

Abstract
Scalable production of rAAV vectors remains a major obstacle to the clinical application of this prototypical gene therapy vector. A recently developed baculovirus-based production protocol (M. Urabe et al., 2002, Hum. Gene Ther. 13, 1935–1943) found limited applications due to the system’s design. Here we report a detailed analysis of the stability of the original baculovirus system components BacRep, BacVP, and transgene cassette-containing BacGFP. All of the baculovirus helpers analyzed were prone to passage-dependent loss-of-function deletions resulting in considerable decreases in rAAV titers. To alleviate the instability and to extend the baculovirus platform to other rAAV serotypes, we have modified both Rep- and Cap-encoding components of the original system. The modifications include a parvoviral phospholipase A2 domain swap allowing production of infectious rAAV8 vectors in vivo. Alternatively, an infectious rAAV8 (or rAAV5) vector incorporating the AAV2 VP1 capsid protein in a mosaic vector particle with AAV8 capsid proteins was produced using a novel baculovirus vector. In this vector, the level of AAV2 VP1 expression is controlled with a “riboswitch,” a self-cleaving ribozyme controlled by toyocamycin in the “ON” mode. The redesigned baculovirus system improves our capacity for rAAV manufacturing by making this production platform more applicable to other existing serotypes.
Keywords: AAV, baculovirus, PLA2
Introduction
The recombinant adeno-associated virus (rAAV) vector emerged recently as one the most versatile gene therapy delivery vehicles. The mainstream utility of rAAV derives in part from the natural plasticity of its structural and regulatory viral components. AAV genomes are widely disseminated in human and non-human primate species, with rapid molecular evolution resulting in the formation of novel, serologically distinct serotypes [1,2]. Taking advantage of the diverse serotypes’ structural relationships, investigators were able to exploit their modular nature by combining specific vector components derived from each serotypes. Using the processes dubbed “pseudotyping” [3] or “cross-packaging” [4], chimeric vectors were constructed that contained AAV2-derived terminal repeats harboring a transgene packaged into capsids of other AAV serotypes. This approach greatly facilitated vector production and therapeutic screening by allowing the same transgene cassette to be packaged for direct comparison of transduction efficiencies of the targeted tissues based specifically on the composition of the viral particle per se. The logical extension of this approach was the generation of chimeric rAAVs using the “transcapsidation” or “cross-dressing” technique whereby the virion consisted of a mosaic of capsid proteins derived from two different AAV serotypes combined at different ratios [5,6]. Such mosaic vectors can exhibit dual receptor-binding characteristics of the parental viruses and, provided optimal stoichiometry of components, may even display a synergistic effect in transduction [6].
Recombinant AAV vector production was further improved by Kotin and colleagues [7], who demonstrated the feasibility of producing these vectors in insect cells using a recombinant baculovirus system. While extremely promising for the production of AAV2, this method remained to be tested for the production of pseudotyped rAAV vectors in a large-scale format. Hereby we report modifications to the original baculovirus-based rAAV production system including enhancement of the helper virus stability and construction of novel baculovirus vectors for rAAV pseudotyping using parvoviral VP1 phospholipase A2 motif swapping as well as AAV2 VP1-based mosaics. The modified system extends the flexibility of rAAV vector production and further promotes the utility of AAV as a clinically applicable gene therapy vector.
Results
As described in the original report [7], we produced rAAV2 vectors by co-infecting insect Sf9 cells with three helper vectors, BacRep, BacVP, and BacGFP, encoding rep, cap, and TR-embedded transgene cassette, respectively. Our initial attempt to produce rAAV2 in this system resulted in titers that were significantly lower than reported by the authors. Consequently, we set out to determine which particular component(s) of the three baculovirus helpers was responsible for the observed lower yields of rAAV2.
Rep Component
Upon replaquing the passage 3 (P3) BacRep, we amplified 10 individual viral stocks of BacRep to generate P1 stocks. For reference, our nomenclature describes the plaque itself as passage zero (P0) and the next generation of baculovirus amplified from the plaque as P1. We infected Sf9 cells with P1 RepBacs and 3 days postinfection analyzed expression of Rep proteins by Western blot. Four of ten BacRep stocks produced relatively little Rep proteins in infected cells (Fig. 1). Titers of rAAV2 vector stocks produced using 10 individual P1 isolates correlated directly with the amount of Rep proteins expressed by the individual helper (not shown). We selected one stock as the best producer among those tested (Fig. 1, lane 5), amplified it, and used it in subsequent stability testing experiments.
FIG. 1.
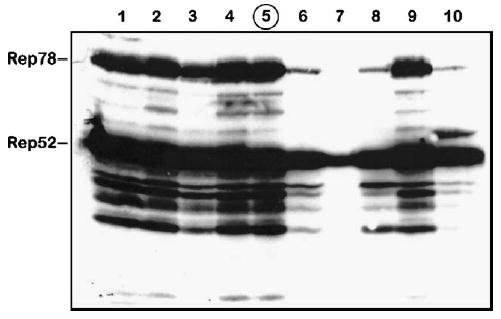
Western blot analysis of Rep proteins expressed in Sf9 cells by individual BacRep baculovirus helper plaque isolates (lanes 1 through 10). Isolate 5 (circled) was selected and propagated for the passage stability test (shown in Fig. 2).
To determine the passaging stability of the selected BacRep, we passaged the helper virus serially up to P5, titered each stock as described by Kool et al. [8], and analyzed the expression of Rep proteins in each viral stock by Western blot after infection of fresh Sf9 cells at a multiplicity of infection (m.o.i.) of 5 (Fig. 2, BacRep). We documented that the amounts of both Rep78 and Rep52 in BacRep-infected cells declined with each passage. There was no preferential loss of either protein but rather a coordinated diminution of both components, suggesting a simple mechanistic explanation of vector instability.
FIG. 2.
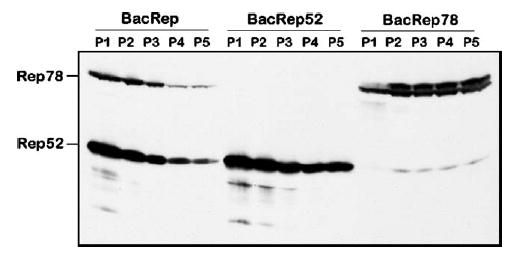
Western blot analysis of Rep proteins expressed in Sf9 cells by BacRep, BacRep52, or BacRep78 baculovirus helpers. Cells were infected with serially passaged baculovirus stocks (P1 through P5) at an m.o.i. of 5.
In the original BacRep helper [7], ΔIE1-driven rep78 and polh-driven rep52 were placed in a head-to-head orientation creating, in effect, a perfect palindrome structure of about 1.2 kb. In the wtAAV genome, these two genes are encoded by two collinear ORFs within one DNA sequence, transcribed into two separate mRNAs from the P5 and P19 promoters. We hypothesized that in the BacRep helper, the palindromic orientation of rep52 and rep78 sequences within the baculovirus genome could result in the formation of an unstable secondary structure leading to recombination and subsequent deletion during replication. To test this hypothesis, we subcloned the rep52 and rep78 genes to derive two separate recombinant baculoviruses (Fig. 3A), BacRep52 and BacRep78, that retained the original expression cassettes, including promoters. We analyzed individual viral stocks, prepared as described above, for the production of Rep52 and Rep78 proteins, selected the best producers, serially passaged them to derive P5, and visualized Rep expression levels by Western blot. Unlike the original BacRep, levels of Rep proteins appeared either to remain constant (Rep78) or to decline only slightly (Rep52) from the first passage stock to the fifth (Fig. 2, BacRep52 and BacRep78). In this experiment, when expressed separately, ΔIE1-driven rep78 and polh-driven rep52 produced comparable amounts of Rep proteins. In addition, BacRep78 produced small amounts of Rep52 derived from mRNA transcribed from the AAV2 P19 promoter, suggesting that the viral P19 sequence retains some residual promoter activity in insect cells.
FIG. 3.
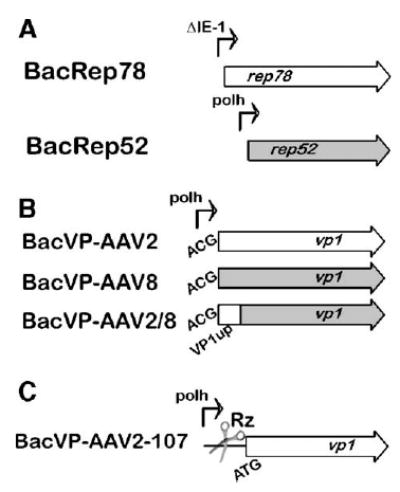
Schematic representation of the baculovirus helper vectors. (A) BacRep78 and BacRep52 as two separate helpers; (B) VP1up domain swapping between AAV2 and AAV8 BacVP helpers; (C) toyocamycin-regulated VP1 AAV2 baculovirus helper cassette. The scissors illustrate the position of a self-cleaving HH Rz control element.
The high stoichiometric ratio of Rep52/Rep78 in favor of the former is critical for the high yield of rAAV [9]. We, therefore, asked whether Rep stoichiometry changes under the conditions of quadruple co-infection with these helper viruses. Seventy-two hours postinfection with various combinations of helper vectors (m.o.i. of 5 each), we analyzed Rep proteins by Western blotting analysis (Fig. 4). Infection with BacRep78 or BacRep52 alone produced ratios that were similar to the original BacRep construct (Fig. 4, lanes 2–4). However, this ratio was shifted slightly in favor of Rep52 (Fig. 4, lane 5) when cells were co-infected with both BacRep78 and BacRep52. Moreover, when two additional baculovirus promoters were introduced (polh in BacVP and p10 in BacGFP in a quadruple co-infection), this ratio shifted in favor of the small Rep (Fig. 4, lane 6), suggesting that three strong viral promoters may compete for available transcription factors and attenuate the ΔIE1 promoter.
FIG. 4.
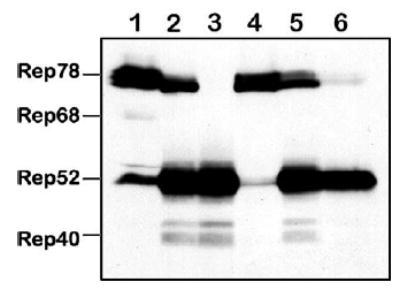
Western blot analysis of Rep proteins expressed in Sf9 cells by BacRep, BacRep52, or BacRep78 baculovirus helpers individually or upon co-infection with other baculovirus helpers (m.o.i. of 5 each). Lane 1, positive control (a lysate from 293 cells transfected with pIM45 [21]; lanes 2 through 6 contain lysates from Sf9 cells infected with, lane 2, BacRep; lane 3, BacRep52; lane 4, BacRep78; lane 5, BacRep78 + BacRep52; lane 6, BacRep78 + BacRep52 + BacVP + BacGFP (the latter vector also contains a strong baculovirus p10 promoter driving the GFP gene inside the transgene cassette [15]).
The redesigned set of vectors requires now a quadruple instead of triple co-infection of Sf9 cells to produce rAAV. In a pilot experiment, we have compared side-by-side yields of rAAV prepared using three vs four helpers (P2 each) at an m.o.i. of 5 each. There was little difference in rAAV titers produced (1.9 × 109 infectious particles (ip)/ml vs 1.4 × 109 ip/ml, respectively).
AAV2 ITR-Flanked Transgene Cassette Component
The palindromic termini of the AAV genome, as well as rAAV derivatives, are notoriously unstable and prone to deletions that render the genome functionally defective. We, therefore, asked whether the ITR-containing component of the helper triumvirate would maintain functional replicative capability for the duration of five consecutive passages. There was a notable loss of the ITR-transgene cassette-containing baculovirus over the five passages. We documented this reduction by assaying rescued TR-containing cassettes replicating in the presence of Rep proteins (Fig. 5A). Titers of rAAV2-GFP, prepared using the respective P1 through P5 BacGFP helpers (m.o.i. of 5 each), closely correlated with the reduction of the ITR-containing sequences (Fig. 5B).
FIG. 5.
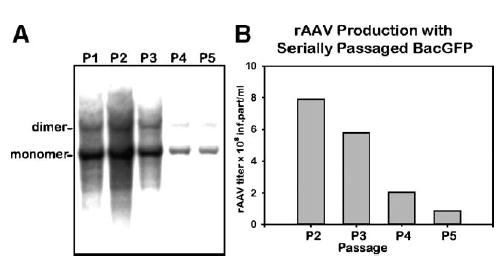
Passaging stability analysis of ITR-containing transgene cassette (BacGFP). (A) Analysis of rescued rAAV cassette. Sf9 cells were infected with BacGFP from consecutive passage stocks (m.o.i. of 5 each) in addition to BacRep (P2, m.o.i. of 5). Forty-eight hours postinfection, DNA was prepared by Hirt DNA extraction, resolved using a 1.2% agarose gel, transferred to a nylon filter, and hybridized with a 32P-labeled GFP probe. (B) Analysis of rAAV2-GFP titers of vector stocks prepared using BacGFP P2 through P5 helpers. Sf9 cells were co-infected with BacVP and BacRep (P2, m.o.i. of 5 each). In addition, cells were co-infected with BacGFP at the indicated passages (m.o.i. 5 of each). Seventy-two hours postinfection, cells were harvested and rAAV infectious titers in crude cell lysates were calculated using GFP fluorescence assay using C12 cells co-infected with Ad5 (m.o.i. of 10) [18].
VP Component
Similarly, we applied the five-passage stability test to the original BacVP viral stock component. As with the other components of this production system, Western blotting analysis demonstrated a notable decline in VP1, VP2, and VP3 capsid proteins expressed by helper vectors from the P1 to P5 (Fig. 6).
FIG. 6.
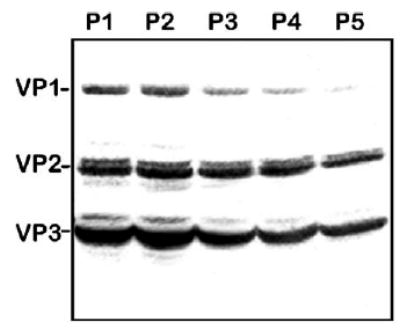
Western blot analysis of AAV2 capsid proteins expressed in Sf9 cells by BacVP helper. Sf9 cells were infected with BacVP (m.o.i. of 5) of consecutive passages, as indicated. Seventy-two hours postinfection, cells were harvested and cell lysates were analyzed by Western blotting as described under Materials and Methods.
The overall utility of the baculovirus AAV production system ultimately resides on its ability to pseudotype an AAV2-ITR transgene cassette with capsid genes of other AAV serotypes. We, therefore, designed BacVP helper vectors to produce AAV5- and AAV8-pseudotyped rAAVs. We designed the constructs to emulate the original pFBDVPm11 construct [7], introducing similar mutations into AAV5 and AAV8 capsid genes encoding VP1 N termini. We screened eight individual plaques of each construct to identify BacVP5 and BacVP8 helper vectors using Western blotting analysis (not shown); we propagated selected clones to P2 and used them in a triple co-infection with BacRep and BacGFP to produce pseudo-typed rAAV5-GFP and rAAV8-GFP.
Titers of the purified rAAV5 and rAAV8 stocks were similar to rAAV2 titers, approaching 5 × 104 DNase I-resistant rAAV particles (drp) per cell. However, as opposed to rAAV2-GFP, the particle-to-infectivity ratios of rAAV5-GFP and rAAV8-GFP were generally higher by 3–4 orders of magnitude (as assayed on HeLa-derived C12 cells upon Ad5 co-infection). The reason for the extremely low infectivity of Sf9-derived serotype 5 and 8 vectors was revealed upon closer investigation of the capsid composition in purified viral particles (Fig. 7). The capsid protein compositions of both 293- and Sf9-derived rAAV2 capsids were similar, with VP1:VP2:VP3 ratios approximating 1:1:10 (Fig. 7A, lanes 1 and 2). However, the amounts of VP1 in Sf9-derived rAAV5 and 8 were considerably lower compared to their 293 counterparts (Fig. 7A, lanes 3, 4, 6, and 7).
FIG. 7.
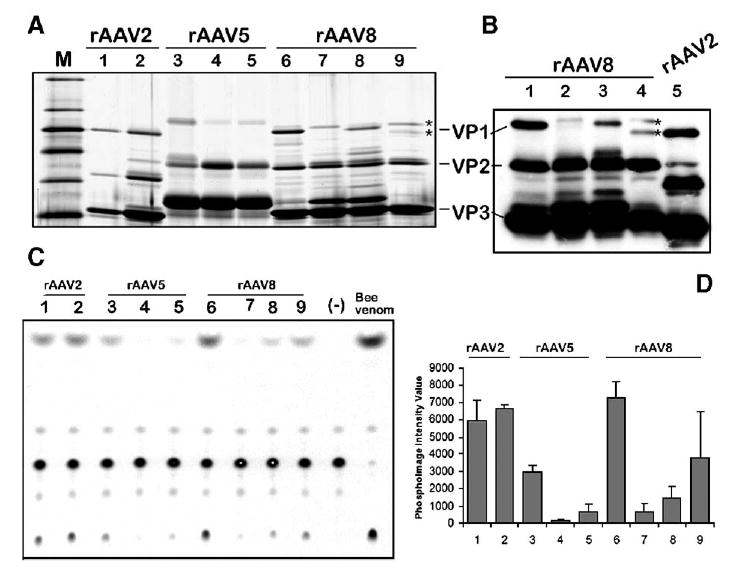
Analysis of the capsid protein VP1 content and the respective VP1up phospholipase A2 activity in rAAV vector stocks produced in 293 cells vs Sf9 cells. (A) Silver stain of polyacrylamide gel analysis (10% PAAG) of purified rAAV stocks prepared in HEK 293 and Sf9 cells. Lane 1, rAAV2-GFP/HEK 293; lane 2, rAAV2-GFP/Sf9; lane 3, rAAV5-GFP/HEK 293; lane 4, rAAV5-GFP/Sf9; lane 5, rAAV2/5-GFP/Sf9; lane 6, rAAV8-GFP/HEK 293; lane 7, rAAV8-GFP/Sf9; lane 8, rAAV2/8-GFP/Sf9; lane 9, mAAV8/2-GFP/Sf9. (B) Western blotting analysis (4–14% gradient PAAG) of rAAV8-GFP vector preparations (samples in lanes 1–4 are the same as in (A), lanes 6–9). For comparison purpose, baculovirus-produced rAAV2-GFP was analyzed next to mosaic mAAV8/2-GFP (compare the mobility of the respective VP1s marked by asterisks in lanes 4 and 5. (C)– Thin-layer chromatography of phospholipase A2 activity of virus produced in 293 cells vs Sf9 cells. The same amounts of rAAV particles (approximately 1010 drp) as in (A) were analyzed by the assay as described under Materials and Methods. Samples in lanes 1–9 correspond to the samples in lanes 1–9 in (A). Lane marked Bee venom contains positive control sample—1 ng of a bee venom phospholipase (Sigma). (D) Average values from two independent phospholipase assays quantified using phosphoimaging analysis. Samples are the same as in (A) and (C). The lower phospholipase activity of rAAV2/293 vs rAAV2/Sf9 reflects the inadvertently smaller amount of particles added to the electrophoresis well and the assay reaction (see A).
Girod et al. [10] have shown that the N terminus of the AAV VP1 capsid protein contains a phospholipase A2 (PLA2) motif that is critical for efficient viral infection. Mutations in this VP1 unique region had no influence on capsid assembly, packaging of viral genomes, or binding to and entry into cells. However, this PLA2 activity is required for endosome exit and viral genome transfer into the nucleus [11]. Therefore, it appeared from our data that the BacVP-AAV5 and BacVP-AAV8 helpers did not provide sufficient VP1 for a fully infectious viral particle. To test whether the shortage of VP1 and, ultimately, low PLA2 activity of the pseudotyped capsids are responsible for the observed infectious titers of these serotypes produced in Sf9 cells, we conducted in vitro phospholipase assays using purified vector preparations (Figs. 7C and 7D). Indeed, while AAV2 prepared in both 293 and Sf9 cells displayed comparable PLA2 activity that correlated with their respective particle-to-infectivity ratios, both AAV5-GFP and AAV8-GFP had significantly lower PLA2 activity when produced in SF9 cells.
PLA2 Domain Swapping
To produce AAV2 in insect cells, Urabe et al. [7] have inventively modified the N terminus of the VP1 ORF. The introduced mutations allow for the proper stoichiometry of the capsid proteins and for the assembly of infectious vector indistinguishable from 293-derived virus. On the other hand, our initial attempt to emulate this approach for the production of pseudotyped AAV5 and AAV8 vectors by introducing similar mutations resulted in assembly of noninfectious viral particles. We, therefore, hypothesized that swapping the portion of the capsid ORF encoding the AAV2 PLA2 domain for the homologous sequence in BacVP-AAV8 might improve the capsid protein stoichiometry in the resultant particles. To this end, we have substituted 134 N-terminal amino acid residues of AAV2 VP1 for the respective domain in AAV8 VP1 (Fig. 3B) using a PCR-mediated protocol. Upon sequence verification, the chimeric BacVP-AAV2/8 helper vector was constructed and a viral stock propagated. The particle titers of rAAV2/8-GFP prepared using this chimeric helper were similar to those of rAAV2, 5, and 8 serotypes produced in Sf9 cells. After purification using the iodixanol/Q-Sepharose protocol, we analyzed the capsid composition by SDS-protein gel electrophoresis (Fig. 7A, lane 8). As anticipated, the amount of AAV2/8 VP1 present within the particle was increased, although the level of this chimeric VP1 was not equivalent to that of AAV8 VP2. Yet, the PLA2 assay confirmed that this partial recovery was sufficient to increase the particles’ phospholipase activity, supporting the original hypothesis (Figs. 7C and 7D).
Production of Pseudotyped rAAV Mosaics
We have investigated an alternative approach to producing infectious pseudotyped AAV5 and AAV8 in which the PLA2 activity is supplemented by the entire AAV2 VP1 capsid protein coexpressed within the same cell. In this scheme, an additional baculovirus vector encoding just AAV2 VP1 is required (BacVP-AAV2-107; Fig. 3C). To control the stoichiometry of the supplementing VP1 driven by the strong baculovirus polyhedron promoter, we utilized a novel gene expression control element—a self-cleaving hammerhead ribozyme (HH Rz) regulated by a small compound, toyocamycin, a nucleoside analog [12]. In the absence of the inducer (toyocamycin-OFF), the cis-acting HH Rz efficiently cleaves the 5′ noncoding sequence of the mRNA, rendering it susceptible to subsequent degradation. In the toyocamycin-ON mode, the latter blocks the degradation by specifically binding to the HH Rz element, enabling the expression of the respective gene (AAV2 VP1). To produce an infectious rAAV8 (or rAAV5), the standard set of baculovirus vectors was supplemented with BacVP-AAV2-107 (m.o.i. of 5) and the resulting rAAV vector constituted mosaics of AAV8 VP1, VP2, and VP3 and AAV2 VP1 (dubbed mAAV8/2-GFP).
This approach has been validated by producing both rAAV5-GFP and rAAV8-GFP (Fig. 7A, lanes 5 and 9). The detailed polyacrylamide gel analysis (PAAG) of the rAAV8-GFP revealed that the vector indeed consisted of the VP1 mosaics derived from AAV8 and AAV2 (Fig. 7B, lanes 1–5, marked by asterisks). The PLA2 in vitro assay confirmed an increased phospholipase activity in the mosaic vector preparation. Curiously, the phospholipase of the AAV2 VP1 mosaic vector had a higher activity compared to the AAV2/8 VP1 chimeric vector, considering the respective amounts of the VP1 proteins.
Transduction of Murine Tissues in Vivo
To test transduction efficiencies of the baculovirus-derived rAAV vectors, we injected 1012 particles of rAAV-GFP preparations into the tail vein of adult mice, using rAAV8-GFP produced in 293 cells as a positive control. Three weeks postinjection, we euthanized the animals, harvested tissues, and visually estimated transduction by the intensity of direct GFP fluorescence. All the analyzed tissues, including liver, cardiac muscle, pancreas, spleen, and lung, were robustly transduced with rAAV8-GFP prepared in 293 cells (for the purpose of clarity, in Fig. 8A only transduction of liver is shown). In contrast, rAAV8-GFP derived from Sf9 cells was essentially noninfectious (Fig. 8B). At the same time, fusion of rAAV2/8-GFP (also Sf9 cell derived) demonstrated increased transduction but not quite as high as rAAV8-GFP/293 (Fig. 8C). Somewhat unexpectedly, the baculovirus-produced mAAV8/2-GFP demonstrated enhanced transduction properties compared to the 293-derived rAAV8-GFP (Figs. 8D, 8E, and 8F).
FIG. 8.
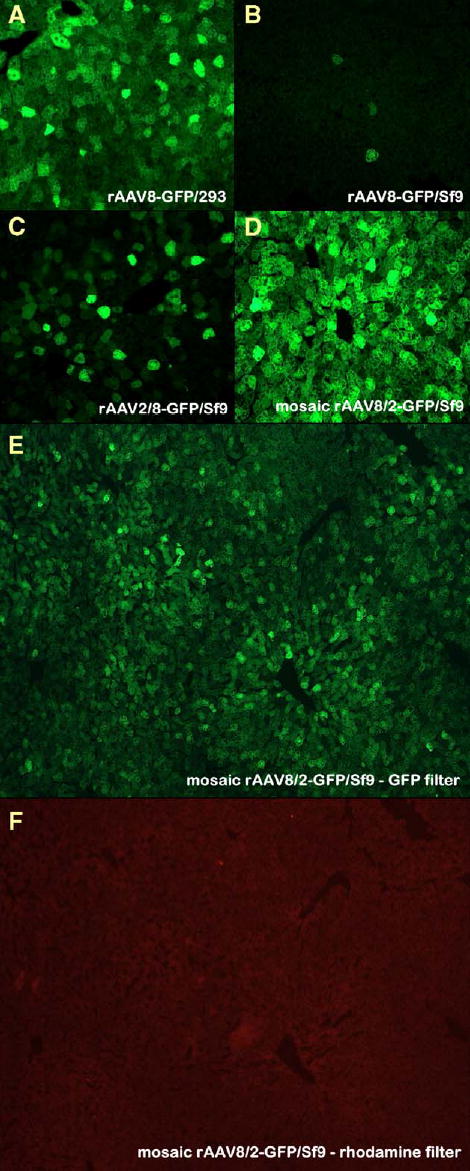
Transduction of murine livers in vivo with rAAV8 or rAAV2/8. Mice were injected with 1012 drp rAAV8-GFP. (A) HEK 293 cell-derived rAAV8-GFP; (B) Sf9 cell-derived rAAV8-GFP; (C) Sf9 cell-produced chimeric rAAV2/8; (D) Sf9 cell-derived mosaic mAAV8/2. (E and F) Specificity of the GFP fluorescence was confirmed by the absence of fluorescence in the same field with a rhodamine filter.
For the mosaic mAAV5/2-GFP, the in vivo transduction efficiencies of the baculo-derived and 293-cell-derived vector were comparable (data not shown). Therefore, both chimeric and mosaic baculovirus-produced vectors were highly infectious in vivo.
Discussion
Difficulties in scaling up rAAV production hinder the advancement of clinical protocols for gene therapy. Therefore, every improvement in production methods, especially related to up-scaling, is welcome in the field. A recently developed baculovirus-based production protocol [7], although potentially promising and elegant, in our hands produced only marginal titers. In addition, rAAV serotype 5 and 8 vectors designed in our laboratory and packaged using baculovirus system were noninfectious. We, therefore, set out to investigate the cause of the baculovirus system variability by testing the stability of each individual component of the system.
Stability of Helper Components
Baculovirus vectors derived via the Bac-to-Bac system are known to display passage-dependent instability [13]. Upon replaquing the original BacRep stock, we discovered that only 6 of 10 individual plaque isolates expressed both Rep52 and Rep78, which was indicative of the inherent instability of the Rep helper construct. By splitting the palindromic orientation of the rep genes and designing two separate helpers expressing Rep52 and Rep78, we have increased the passaging stability of the vector to P5.
Two other components of the original helper set also appeared to be unstable during continuous passaging, with the rAAV-ITR vector displaying a declining quality as early as at P3. When propagating ITR-containing rAAV vector plasmids in Escherichia coli, investigators utilize recombination pathway-deficient bacterial strains, such as SURE, to maintain the integrity of inverted terminal palindromic structures. To our knowledge, no such equivalent exists among insect cell lines. The stability of the ITR-containing helper after P2, therefore, appears to be a limiting factor for scaling up the system. However, even with such limitation, the total yield of P2 baculovirus vectors is sufficient to infect up to 300 L of Sf9 cells in suspension culture with an m.o.i. of 5 to produce rAAV. The agility of the system was recently supported by Meghrous et al., who demonstrated the scaling of rAAV2 baculovirus-based production up to 20 L in a bioreactor yielding 7.1 × 1015 rAAV particles [14].
Using the Baculovirus System for rAAV Pseudotyping
The utility of the production system depends largely on the flexibility of its components to package (pseudotype) a particular rAAV cassette into other AAV serotype capsids. Vectors of other serotypes can achieve a higher transduction of a targeted tissue resulting in a reduced therapeutic vector dose. Initially, we were unsuccessful in designing BacVP-AAV5 and BacVP-AAV8 helper vectors by emulating the BacVP-AAV2 capsid helper and placing the noncanonical ACG start codon within the short sequence context of a 9-nucleotide element derived from upstream of the VP2 ACG codon [15]. Apparently, this short sequence is not adequate for the efficient initiation of translation as both rAAV serotypes 5 and 8 (Fig. 7A) contained very little VP1. To alleviate the deficiency, we have redesigned the vector by swapping the respective VP1up domains between AAV2 and AAV8 helpers. The resulting chimeric rAAV2/8 partially reconstituted the levels of VP1 protein and, as a result, increased PLA2 activity in vitro and infectivity in vivo. This approach of swapping VP1up domains was independently substantiated by Urabe et al. [15], who described the rescue of infectivity of insect cell-derived rAAV type 5 vector by swapping with larger portions of the AAV2 VP1 N terminus.
A new approach dubbed “transcapsidation” or “cross-dressing” has been recently developed to mix and match capsid proteins from different serotypes within one virion [5,6]. These virions consist of a mosaic of proteins derived from two different AAV serotypes and exhibit receptor binding characteristics of the parental viruses [6]. We chose to exploit this approach for the purpose of substituting the entire AAV2 VP1 to compensate for the defective PLA2 component in the baculovirus system-derived rAAV8 (or rAAV5). This could have been achieved by adding another helper BacVP encoding all three AAV2 capsid proteins. The resulting mosaic vector would have consisted of the about equal ratios of AAV2 and AAV8 (or AAV5) VP1, VP2, and VP3 capsid proteins. However, we wanted to preserve the intrinsic tropism of the rAAV serotypes without introducing the heparin affinity of AAV2. This was achieved by producing a mosaic of AAV8 (or AAV5) VP1, VP2, and VP3 with just the AAV2 VP1, the latter encoded by a newly designed baculovirus vector BacVP-AAV2-107 (Fig. 3C).
The new helper vector design was driven by the requirement to maintain proper capsid protein stoichiometry in the infectious particle. The initiation codon of the VP1 ORF was reverted back to canonical ATG, while the silent mutations eliminating the splice acceptor site [15] were retained. To control the level of VP1 synthesis driven by the strong baculovirus polyhedron promoter, we chose a novel portable riboswitch element—a toyocamycin-regulated HH Rz [12]. In the absence of the inducer, there was no VP1 synthesized and the resulting rAAV8 vector was not infectious (data not shown). Blocking the mRNA degradation by toyocamycin resulted in the synthesis of AAV2 VP1 (clearly resolved by PAAG as AAV2 and AAV8 VP1 orthologs have distinct mobilities, as shown in Fig. 7B).
Earlier, Rabinowitz et al. have shown that mosaic vectors at parent capsid ratios of 19:1 displayed the transduction properties of the prevailing parent [6]. There is no evidence of either AAV2 VP2 or AAV2 VP3 being integrated into mosaics (Fig. 7B), indicating that the vectors we have designed incorporated AAV2 VP1 only (about 2–3 molecules per capsid). Such mosaic vectors, therefore, are expected to display the tropism similar to vectors serotypes produced in 293 cells.
The formation of the rAAV2/rAAV8 mosaic appeared to be more efficient as opposed to rAAV2/rAAV5. This could be explained by degrees of homology of the respective serotypes: AAV2 and AAV8 belong to the same capsid interaction class A, while AAV2 and AAV5—to the less efficient interaction class B [6]. Therefore, to produce an infectious vector of a less related serotype such as AAV5, the PLA2 domain swap might be the more preferable approach.
In in vivo experiments, the redesigned chimeric rAAV2/8-GFP appeared to be targeted mainly to the liver, unlike the mammalian cell-derived rAAV8-GFP, which transduced indiscriminately all the tissues tested. Currently, we cannot identify whether this hepatocyte-specific transduction resulted from the overall reduced VP1 PLA2 activity or from the change in vector tropism. This possibility will be the subject of further experiments. Unexpectedly, the mosaic mAAB8/2-GFP appeared to have somewhat stronger in vivo transduction efficiency compared to the 293-derived vector. These synergistic effects of the cross-dressed virions were documented earlier for rAAV1 and rAAV2 (or rAAV3) mosaics [6]. It is conceivable that incorporation of the AAV2 VP1 ortholog into the AAV8 capsid modulated the overall topology of the pore-forming pentamer structure [16], promoting the unfolding and passage of the VP1up PLA2 domain outside, thereby enhancing the escape of the vector from the endosome. Alternatively, the mosaic particles had greater uncoating efficiency due to less stable interfacial contacts resulting from the incorporation of the AAV2 VP1. Experiments addressing these mechanisms are under way.
The experiments described herein further improve and extend the application of baculovirus for the production of AAV vectors. These results demonstrate that the VP1up domains of the AAV viruses are completely modular and could be replaced with homologous domains from other parvoviral capsids. It is conceivable that such interchangeable PLA modules could be utilized as universal building blocks for a novel, highly efficacious vector platform combining serotypes’ tropism diversity with superior transduction rates. The novel helper vector described here allows for more flexibility in selecting strategies for the production of infectious pseudotyped mosaic vectors. Subsequent optimization of the toyocamycin concentrations and baculovirus helper m.o.i. may increase both the AAV2 VP1 content and the infectivity of the mosaic vectors. The redesigned baculovirus system presented here will enhance our capacity for rAAV production, making the AAV platform more amenable to large-scale clinical manufacturing.
Materials and Methods
Construction and production of recombinant baculovirus
Recombinant plasmids were constructed using standard molecular biology techniques. Gene fusions were performed using an overlap PCR protocol using Pfu Turbo polymerase (Stratagene, La Jolla, CA, USA). The hammerhead ribozyme N107 [12] was reconstructed using Klenow polymerase-mediated extension of partially overlapping synthetic oligonucleotides. Recombinant baculoviruses were constructed using the Bac-to-Bac system (Gibco BRL). DH10Bac competent cells containing the baculovirus genome were transformed with the pFastBac transfer plasmids containing the AAV component insert. Bacmid DNA purified from recombination-positive white colonies was transfected into Sf9 cells using TransIT Insecta reagent (Mirus). Three days posttransfection, medium containing baculovirus (pooled viral stock) was harvested and a plaque assay was conducted to prepare independent plaque isolates. Routinely, eight individual plaques were propagated to P1 to assay for the expression of the transgene or the ability of the transgene cassette to rescue and replicate as rAAV genome. Selected clones were propagated to P2, titered, and used for large-scale rAAV preparations (described below). Baculovirus titers were determined by plaque assay following the Bac-to-Bac system manual. Serial passaging was conducted as described by Kool et al. [8] using an m.o.i. of 0.1.
Production of rAAV vectors
Serum-free medium-adapted Sf9 cells were used for large-scale rAAV preparations. Sf9 cells at densities of 2 × 106 cells/ml were co-infected with BacRep, BacVP, and BacGFP at an m.o.i. of 5 each (unless indicated otherwise). Alternatively, cells were co-infected with BacRep52 and BacRep78 (m.o.i. of 5 each) to replace the BacRep virus. The mosaic vectors were produced by adding BacVP-AAV2-107 to the standard co-infection mixture (m.o.i. of 5) and toyocamycin (Berry & Associates, Inc., Ann Arbor, MI, USA) at 0.5 μM. Sixty hours postinfection, cells were harvested and processed as described earlier [15]. Vectors were purified by iodixanol gradient centrifugation and column chromatography as described [17,18]. Virus was then concentrated and the buffer was exchanged in three cycles to lactated Ringer’s using centrifugal spin concentrators (Apollo, 150-kDa cut-off, 20-ml capacity) (CLP). Physical and infectious rAAV particle titers were determined as described by Potter et al. [19].
Western blot analysis
Sf9 cells (3 × 106) were seeded in 6-cm dishes. Three days postinfection, cells were harvested and lysed in 100 μl of buffer containing 50 mM Tris, pH 7.6, 120 mM NaCl, 1% Nonidet P-40, 10% glycerol, 2 mM Na3VO4, 1 mM PMSF, 10 mM NaP2O7, 40 μg/ml leupeptin, 5 μg/ml aprotinin, 100 μM NaF, 1 mM EDTA, 1 mM EGTA, 1 μg/ml pepstatin. After incubation on ice for 1 h, cell lysates were centrifuged at 12,000 rpm for 10 min. Clarified samples were separated by using SDS/10% polyacrylamide gel electrophoresis, transferred to a PVDF membrane, and probed with the anti-AAV2 capsid monoclonal antibody B1 (1:2000; American Research Products), which also recognizes the AAV1 and AAV5 capsid proteins [20] as well as the AAV8 capsid, or with anti-Rep monoclonal antibodies (clone 1F11.8, 1:2000 dilution), depending on the context of the experiment. Detection was carried out using horse-radish peroxidase-conjugated sheep anti-mouse (Amersham Biosciences) at 1:5000 and the ECL Western Detection kit.
Phospholipase assay
The PLA mixed-micelles assay was conducted as described previously [11]. Specifically, 1010 purified DNase I-resistant rAAV particles were pretreated for 2 min at 70°C in 40 mM Tris, pH 8.0, in a final volume of 17 μl. The assay was carried out in a total reaction volume of 50 μl containing the heat-treated virus in 100 mM Tris-HCl, pH 8.0, 10 mM CaCl2, 100 mM NaCl, 1 mM Triton X-100, 40 μM phosphatidylcholine, with 0.0625 μCi [14C]phosphatidylcholine. The reactions were incubated at 37°C for 30 min. The products were extracted with chloroform:methanol:4 M KCl (2:1:1). After centrifugation the products were separated by silica gel thin-layer chromatography with chloroform:methanol:water (65:35:4). The products were quantified by phosphoimaging analysis.
In vivo experiments
Animals were cared for in accordance with the principles of the Guide to the Care and Use of Experimental Animals. Vector (1012 drp of rAAV8-GFP prepared in 293 or Sf9 cells or rAAV2/8-GFP from Sf9 cells) was injected into the tail veins of C57BL/6 mice. Two weeks postinjection, mice were euthanized and tissues harvested for GFP visualization by fluorescence microscopy. Following fixation in 10% neutral-buffered formalin overnight, samples were incubated in 30% sucrose in PBS (pH 7.4) for 24 h and then embedded in OCT medium (Fisher). Cryosections (4 μm) were dried overnight at room temperature and then washed with TBS buffer followed by incubation with rabbit anti-GFP-Alexa Fluor 488 conjugate (Molecular Probes, Eugene, OR, USA) at 1:100 dilution for 1 h at room temperature. Sections were washed in buffer and mounted (Vectashield with DAPI, Vector Laboratories, Burlingame, CA, USA). Slides were viewed on a Zeiss Axioskop with a GFP filter (Chroma, 41028) and representative digital images taken from each animal at the same exposure settings using an Axiocam microscope. Autofluorescence was evaluated in the same field with a rhodamine filter (Zeiss Filter set 14, 510-560/590) and was negligible (shown in Fig. 8F).
Acknowledgments
This study was supported in part by National Institutes of Health Grants NIDDK-R01 DK62302, NHLBI-P50-HL59412, P01-DK58327, and JDFI Center Grant. S.Z., N.M., B.J.B., and R.O.S. are inventors on patents related to recombinant AAV technology and own equity in a gene therapy company (AGTC) that is commercializing AAV for gene therapy applications.
References
- 1.Gao G, et al. Clades of adeno-associated viruses are widely disseminated in human tissues. J Virol. 2004;78:6381 – 6388. doi: 10.1128/JVI.78.12.6381-6388.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gao G, et al. Adeno-associated viruses undergo substantial evolution in primates during natural infections. Proc Natl Acad Sci USA. 2003;100:6081 – 6086. doi: 10.1073/pnas.0937739100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hildinger M, et al. Hybrid vectors based on adeno-associated virus serotypes 2 and 5 for muscle-directed gene transfer. J Virol. 2001;75:6199 – 6203. doi: 10.1128/JVI.75.13.6199-6203.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rabinowitz JE, et al. Cross-packaging of a single adeno-associated virus (AAV) type 2 vector genome into multiple AAV serotypes enables transduction with broad specificity. J Virol. 2002;76:791 – 801. doi: 10.1128/JVI.76.2.791-801.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hauck B, Chen L, Xiao W. Generation and characterization of chimeric recombinant AAV vectors. Mol Ther. 2003;7:419 – 425. doi: 10.1016/s1525-0016(03)00012-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rabinowitz JE, et al. Cross-dressing the virion: the transcapsidation of adeno-associated virus serotypes functionally defines subgroups. J Virol. 2004;78:4421 – 4432. doi: 10.1128/JVI.78.9.4421-4432.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Urabe M, Ding C, Kotin RM. Insect cells as a factory to produce adeno-associated virus type 2 vectors. Hum Gene Ther. 2002;13:1935 – 1943. doi: 10.1089/10430340260355347. [DOI] [PubMed] [Google Scholar]
- 8.Kool M, van den Berg PM, Tramper J, Goldbach RW, Vlak JM. Location of two putative origins of DNA replication of Autographa californica nuclear polyhedrosis virus. Virology. 1993;192:94 – 101. doi: 10.1006/viro.1993.1011. [DOI] [PubMed] [Google Scholar]
- 9.Xiao X, Li J, Samulski RJ. Production of high-titer recombinant adeno-associated virus vectors in the absence of helper adenovirus. J Virol. 1998;72:2224 – 2232. doi: 10.1128/jvi.72.3.2224-2232.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Girod A, et al. The VP1 capsid protein of adeno-associated virus type 2 is carrying a phospholipase A2 domain required for virus infectivity. J Gen Virol. 2002;83:973 – 978. doi: 10.1099/0022-1317-83-5-973. [DOI] [PubMed] [Google Scholar]
- 11.Zadori Z, et al. A viral phospholipase A2 is required for parvovirus infectivity. Dev Cell. 2001;1:291 – 302. doi: 10.1016/s1534-5807(01)00031-4. [DOI] [PubMed] [Google Scholar]
- 12.Yen L, et al. Exogenous control of mammalian gene expression through modulation of RNA self-cleavage. Nature. 2004;431:471 – 476. doi: 10.1038/nature02844. [DOI] [PubMed] [Google Scholar]
- 13.Pijlman GP, van Schijndel JE, Vlak JM. Spontaneous excision of BAC vector sequences from bacmid-derived baculovirus expression vectors upon passage in insect cells. J Gen Virol. 2003;84:2669 – 2678. doi: 10.1099/vir.0.19438-0. [DOI] [PubMed] [Google Scholar]
- 14.Meghrous J, et al. Production of recombinant adeno-associated viral vectors using a baculovirus/insect cell suspension culture system: from shake flasks to a 20-L bioreactor. Biotechnol Prog. 2005;21:154 – 160. doi: 10.1021/bp049802e. [DOI] [PubMed] [Google Scholar]
- 15.Urabe M, Nakakura T, Ozawa K, Kotin RM. Production of recombinant adeno-associated virus type 5 in insect cells. Mol Ther. 2004;9:S160. doi: 10.1128/JVI.80.4.1874-1885.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kronenberg S, Bottcher B, von der Lieth CW, Bleker S, Kleinschmidt JA. A conformational change in the adeno-associated virus type 2 capsid leads to the exposure of hidden VP1 N termini. J Virol. 2005;79:5296 – 5303. doi: 10.1128/JVI.79.9.5296-5303.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zolotukhin S, et al. Production and purification of serotype 1, 2, and 5 recombinant adeno-associated viral vectors. Methods. 2002;28:158 – 167. doi: 10.1016/s1046-2023(02)00220-7. [DOI] [PubMed] [Google Scholar]
- 18.Zolotukhin S, et al. Recombinant adeno-associated virus purification using novel methods improves infectious titer and yield. Gene Ther. 1999;6:973 – 985. doi: 10.1038/sj.gt.3300938. [DOI] [PubMed] [Google Scholar]
- 19.Potter M, Chesnut K, Muzyczka N, Flotte T, Zolotukhin S. Streamlined large-scale production of recombinant adeno-associated virus (rAAV) vectors. Methods Enzymol. 2002;346:413 – 430. doi: 10.1016/s0076-6879(02)46069-7. [DOI] [PubMed] [Google Scholar]
- 20.Wobus CE, et al. Monoclonal antibodies against the adeno-associated virus type 2 (AAV-2) capsid: epitope mapping and identification of capsid domains involved in AAV-2-cell interaction and neutralization of AAV-2 infection. J Virol. 2000;74:9281 – 9293. doi: 10.1128/jvi.74.19.9281-9293.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McCarty DM, Christensen M, Muzyczka N. Sequences required for coordinate induction of adeno-associated virus p19 and p40 promoters by Rep protein. J Virol. 1991;65:2936 – 2945. doi: 10.1128/jvi.65.6.2936-2945.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]