zhao H, Yang L, Powell J, Wilson WH, Jaffe ES, Simon R, Klausner RD, Staudt LM. J Exp Med. 2003a;198:851. doi: 10.1084/jem.20031074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenwald A, Wright G, Wiestner A, Chan WC, Connors JM, Campo E, Gascoyne RD, Grogan TM, Muller-Hermelink HK, Smeland EB, Chiorazzi M, Giltnane JM, Hurt EM, Zhao H, Averett L, Henrickson S, Yang L, Powell J, Wilson WH, Jaffe ES, Simon R, Klausner RD, Montserrat E, Bosch F, Greiner TC, Weisenburger DD, Sanger WG, Dave BJ, Lynch JC, Vose J, Armitage JO, Fisher RI, Miller TP, LeBlanc M, Ott G, Kvaloy S, Holte H, Delabie J, Staudt LM. Cancer Cell. 3:185. doi: 10.1016/s1535-6108(03)00028-x. [DOI] [PubMed] [Google Scholar]
- Savage KJ, Monti S, Kutok JL, Cattoretti G, Neuberg D, De Leval L, Kurtin P, Dal Cin P, Ladd C, Feuerhake F, Aguiar RC, Li S, Salles G, Berger F, Jing W, Pinkus GS, Habermann T, Dalla-Favera R, Harris NL, Aster JC, Golub TR, Shipp MA. Blood. 2003;102:3871. doi: 10.1182/blood-2003-06-1841. [DOI] [PubMed] [Google Scholar]
- Schauer SL, Wang Z, Sonenshein GE, Rothstein TL. J Immunol. 1996;157:81. [PubMed] [Google Scholar]
- Schlosser I, Holzel M, Murnseer M, Burtscher H, Weidle UH, Eick D. Nucleic Acids Res. 2003;31:6148. doi: 10.1093/nar/gkg794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab U, Stein H, Gerdes J, Lemke H, Kirchner H, Schaadt M, Diehl V. Nature. 1982;299:65. doi: 10.1038/299065a0. [DOI] [PubMed] [Google Scholar]
- Seto M, Yamamoto K, Iida S, Akao Y, Utsumi KR, Kubonishi I, Miyoshi I, Ohtsuki T, Yawata Y, Namba M, et al. Oncogene. 1992;7:1401. [PubMed] [Google Scholar]
- Seyfert VL, Allman D, He Y, Staudt LM. Oncogene. 1996;12:2331. [PubMed] [Google Scholar]
- Shaffer AL, Lin KI, Kuo TC, Yu X, Hurt EM, Rosenwald A, Giltnane JM, Yang L, Zhao H, Calame K, Staudt LM. Immunity. 2002;17:51. doi: 10.1016/s1074-7613(02)00335-7. [DOI] [PubMed] [Google Scholar]
- Shaffer AL, Rosenwald A, Hurt EM, Giltnane JM, Lam LT, Pickeral OK, Staudt LM. Immunity. 2001;15:375. doi: 10.1016/s1074-7613(01)00194-7. [DOI] [PubMed] [Google Scholar]
- Shaffer AL, Rosenwald A, Staudt LM. Nat Rev Immunol. 2002;2:920. doi: 10.1038/nri953. [DOI] [PubMed] [Google Scholar]
- Shaffer AL, Shapiro-Shelef M, Iwakoshi NN, Lee A-H, Qian S-B, Zhao H, Yu X, Yang L, Tan BK, Rosenwald A, Hurt EM, Petroulakis E, Sonenberg N, Yewdell JW, Calame K, Glimcher LH, Staudt LM. Immunity. 2004;21:81. doi: 10.1016/j.immuni.2004.06.010. [DOI] [PubMed] [Google Scholar]
- Shaffer AL, Yu X, He Y, Boldrick J, Chan EP, Staudt LM. Immunity. 2000;13:199. doi: 10.1016/s1074-7613(00)00020-0. [DOI] [PubMed] [Google Scholar]
- Shapiro-Shelef M, Lin KI, McHeyzer-Williams LJ, Liao J, McHeyzer-Williams MG, Calame K. Immunity. 2003;19:607. doi: 10.1016/s1074-7613(03)00267-x. [DOI] [PubMed] [Google Scholar]
- Sherr CJ, McCormick F. Cancer Cell. 2002;2:103. doi: 10.1016/s1535-6108(02)00102-2. [DOI] [PubMed] [Google Scholar]
- Shipp M. N Engl J Med. 1993;329:987. doi: 10.1056/NEJM199309303291402. [DOI] [PubMed] [Google Scholar]
- Shipp MA, Ross KN, Tamayo P, Weng AP, Kutok JL, Aguiar RCT, Gaasenbeek M, Angelo M, Reich M, Pinkus GS, Ray TS, Koval MA, Last KW, Norton A, Lister TA, Mesirov J, Neuberg DS, Lander ES, Aster JC, Golub TR. Nature Medicine. 2002;8:68. doi: 10.1038/nm0102-68. [DOI] [PubMed] [Google Scholar]
- Sicinski P, Donaher JL, Parker SB, Li T, Fazeli A, Gardner H, Haslam SZ, Bronson RT, Elledge SJ, Weinberg RA. Cell. 1995;82:621. doi: 10.1016/0092-8674(95)90034-9. [DOI] [PubMed] [Google Scholar]
- Skinnider BF, Elia AJ, Gascoyne RD, Trumper LH, von Bonin F, Kapp U, Patterson B, Snow BE, Mak TW. Blood. 2001;97:250. doi: 10.1182/blood.v97.1.250. [DOI] [PubMed] [Google Scholar]
- Sorlie T, Tibshirani R, Parker J, Hastie T, Marron JS, Nobel A, Deng S, Johnsen H, Pesich R, Geisler S, Demeter J, Perou CM, Lonning PE, Brown PO, Borresen-Dale AL, Botstein D. Proc Natl Acad Sci U S A. 2003;100:8418. doi: 10.1073/pnas.0932692100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staudt LM. N Engl J Med. 2003;348:1777. doi: 10.1056/NEJMra020067. [DOI] [PubMed] [Google Scholar]
- Sui L, Zhang W, Liu Q, Chen T, Li N, Wan T, Yu M, Cao X. Biochem Biophys Res Commun. 2003;304:393. doi: 10.1016/s0006-291x(03)00601-6. [DOI] [PubMed] [Google Scholar]
- Timmerman JM, Czerwinski DK, Davis TA, Hsu FJ, Benike C, Hao ZM, Taidi B, Rajapaksa R, Caspar CB, Okada CY, van Beckhoven A, Liles TM, Engleman EG, Levy R. Blood. 2002;99:1517. doi: 10.1182/blood.v99.5.1517. [DOI] [PubMed] [Google Scholar]
- Tonnelle C, D'Ercole C, Depraetere V, Metras D, Boubli L, Fougereau M. Int Immunol. 1997;9:407. doi: 10.1093/intimm/9.3.407. [DOI] [PubMed] [Google Scholar]
- Tsai RY, McKay RD. Genes Dev. 2002;16:2991. doi: 10.1101/gad.55671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukamoto H, Nagasawa K, Ueda Y, Mayumi T, Furugo I, Tsuru T, Niho Y. Immunology. 1992;77:621. [PMC free article] [PubMed] [Google Scholar]
- Tunyaplin C, Shaffer AL, Angelin-Duclos CD, Yu X, Staudt LM, Calame KL. J Immunol. 2004;173:1158. doi: 10.4049/jimmunol.173.2.1158. [DOI] [PubMed] [Google Scholar]
- Tuscano JM, Druey KM, Riva A, Pena J, Thompson CB, Kehrl JH. Blood. 1996;88:1359. [PubMed] [Google Scholar]
- Tzankov A, Pehrs AC, Zimpfer A, Ascani S, Lugli A, Pileri S, Dirnhofer S. J Clin Pathol. 2003;56:747. doi: 10.1136/jcp.56.10.747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velders GA, Kluin-Nelemans JC, De Boer CJ, Hermans J, Noordijk EM, Schuuring E, Kramer MH, Van Deijk WA, Rahder JB, Kluin PM, Van Krieken JH. J Clin Oncol. 1996;14:1269. doi: 10.1200/JCO.1996.14.4.1269. [DOI] [PubMed] [Google Scholar]
- Withers DA, Harvey RC, Faust JB, Melnyk O, Carey K, Meeker TC. Mol Cell Biol. 1991;11:4846. doi: 10.1128/mcb.11.10.4846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witzig TE, Gordon LI, Cabanillas F, Czuczman MS, Emmanouilides C, Joyce R, Pohlman BL, Bartlett NL, Wiseman GA, Padre N, Grillo-Lopez AJ, Multani P, White CA. J Clin Oncol. 2002;20:2453. doi: 10.1200/JCO.2002.11.076. [DOI] [PubMed] [Google Scholar]
- Wong CW, Privalsky ML. Mol Cell Biol. 1998;18:5500. doi: 10.1128/mcb.18.9.5500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood KM, Roff M, Hay RT. Oncogene. 1998;16:2131. doi: 10.1038/sj.onc.1201735. [DOI] [PubMed] [Google Scholar]
- Wright G, Tan B, Rosenwald A, Hurt EH, Wiestner A, Staudt LM. Proc Natl Acad Sci U S A. 2003;100:9991. doi: 10.1073/pnas.1732008100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye BH, Cattoretti G, Shen Q, Zhang J, Hawe N, de Waard R, Leung C, Nouri-Shirazi M, Orazi A, Chaganti RS, Rothman P, Stall AM, Pandolfi PP, Dalla-Favera R. Nat Genet. 1997;16:161. doi: 10.1038/ng0697-161. [DOI] [PubMed] [Google Scholar]
- Zarate-Osorno A, Medeiros LJ, Longo DL, Jaffe ES. Am J Surg Pathol. 1992;16:885. doi: 10.1097/00000478-199209000-00007. [DOI] [PubMed] [Google Scholar]
- Zeller KI, Jegga AG, Aronow BJ, O'Donnell KA, Dang CV. Genome Biol. 2003;4:R69. doi: 10.1186/gb-2003-4-10-r69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu D, McCarthy H, Ottensmeier CH, Johnson P, Hamblin TJ, Stevenson FK. Blood. 2002;99:2562. doi: 10.1182/blood.v99.7.2562. [DOI] [PubMed] [Google Scholar]