

Summary
Both the Arp2/3 complex and cofilin are believed to be important for the generation of protrusive force at the leading edge; however, their relative contributions have not been explored in vivo. Our results with living cells show that cofilin enters the leading edge immediately before the start of lamellipod extension, slightly earlier than Arp2/3, which begins to be recruited slightly later as the lamellipod is extended. Blocking either the Arp2/3 complex or cofilin function in cells results in failure to extend broad lamellipods and inhibits free barbed ends, suggesting that neither factor on its own can support actin polymerization-mediated protrusion in response to growth factor stimulation. High-resolution analysis of the actin network at the leading edge supports the idea that both the severing activity of cofilin and the specific branching activity of the Arp2/3 complex are essential for lamellipod protrusion. These results are the first to document the relative contributions of cofilin and Arp2/3 complex in vivo and indicate that cofilin begins to initiate the generation of free barbed ends that act in synergy with the Arp2/3 complex to create a large burst in nucleation activity.
Keywords: Cofilin, Arp2/3, Cytoskeleton, Actin
Introduction
Cell motility is an important aspect of many cell-driven processes, such as wound healing, inflammatory response, embryonic development and tumor metastasis. The ability of cells to extend a protrusion and move in a given direction largely depends on actin polymerization in the protruding lamellipod of the cell (Abercrombie et al., 1970; Lauffenburger and Horwitz, 1996; Mitchison and Cramer, 1996; Pollard and Borisy, 2003). The initial protrusion event is thought to depend on factors that directly upregulate localized actin polymerization (Condeelis, 2001; Pollard et al., 2000). Later events, such as the traction of the cell body in the direction of protrusion, are thought to be mediated by myosin motors (Lee et al., 1993; Lauffenburger and Horwitz, 1996; Svitkina and Borisy, 1999; Svitkina et al., 1997).
Actin polymerization and depolymerization transients are tightly regulated in cells, and in a typical resting cell, most of the actin filaments are capped, and thus unavailable for polymerization (Pollard et al., 2000; Condeelis, 2001). The development of protrusions following a stimulus requires the rapid generation of filaments with free barbed ends in a controlled and localized manner, at the leading edge of the advancing lamellipod (Handel et al., 1990; Symons and Mitchison, 1991; Chan et al., 1998; Welch and Mullins, 2002). Two factors that appear to play an essential role in regulating actin polymerization are the Arp2/3 complex and ADF/cofilin, thereafter referred to as cofilin (Pollard and Borisy, 2003). First identified in Acanthamoeba (Machesky et al., 1994), and further characterized as the first genuine nucleator of actin polymerization in vivo (Welch et al., 1997b; Welch et al., 1998), the Arp2/3 complex is now recognized as one of the major modulators of actin polymerization in cell protrusion (Welch and Mullins, 2002; Higgs and Pollard, 2001). On the basis of its ability to nucleate branched actin filaments in vitro (Mullins et al., 1998; Blanchoin et al., 2000; Pantaloni et al., 2000; Amann and Pollard, 2001; Ichetovkin et al., 2000) and to distribute at Y-branches within the filament network at the leading edge in cells (Bailly et al., 1999; Svitkina and Borisy, 1999), it has been proposed that the complex is the motor that drives actin polymerization in motile cells by comprehensively nucleating new filaments from the pre-existing network (Pollard and Borisy, 2003). Cofilin, on the other hand, is generally believed to contribute to the turnover of actin filaments, thus providing the actin monomers that fuel the leading edge advance (Pollard and Borisy, 2003; Bamburg, 1999). And indeed, cofilin has been shown to contribute to the turnover of actin filaments in vivo (Carlier et al., 1997; Lappalainen and Drubin, 1997; Rosenblatt et al., 1997), in accordance with its in vitro ability to depolymerize F-actin by increasing the off rate at the pointed end (Carlier et al., 1997) or by direct severing of actin filaments (Maciver et al., 1991; Du and Frieden, 1998; Chan et al., 2000; Ichetovkin et al., 2000).
Although the subsequently derived ‘dentritic’ model of actin nucleation is most probably applicable to fast crawling cells such as fish keratocytes (Svitkina and Borisy, 1999), there is growing evidence that different cell types may utilize slightly different mechanisms. Recent work has shown that in vitro, cofilin and Arp2/3 can directly and synergistically cooperate to increase actin polymerization (Ichetovkin et al., 2002). Also, the view that cofilin is merely a provider of actin monomers at the leading edge has been complicated by recent data showing an important role for cofilin in defining cell polarity (Dawe, 2003; Bailly and Jones, 2003) (Ghosh et al., 2004). Therefore, although the Arp2/3 complex and cofilin are likely to remain main players in actin-mediated protrusive force, their interactions with the cytoskeleton might be more complex than initially thought.
Previous studies have shown that, following growth factor stimulation, both Arp2/3 and cofilin are present at the leading edge of lamellipods in rat metastatic carcinoma (MTLn3) cells, i.e. the site of active actin polymerization and protrusion (Bailly et al., 1999; Chan et al., 2000). We have also shown that inhibiting Arp2/3 function by using function-blocking antibodies inhibits actin polymerization and branching in vitro, and blocks lamellipod protrusion in vivo (Bailly et al., 2001). Similarly, cells in which cofilin activity is blocked, either following microinjection of function-blocking antibodies (Chan et al., 2000) or inactivation of cofilin by increasing its phosphorylation level using the kinase domain of LIM-kinase1 (Zebda et al., 2000), are unable to extend a lamellipod following epidermal growth factor (EGF) stimulation. The semiquantitative measurements performed at the time indicate that the mechanism by which lamellipod extension is abolished in each case might be different. The transient increase in actin nucleation sites (free barbed ends) normally generated at the leading edge of the cells following EGF stimulation appeared completely annihilated when cofilin function was altered (Chan et al., 2000; Zebda et al., 2000). Blocking Arp2/3 activity, however, significantly but only partially reduced the number of free barbed ends generated after stimulation (Bailly et al., 2001).
Despite these results supporting the hypothesis that cofilin may be the major provider of free barbed ends after growth factor stimulation (Condeelis, 2001), we were concerned that they might not reflect the in vivo situation completely accurately. In these previous studies, the free barbed ends were measured using a time-resolved fixed-cell assay, in which live cells are permeabilized and labeled actin monomers are allowed to incorporate into filaments, wherever free barbed ends are available in the cells (Symons and Mitchison, 1991; Bailly et al., 2001). Recently, we have developed a new live cell method that avoids some of the disadvantages of the fixed-cell barbed end assay, such as permeabilization and fixation (Lorenz et al., 2004). In this method, live cells that express GFP-βactin are stimulated to initiate lamellipod protrusion and time-lapsed to measure the increase of fluorescence at the leading edge, which is due to GFP-βactin accumulation. From this measurement, the initial rate of GFP-βactin accumulation is calculated. This rate is directly proportional to the number of barbed ends formed, thus this assay allows a relative barbed end measurement in live cells. We have used this method here to examine the relative contributions of the Arp2/3 complex and cofilin to barbed end formation in vivo. In addition, we performed a detailed analysis of the ultra-structure of the actin network at the leading edge of cells after blocking either Arp2/3 or cofilin activity, which allowed us to decipher the relative contributions of the two to the establishment of the optimal organization of the actin network for maximum protrusive force. We propose thereafter a model for stimulated protrusion that incorporates our new data on the interaction between Arp2/3 and cofilin, and we discuss its relationship to the classic dendritic model of protrusion (Pollard and Borisy, 2003).
Materials and Methods
Cell culture and cell lines
MTln3 (rat adenocarcinoma) cells were cultured in α-MEM (Gibco) with 5% FBS, as described previously (Segall et al., 1996; Bailly et al., 1998b). Cells were plated at low density in complete medium for at least 24 hours. Before EGF stimulation, cells were starved for 3 hours in Leibovitz’s L15 medium (Gibco) containing 0.35% BSA. For stimulation, cells were treated with a final concentration of 5 nM murine epidermal growth factor (Life Technologies) in starvation medium for the time indicated. MTLn3 stable cells expressing GFP-βactin have been described previously (Lorenz et al., 2004). Arp3-CFP/YFP and cofilin-GFP/CFP/YFP constructs were made by subcloning the full-length human arp3 sequence (from an Arp3-GFP construct) (Welch et al., 1997a), and the full-length rat cofilin sequence (from a pCDNA3 construct) (Zebda et al., 2000) into pEGFP-N1, pECFP-N1 and pEYFP-N1 vectors (Clontech), respectively. The cells were transfected using Lipofectamine 2000 (Life Technologies).
Antibodies
The function-blocking anti-cofilin antibody (Ab286) was generated against a peptide that covers both the actin binding site and the phosphatidylinositol 4,5-bisphosphate binding site of cofilin (Chan et al., 2000). The function-blocking anti-p34 antibody (Ab360) was generated against a peptide of the p34 protein of the Arp2/3 complex (Bailly et al., 2001). Both antibodies are polyclonal and were generated in rabbits. In each case, IgG fractions were isolated using T-Gel (Pierce). Antibodies were further purified by affinity chromatography against the peptide immunogen. Nonimmune rabbit IgG was purchased from Sigma and purified on T-Gel. For the EM studies, 5 nm gold conjugated goat anti-biotin antibodies were purchased from Ted Pella.
Microinjection
Microinjections were performed as described (Chan et al., 2000) using an Eppendorf semiautomatic microinjection system and Eppendorf femtotips II. The antibody concentration was 2 mg/ml in PBS for anti-p34 and control IgG, and 3 mg/ml in PBS for anti-cofilin. Approximately 10% of the cell volume was injected into each cell. For the light microscopy study, Texas Red dextran (10,000 MW, Molecular Probes) at a final concentration of 0.8 mg/ml was co-injected with antibodies. For electron microscopy, cells were microinjected with a solution containing anti-p34 (at 2 mg/ml) or anti-cofilin antibody (3 mg/ml) in PBS, as well as 2.5 mg/ml monomeric biotin-actin (Cytoskeleton) and 0.8 mg/ml FITC-dextran (Molecular Probes). Cells were microinjected 2 hours into the starvation process and were allowed to recover for 1 hour after microinjection and before addition of EGF.
Kinetics of F-actin/barbed ends accumulation at the leading edge
Live MTLn3-GFP βactin cells were time-lapsed on an Olympus IX70 microscope using constant settings with 60× NA 1.4 infinity-corrected optics coupled to a computer-driven cooled CCD camera using IP lab spectrum software (VayTek). Images were taken every 10 seconds with constant exposure times. Both a heated stage and an objective heater were used at 37°C. Images were captured below the saturation level of the camera. At the beginning of each time-lapse, an image was taken in the rhodamine channel with a fixed exposure time to measure relative amounts of Texas Red dextran. The mean fluorescence intensity of each cell was measured by linearly converting digitized image using NIH image (program developed by the National Institute of Health, available on the internet) and analyzing the images in the same program. Cells were grouped according to their mean fluorescent intensity into four groups of increasing intensity for further analysis. To measure fluorescent edge intensities, images captured by time-lapse were linearly converted using NIH image and analyzed using a software macro. First, cells were traced by fluorescence threshold. The macro then automated the collection of pixel intensity in a perimeter of the cell starting 1.1 μm outside the cell and extending 4.18 μm into the cell in 0.22 μm steps. Because lamellipods are flat and of uniform thickness in MTLn3 cells, there is negligible contribution of the variation of thickness to fluorescence intensity (Bailly et al., 1998a; Bailly et al., 1998b; Chan et al., 1998; Rotsch et al., 2001). In all measurement, a background subtraction was included. For leading edge intensity measurements (Fig. 3D), the values of the steps 0.22 μm, 0.44 μm and 0.66 μm inside the cell were averaged and plotted as a function of time. The cell area in each image is supplied by the macro. As described previously (Lorenz et al., 2004), the number of new barbed ends formed at the leading edge is directly proportional to the initial rate of GFP-βactin incorporation: the initial rate of GFP-βactin incorporation is the first derivative of the edge intensity increase over time [d(GFP-βactin concentration)/dt] and is proportional to N, the number of barbed ends. Here, we calculated the slope of the initial edge intensity increase, which is an approximation of the first derivative, as a measure of newly formed barbed ends.
Fig. 3.
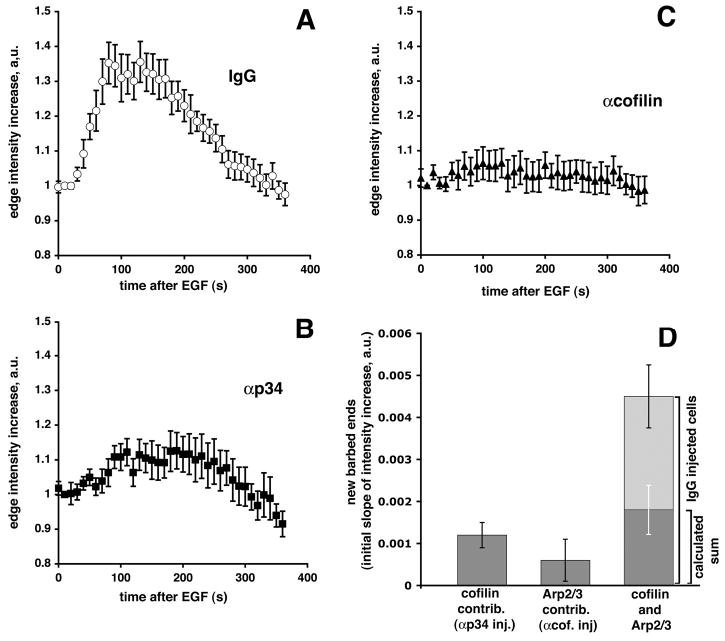
Inhibition of Arp 2/3 or cofilin function causes a reduction in barbed ends. Cells were time-lapsed after addition of EGF and the fluorescent edge intensity (GFP-βactin) was measured every 10 seconds and barbed ends calculated (see Materials and Methods). (A) Fold GFP-actin edge intensity increase after addition of EGF for nonimmune IgG-injected cells (n=15); (B) for anti-p34-injected cells (n=18); (C) for anti-cofilin-injected cells (n=17) (error bars: ±s.e.m.). (D) The relative increase in barbed ends at the leading edge after stimulation. Barbed ends were calculated by determining the slopes of the initial fluorescence intensity increase (10–100 seconds after EGF stimulation) as a measure of the rate of GFP-βactin incorporation (Lorenz et al., 2004). The first bar shows barbed ends present after injection of anti-p34, i.e. essentially the cofilin contribution to barbed ends. The second bar indicates barbed ends present after injection of anti-cofilin, i.e. the Arp2/3 contribution to barbed ends. The last bar shows barbed ends present in control IgG-injected cells (full length of the bar). The overlay (dark area on the bar) corresponds to the calculated sum of cofilin and Arp2/3 contributions as displayed in the first two bars. To estimate error bars for D, slopes were determined for straight lines that were calculated using the data in A-C±s.e.m.
Kinetics of Arp3 and cofilin recruitment to the leading edge
MTLn3 cells were transiently transfected with fluorescent constructs and analyzed after 24 hours. Images were recorded on a Zeiss Axiovert 100M equipped with a 63× Plan Apochromat 1.4 NA objective, a temperature control chamber, and driven by the OpenLab software (Improvision). Images were recorded every 5 seconds using a customized automation. For quantitation purposes, both GFP or YFP constructs were used indistinctively, and the acquired images were analyzed in NIH Image using previously described macros (Chan et al., 2000), which record the mean cortical fluorescence intensity within a 0.7 μm depth zone at the leading edge. The same macros record the cell area, as an internal control for the response to EGF. In some instances, cells were transfected with a mix of CFP-Arp3 and YFP-cofilin DNA, and images were acquired using a short band-pass FRET-specific filter to generate images of the Arp3 and cofilin localization within the same cell without any cross-talk from one channel to the other.
Electron microscopy
MTLn3 cells were grown on 5 mm glass coverslips, starved and microinjected with antibody and biotin-actin. After recovery time and stimulation with EGF, they were permeabilized and processed for rapid freeze/freeze dry/rotary shadow as previously described (Bailly et al., 1999). After fixation, cells were incubated with 5 nm gold-coupled anti-biotin antibodies, diluted between 0.5-1 OD520nm final concentration on cells. Coverslips were mounted on a rapid freezing apparatus (CF100; Life Cell Corp.) and frozen by slamming them into a liquid-nitrogen-cooled copper block. Samples were transferred to the specimen mount of a freeze-fracture apparatus (CFE-50; Cressington) and rotary shadowed at a 45° angle with 1.2–1.3 nm tantalum-tungsten, and 2.5 nm carbon at 90°. The replicas were separated from the glass coverslips with 25% hydrofluoric acid, washed into distilled water and picked up onto the surface of formvar-coated copper grids. Samples were examined using a JEOL 1010 transmission electron microscope at 80 kV. For quantitation purposes, the negatives were scanned, coded by number and further analyzed blindly in NIH Image. Filament lengths were assessed as previously described (Bailly et al., 1999). Briefly, filaments with one free end terminating at the leading edge were traced back to their origin at the intersection with another filament. The incidence angle at the membrane was measured as the angle between the filaments and the normal to the leading edge using the definition provided by Maly and Borisy (Maly and Borisy, 2001). The angles were measured using NIH Image angle measurement function by tracing manually only free end filaments terminating at the extreme edge of the lamellipod (i.e. the ones that would be directly abutting the membrane).
Results
Temporal recruitment of cofilin and Arp3 to the leading edge after EGF stimulation
Stimulation of carcinoma cells (MTLn3) with EGF triggers a rapid peripheral lamellipod extension, the kinetics of which have been extensively characterized (Segall et al., 1996; Bailly et al., 1998b). To determine how fast cofilin and Arp2/3 are recruited to the edge of the lamellipods of MTLn3 carcinoma cells after EGF stimulation, we transiently expressed fluorescent versions of full-length Arp3 and cofilin in these cells. In low-to-medium expressers, the recombinant proteins localized within the cytoplasm and the leading edge in the same manner as their respective endogenous counterparts, and did not appear to affect cell behavior (Fig. 1A, data not shown) (Chan et al., 2000; Bailly et al., 1999). Recruitment of Arp3 and cofilin to the leading edge after EGF stimulation was measured by quantitating the changes in fluorescent edge intensity as a function of time (Fig. 1B,C), whereas lamellipod extension was evaluated at the same time as the increase in total cell area (Bailly et al., 1998b). Both cofilin and Arp3 were recruited to the leading edge after EGF stimulation, and both proteins reached maximum intensity around 100 seconds after EGF stimulation. As has been shown previously, barbed ends are generated rapidly in carcinoma MTLn3 cells after EGF stimulation. This accumulation of barbed ends in the leading edge compartment reaches a maximum around 40 seconds after EGF stimulation, both measured in fixed as well as in live cells (Lorenz et al., 2004). Because the aim of this study is to examine the contributions of cofilin and Arp2/3 to barbed end generation and protrusion, we focused on the early phase of recruitment of these two proteins to the leading edge, the window of time in which the burst of barbed ends occurs. During this phase, cofilin began entering the leading edge immediately before the start of lamellipod extension (within 40 seconds after stimulation; see Fig. 1A,C), while Arp3 recruitment was slightly slower and occurred as the lamellipod extended, with a significant accumulation beginning at 50 seconds after EGF stimulation (Fig. 1A,B). To show the faster initial accumulation of cofilin within the first 60 seconds of EGF stimulation, we calculated the rate of cofilin and Arp3 accumulation at the leading edge during the initial lamellipod protrusion and are displaying this as cofilin-GFP or Arp3-GFP intensity increase in Fig. 1D. This analysis showed that initially, cofilin was recruited to the leading edge twice as fast as Arp2/3.
Fig. 1.
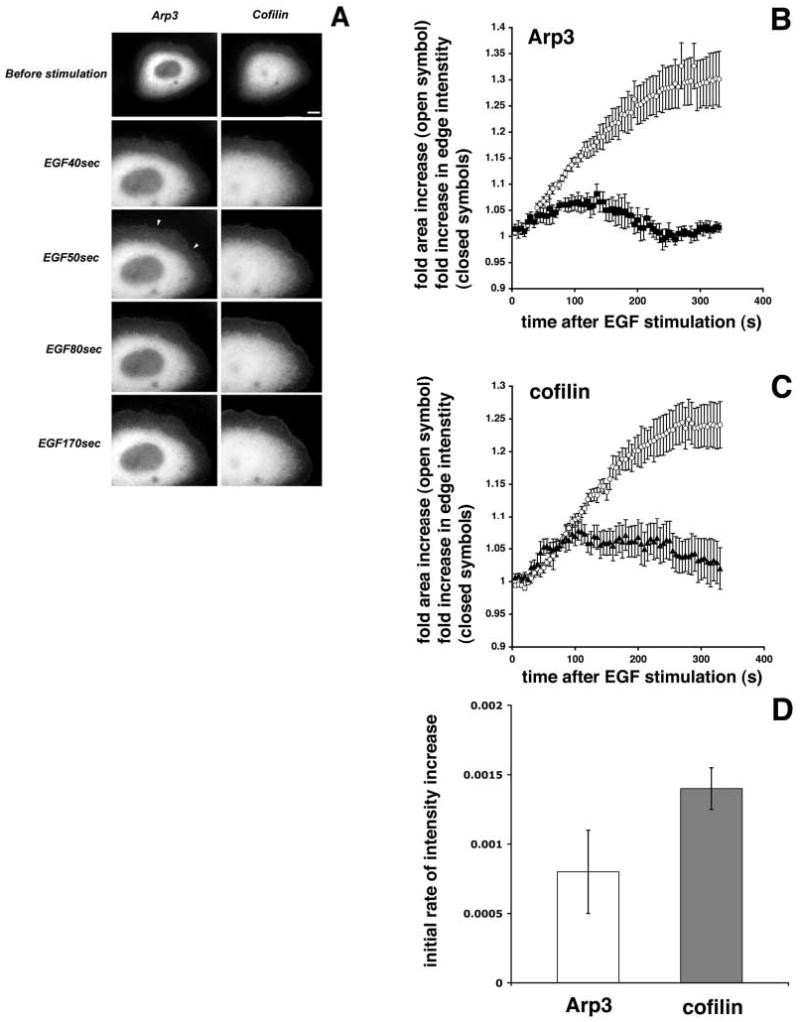
Cofilin recruitment to the leading edge of the lamellipod in the initial phase after EGF stimulation is initiated faster than Arp2/3 recruitment. (A) CFP-Arp3 and YFP-cofilin distribution in the same cell before and after EGF stimulation. Arrowheads indicate accumulation of GFP-Arp3 at the leading edge. Bar, 10 μm. (B-D) Cells transfected with GFP-cofilin or GFP-Arp3 were time-lapsed after the addition of EGF, followed by measurement of the fluorescent edge intensity and cell area throughout the time-course. The area was plotted as fold area increase over time (B and C, open circles). The fluorescent edge intensity in a 0.7 μm depth from the edge of the membrane was plotted as mean fold fluorescent intensity increase over time (B: filled squares, GFP-Arp3, n=7; C: filled triangles, GFP-cofilin, n=9). Error bars indicate the standard error of the mean (s.e.m.). (D) To calculate the initial rates of intensity increase for Arp3 and cofilin, data points for the intensity plots in B and C from 20 to 60 seconds were fitted to straight lines and the slopes of these lines were taken as the initial rates of increase. To estimate errors, the slope calculations were conducted with the data from B and C plus or minus s.e.m.
The Arp2/3 and cofilin pathways appear to be synergistic in vivo
Previous studies have shown that EGF-stimulated lamellipod protrusions are dependant on actin polymerization at the leading edge (Chan et al., 1998) and that both the Arp2/3 complex and cofilin appear to be significantly involved in the generation of free barbed ends for actin polymerization at the leading edge after EGF stimulation (Bailly et al., 2001; Zebda et al., 2001; Chan et al., 2000). However, the relative contributions of the Arp2/3 complex and cofilin have not been determined, nor has their role in generating free barbed ends been analyzed in live cells. To determine the contribution of Arp2/3 and cofilin to the nucleation transient at the leading edge of living cells following growth factor stimulation, we used MTLn3 cells that express constitutively normal levels of GFP-βactin (Lorenz et al., 2004). Using these cells, we have recently shown that the recruitment of fluorescent actin to the leading edge can be used as a reliable marker to evaluate free barbed ends appearing after stimulation in live cells (Lorenz et al., 2004). First, to verify that the blocking antibodies were functional in these cells and that saturating amounts of antibody were injected, we conducted dose-response experiments (Fig. 2). Along with function blocking antibodies, cells were co-injected with Texas Red-labeled dextran, and the relative fluorescence intensity of the dextran was measured for each cell before time lapsing. Cells were then grouped into sets of similar fluorescent intensities and the average area of lamellipod extension 200 seconds after EGF stimulation was plotted for each set. Cells with low dextran fluorescent intensities, and thus low antibody concentrations, had lamellipod extensions similar to control uninjected cells (first two bars, Fig. 2R,S). Cells with higher dextran fluorescent intensities, and thus higher antibody concentrations (last three bars, Fig. 2R,S) did not extend broad lamellipods and only exhibited very minor protrusions of small structures (Fig. 2K,O arrowheads; Fig. 2L,P red regions and Fig. 4, arrowheads). In fact, lamellipod protrusion decreased with increasing amounts of antibodies until a maximal inhibition, at which point injection of more antibody had no greater effect on protrusive activity (Fig. 2R,S). This indicates that the effect of microinjecting antibodies on broad lamellipod protrusion was saturable, and that inhibition of either Arp2/3 or cofilin separately was sufficient to prevent significant broad lamellipod extension after EGF stimulation.
Fig. 2.
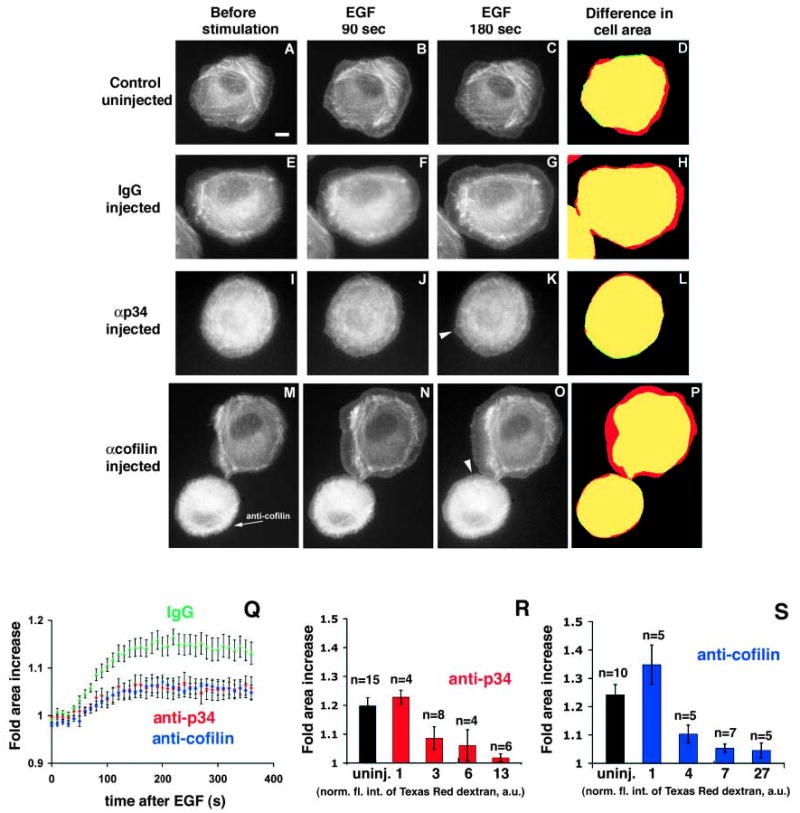
Both cofilin and Arp2/3 are required for lamellipod extension. GFP-βactin-expressing MTLn3 cells were either not injected (A-D), or microinjected with control nonimmune rabbit IgG (E-H), with anti-p34 function-blocking antibody (I-L), or with anti-cofilin function-blocking antibody (M-P, injected cell indicated with arrow). Cells were stimulated with EGF and time-lapsed to follow lamellipod extension and accumulation of F-actin at the leading edge. A, E, I and M show cells before the addition of EGF; B, F, J and N 90 seconds after EGF; and C, G, K and O 180 seconds after EGF. D, H, L and P show the difference in cell area before and after EGF (cell area before EGF: green; after EGF: red; overlap of areas: yellow). Some small protruding areas are indicated with arrowheads in K and O. Bar in A, 10 μm. (Q) Quantitation of cell area during lamellipod extension. Cell areas were measured every 10 seconds during time-lapse. Q shows the fold-increase in area after EGF stimulation of control nonimmune IgG-injected cells (n=15), anti-p34 (n=18) and anti-cofilin (n=17) antibody injected cells. (R) Dose-response for anti-p34 microinjected cells. Cells were co-injected with antibody and Texas Red-labeled dextran. Texas Red fluorescence was quantified as an indicator of relative amounts of antibody injected per cell, and measurements of cells were combined into four groups according to the fluorescent intensity of the cell (shown as normalized fluorescence intensity of Texas Red dextran, arbitrary units, a.u., below each column). The dose-response for each group is shown as average fold area increase 200 seconds after the addition of EGF, at which time cells have maximally extended. Measurements for uninjected cells are shown in the first column for comparison. (S) Dose-response for anti-cofilin injected cells. Error bars indicate ±s.e.m. In each case, both for R and S, there is no statistic difference between the last three columns, whereas the difference comparing the last three columns to the first two columns is statistically significant.
Fig. 4.
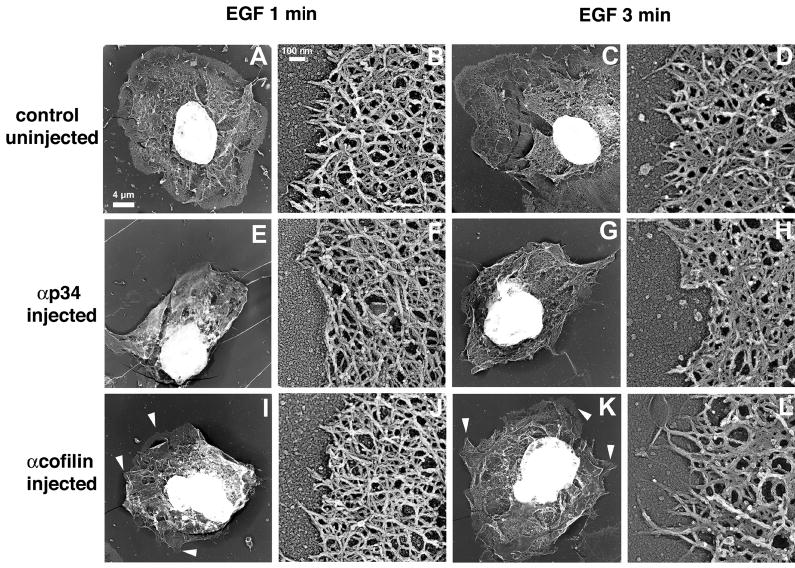
Blocking Arp2/3 function disrupts the actin network structure at the leading edge. Cells were microinjected with antibodies (E-H, anti-p34; I-L, anti-cofilin) or left untreated (A-D). After stimulation for 1 or 3 minutes with EGF, the samples were processed for replica electron microscopy. Low-magnification images are shown in A, C, E, G, I and K (Bar, 4 μm). High-magnification images (B,D,F,H,J,L) show typical leading edge areas or closest structure when leading edges were absent (Bar, 0.1 μm). Arrowheads indicate small protrusions, as opposed to broad lamellipods.
For the graph in Fig. 2Q as well as further analysis in Fig. 3, only data for those cells that had moderate-to-high dextran fluorescent intensities was used. These cells were thus near the saturation point of antibody to antigen (normalized fluorescent intensities 3–13 for anti-p34 and normalized fluorescent intensities 4–27 for anti-cofilin).
When uninjected or control IgG-injected GFP-βactin MTln3 cells were stimulated with EGF (Fig. 2A-H,Q), they extended broad lamellipods rapidly within a few minutes (the area increase at 180 seconds is indicated in red in Fig. 2D,H). They also showed extensive GFP-βactin accumulation at the leading edge within 90 seconds after EGF addition (Fig. 2B,F). As expected, neither cells microinjected with the anti-p34 function-blocking antibody (Fig. 2I-L) (Bailly et al., 2001) nor with anti-cofilin function-blocking antibody (Fig. 2M-O) (Chan et al., 2000) extended broad lamellipods, nor did they show normal accumulation of GFP-βactin at the leading edge (Fig. 2K,O and Fig. 3).
The kinetics of accumulation of GFP-βactin at the leading edge were then analyzed to determine the effects of the antibodies on accumulation of barbed ends. After time-lapse of EGF-stimulated cells, the fluorescent edge intensity of cells due to GFP-βactin incorporation was measured (shown as fold edge intensity increase versus time in Fig. 3A-C). Cells injected with control IgG had a large increase of GFP-βactin at the leading edge, with a maximum incorporation around 100 seconds after EGF stimulation (Fig. 3A). Cells injected with anti-p34 (Fig. 3B) or anti-cofilin antibodies (Fig. 3C) had very little increase in fluorescent edge intensity after EGF stimulation. The rate of GFP-βactin incorporation at the leading edge is proportional to free barbed end formation (Lorenz et al., 2004). On the basis of this observation, we calculated the slope of the initial edge intensity increase as a measure of newly formed barbed ends (Fig. 3D). This indicates that anti-p34-injected cells had a 3.5-fold decrease in barbed ends compared with IgG-injected control cells, but retained a significant amount of barbed ends (about 30% of the control). Anti-cofilin-injected cells had a sevenfold decrease in barbed ends, resulting in few free barbed ends (less than 15% of control). Altogether, this supports the idea that cofilin and Arp2/3 are two main direct providers of free barbed ends after EGF stimulation. If we take the relative number of barbed ends in cells microinjected with anti-p34 mostly as a representation of the cofilin contribution to barbed ends (since these cells have Arp2/3 activity blocked), and conversely, the relative number of barbed ends in cells microinjected with anti-cofilin as the Arp2/3 complex contribution to barbed end generation, we can calculate the theoretical ‘synergy limit’, or calculated sum, of these two separate contributions (Fig. 3D). The actual barbed end number measured in IgG-injected control cells was much higher than this calculated limit, consistent with a significant synergy of Arp2/3 complex and cofilin activities in barbed end generation in vivo, as observed with purified proteins in vitro (Ichetovkin et al., 2002).
Inhibition of Arp2/3 but not cofilin changes the incidence angle of filaments at the membrane in vivo
The actin network responsible for lamellipod protrusion at the leading edge of cells consists of a highly regular, branched network of actin filaments, which is thought to produce the mechanical force for cell protrusion. We have shown that extension of broad lamellipods is inhibited in cells where either Arp2/3 complex or cofilin function is blocked. The inhibition of lamellipod extension in anti-cofilin antibody-injected cells could be explained by the very low levels of barbed ends (less than 15% of control, Fig. 3D). However, there was a significant residual level of free barbed ends (30% of control, Fig. 3D) in cells that had received the anti-p34 antibody. Therefore, we reasoned that a more complex mechanism could be at work in these cells that prevented the residual actin polymerization from translating into an efficient protrusive force.
The structure of the actin network at the leading edge of MTLn3 cells has been extensively characterized at high resolution, and is consistent with the protrusive force being generated by actin polymerization (Bailly et al., 1999; Svitkina and Borisy, 1999). To determine if the morphology of this specialized network structure could be affected by blocking Arp2/3 and cofilin functions, we used high-resolution replica electron microscopy to perform a qualitative and quantitative analysis of the leading edge structure after stimulation in cells where Arp2/3 complex or cofilin activities had been blocked (Figs 4–6; Tables 1 and 2). Stimulated control cells exhibited broad circumferential lamellipods, containing a characteristic dense actin network (Fig. 4A,C) (Bailly et al., 1999). An example of such a broad lamellipod is shown in Fig. 4A, consisting of a dense actin network, several micrometers thick, at the cell edge, that extends circumferentially along the entire perimeter of the cell. At higher magnification, the leading edge area in the control cells showed the typical dendritic actin array observed previously, with many filaments pointing out of the network towards the membrane (Fig. 4B,D) (Bailly et al., 1999). Cells injected with either anti-p34 or anti-cofilin-blocking antibodies displayed a smaller area extension than control cells after EGF stimulation, which resulted from a failure to extend broad lamellipods (Fig. 4E-L). As reported previously for light microscopy studies (Chan et al., 2000), cells that had received the anti-cofilin antibodies appeared to have a less disturbed general shape and a cell morphology more typical of unstimulated cells, with small protrusions (Fig. 4I,K arrowheads). However, these cells never displayed the large, broad lamellipods of the control cells (Fig. 4A,C). Filament networks near the membrane and within small protrusions of anti-cofilin-injected cells appeared quite normal, with a dense array of actin filaments (Fig. 4J,L). However, most cells injected with the anti-p34 antibodies displayed a disturbed cytoskeleton with very few filaments pointing outwards (Fig. 4F,H).
Fig. 6.
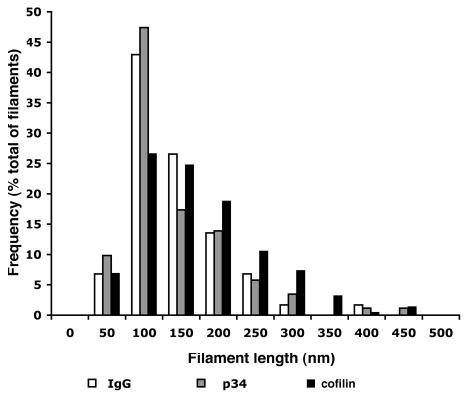
The filament length distribution is altered in anti-cofilin treated cells. The length of filaments with free ends at the leading edge 1 minute after EGF stimulation was measured. The frequency of filaments in each category was calculated for a total of 177 filaments for the IgG control (19 cells), 173 filaments for anti-p34 (17 cells) and 218 filaments for anti-cofilin samples (20 cells). Corresponding means and standard error of the mean are provided in Table 2.
Table 1.
Incidence angles at the leading edge after 1 minute of EGF stimulation
| Angle (°) | No. of filaments | |
|---|---|---|
| Control uninjected | 31.4±0.5 | 2007 |
| Nonimmune IgG injected | 38.1±1.3 | 491 |
| Anti-p34 injected | 47.8±2.0 | 355 |
| Anti-cofilin injected | 34.8±1.4 | 382 |
All values are expressed as mean±s.e.m. and are derived from the analysis of filaments with one free end terminating at the extreme edge of the lamellipod. Data shows pooled results from at least ten cells per category. The incidence angle at the membrane was measured manually as the angle between the filaments and the normal to the leading edge (according to the definitions provided by Maly and Borisy, 2001). ANOVA statistical analysis was performed on all four groups of cells, resulting in a P<0.0001 for significant differences. A subsequent Tukey multiple comparison test (95% confidence interval) showed that the following pairs had a significant difference in their angle values: control versus IgG, control versus anti-p34, IgG versus anti-p34, and anti-cofilin versus anti-p34. No significant difference was found between control versus anti-cofilin and IgG versus anti-cofilin.
Table 2.
Filament length at the leading edge after 1 minute of EGF stimulation
| Filament length (nm) | No. of filaments | |
|---|---|---|
| Nonimmune IgG injected | 118±5 | 176 |
| Anti-p34 injected | 115±6 | 173 |
| Anti-cofilin injected | 158±6 | 218 |
All values are expressed as mean±s.e.m. The filament length was measured on filaments with one free end pointing outwards towards the leading edge (Bailly et al., 1999) and is expressed in nm. Data shows pooled results from at least ten cells per category.
ANOVA statistical analysis was performed on all three groups of cells resulting in a P<0.0001 for significant differences. A subsequent Tukey multiple comparison test (95% confidence interval) showed that the following pairs had a significant difference in their filament length values: nonimmune IgG versus anti-cofilin and anti-p34 versus anti-cofilin.
It has been proposed that the polymerization of actin filaments right underneath the plasma membrane is the motor for the propulsive force that pushes the membrane outwards and generates the protrusion (Maly and Borisy, 2001; Mogilner and Oster, 2003). According to this model, the rate of membrane advance of the leading edge depends on the rate of filament elongation and the incidence angle, which is the angle of the filament with respect to the normal to the leading edge. This model is consistent with the dendritic array observed at the leading edge of rapidly moving cells, where most branches occur at an angle around 70° (Svitkina and Borisy, 1999) and where barbed ends uniformly face towards the plasma membrane on filaments that make a 35° angle with the membrane (Small et al., 1995; Maly and Borisy, 2001). The model predicts that changes in the incidence angle of the filaments with the membrane will directly affect the protrusion and that the ideal incidence angle for protrusion of ±35° depends on the proper, dense actin network containing 70° branch-points due to Arp2/3. Recent work by Verkhovsky et al. (Verkhovsky et al., 2003) has also shown that this geometry of angles holds up when the diagonal actin meshwork near the membrane was evaluated with an enhanced phase contrast microscopy technique. Because anti-p34 injected cells did not protrude, had obvious disruptions of the dendritic actin network and appeared to have altered filament orientations towards the membrane, we analyzed the incidence angle and the length of the filaments at the leading edge of the cell after EGF stimulation to determine the extent of perturbation of the actin network in cells where the Arp2/3 complex or cofilin activity had been blocked. While incidence angles measured for control cells as well as anti-cofilin-injected cells were near the predicted value of ±35° (Maly and Borisy, 2001), the incidence angle measured in the cells having received the anti-p34 antibody deviated significantly from that value (Fig. 5; Table 1). The higher value of the angle confirmed our observations that most filaments were running more parallel to the membrane rather than abutting at the optimal angle, thus being less productive in pushing the membrane.
Fig. 5.
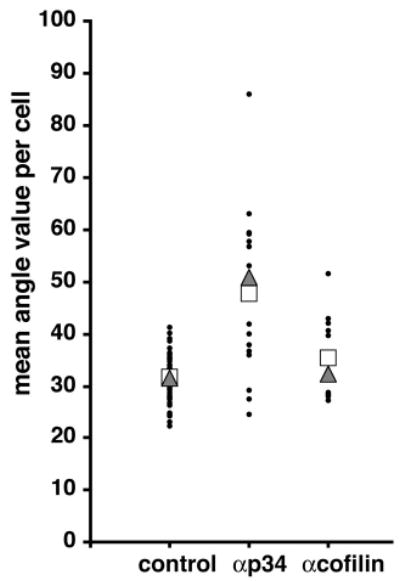
The filament incidence angles at the membrane are altered in anti-p34 injected cells. The value of the incidence angle of free end filaments at the leading edge 1 minute after stimulation was measured. Values plotted are mean angle value per cell (filled circle), median (triangle) and mean (square). (See Table 1 for statistics.)
We have shown previously that the average filament length at the leading edge is significantly decreased after EGF stimulation, coincidently with the peak of nucleation activity and cofilin recruitment to the leading edge, supporting a role for a cofilin-mediated severing activity in the transient generation of free barbed ends (Chan et al., 2000; Bailly et al., 1999). Following from this hypothesis, blocking cofilin activity in cells should prevent the severing-mediated shortening of filament length after stimulation. Thus, we measured filament length, as described previously (Bailly et al., 1999; Bear et al., 2002), in cells where Arp2/3 or cofilin activity had been blocked. We observed that, in cells having received the anti-cofilin antibody, filaments were significantly longer (Fig. 6; Table 2), which is consistent with the expected blocking of the severing/depolymerization function of cofilin in these cells.
Discussion
Lamellipod protrusion in carcinoma cells can be initiated by the addition of EGF, causing a rapid, synchronous and broad lamellipod extension (Segall et al., 1996; Bailly et al., 1998b). This protrusion is believed to be driven by localized actin polymerization at the leading edge of the lamellipod, which pushes the plasma membrane outward. This is paralleled by a sharp localized increase in the nucleation sites for actin polymerization (free barbed ends) within the first 1 μm of the leading edge, which reaches its maximum around 40 seconds after growth factor stimulation and then steadily drops back to residual prestimulus levels (Lorenz et al., 2004; Bailly et al., 1999). Hence we refer to that region at the leading edge of the lamellipod as the actin nucleation zone (Chan et al., 1998; DesMarais et al., 2002). We have shown previously that this represents a specialized compartment within the cell, enriched in tropomyosin-free filaments (DesMarais et al., 2002), which are thus potentially susceptible to Arp2/3 and cofilin binding (Blanchoin et al., 2001; DesMarais et al., 2002).
Relative contribution of the Arp2/3 complex and cofilin to the generation of barbed ends in the nucleation zone
We have confirmed here that cofilin and the Arp2/3 complex are two main contributors to the generation of the barbed end transient in cells after EGF stimulation, as inhibition of either protein leads to a large reduction in barbed end production. While both cofilin and Arp2/3 are recruited to the membrane, with maximal accumulation around 100 seconds of EGF stimulation, they show slightly different rates of recruitment in the very early phase of recruitment, the first 60 seconds after EGF stimulation. This is significant, as it has been shown both in vitro and in vivo that barbed end generation after EGF stimulation in MTLn3 carcinoma cells sharply peaks around 40 seconds after EGF stimulation (Lorenz et al., 2004), thus in this time-frame of fast kinetics even a difference of 10–20 seconds can be important. Our data shows that cofilin recruitment to the leading edge in the initial phase of recruitment is slightly faster compared with Arp2/3, suggesting that the cofilin function that contributes to barbed end generation is initiated faster. This is directly followed by recruitment of Arp2/3, suggesting a slightly slower initiation of its contribution to barbed ends. This is consistent with a model in which cofilin would begin to sever the actin filaments at the leading edge immediately before Arp2/3 can efficiently generate a highly branched network of newly polymerized actin filaments.
The severe inhibition of the barbed end transient to basal levels by cofilin function-blocking antibodies suggests that cofilin function is essential for barbed end nucleation, as Arp2/3 in cells with inhibited cofilin function is not able to generate a significant burst in barbed ends. It is unlikely that the Arp2/3 complex would depend only on cofilin to provide free monomers for actin polymerization, as there are sufficient amounts of G-actin (about 75 μM) available at the onset of protrusion in these EGF responsive cells (Chan et al., 2000; Zebda et al., 2000). Thus, this implicates cofilin’s severing activity in barbed end formation. The moderate inhibition of barbed ends due to Arp2/3 function-blocking antibody suggests that Arp2/3 also plays an important role in barbed end generation, as cofilin function alone in cells microinjected with anti-p34 antibody is not sufficient to generate the large burst in barbed ends seen in control cells. Although we cannot rule out the intervention of a third pathway, e.g. uncapping, our data suggest that its contribution to the barbed end transient is likely to be limited or mostly dependent on cofilin and/or Arp2/3 activity, as anti-cofilin antibody injections almost eliminated barbed end generation completely.
Synergy of Arp2/3 and cofilin for maximal generation of barbed ends and protrusive force
In a previous in vitro study it has been shown that either cofilin severing activity or Arp2/3 branching activity alone causes a moderate increase in actin polymerization in a light-microscope actin filament assay. However, the presence of both proteins together causes a much larger burst in actin polymerization than the sum of the individual contributions, indicating synergy between the two proteins (Ichetovkin et al., 2002). This is similar to our current data where we observed that in vivo, when the contribution of either cofilin or Arp2/3 was measured in isolation, by blocking the function of the respective other protein, either cofilin or Arp2/3 only showed minor contributions to barbed end formation. Also, the sum of these contributions was much less than the amount of barbed ends generated in control cells in which both cofilin and Arp2/3 were active, again suggesting synergy between the two. We propose that both in vivo and in vitro cofilin and Arp2/3 cooperate and act synergistically in driving actin polymerization.
We think that this synergy might be the result of, first, the ability of cofilin to sever older, ADP-containing actin filaments, producing free barbed ends in the process and thus producing more new, ATP-actin filaments. Second, in vitro the Arp2/3 complex has a preference for branching on the ATP-actin-containing newly polymerized filaments, and not the older, ADP-actin-containing core of the filaments (Ichetovkin et al., 2002). This branching would be biased towards the barbed end of elongating filaments, as shown in vitro (Pantaloni et al., 2000; Ichetovkin et al., 2002), an ideal geometry for pushing against the membrane. Thus, we think that for maximal actin polymerization, both in vivo and in vitro, the cofilin severing activity could increase the number of barbed ends in a cross-linked network of actin filaments and thus amplify the number of ATP-cap-containing filament ends. This increase in ATP-capped filaments then could allow for maximal dendritic nucleation of actin mediated by the Arp2/3 complex. This model indicates that for maximal protrusive force generated by actin polymerization both cofilin-severing and Arp2/3-branching activities are required.
Arp2/3 function is necessary to generate the proper actin filament geometry for protrusion
The analysis of the ultra structure of the actin network at the leading edge in cells where the function of the Arp2/3 complex and cofilin have been altered provides further information on how the two contribute towards maximizing actin polymerization for protrusive force. While blocking the Arp2/3 complex activity did not seem to significantly alter the average length of the filaments within the nucleation zone, significantly longer filaments were present in cells where cofilin function had been blocked. This is in direct agreement with our previous prediction that the shortening of filament length that is observed after EGF stimulation is due to the severing activity of cofilin (Bailly et al., 1999; Chan et al., 2000). In addition, the present study indicates that inhibition of Arp2/3 function, but not that of cofilin, alters the structure of the dendritic actin network at the leading edge of protruding cells. Consistent with its dramatic effect on branching in vitro (Bailly et al., 2001), the anti-p34 blocking antibody appeared to alter the ability of the Arp2/3 complex to generate the typical 70° angle branches within the actin network. The dense actin network containing 70° Arp2/3-dependent branch points is predicted to be necessary to allow filaments to form incidence angles with the membrane of 35° for maximal protrusive force. Thus, the incidence angle can be used as an indicator of the formation of a normal dendritic structure in the lamellipod. In control cells, the average incidence angle is close to the predicted value of 35°, presumably allowing for maximal protrusive force (Maly and Borisy, 2001; Mogilner and Oster, 2003), but a much higher angle is present in cells having received the Arp2/3 blocking antibody. As a result, many of the elongating filaments extend at a steeper angle to the membrane of the leading edge in these cells rather than abutting towards the membrane, and consequently are unable to generate an efficient protrusive force. This can explain why cells in which the Arp2/3 function has been blocked are unable to support even a partial protrusion, while still showing a significant barbed end transient in response to growth factor stimulation. These results stress the importance of the structural role of the Arp2/3 complex in the lamellipod of cells, where its specific branching pattern is required to set up the optimal geometry of actin filaments at the leading edge adjacent to the plasma membrane for maximal protrusive force.
A model for growth factor-stimulated lamellipod extension
We provide here the first functional evidence for a synergistic interaction between cofilin and the Arp2/3 complex in the generation of protrusion after growth factor stimulation in live cells. Combined with our previous studies (Chan et al., 1998; Bailly et al., 1999; Chan et al., 2000; Zebda et al., 2000; Bailly et al., 2001; DesMarais et al., 2002) and biochemical evidence (Ichetovkin et al., 2000; Bailly et al., 2001; Ichetovkin et al., 2002), the data reported support a modification of the ‘dendritic model’ for constitutive movement of cells (Mullins et al., 1998; Svitkina and Borisy, 1999) (Fig. 7), which applies in the case of stimulated protrusion. As discussed previously (Condeelis, 2001; Pollard et al., 2000), to extend a protrusion in response to a stimulus, resting cells need to rapidly generate free barbed ends for actin polymerization at the membrane. We hypothesize that in the stimulated protrusion model cofilin-mediated severing of pre-existing filaments initiates the burst in barbed ends. The cofilin-generated free barbed ends rapidly polymerize, as the monomer pool is abundant in a cell that was previously resting. This model is supported by experiments in which local cofilin activation by uncaging was sufficient to initiate protrusion and define the location and direction of protrusion in vivo (Ghosh et al., 2004) and by experiments showing evidence that the actin-filament-severing activity of cofilin is crucial for protrusions such as growth cones and neurite extensions (Endo et al., 2003).
Fig. 7.
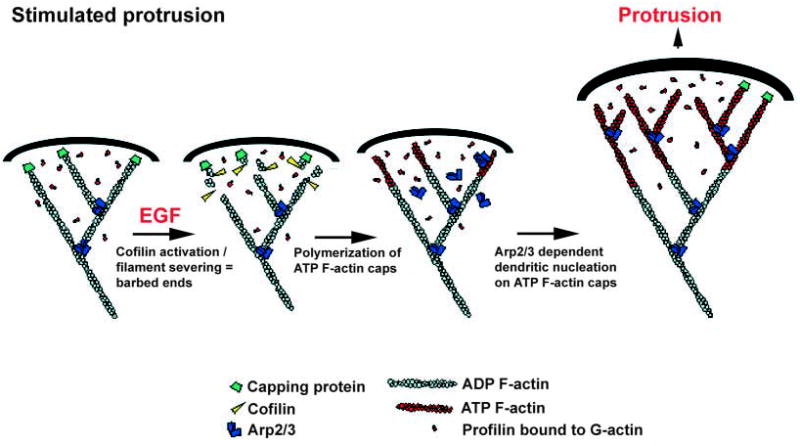
A model for stimulated protrusion. Upon EGF stimulation, cofilin severing initiates a burst in free barbed ends. Polymerization at these barbed ends leads to the generation of ATP-actin-rich filaments at the leading edge. These filaments promote the nucleation and branching activity of the Arp2/3 complex, leading to the formation of a dense actin network adjacent to the plasma membrane, which facilitates cell protrusion.
In this model, the filaments newly polymerized from cofilin-generated barbed ends are ATP-rich filaments that promote the nucleation and branching activity of the Arp2/3 complex. The new filaments are structurally organized by the activity of the Arp2/3 complex, which ensures the proper angle is generated towards the membrane to achieve optimal protrusive force. As the initial protrusion is transformed into a more constitutive movement, G-actin levels would fall to the point where the severing activity of cofilin would replenish the actin monomer pool. With this transition, the cell would enter the motility phase corresponding to the classical ‘dendritic model’ (Pollard and Borisy, 2003; Pollard et al., 2001).
As more regulators of the Arp2/3 complex are unveiled (cortactin, coronin, tropomyosin,…), it is probable that there will be more than one adaptation of the dendritic model to suit different motility situations and differently shaped protrusions. Our work here stresses the importance of cofilin as a direct provider of free barbed ends in cells. The fundamental structural role of the Arp2/3 complex in making branched arrays for pushing may be common to all pathways. It is clear that to generate a successful protrusion, one needs both free barbed ends and a proper geometrical organization of the actin network. The challenge will now be to decipher how cofilin and Arp2/3 function are regulated cooperatively in this system, and to integrate other partners that regulate Arp2/3 function into the fundamental structural role played by Arp2/3.
Acknowledgments
The authors thank Noureddine Zedba for technical help and advice in making the fluorescent cofilin and Arp3 constructs, Michael Cammer for help with image acquisition and analysis and Robert Eddy for drawing the model in Fig. 7. This work was supported by grants from the Wellcome Trust, the University College London Central Research Fund and the Special Trustees of Moorfields Eye Hospital (M.B.), and by NIH grant GM38511 (J.C.).
References
- Abercrombie M, Heaysman JE, Pegrum SM. The locomotion of fibroblasts in culture. I Movements of the leading edge. Exp Cell Res. 1970;59:393–398. doi: 10.1016/0014-4827(70)90646-4. [DOI] [PubMed] [Google Scholar]
- Amann KJ, Pollard TD. Direct real-time observation of actin filament branching mediated by Arp2/3 complex using total internal reflection fluorescence microscopy. Proc Natl Acad Sci USA. 2001;98:15009–15013. doi: 10.1073/pnas.211556398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailly M, Jones GE. Polarised migration: cofilin holds the front. Curr Biol. 2003;13:R128–R130. doi: 10.1016/s0960-9822(03)00072-1. [DOI] [PubMed] [Google Scholar]
- Bailly M, Condeelis JS, Segall JE. Chemoattractant-induced lamellipod extension. Microsc Res Tech. 1998a;43:433–443. doi: 10.1002/(SICI)1097-0029(19981201)43:5<433::AID-JEMT9>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Bailly M, Yan L, Whitesides GM, Condeelis JS, Segall JE. Regulation of protrusion shape and adhesion to the substratum during chemotactic responses of mammalian carcinoma cells. Exp Cell Res. 1998b;241:285–299. doi: 10.1006/excr.1998.4031. [DOI] [PubMed] [Google Scholar]
- Bailly M, Macaluso F, Cammer M, Chan A, Segall JE, Condeelis JS. Relationship between Arp2/3 complex and the barbed ends of actin filaments at the leading edge of carcinoma cells after epidermal growth factor stimulation. J Cell Biol. 1999;145:331–345. doi: 10.1083/jcb.145.2.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailly M, Ichetovkin I, Grant W, Zebda N, Machesky LM, Segall JE, Condeelis J. The F-actin side binding activity of the Arp2/3 complex is essential for actin nucleation and lamellipod extension. Curr Biol. 2001;11:620–625. doi: 10.1016/s0960-9822(01)00152-x. [DOI] [PubMed] [Google Scholar]
- Bamburg JR. Proteins of the ADF/cofilin family: essential regulators of actin dynamics. Annu Rev Cell Dev Biol. 1999;15:185–230. doi: 10.1146/annurev.cellbio.15.1.185. [DOI] [PubMed] [Google Scholar]
- Bear JE, Svitkina TM, Krause M, Schafer DA, Loureiro JJ, Strasser GA, Maly IV, Chaga OY, Cooper JA, Borisy GG, et al. Antagonism between Ena/VASP proteins and actin filament capping regulates fibroblast motility. Cell. 2002;109:509–521. doi: 10.1016/s0092-8674(02)00731-6. [DOI] [PubMed] [Google Scholar]
- Blanchoin L, Amann KJ, Higgs HN, Marchand JB, Kaiser DA, Pollard TD. Direct observation of dendritic actin filament networks nucleated by Arp2/3 complex and WASP/Scar proteins. Nature. 2000;404:1007–1011. doi: 10.1038/35010008. [DOI] [PubMed] [Google Scholar]
- Blanchoin L, Pollard TD, Hitchcock-DeGregori SE. Inhibition of the Arp2/3 complex-nucleated actin polymerization and branch formation by tropomyosin. Curr Biol. 2001;11:1300–1304. doi: 10.1016/s0960-9822(01)00395-5. [DOI] [PubMed] [Google Scholar]
- Carlier MF, Laurent V, Santolini J, Melki R, Didry D, Xia GX, Hong Y, Chua NH, Pantaloni D. Actin depolymerizing factor (ADF/cofilin) enhances the rate of filament turnover: implication in actin-based motility. J Cell Biol. 1997;136:1307–1322. doi: 10.1083/jcb.136.6.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan AY, Raft S, Bailly M, Wyckoff JB, Segall JE, Condeelis JS. EGF stimulates an increase in actin nucleation and filament number at the leading edge of the lamellipod in mammary adenocarcinoma cells. J Cell Sci. 1998;111:199–211. doi: 10.1242/jcs.111.2.199. [DOI] [PubMed] [Google Scholar]
- Chan AY, Bailly M, Zebda N, Segall JE, Condeelis JS. Role of cofilin in epidermal growth factor-stimulated actin polymerization and lamellipod protrusion. J Cell Biol. 2000;148:531–542. doi: 10.1083/jcb.148.3.531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condeelis J. How is actin polymerization nucleated in vivo? Trends Cell Biol. 2001;11:288–293. doi: 10.1016/s0962-8924(01)02008-6. [DOI] [PubMed] [Google Scholar]
- Dawe HR, Minamide LS, Bamburg JR, Cramer LP. ADF/cofilin controls cell polarity during fibroblast migration. Curr Biol. 2003;13:252–257. doi: 10.1016/s0960-9822(03)00040-x. [DOI] [PubMed] [Google Scholar]
- DesMarais V, Ichetovkin I, Condeelis J, Hitchcock-DeGregori SE. Spatial regulation of actin dynamics: a tropomyosin-free, actin-rich compartment at the leading edge. J Cell Sci. 2002;115:4649–4660. doi: 10.1242/jcs.00147. [DOI] [PubMed] [Google Scholar]
- Du J, Frieden C. Kinetic studies on the effect of yeast cofilin on yeast actin polymerization. Biochem. 1998;37:13276–13284. doi: 10.1021/bi981117r. [DOI] [PubMed] [Google Scholar]
- Endo M, Ohashi K, Sasaki Y, Goshima Y, Niwa R, Uemura T, Mizuno K. Control of growth cone motility and morphology by LIM kinase and Slingshot via phosphorylation and dephosphorylation of cofilin. J Neurosci. 2003;23:2527–2537. doi: 10.1523/JNEUROSCI.23-07-02527.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh M, Song X, Mouneimne G, Sidani M, Lawrence DS, Condeelis JS. Cofilin promotes actin polymerization and defines the direction of cell motility. Science. 2004;304:743–746. doi: 10.1126/science.1094561. [DOI] [PubMed] [Google Scholar]
- Handel SE, Hendry KA, Sheterline P. Microinjection of covalently cross-linked actin oligomers causes disruption of existing actin filament architecture in PtK2 cells. J Cell Sci. 1990;97:325–333. doi: 10.1242/jcs.97.2.325. [DOI] [PubMed] [Google Scholar]
- Higgs HN, Pollard TD. Regulation of actin filament network formation through ARP2/3 complex: activation by a diverse array of proteins. Annu Rev Biochem. 2001;70:649–676. doi: 10.1146/annurev.biochem.70.1.649. [DOI] [PubMed] [Google Scholar]
- Ichetovkin I, Han J, Pang KM, Knecht DA, Condeelis JS. Actin filaments are severed by both native and recombinant dictyostelium cofilin but to different extents. Cell Motil Cytoskeleton. 2000;45:293–306. doi: 10.1002/(SICI)1097-0169(200004)45:4<293::AID-CM5>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Ichetovkin I, Grant W, Condeelis J. Cofilin produces newly polymerized actin filaments that are preferred for dendritic nucleation by the Arp2/3 complex. Curr Biol. 2002;12:79–84. doi: 10.1016/s0960-9822(01)00629-7. [DOI] [PubMed] [Google Scholar]
- Lappalainen P, Drubin DG. Cofilin promotes rapid actin filament turnover in vivo. Nature. 1997;388:78–82. doi: 10.1038/40418. [DOI] [PubMed] [Google Scholar]
- Lauffenburger DA, Horwitz AF. Cell migration: a physically integrated molecular process. Cell. 1996;84:359–369. doi: 10.1016/s0092-8674(00)81280-5. [DOI] [PubMed] [Google Scholar]
- Lee J, Ishihara A, Theriot JA, Jacobson K. Principles of locomotion for simple-shaped cells. Nature. 1993;362:167–171. doi: 10.1038/362167a0. [DOI] [PubMed] [Google Scholar]
- Lorenz M, DesMarais V, Macaluso F, Singer R, Condeelis J. Correlation between barbed ends, actin polymerization and motility in live cells after EGF-stimulation. Cell Motil Cytoskeleton. 2004;57 :207–217. doi: 10.1002/cm.10171. [DOI] [PubMed] [Google Scholar]
- Machesky LM, Atkinson SJ, Ampe C, Vandekerckhove J, Pollard TD. Purification of a cortical complex containing two unconventional actins from Acanthamoeba by affinity chromatography on profilin-agarose. J Cell Biol. 1994;127:107–115. doi: 10.1083/jcb.127.1.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maciver SK, Zot HG, Pollard TD. Characterization of actin filament severing by actophorin from Acanthamoeba castellanii. J Cell Biol. 1991;115:1611–1620. doi: 10.1083/jcb.115.6.1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maly IV, Borisy GG. Self-organization of a propulsive actin network as an evolutionary process. Proc Natl Acad Sci USA. 2001;98:11324–11329. doi: 10.1073/pnas.181338798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchison TJ, Cramer LP. Actin-based cell motility and cell locomotion. Cell. 1996;84:371–379. doi: 10.1016/s0092-8674(00)81281-7. [DOI] [PubMed] [Google Scholar]
- Mogilner A, Oster G. Force generation by actin polymerization II: the elastic ratchet and tethered filaments. Biophys J. 2003;84:1591–1605. doi: 10.1016/S0006-3495(03)74969-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullins RD, Heuser JA, Pollard TD. The interaction of Arp2/3 complex with actin: nucleation, high affinity pointed end capping, and formation of branching networks of filaments. Proc Natl Acad Sci USA. 1998;95:6181–6186. doi: 10.1073/pnas.95.11.6181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantaloni D, Boujemaa R, Didry D, Gounon P, Carlier MF. The Arp2/3 complex branches filament barbed ends: functional antagonism with capping proteins. Nat Cell Biol. 2000;2:385–391. doi: 10.1038/35017011. [DOI] [PubMed] [Google Scholar]
- Pollard TD, Borisy GG. Cellular motility driven by assembly and disassembly of actin filaments. Cell. 2003;112:453–465. doi: 10.1016/s0092-8674(03)00120-x. [DOI] [PubMed] [Google Scholar]
- Pollard TD, Blanchoin L, Mullins RD. Molecular mechanisms controlling actin filament dynamics in nonmuscle cells. Annu Rev Biophys Biomol Struct. 2000;29:545–576. doi: 10.1146/annurev.biophys.29.1.545. [DOI] [PubMed] [Google Scholar]
- Pollard TD, Blanchoin L, Mullins RD. Actin dynamics. J Cell Sci. 2001;114:3–4. doi: 10.1242/jcs.114.1.3. [DOI] [PubMed] [Google Scholar]
- Rosenblatt J, Agnew BJ, Abe H, Bamburg JR, Mitchison TJ. Xenopus actin depolymerizing factor/cofilin (XAC) is responsible for the turnover of actin filaments in Listeria monocytogenes tails. J Cell Biol. 1997;136:1323–1332. doi: 10.1083/jcb.136.6.1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotsch C, Jacobson K, Condeelis J, Radmacher M. EGF-stimulated lamellipod extension in adenocarcinoma cells. Ultramicroscopy. 2001;86:97–106. doi: 10.1016/s0304-3991(00)00102-9. [DOI] [PubMed] [Google Scholar]
- Segall JE, Tyerech S, Boselli L, Masseling S, Helft J, Chan A, Jones J, Condeelis J. EGF stimulates lamellipod extension in metastatic mammary adenocarcinoma cells by an actin-dependent mechanism. Clin Exp Metastasis. 1996;14:61–72. doi: 10.1007/BF00157687. [DOI] [PubMed] [Google Scholar]
- Small JV, Herzog M, Anderson K. Actin filament organization in the fish keratocyte lamellipodium. J Cell Biol. 1995;129:1275–1286. doi: 10.1083/jcb.129.5.1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svitkina TM, Borisy GG. Arp2/3 complex and actin depolymerizing factor/cofilin in dendritic organization and treadmilling of actin filament array in lamellipodia. J Cell Biol. 1999;145:1009–1026. doi: 10.1083/jcb.145.5.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svitkina TM, Verkhovsky AB, McQuade KM, Borisy GG. Analysis of the actin-myosin II system in fish epidermal keratocytes: mechanism of cell body translocation. J Cell Biol. 1997;139:397–415. doi: 10.1083/jcb.139.2.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symons MH, Mitchison TJ. Control of actin polymerization in live and permeabilized fibroblasts. J Cell Biol. 1991;114:503–513. doi: 10.1083/jcb.114.3.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkhovsky AB, Chaga OY, Schaub S, Svitkina TM, Meister JJ, Borisy GG. Orientational order of the lamellipodial actin network as demonstrated in living motile cells. Mol Biol Cell. 2003;14:4667–4675. doi: 10.1091/mbc.E02-10-0630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch MD, Mullins RD. Cellular control of actin nucleation. Annu Rev Cell Dev Biol. 2002;18:247–288. doi: 10.1146/annurev.cellbio.18.040202.112133. [DOI] [PubMed] [Google Scholar]
- Welch MD, DePace AH, Verma S, Iwamatsu A, Mitchison TJ. The human Arp2/3 complex is composed of evolutionarily conserved subunits and is localized to cellular regions of dynamic actin filament assembly. J Cell Biol. 1997a;138:375–384. doi: 10.1083/jcb.138.2.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch MD, Iwamatsu A, Mitchison TJ. Actin polymerization is induced by Arp2/3 protein complex at the surface of Listeria monocytogenes. Nature. 1997b;385:265–269. doi: 10.1038/385265a0. [DOI] [PubMed] [Google Scholar]
- Welch MD, Rosenblatt J, Skoble J, Portnoy DA, Mitchison TJ. Interaction of human Arp2/3 complex and the Listeria monocytogenes ActA protein in actin filament nucleation. Science. 1998;281:105–108. doi: 10.1126/science.281.5373.105. [DOI] [PubMed] [Google Scholar]
- Zebda N, Bernard O, Bailly M, Welti S, Lawrence DS, Condeelis JS. Phosphorylation of ADF/cofilin abolishes EGF-induced actin nucleation at the leading edge and subsequent lamellipod extension. J Cell Biol. 2000;151:1119–1128. doi: 10.1083/jcb.151.5.1119. [DOI] [PMC free article] [PubMed] [Google Scholar]