

Abstract
The G-protein α subunit, α13, regulates cell growth and differentiation through the monomeric Rho GTPase. α13 activates Rho through direct stimulation of the guanine nucleotide exchange factor p115RhoGEF, which contains a regulator of G-protein signaling homology domain (RH) in its N-terminus. Through its RH domain p115RhoGEF also functions as a GAP for Gα13. The mechanism for the Gα13/p115RhoGEF interaction is not well understood. Here, we determined specific α13 residues important for its interaction with p115RhoGEF. GST-pull downs and co-immunoprecipitation assays revealed that individually mutating α13 residues Lys204, Glu229, or Arg232 to opposite charge residues disrupts the interaction of activated α13 with the RH domain of p115RhoGEF or full-length p115RhoGEF. We further demonstrate that mutation of Glu229, and to a lesser extent Lys204 or Arg232, disrupts the ability of activated α13 to induce the recruitment of p115RhoGEF to the plasma membrane (PM) and to activate Rho-mediated SRE-luciferase gene transcription. Interestingly, an α13 mutant where a conserved Gly was mutated to a Ser (G205S) retained its ability to bind to p115RhoGEF, induce p115RhoGEF recruitment to the PM, and activate Rho-dependent signaling, even though identical Gly to Ser mutations in other α disrupt their interaction with RGS proteins. These results demonstrate that whereas several features of a typical α/RGS interaction are preserved in the α13/p115RhoGEF interaction, there are also significant differences.
Keywords: signal transduction, heterotrimeric G protein, Rho GTPase, RGS protein, guanine-nucleotide exchange factor
INTRODUCTION
Heterotrimeric G-proteins1 link G-protein-coupled receptors to numerous intracellular signaling pathways. The α12 family of G protein α subunits function as regulators of cell growth and differentiation. Several studies have shown that both α12 and α13 can act as very powerful transforming agents (Jiang et al., 1993; Voyno-Yasenetskaya et al., 1994b; Xu et al., 1993). In addition, Drosophila embryos carrying mutations in the concertina (cta) gene, the Drosophila homologue for α12/13, fail to undergo proper gastrulation, and lack of α13 in mice results in embryonal death (day 9.5) due to severely impaired angiogenesis (Offermanns, 2000; Parks & Wieschaus, 1991).
In the past years, the members of the α12 family have been shown to function as regulators of a variety of cellular signaling pathways. α12 and α13 can activate the Na+/H+ exchanger (Voyno-Yasenetskaya et al., 1994a), the c-Jun NH2-terminal kinase (Prasad et al., 1995), extracellular-signal–regulated kinases (ERK) (Voyno-Yasenetskaya et al., 1996), tyrosine kinases (Mao et al., 1998a; Shi et al., 2000), serum response element-mediated gene transcription (Mao et al., 1998b), stress fiber and focal adhesion formation (Buhl et al., 1995; Gohla et al., 1999), neurite retraction (Katoh et al., 1998; Kranenburg et al., 1999), and apoptosis (Althoefer et al., 1997; Berestetskaya et al., 1998). Several of these pathways are mediated by the monomeric Rho GTPase, a member of the Ras superfamily of GTPases. α13 activates Rho through direct stimulation of the guanine nucleotide exchange factor p115RhoGEF (Hart et al., 1998; Kozasa et al., 1998). Like all G-proteins, Rho cycles between the inactive GDP bound and active GTP bound form. RhoGEFs contain a Dbl-homology (DH) domain, which activates Rho by catalyzing the exchage of GDP for GTP. p115RhoGEF, together with leukemia associated RhoGEF (LARG) and PDZ-RhoGEF, comprise a subfamily of GEFs that contain a regulator of G-protein signaling homology (RH) domain. There are over 30 known regulator of G-protein signaling (RGS) proteins, almost all of which share the ability to switch off heterotrimeric G-proteins by functioning as GTPase activating proteins (GAP) for G protein α subunits (Hollinger & Hepler, 2002). The RH domain of p115RhoGEF serves as a binding site for α12/13 and acts as a GAP specifically for the α12 family of proteins. However, it is only α13 that can activate p115RhoGEF’s exchange activity on Rho. Thus, p115RhoGEF carries a dual function in its interaction with α13 through its ability to catalyze the deactivation of α13 by accelerating α13-GTP hydrolysis and also to mediate α13 induced Rho activation.
The mechanism through which α13 is regulated by and activates p115RhoGEF is not clearly defined. Mutagenic analysis of p115RhoGEF has revealed a complex model where several regulatory mechanisms seem to be involved (Bhattacharyya & Wedegaertner, 2000; Bhattacharyya & Wedegaertner, 2003a; Bhattacharyya & Wedegaertner, 2003b; Chen et al., 2003; Wells et al., 2002a; Wells et al., 2001; Wells et al., 2002b). Though the RH domain of p115RhoGEF shares low sequence identity with known RGS proteins, the crystal structure of the RH domain of p115RhoGEF revealed that the core of this domain shared significant structural similarity with the corresponding regions of RGS4 and RGS9 (Chen et al., 2001). The crystal structure of the RGS4/αi1 complex has been solved and has revealed residues in the Switch I and Switch II segments of αi1, which are known to undergo conformational changes upon GTP hydrolysis, to be the contact sites for RGS proteins (Tesmer et al., 1997). The similarities between p115RH and other RGS proteins suggest that elements of the α/RGS interaction may be preserved in the interaction of α13 with p115RhoGEF. Therefore, based on the crystal structure of the RGS4/αi1 complex, studies on other α/RGS pairs, and sequence alignments of αi1 with α13, we targeted four specific residues of α13 for mutagenesis. Analysis of these α13 mutants revealed that mutating Glu229 to Lys in α13 completely disrupted the ability of α13 to interact with p115RhoGEF, induce PM recruitment of p115RhoGEF, and stimulate Rho-dependent signaling. We also demonstrate that whereas mutating Lys204 to Glu and Arg232 to Glu disrupted the α13/p115RhoGEF interaction, as revealed by our GST-p115RH pulldown and co-immunoprecipitation assays, these mutations did not fully disrupt α13 induced PM translocation of p115RhoGEF and Rho-dependent signaling. Thus, the α13/p115RhoGEF interaction is generally similar to a typical α/RGS interaction. However, in contrast to other α/RGS interactions, the substitution of a conserved glycine for a serine (G205S), termed an RGS-insensitive mutation in other α subunits (DiBello et al., 1998), had no effect on the ability of α13 to interact with and activate p115RhoGEF. In addition, we present the surprising result that the double mutations K204E/E229K and E229K/R232E partially rescue function of α13.
RESULTS
Identification of α13 residues that are critical for the interaction with p115RhoGEF
The structural similarity between the core domain of p115RH and RGS4 suggests that the α13/p115RhoGEF interaction might mimic the αi1/RGS4 interaction. The crystal structure of the αi1/RGS4 complex identified αi1 residues Thr182, Glu207, and Lys210 as important contact sites for RGS4, and Glu207 also appears to play a critical role in orienting the side chains of Thr182 and Lys210 (Tesmer et al., 1997). These residues correspond to α13 Lys204, Glu229, and Arg232, respectively, as revealed by sequence alignments of the two proteins (Fig. 1). To determine residues in Gα13 that define its interaction with p115RhoGEF, we mutated the above α13 residues to an opposite charge (K204E, E229K, and R232E) and also to alanines (K204A, E229A, and R232A). In addition, a conserved glycine at position 205 was substituted for a serine (G205S) since this mutation has been shown to disrupt the interaction of Gpa1 (yeast Gα), αq, αi, and αo, with RGS proteins (DiBello et al., 1998; Lan et al., 1998).
Fig. 1. Alignment of α13 with αi1.
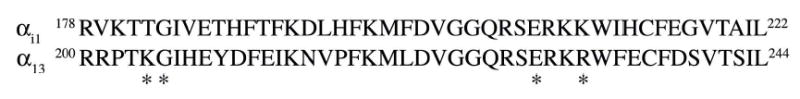
The amino acids 179-222 of αi1 and 200-244 of α13 are shown. α13 residues that were mutated in this study have been marked by asterisks.
HEK293 cell lysates expressing HA-tagged α13wt and mutants were incubated with purified p115RH fused to GST (GST-p115RH) on glutathione agarose beads, in the reversibly activate subunits by binding presence or absence of AlF−4. AlF−4 is known to α to α-GDP and inducing a conformation similar to that of the transition state for GTP-hydrolysis (Coleman et al., 1994). The ability of α13wt and mutants to bind to GST-p115RH was determined by immunoblotting with an anti-HA antibody. In the presence of AlF−4, GST-p115RH was able to efficiently pull down α13wt (Fig. 2A). Interestingly, α13G205S was also strongly pulled down by GST-p115RH in the presence of AlF−4 (Fig. 2A, left panel). Thus, the G205S mutation, though it disrupts RGS interactions for several α subunits, had no effect on the ability of α13 to bind GST-p115RH, indicating differences between the α13/p115RH interaction and other α/RGS interactions. GST-p115RH very weakly pulled down AlF−4 activated α13K204E suggesting that this mutant is severely defective in its interaction with GST-p115RH. α13E229K was not detected in the pulldown in the presence of AlF−4, indicating that it is unable to interact with GST-p115RH. Also, the α13R232E mutant has a strongly reduced ability to interact with GST-p115RH as revealed by the very small amount of AlF−4 activated α13R232E pulled down by GST-p115RH (Fig. 2A, left panel). In addition, the double mutants α13K204E/E229K, α13K204E/R232E, α13E229K/R232E and a construct containing all three mutations (α13T) were also not pulled down by GST-p115RH (Fig. 2A, middle panel). To further understand the structural determinants for the α13/p115RH interaction, we also investigated the effects of mutating α13 residues Lys204, Glu229, and Arg232 to alanines. Interestingly, α13E229A and α13R232A were efficiently pulled down by GST-p115RH whereas α13K204A showed a very weak pulldown (Fig. 2A, right panel). Thus, the primary role for Glu229 and Arg232 seems to be charge complementarity whereas for Lys204 both charge and size seem to be important.
Fig. 2. Interaction of AlF−4 activated α13 mutants with GST-p115RH.
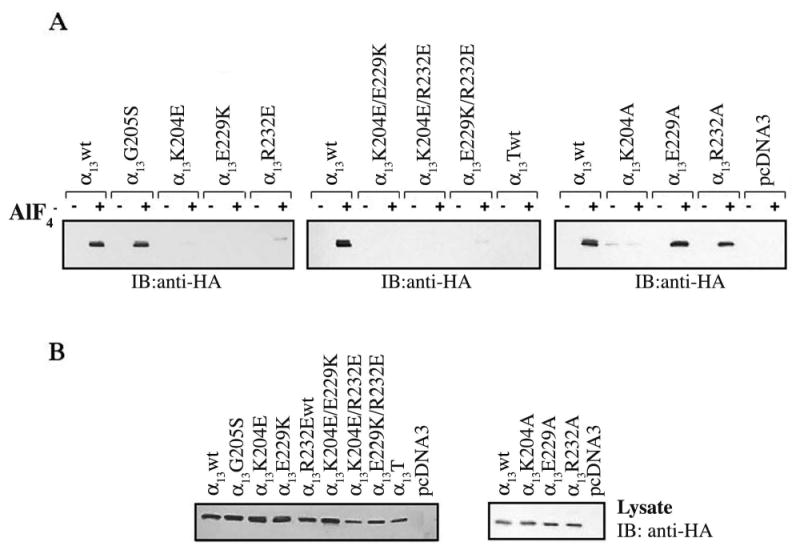
HEK293 cells were transfected with 2 μg of plasmids encoding for HA-α13wt or the indicated mutants and 1μg of pcDNA3. Cells were lysed 48 h after transfection, and cellular lysates were incubated with 10 μg of GST-p115RH immobilized on glutathione agarose beads in the presence (+) or absence (−) of AlF−4 as indicated. (A) Interaction of AlF−4 activated, α13 mutants with GST-p115RH fusion protein was visualized by immunoblotting with an anti-HA monoclonal antibody (12CA5). (B) Cellular lysates were immunoblotted with an anti-HA monoclonal antibody to compare expression levels for α13 mutants. Results shown are representative of at least three independent experiments.
All of the opposite charge mutants were also introduced in the constitutively activated form of α13, where a conserved glutamine (Q226) has been mutated to a leucine (α13QL) (Voyno-Yasenetskaya et al., 1994a). Constitutively active α13QL and α13G205S □□ were efficiently pulled down by GST-p115RH (Fig. 3A). α13K204E QL and α13R232E QL were weakly pulled down by GST-p115RH whereas α13E229K QL was not detected in the pulldown (Fig. 3A). Thus, the constitutively activated point mutants mimic the behavior of their AlF−4 activated forms in their interaction with GST-p115RH. Interestingly, even though α13E229K QL shows no interaction with GST-p115RH, the double mutants α13K204E/E229K QL and α13E229K/R232E QL were significantly pulled down by GST-p115RH. Thus, the charge reversal mutations K204E or R232E partially rescue the GST-p115RH binding defect of α13E229K QL, but not of AlF−4-activated α13E229K. α13K204E/R232E QL is not pulled down by GST-p115RH, consistent with its AlF−4 activated form. The triple mutant varied in its ability to interact with GST-p115RH, suggesting that the triple mutations may affect the stability of this construct. These results with double mutants (Fig. 3) are consistent with the possibility that Glu229, like Glu207 in αi1 (Tesmer et al., 1997), functions, at least partly to stabilize other key residues.
Fig. 3. Interaction of constitutively active α13 mutants with GST-p115RH.
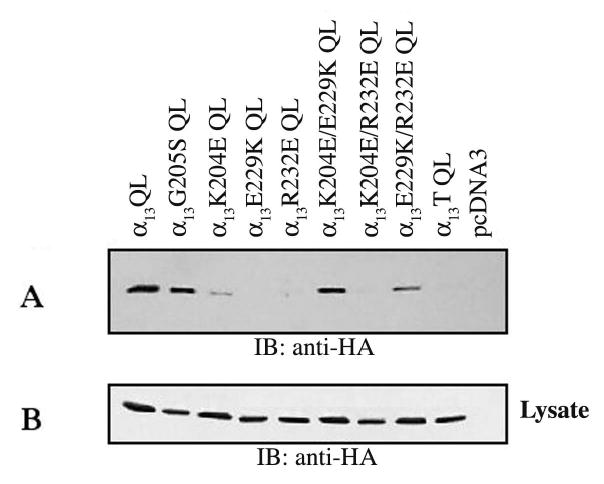
HEK293 cells were transfected with the indicated α13QL constructs (2μg) and pcDNA3 (1μg). GST-p115RH (10μg) immobilized on glutathione agarose beads was used to pulldown α13QL or mutants. (A) α13QL or mutant pulldowns were detected by immunoblotting with anti-HA antibody. (B) Equal expression of α13QL mutants was determined by immunoblotting of cellular lysates with an anti-HA antibody. Results shown are representative of at least three independent experiments.
α 13 mutants that are defective in binding to GST-p115RH are also defective in their interaction with full length p115RhoGEF
To determine whether our α13 mutations also prevented the interaction of α13 with full-length p115RhoGEF, co-immunoprecipitations were performed. HEK293 cells were co-transfected with HA-tagged α13wt and mutants, along with a Myc-tagged p115RhoGEF, and immunoprecipitations with an anti-HA antibody were carried out to determine the ability of α13 mutants to interact with p115RhoGEF (Fig. 4). p115RhoGEF was detected in the immunoprecipitation with AlF−4 activated α13wt and α13G205S, but not with AlF−4 activated α13K204E, α13E229K, and α13R232E (Fig. 4A, left panel). Thus, consistent with the GST-p115RH pulldown assays, the G205S mutation has no effect on the ability of α1□ to interact with p115RhoGEF, whereas K204E, E229K, and R232E mutations disrupt the interaction. Also, as observed with the GST-p115RH interaction assays, no p115RhoGEF was detected in the immunoprecipitations with the double and triple mutants (Fig. 4A, middle panel), indicating that they are not able to bind to p115RhoGEF in the presence of AlF−4. p115RhoGEF was not co-immunoprecipitated with AlF−4 activated α13K204A, consistent with the GST-p115RH interaction assays, (Fig. 4A, right panel), but p115RhoGEF was detected in the immunoprecipitations with AlF−4 activated α13E229A and α13R232A, though to reduced levels in comparison to α13wt (Fig. 4A, right panel).
Fig. 4. Interaction of α13 mutants with p115RhoGEF.
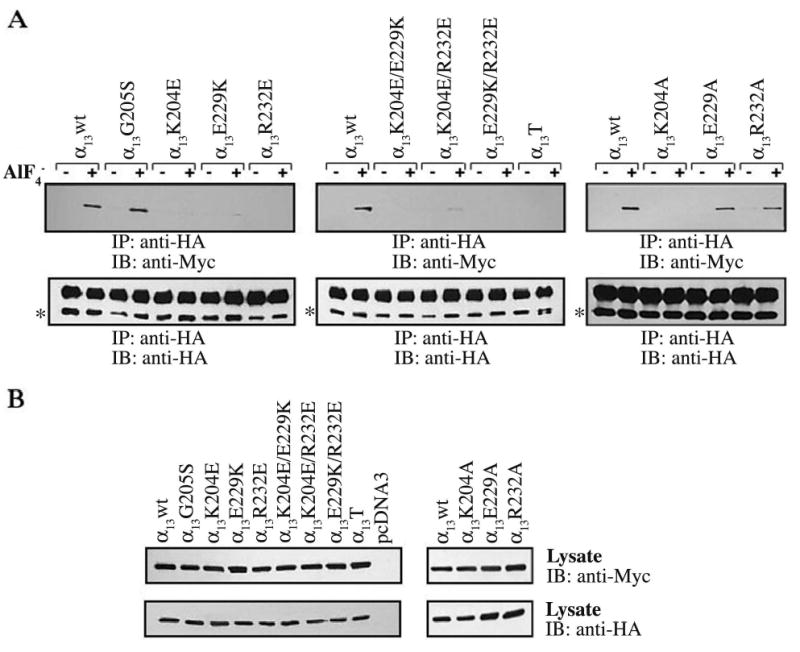
HEK293 cells were transfected with plasmids encoding for Myc epitope-tagged p115RhoGEF and the indicated HA epitope-tagged α13 constructs. 48 h after transfection cells were lysed and, as indicated, incubated in the presence (+) or absence (−) of AlF−4. α13 or its mutants were precipitated with an anti-HA monoclonal antibody. (A) Immunoprecipitated proteins were separated by SDS-PAGE and immunoblotted with a monoclonal antibody specific for the Myc-epitope tag to determine the ability to α13 mutants to bind p115RhoGEF (upper panel). Immunoprecipitated samples were also subjected to immunoblotting with anti-HA antibody to show equal amounts of immunoprecipitated HA-α13 (lower panel). In the lower panel, the asterisks indicate HA-α13 proteins, while the upper band corresponds to the antibody heavy chain. (B) Cell lysates were immunoblotted with an anti-Myc (upper panel) and anti-HA (lower panel) antibody to determine expression levels of Myc-p115RhoGEF and HA-α13 constructs, respectively. Results shown are representatives of at least three independent experiments.
Activated α 13E229K fails to promote PM translocation of p115RhoGEF
G-protein α subunits are found in tight association with the plasma membrane (Bhattacharyya & Wedegaertner, 2000; Neubig, 1994). p115RhoGEF is a cytoplasmic protein and it has been shown that α13QL or GPCR activated α13 induces the translocation of p115RhoGEF from the cytoplasm to the plasma membrane (Bhattacharyya & Wedegaertner, 2000). Upon GPCR activation, Rho has also been shown to translocate from the cytoplasm to the plasma membrane (Fleming et al., 1996; Kranenburg et al., 1997). Thus, α13 induced p115RhoGEF translocation to the plasma membrane may be an important step in Rho activation. To determine how disruption of the α13/p115RhoGEF interaction affects α13 induced recruitment of p115RhoGEF to the PM, HEK293 cells were co-transfected with the indicated constitutively active (QL) HA-tagged α13 constructs and a GFP-tagged full-length p115RhoGEF. All constitutively active point mutants and the double mutants α13K204E/E229K QL and α13E229K/R232E QL were found in association with the PM, as indicated by the sharp staining at the cell periphery and lack of a nuclear shadow (Fig. 5A–H, Anti-HA). The PM localization of these constructs suggests that the structural integrity of these mutants is intact. On the other hand, α13K204E/R232E QL was characterized by a diffuse staining of the cytoplasm and the presence of a strong nuclear shadow (data not shown) whereas the triple mutant varied in its ability to localize to the PM (data not shown), corresponding to its varied ability to bind GST-p115RH. This inability to localize properly to the PM suggests that the double mutation α13K204E/R232E and the triple mutation are detrimental to the structural folding and stability of α13.
Fig. 5. Subcellular localization of p115RhoGEF in cells co-expressing constitutively active α13 mutants.
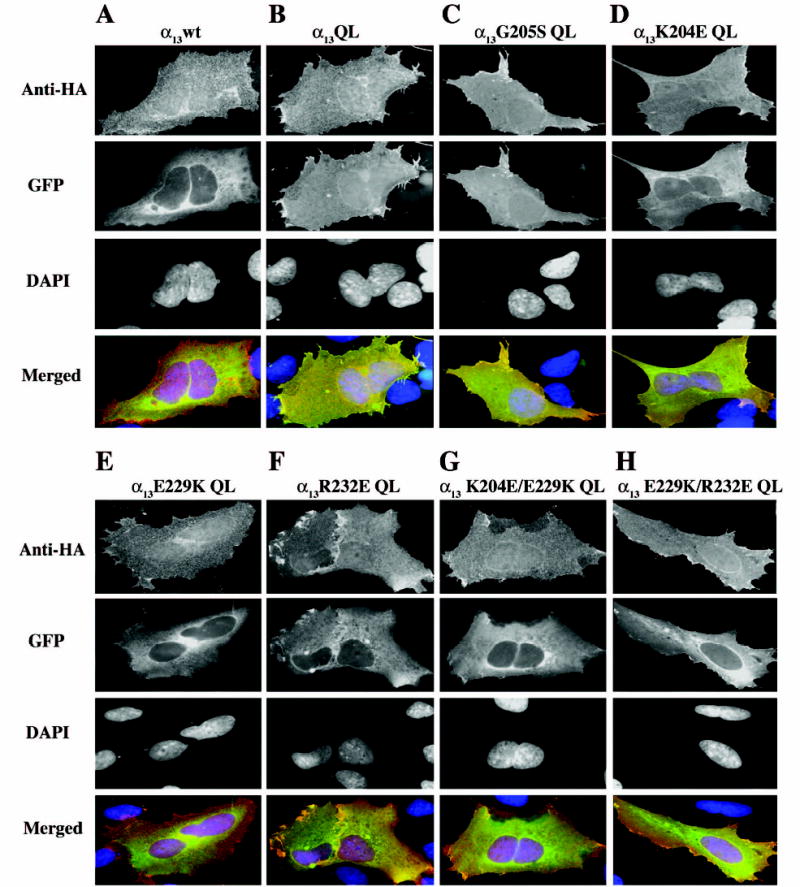
HEK293 cells were transiently co-transfected with 0.5μg each expression plasmid for the indicated α13 constructs and 0.1 μg DNA encoding p115RhoGEF-GFP. 48 h after transfection, cells were fixed and stained with a monoclonal anti-HA antibody, followed by Alexa-594-conjugated anti-mouse antibody, to stain for the HA epitope tagged α13 constructs (anti-HA panels). p115RhoGEF’s localization was determined by visualization of the intrinsic fluorescence of GFP (GFP panels), and DAPI staining was used to identify nuclei (DAPI panels). Three color merged images show α13 (red), p115RhoGEF-GFP (green) and nuclei (blue) (Merged panels). A representative image of more than 100 cells analyzed in three separate experiments is shown.
As previously shown, when co-expressed with α13wt, p115RhoGEF displayed a diffuse cytoplasmic staining (Fig. 5A, GFP) (Bhattacharyya & Wedegaertner, 2000). α13QL, on the other hand, induced the translocation of p115RhoGEF to the PM, as evidenced by the sharp membrane staining and the lack of a nuclear shadow (Fig. 5B, GFP). Similar to α13QL, the α13G205S QL mutant strongly promoted PM recruitment of p115RhoGEF, consistent with its ability to bind p115RhoGEF (Fig. 5C, GFP). The lack of nuclear shadow and sharp membrane staining of p115RhoGEF when co-expressed with α13K204E QL demonstrate that this mutant also promotes the PM recruitment of p115RhoGEF (Fig. 5D, GFP). When co-expressed with α13E229K QL, p115RhoGEF remained largely cytoplasmic, as indicated by the cytoplasmic staining decreasing in intensity towards the cell’s periphery, and the strong nuclear shadow (Fig. 5E, GFP). In addition, p115RhoGEF displayed both diffuse cytoplasmic staining and some sharp staining at the cell periphery when co-expressed with α13R232E QL (Fig. 5F, GFP). The double mutants α13K204E/E229K QL and α13E229K/E232E QL also show a reduced ability to promote PM recruitment of p115RhoGEF, as indicated by the largely cytoplasmic staining of p115RhoGEF, but in contrast to the complete cytoplasmic staining of p115RhoGEF when co-expressed with α13E229K QL, p115RhoGEF displayed some sharp staining at the cell periphery when co-expressed with either of the two double mutants (Fig. 5G–H, GFP). Thus, consistent with our GST-pulldowns and co-immunoprecipitation assays, active α13 mutants that are not fully defective in their ability to bind p115RhoGEF also retain some of their ability to induce its PM recruitment.
α 13 mutants are deficient in inducing Rho-mediated signaling
Since p115RhoGEF functions as the link between Gα13 and RhoA we wanted to determine whether the mutations in α13 that disrupted binding to p115RhoGEF in our GST-pulldown and immunoprecipitation assays also disrupt α13 induced Rho-mediated signaling. Activated G proteins αq/11, α12, and α13 have been shown to stimulate serum response element (SRE) mediated gene transcription through RhoA-dependent activation of the serum response factor (SRF) (Mao et al., 1998b). The ability of constitutively active α13 mutants to activate Rho-dependent signaling was determined using the SRE-mediated luciferase gene transcription assay, where HEK293 cells were transiently transfected with the constitutively active α13 or mutants and a reporter plasmid that expresses the luciferase gene under the control of SRE. As previously shown, α13QL stimulated SRE-mediated luciferase gene transcription (Fig. 6) (Bhattacharyya & Wedegaertner, 2000). Co-expression of Clostridium botulinum C3 transferase, which has been shown to inhibit RhoA, completely blocked α13QL induced SRE-mediated gene transcription, indicating that active α13 is acting through RhoA (Mao et al., 1998c). Consistent with the GST-p115RH pulldown, immunoprecipitation, and immunofluorescence studies, the G205S mutation had no effect on α13QL’s ability to activate Rho mediated SRE-luciferase gene transcription, delineating differences in the α13/p115RhoGEF interaction from other α/RGS interactions (Fig. 6A). The α13K204E QL and α13R232E QL mutants showed no statistical difference in their ability to stimulate SRE-luciferase gene transcription in comparison to α13QL whereas α13E229K QL was strongly deficient (Fig. 6A). 10 ng of each construct was transfected because this level of α13QL expression provided a robust signal yet, importantly, was in the linear range of the response. The double mutants retained their ability to induce SRE mediated luciferase gene transcription, corresponding to their behavior in the binding assays and immunofluorescence studies (Fig. 6A). To more closely examine whether there were any signaling defects in the p115RhoGEF binding-impaired α13K204E QL and α13R232E QL mutants, these mutants were expressed a varying levels (Fig. 6B). The α13K204E QL and α13R232E QL mutants were deficient in their ability to stimulate SRE-luciferase gene transcription in comparison to α13QL only when lower amounts (2.5 or 5 ng) were transfected (Fig. 6B). Thus, consistent with the binding assays, all three mutants are defective in stimulating Rho-dependent signaling, but α13E229K QL is most deficient in comparison to α13K204E QL, and α13R232E QL.
Fig. 6. SRE-mediated gene transcription by constitutively active α13 mutants.
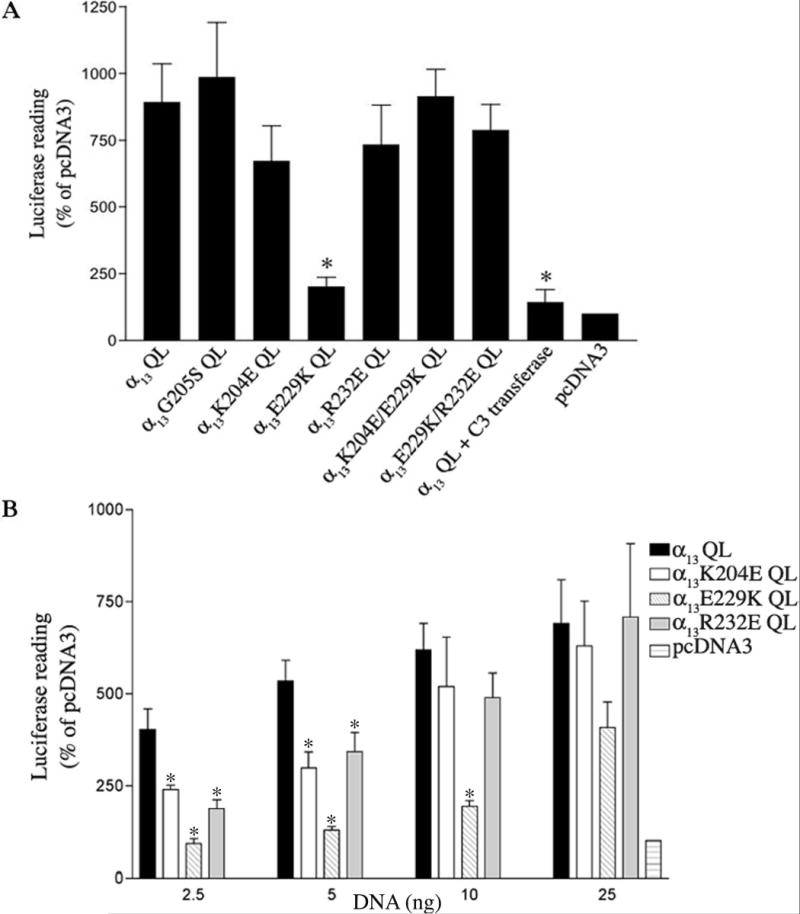
(A) pSRE-Luc (0.05μg) and pCMV-βgal (0.1 μg) were cotransfected into HEK293 cells with either 0.85 μg of pcDNA3 (vector) alone, or with 0.84 μg pcDNA3 and 10 ng of the indicated α13 constructs. 25 ng C3-transferase was also co-expressed with α13QL as indicated. (B) HEK293 cells were cotransfected with 2.5 to 25 ng of α13 constructs as indicated, and pSRE-Luc, pCMV-βgal, and pcDNA3. Cells were lysed 24 h after transfection, and assayed for luciferase activity and β-galactosidase activity. Luciferase activity was normalized by the β-galactosidase activity present in each lysate. Results are presented as percentage activity with respect to control cells (vector) and are the means +/− S.E. of results obtained from (A) 3 independent experiments performed in duplicate (N=6) or (B) 2 independent experiments performed in duplicates (N=4). The statistical difference between the indicated mutant and constitutively active α13QL is indicated by asterisks (*p<0.05).
α 13 point mutations do not affect heterotrimer formation or interaction with PP5-TPR
In its inactive state the GDP-bound α subunit is found as a heterotrimeric complex with βγ subunits (Hamm, 1998). G-protein coupled receptor activation results in the dissociation of the GTP bound α subunit from the βγ dimers, and the subsequent activation of their respective effectors. To ensure that our mutations did not affect the proper structural folding and stability of α13, we first looked at the ability of these mutants to form heterotrimers. HEK293 cells stably expressing N-terminal Myc-His tagged β1γ □ dimers were transfected with α13wt and the indicated constructs. The hexahistidine tag on β1 allowed for the pulldowns of heterotrimeric complexes on Ni-NTA magnetic beads. Immunoblotting with an anti-HA antibody revealed that α13wt bound to Myc-His β1γ □. A control experiment showed that αq was effectively pulled down by β1γ □, whereas a mutant of αq (αqIE), previously reported to be β1γ □-binding defective, did not show any binding (Fig. 7) (Evanko et al., 2000). The point mutants α13G205S, α13K204E, α13E229K, α13R232E and the double mutants α13K204E/E229K and α13E229K/R232E also bound to Myc-His β1γ □ dimers, indicating that these mutations do not affect the structural integrity of α13. On the other hand, α13K204E/R232E and the triple mutant were deficient in their binding ability (data not shown).
Fig. 7. Binding of α13 mutants to β1γ 2 dimers.
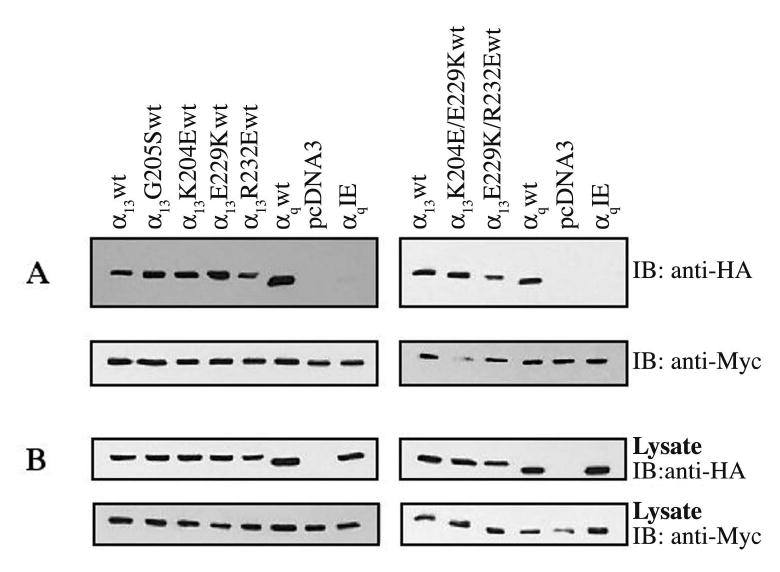
α13 mutants (2μg) were transfected in HEK293 cells stably expressing Myc-His tagged β1γ 2 dimers.48 h after transfection cells were lysed, and βγ dimers were pulled down using Ni-NTA beads. (A) Immunoblotting with an anti-HA antibody was used to determine binding of α13 constructs to Myc-His tagged β1γ 2dimers (upper panel). Samples were also subjected to immunoblotting with an anti-Myc antibody showing equal pulldowns of β1γ 2dimers (lower panels). (B) Cell lysates were immunoblotted with an anti-HA (upper panel) and anti-Myc antibody (lower panel) to determine expression levels of HA-α13 constructs and Myc-His tagged β1γ 2dimers, respectively.
Next we wanted to determine whether our mutants could interact with α13 effectors other that p115RhoGEF. It has been demonstrated that activated α13 interacts with Ser/Thr protein phosphatase type 5 (PP5) through its tetratricopeptide repeat domain (TPR) (Yamaguchi et al., 2002; Yamaguchi et al., 2003). The purified TPR domain of PP5 fused to GST (GST-TPR) on glutathione agarose beads was incubated with COS-7 cells lysates expressing constitutively activated α13 or mutants. As expected, α13 QL was pulled down by GST-TPR, whereas in a control experiment αqQL did not show any binding, confirming that this interaction is specific for α13 (Yamaguchi et al., 2002). The point mutants α13K204E QL, α13E229K QL, α13R232E QL also bound to GST-TPR indicating that these mutations do not affect the ability of α13 to interact with an effector other than p115RhoGEF.
DISCUSSION
Based on sequence alignment of α13 with αi1, and the crystal structure of αi1/RGS4 complex, we predicted α13 residues Lys204, Glu229, and Arg232 to be important for the α13/p115RhoGEF interaction (Tesmer et al., 1997). Herein we demonstrate that mutating each of these three amino acids to opposite charge residues disrupts the interaction of activated α13 with the RH domain of p115RhoGEF or full-length p115RhoGEF. We also demonstrate that it is only amino acid Glu229 in α13, that when mutated to an opposite charge lysine, strongly disrupts the ability of activated α13 to induce the recruitment of p115RhoGEF to the PM and to activate Rho-dependent signaling. Though α13K204E and α13R232E are deficient in binding p115RhoGEF, they induce at least partial recruitment of p115RhoGEF to the PM. Moreover, both α13K204E and α13R232E retain some ability to activate Rho-dependent signaling. Another mutation, G205S, has no effect on the ability of α13 to interact with p115RhoGEF, even though cognate Gly to Ser mutations in several other α subunits have been shown to confer resistance to interacting with RGS proteins. Consistent with previous studies focusing on mutational analyses of p115RhoGEF, our results indicate that the α13/p115RhoGEF interaction shares common features with other known α/RGS interactions but also has notable differences (Bhattacharyya & Wedegaertner, 2000; Bhattacharyya & Wedegaertner, 2003a; Bhattacharyya & Wedegaertner, 2003b; Chen et al., 2003; Chen et al., 2001; Nakamura et al., 2004).
The α13 mutations K204E and R232E cause decreased interaction with p115RhoGEF (Fig. 2–4), and these results are consistent with the critical role demonstrated for αi1 residues at identical positions in mediating interaction with RGS4 (Tesmer et al., 1997). Lys204 and Arg232 in α13 correspond to α i1 amino acids Thr182 and Lys210, respectively. Both Thr182 and Lys210 of α i1 make extensive contacts with RGS4 residues (Tesmer et al., 1997). Our results showing greatly decreased interaction of α13K204E and α13R232E with p115RhoGEF suggest that α13 and αi1 utilize similar surfaces to interact with p115RhoGEF and RGS4, respectively. Nonetheless, Lys204 and Arg232 in α13 are structurally distinct from αi1 residues Thr182 and Lys210, particularly Lys204 versus Thr182, and thus the specific contacts in α13/p115RhoGEF and αi1/RGS4 will be distinct. An interesting difference between Lys204 and Arg232 is that mutation to an opposite charge glutamic acid, as in α13K204E and α13R232E, strongly disrupts interaction with the RH domain of p115RhoGEF or full-length p115RhoGEF, but when mutated to alanine only α13K204A loses interaction with the RH domain of p115RhoGEF; α13R232A retains efficient activation-dependent GST pull down with the RH domain of p115RhoGEF (Fig. 2).
Although α13K204E and α13R232E were strongly deficient in their ability to interact with p115RhoGEF, both constitutively active forms, α13K204E QL and α13R232E QL, displayed only a partial defect in Rho-dependent signaling (Fig. 6). Moreover, α13K204E QL retained the ability to promote PM recruitment of p115RhoGEF, while α13R232E QL was partially impaired (Fig. 5). It is possible that weak interaction of these mutants with p115RhoGEF inside cells may be sufficient to translocate p115RhoGEF to the PM and to activate Rho-dependent signaling.
The lack of effect of the G205S mutation in α13 is distinct from the RGS insensitive phenotype observed for identical Gly to Ser mutations in most other Gα (DiBello et al., 1998; Lan et al., 1998), and thus, results with α13G205S provide further evidence for differences between the α13/p115RhoGEF interaction and other canonical Gα/RGS interactions. However, the α13/p115RhoGEF pair is not the only case in which a Gly to Ser mutation at this Gα position fails to affect the interaction; a recent report demonstrated that such a Gly to Ser mutant of αq, αqG188S, retains interaction with the RH domain of GRK2 even though it loses interaction with RGS2 (Sterne-Marr et al., 2003).
The results presented here suggest that Glu229 is particularly crucial for α13 to bind to and activate p115RhoGEF. Like α13K204E and α13R232E, α13E229K loses activation-dependent interaction with p115RhoGEF (Fig. 2–4) but, in contrast to constitutively active α13K204E QL and α13R232E QL, α13E229K QL is strongly deficient in recruiting p115RhoGEF to the PM and in activating Rho signaling (Fig. 5 and 6). A glutamic acid at positions identical to 229 in α13 is conserved in all Gα, and the identical Glu207 in α i1 makes critical intramolecular contacts and numerous contacts with residues in RGS4 (Tesmer et al., 1997). Although α13E229K was disrupted in all p115RhoGEF interaction assays, including PM recruitment of p115RhoGEF and activation of Rho signaling, it retained the ability to properly localize at the PM and to interact with β1γ 2 and the TPR domain of PP5 in pull down assays (Fig. 7 and 8), suggesting that the E229K mutation does not cause a global disruption of proper folding of α13. In contrast to α13E229K, the α13E229A mutant retained interaction with p115RhoGEF (Fig. 2 and 4) suggesting that Glu229 functions predominantly in charge complementarity interactions.
Fig. 8. Interaction of constitutively active α13 mutants with GST-TPR.
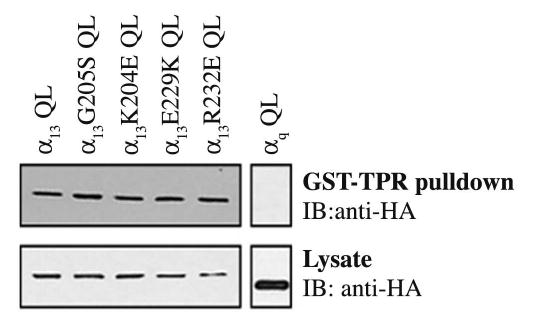
COS-7 cells were transfected with the indicated α13QL constructs (4.5 μg) and pcDNA3 (4.5 μg). α13QL or mutants were pulled down with 10 μg of GST-TPR immobilized on glutathione agarose beads. α13QL or mutant pulldowns were detected by immunoblotting with anti-HA antibody (upper panel). Cell lysates were immunoblotted with an anti-HA antibody to determine equal expression of α13QL mutants (lower panel). Results shown are representative of at least three independent experiments.
A potential role for Glu229 of α13 in stabilizing and orienting other p115RhoGEF-interacting amino acids of α13 is revealed by the gain of function of the constitutively active double mutants α13K204E/E229K QL and α13E229K/R232E QL. Such a role is consistent with the structure of the αi1/RGS4 complex, in which Glu207 makes contacts with the side chains of other RGS4-interacting residues of αi1 (Tesmer et al., 1997). Introduction of the charge reversal mutations K204E or R232E into α13E229K QL partially restored the ability of α13E229K QL to interact with GST-p115RH, to recruit p115RhoGEF to the PM, and to activate Rho signaling. Rescue of binding to GST-p115RH was only observed in the α13QL background; AlF−4-activated α13K204E/E229K and α13E229K/R232E did not interact with GST-p115RH or co-immunoprecipate with p115RhoGEF. The reason for this difference is unclear. Slight structural differences in mutationally activated versus AlF−4-activated α13 or a role for potential binding sites on p115RhoGEF outside of the RH domain (Chen et al., 2003; Wells et al., 2002b) may contribute to an explanation. Nonetheless, the results with α13K204E/E229K QL and α13E229K/R232E QL are consistent with charge-charge interactions among α13 amino acids at the p115RhoGEF binding interface, with Glu229 playing a major role. Confirmation of this speculation and identification of the exact role for α13 amino acids tested in this study awaits structural determination of an α13/p115RhoGEF complex.
α13 regulates the activity of several molecules, in addition to the RhoGEFs. Its role in regulating cell growth and differentiation seems to be mediated at least partially by its interaction with p115RhoGEF, or PDZ-RhoGEF or LARG, and the subsequent activation of Rho. Several additional pathways might be involved in the α13 mediated regulation of cell growth and differentiation since α13, as previously demonstrated, is a very powerful transformant, whereas Rho is a weak one (Symons, 1996). Radixin, a member of the ERM family of proteins, and cadherins have also been linked to the transforming ability of α13 (Meigs et al., 2002; Meigs et al., 2001; Vaiskunaite et al., 2000). Other proteins yet to be identified may also play a role. The identification of α13 mutants defective in their interaction with p115RhoGEF, ability to activate Rho and induce recruitment of p115RhoGEF to the PM could provide significant insight into the mechanisms through which α13 interacts with other effectors and induces transformation.
MATERIALS AND METHODS
Cell Culture and Transfection
HEK293 cells were obtained from A. Marchese (Thomas Jefferson University, Philadelphia, PA). HEK293 and COS-7 were maintained in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum, and penicillin-streptomycin. HEK293 cells stably expressing Myc-His epitope tagged β1γ 22 were propagated in DMEM with 10% fetal bovine serum and G418. Unless otherwise noted cells were plated in 6 cm or 6-well plates 24 h prior to transfection. Cells were transfected with either 1μg of DNA/well of 6-well plate or 3 μg DNA in a 6 cm plate using FuGENE 6 (Roche Diagnostics, Indianapolis, IN), according to manufacturer’s protocol.
Expression Plasmids
The HA epitope (DVPDYA)-tagged pcDNA3HA α13wt and pcDNA3HA α13QL were gifts from J.S.Gutkind (Fukuhara et al., 1999). pGEX-4T-2 PP5-TPR was a gift from M.Negishi (Kyoto University, Kyoto, Japan) (Yamaguchi et al., 2002). The N-terminal Myc epitope (MEQKLISEED)-tagged pcDNA3Myc p115RhoGEF, and N-terminal Myc-His-epitope tagged β1 in pcDNA3 have been described (Bhattacharyya & Wedegaertner, 2000; Bhattacharyya & Wedegaertner, 2003a; Takida & Wedegaertner, 2003). The Stratagene QuickChange site-directed mutagenesis kit or Sequential PCR was used to create the K204E, E229K, R232E, K204E/E229K, K204E/R232E, E229K/R232E mutants in both α13wt and α13QL, and the K204A, E229A, and R232A mutants in α13wt. For the generation of the RGS homology domain of p115RhoGEF (p115RH) fused to GST, a fragment of p115RhoGEF containing residues 1-252 was amplified with forward and reverse primers containing a 5’ EcoRI site and a 3’ SalI site for subcloning into pGEX-5x-1. The correct DNA sequence of the mutants was confirmed by DNA sequencing of the entire open reading frame (Kimmel Cancer Institute Nucleic Acid Facility). The reporter plasmid that expresses the luciferase gene under the control of serum response element (SRE), termed pSRE-Luc, was purchased from Stratagene (La Jolla, CA). The plasmids carrying the HA epitope-tagged activated RhoA (pcDNA3 RhoV14) or the β-galactosidase gene (pCMV-β-gal) were obtained from P. Tsichlis (Tufts University, Boston, MA). The Myc epitope tagged C3-transferase (pEF-Myc-C3-transferase) was a gift from A. Hall (University College, London, U.K.)
Protein Purification
p115RH (1-252) fused to GST was expressed in transformed E.coli cells (BL21 strain). The TPR domain of PP5 fused to GST was expressed in transformed E.coli cells (DH5α strain). Cells were grown in LB media at 37°C to A600~ 0.7 and induced with 0.5 mM isopropyl-1-thiogalactopyranoside at 37°C for 2 h. Cells were pelleted by centrifugation and lysed by sonication in PBS with 50 μg/ml PMSF, 1X Complete protease inhibitor cocktail (Roche), 2 mg/ml lysozyme, 1% Triton X-100, and 2 mM DTT. Next, suspensions were centrifuged at 15,000 rpm for 30 min at 4°C. The supernatants were then incubated with glutathione agarose beads for 1 h at 4°C. Beads were washed 3 times with Buffer A (PBS, 1% Triton X-100, 50 μg/ml PMSF, and 1 mg/ml leupeptin and aprotinin) and 3 times with Buffer B (PBS, 50 μg/ml PMSF, and 1 mg/ml leupeptin and aprotinin). Following washes, beads were re-suspended in Buffer B. The expressed proteins were resolved by SDS-PAGE. Protein concentration was determined by comparison with bovine serum albumin standards after staining with Comassie brilliant blue.
Immunoprecipitations and GST binding assays
For co-immunoprecipitations and GST-p115RH interaction assays, HEK293 cells in 6 cm plates were transfected with the indicated constructs for 48 h. For GST-TPR assays COS-7 cells in 10 cm plates were transfected with the indicated constructs for 24 h. Cells were washed twice with ice cold PBS and lysed with 0.5 ml of lysis buffer (50 mM HEPES, 150 mM NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 1 mM EDTA, 2.5 mM MgCL2, 1 mM dithiothreitol, 25 mM glycerophosphate, 1 mM phenylmethylsulfonyl fluoride, and 5 μg/ml leupeptin and aprotinin). For the AlF−4 experiments each cell lysate was divided into two aliquots. One aliquot was treated with 50 μM AlCl3, 10 mM MgCl2, and 10 mM NaF (+AlF−4) and the other was left untreated (−AlF−4). 1 h after lysis at 4°C, cells lysates were centrifuged at 13000rpm for 3 min. 5 μg of an anti-HA epitope (12CA5) mouse monoclonal antibody (Roche Applied Sciences, Indianapolis, IN) was added, and the supernatants were tumbled for 2 h at 4°C. Next, 20 μl of Protein A/G Plus agarose (Santa Cruz Biotechnology, Santa Cruz, CA) were added, and samples were tumbled overnight at 4°C. GST-pulldown experiments were performed with 10 μg of GST-p115RH (1-252) or 10 μg of GST-TPR immobilized on glutathione agarose beads for 2 h at 4°C. Beads were then washed 3 times with lysis buffer. Bound proteins were eluted in 50 μl of SDS sample buffer and boiled for 5 min. Co-immunoprecipitation and GST-pulldown samples were subjected to SDS-PAGE, transferred to PVDF and probed with 0.1 μg/ml of an anti-Myc epitope (9E10) mouse monoclonal antibody (Covance, Berkeley, CA) or with 0.5 μg/ml 12CA5, followed by horseradish peroxidase-conjugated anti-mouse antibody (Promega, Madison, WI). The blots were visualized using Supersignal West Pico (Pierce Chemical, Rockford, IL)
Ni-NTA pulldown of β1
HEK293 cells stably expressing N-terminal Myc-His tagged β1γ 2 were transfected in 6-cm plates with the indicated Gα13 or Gαq constructs. 36 h after transfection, cells were washed once with ice cold PBS and lysed in 0.5 ml of lysis buffer (20 mM HEPES, pH 7.5, 100 mM NaCl, 0.7% Triton X-100, 1 mM EDTA, 5 mM MgCl2, 1 mM DTT, 10 mM immidazole, 0.5 mM phenylmethylsulfonyl fluoride, and 2 μg/ml leupeptin and aprotinin). After 1 h of lysis at 4°C, nuclei and insoluble material were removed by centrifugation at 13000 rpm for 3 min. 10 μl of Ni-NTA magnetic agarose beads (QIAGEN, Valencia, CA) were added to the clarified lysates and samples were tumbled for 2 h at 4° C. Next, the samples were placed on the QIAGEN 12-tube Magnet to pellet the beads. The supernatants were discarded and beads were washed three times with lysis buffer containing 20 mM immidazole. Next, the beads were incubated with lysis buffer containing 250 mM immidazole for 3 min to elute β1γ 2 and proteins bound to them. Eluates were separated by 10% SDS-PAGE, transferred to PVDF, and immunoblotted with the appropriate antibodies as indicated in figure legends.
Immunofluorescence microscopy
24 h after transfection in 6-well plates, HEK293 cells were split onto coverslips and then grown for an additional 24 h. Cells were fixed with 3.7% formaldehyde in PBS for 15 min, washed three times with PBS, and permeabilized by incubation in blocking buffer containing TBS (50 mM Tris-Hcl, pH 7.5, 150 mM NaCl), 1 % Triton X-100, and 2.5 % nonfat milk. Cells were then incubated with 12CA5 mouse monoclonal antibody (5μg/ml) in blocking buffer for 1 h. Following five washes with blocking buffer, cells were incubated in a 1:100 dilution of Alexa Fluor 594 goat anti-mouse (Molecular Probes, Eugene, OR) secondary antibody for 30 min. The cells were then washed five times with TBS/1% Triton X-100 and once with PBS. 1 ml of warm PBS containing 0.1μg/ml of DAPI (Molecular Probes, Eugene, OR) was added to the cells for five minutes. The coverslips were washed with PBS, rinsed with distilled water and mounted on glass slides with 20 μl of Prolong Antifade reagent (Molecular Probes, Eugene, OR).
Representative images were acquired using an Olympus BX-61 microscope with an ORCA-ER (Hamamatsu, Bridgewater, NJ) cooled charge-coupled device camera controlled by Slidebook version 4.0 (Intelligent Imaging Innovations, Denver, CO). Images were processed with Adobe Photoshop.
SRE-mediated luciferase gene transcription assay
HEK293 cells were plated in 6-well plates in serum supplemented DMEM for 24 h. Cells were then switched to serum free DMEM and transfected with 0.05 μg of pSRE-Luc, 0.1 μg pCMV-β-gal, pcDNA3 and α13 constructs as indicated. 24 h after transfection cells were washed with ice-cold PBS and lysed using reporter lysis buffer according to the manufacturer’s protocol (Promega, Madison, WI). 20 μl of the lysates were mixed with 50 μl of the luciferase substrate (Promega, Madison, WI) at room temperature. Luciferase activity was determined by measuring luminescence intensity. The β-galactosidase activities were measured by the colorimetric method and were used to normalize transfection efficiency. All assays were performed in duplicates.
Acknowledgments
This work was supported by grant GM62884 (P.W.) from the NIH. P.W. is an Established Investigator of the American Heart Association.
Footnotes
The abbreviations used are: G protein, guanine nucleotide-binding protein; GPCR, G protein-coupled receptor; RGS, regulator of G protein signaling; RH, regulator of G protein signaling homology domain; GEF, guanine-nucleotide exchange factor; LARG, leukemia-associated RhoGEF; GAP, GTPase activating protein; SRE, serum response element; PM, plasma membrane; GRK, GPCR kinase; GFP, green fluorescent protein; HA, hemagglutinin; Ni-NTA, nickel-nitrilotriacetic acid.
D. S. Evanko, M. M. Thiyagarajan, S. Takida, and P.B. Wedegaertner, unpublished results.
References
- Althoefer H, Eversole-Cire P, Simon MI. J Biol Chem. 1997;272:24380–6. doi: 10.1074/jbc.272.39.24380. [DOI] [PubMed] [Google Scholar]
- Berestetskaya YV, Faure MP, Ichijo H, Voyno-Yasenetskaya TA. J Biol Chem. 1998;273:27816–23. doi: 10.1074/jbc.273.43.27816. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya R, Wedegaertner PB. J Biol Chem. 2000;275:14992–9. doi: 10.1074/jbc.M000415200. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya R, Wedegaertner PB. Biochem J. 2003a;371:709–20. doi: 10.1042/BJ20021897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya R, Wedegaertner PB. FEBS Lett. 2003b;540:211–6. doi: 10.1016/s0014-5793(03)00267-9. [DOI] [PubMed] [Google Scholar]
- Buhl AM, Johnson NL, Dhanasekaran N, Johnson GL. J Biol Chem. 1995;270:24631–4. doi: 10.1074/jbc.270.42.24631. [DOI] [PubMed] [Google Scholar]
- Chen Z, Singer WD, Wells CD, Sprang SR, Sternweis PC. J Biol Chem. 2003;278:9912–9. doi: 10.1074/jbc.M212695200. [DOI] [PubMed] [Google Scholar]
- Chen Z, Wells CD, Sternweis PC, Sprang SR. Nat Struct Biol. 2001;8:805–9. doi: 10.1038/nsb0901-805. [DOI] [PubMed] [Google Scholar]
- Coleman DE, Berghuis AM, Lee E, Linder ME, Gilman AG, Sprang SR. Science. 1994;265:1405–1412. doi: 10.1126/science.8073283. [DOI] [PubMed] [Google Scholar]
- DiBello PR, Garrison TR, Apanovitch DM, Hoffman G, Shuey DJ, Mason K, Cockett MI, Dohlman HG. J Biol Chem. 1998;273:5780–4. doi: 10.1074/jbc.273.10.5780. [DOI] [PubMed] [Google Scholar]
- Evanko DS, Thiyagarajan MM, Wedegaertner PB. J Biol Chem. 2000;275:1327–36. doi: 10.1074/jbc.275.2.1327. [DOI] [PubMed] [Google Scholar]
- Fleming IN, Elliott CM, Exton JH. J Biol Chem. 1996;271:33067–73. doi: 10.1074/jbc.271.51.33067. [DOI] [PubMed] [Google Scholar]
- Fukuhara S, Murga C, Zohar M, Igishi T, Gutkind JS. J Biol Chem. 1999;274:5868–79. doi: 10.1074/jbc.274.9.5868. [DOI] [PubMed] [Google Scholar]
- Gohla A, Offermanns S, Wilkie TM, Schultz G. J Biol Chem. 1999;274:17901–7. doi: 10.1074/jbc.274.25.17901. [DOI] [PubMed] [Google Scholar]
- Hamm HE. J Biol Chem. 1998;273:669–72. doi: 10.1074/jbc.273.2.669. [DOI] [PubMed] [Google Scholar]
- Hart MJ, Jiang X, Kozasa T, Roscoe W, Singer WD, Gilman AG, Sternweis PC, Bollag G. Science. 1998;280:2112–4. doi: 10.1126/science.280.5372.2112. [DOI] [PubMed] [Google Scholar]
- Hollinger S, Hepler JR. Pharmacol Rev. 2002;54:527–59. doi: 10.1124/pr.54.3.527. [DOI] [PubMed] [Google Scholar]
- Jiang H, Wu D, Simon MI. FEBS Lett. 1993;330:319–22. doi: 10.1016/0014-5793(93)80896-3. [DOI] [PubMed] [Google Scholar]
- Katoh H, Aoki J, Yamaguchi Y, Kitano Y, Ichikawa A, Negishi M. J Biol Chem. 1998;273:28700–7. doi: 10.1074/jbc.273.44.28700. [DOI] [PubMed] [Google Scholar]
- Kozasa T, Jiang X, Hart MJ, Sternweis PM, Singer WD, Gilman AG, Bollag G, Sternweis PC. Science. 1998;280:2109–11. doi: 10.1126/science.280.5372.2109. [DOI] [PubMed] [Google Scholar]
- Kranenburg O, Poland M, Gebbink M, Oomen L, Moolenaar WH. Journal of Cell Science. 1997;110:2417–27. doi: 10.1242/jcs.110.19.2417. [DOI] [PubMed] [Google Scholar]
- Kranenburg O, Poland M, van Horck FP, Drechsel D, Hall A, Moolenaar WH. Molecular Biology of the Cell. 1999;10:1851–7. doi: 10.1091/mbc.10.6.1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan KL, Sarvazyan NA, Taussig R, Mackenzie RG, DiBello PR, Dohlman HG, Neubig RR. J Biol Chem. 1998;273:12794–7. doi: 10.1074/jbc.273.21.12794. [DOI] [PubMed] [Google Scholar]
- Mao J, Xie W, Yuan H, Simon MI, Mano H, Wu D. EMBO Journal. 1998a;17:5638–46. doi: 10.1093/emboj/17.19.5638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao J, Yuan H, Xie W, Simon MI, Wu D. J Biol Chem. 1998b;273:27118–23. doi: 10.1074/jbc.273.42.27118. [DOI] [PubMed] [Google Scholar]
- Mao J, Yuan H, Xie W, Wu D. Proc Natl Acad Sci U S A. 1998c;95:12973–6. doi: 10.1073/pnas.95.22.12973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meigs TE, Fedor-Chaiken M, Kaplan DD, Brackenbury R, Casey PJ. J Biol Chem. 2002;277:24594–600. doi: 10.1074/jbc.M201984200. [DOI] [PubMed] [Google Scholar]
- Meigs TE, Fields TA, McKee DD, Casey PJ. Proc Natl Acad Sci U S A. 2001;98:519–24. doi: 10.1073/pnas.021350998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura S, Kreutz B, Tanabe S, Suzuki N, Kozasa T. Mol Pharmacol. 2004;66:1029–34. doi: 10.1124/mol.104.002287. [DOI] [PubMed] [Google Scholar]
- Neubig RR. FASEB J. 1994;8:939–946. doi: 10.1096/fasebj.8.12.8088459. [DOI] [PubMed] [Google Scholar]
- Offermanns S. Rev Physiol Biochem Pharmacol. 2000;140:63–133. doi: 10.1007/BFb0035551. [DOI] [PubMed] [Google Scholar]
- Parks S, Wieschaus E. Cell. 1991;64:447–58. doi: 10.1016/0092-8674(91)90652-f. [DOI] [PubMed] [Google Scholar]
- Prasad MV, Dermott JM, Heasley LE, Johnson GL, Dhanasekaran N. J Biol Chem. 1995;270:18655–9. doi: 10.1074/jbc.270.31.18655. [DOI] [PubMed] [Google Scholar]
- Shi CS, Sinnarajah S, Cho H, Kozasa T, Kehrl JH. J Biol Chem. 2000;275:24470–24476. doi: 10.1074/jbc.M908449199. [DOI] [PubMed] [Google Scholar]
- Sterne-Marr R, Tesmer JJ, Day PW, Stracquatanio RP, Cilente JA, O'Connor KE, Pronin AN, Benovic JL, Wedegaertner PB. J Biol Chem. 2003;278:6050–8. doi: 10.1074/jbc.M208787200. [DOI] [PubMed] [Google Scholar]
- Symons M. Trends Biochem Sci. 1996;21:178–81. [PubMed] [Google Scholar]
- Takida S, Wedegaertner PB. J Biol Chem. 2003;278:17284–90. doi: 10.1074/jbc.M213239200. [DOI] [PubMed] [Google Scholar]
- Tesmer JJ, Berman DM, Gilman AG, Sprang SR. Cell. 1997;89:251–61. doi: 10.1016/s0092-8674(00)80204-4. [DOI] [PubMed] [Google Scholar]
- Vaiskunaite R, Adarichev V, Furthmayr H, Kozasa T, Gudkov A, Voyno-Yasenetskaya TA. J Biol Chem. 2000;275:26206–12. doi: 10.1074/jbc.M001863200. [DOI] [PubMed] [Google Scholar]
- Voyno-Yasenetskaya T, Conklin BR, Gilbert RL, Hooley R, Bourne HR, Barber DL. J Biol Chem. 1994a;269:4721–4724. [PubMed] [Google Scholar]
- Voyno-Yasenetskaya TA, Faure MP, Ahn NG, Bourne HR. J Biol Chem. 1996;271:21081–7. doi: 10.1074/jbc.271.35.21081. [DOI] [PubMed] [Google Scholar]
- Voyno-Yasenetskaya TA, Pace AM, Bourne HR. Oncogene. 1994b;9:2559–65. [PubMed] [Google Scholar]
- Wells C, Jiang X, Gutowski S, Sternweis PC. Methods Enzymol. 2002a;345:371–82. doi: 10.1016/s0076-6879(02)45030-6. [DOI] [PubMed] [Google Scholar]
- Wells CD, Gutowski S, Bollag G, Sternweis PC. J Biol Chem. 2001;276:28897–905. doi: 10.1074/jbc.M102913200. [DOI] [PubMed] [Google Scholar]
- Wells CD, Liu MY, Jackson M, Gutowski S, Sternweis PM, Rothstein JD, Kozasa T, Sternweis PC. J Biol Chem. 2002b;277:1174–81. doi: 10.1074/jbc.M105274200. [DOI] [PubMed] [Google Scholar]
- Xu N, Bradley L, Ambdukar I, Gutkind JS. Proc Natl Acad Sci USA. 1993;90:6741–6745. doi: 10.1073/pnas.90.14.6741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi Y, Katoh H, Mori K, Negishi M. Curr Biol. 2002;12:1353–8. doi: 10.1016/s0960-9822(02)01034-5. [DOI] [PubMed] [Google Scholar]
- Yamaguchi Y, Katoh H, Negishi M. J Biol Chem. 2003;278:14936–9. doi: 10.1074/jbc.M301409200. [DOI] [PubMed] [Google Scholar]