

Abstract
At a certain point in development, axons in the mammalian central nervous system lose their ability to regenerate after injury. Using the optic nerve model, we show that this growth failure coincides with two developmental events: the loss of Bcl-2 expression by neurons and the maturation of astrocytes. Before postnatal day 4, when astrocytes are immature, overexpression of Bcl-2 alone supported robust and rapid optic nerve regeneration over long distances, leading to innervation of brain targets by day 4 in mice. As astrocytes matured after postnatal day 4, axonal regeneration was inhibited in mice overexpressing Bcl-2. Concurrent induction of Bcl-2 and attenuation of reactive gliosis reversed the failure of CNS axonal re-elongation in postnatal mice and led to rapid axonal regeneration over long distances and reinnervation of the brain targets by a majority of severed optic nerve fibers up to 2 weeks of age. These results suggest that an early postnatal downregulation of Bcl-2 and post-traumatic reactive gliosis are two important elements of axon regenerative failure in the CNS.
Keywords: Astrocyte intermediate filament, GFAP, Vimentin, Bcl-2, Axon regeneration
Introduction
At a certain point in development, axons in the mammalian central nervous system (CNS) lose their ability to regenerate after injury. The mechanisms of this growth failure are unclear. According to the prevailing view, CNS regenerative failure reflects both the intrinsic inability of adult CNS neurons to survive and reinitiate axonal growth and the lack of a permissive environment for such growth (Bregman, 1998; Chen and Tonegawa, 1998; Goldberg and Barres, 2000; Horner and Gage, 2000). Proposed potential growth obstacles within the CNS include myelin-associated inhibitory molecules, glial scarring after injury, and lack of growth-promoting substrates and neurotrophic factors (Fournier and Strittmatter, 2001; Goldberg and Barres, 2000; Morgenstern et al., 2002). Successful regeneration in the adult CNS may require manipulating both the intrinsic features of injured neurons and the CNS environment. Despite the recent identification of growth-inhibitory molecules and their receptors (McGee and Strittmatter, 2003), two pertinent questions remain unanswered. What are the essential intrinsic and environmental components for promoting CNS regeneration, and how can the axonal growth potential lost during development be restored in the mature CNS?
Optic nerve injury is a standard model for studying CNS regeneration. Rodent retinal ganglion cells (RGCs), whose axons form the optic nerve, normally cannot regenerate their axons through an injured optic nerve. When provided with permissive substrate or given a novel treatment, only a small population of severed axons are induced to regenerate; the regeneration is slow, and the axons stop extending before they reach their targets (Li et al., 2003; Yin et al., 2003). Rodent RGCs lose their intrinsic ability for axon elongation before birth (Chen et al., 1995; Goldberg et al., 2002). Developing RGCs have two distinct stages of axonal growth – elongation and arborization – distinguished by contrasting rates of axon extension (Goldberg and Barres, 2000; Jhaveri et al., 1991). Embryonic RGC axons elongate about 10 times faster than mature ones (0.5–2.0 mm/d versus 40–60 μm/d). This difference has been attributed primarily to the maturational change in the intrinsic property of neurons and contributes critically to the failure of optic nerve regeneration (Goldberg et al., 2002).
RGCs lose their intrinsic capacity for axonal elongation at embryonic day 18 (E18), before the onset of growth inhibition in the CNS environment (Chen and Tonegawa, 1998). Such a developmental scheme offers an opportunity to characterize and uncover independently the intrinsic and environmental elements in optic nerve regenerative failure. Naturally, the first important question is what determines the intrinsic regenerative capacity of RGC axons. Previously we showed that one determining factor is the anti-apoptotic gene Bcl-2 (Chen et al., 1997). In lower vertebrates (e.g. fish and frog), CNS neurons upregulate Bcl-2 after injury and readily regenerate axons and re-establish topographically organized connections (Cristino et al., 2000). In mammals, Bcl-2 expression parallels axon extension during development. Bcl-2 expression declines in the developing CNS as neurons lose their ability to grow new axons but is maintained at a high level in adult neurons of the peripheral nervous system, where axons regenerate robustly throughout life (Merry and Korsmeyer, 1997; Merry et al., 1994). In the absence of Bcl-2, the ability of embryonic neurons to elaborate axons is reduced, even in the presence of potent neuronal growth factors (Chen et al., 1997; Hilton et al., 1997). By contrast, overexpression of Bcl-2 enhances neurite outgrowth in several neuronal cell lines and promotes the regeneration of RGC axons into a permissive brain environment in culture (Chen et al., 1997; Oh et al., 1996; Zhang et al., 1996). These findings suggest that Bcl-2 may act cell autonomously to support the intrinsic regenerative capacity of CNS axons.
Recent evidence, however, indicates that Bcl-2 alone is not sufficient to support CNS axonal regeneration in vivo. Overexpression of Bcl-2 in transgenic mice (Bcl-2tg) failed to promote optic nerve regrowth in mice at postnatal day 5 (P5) or in adult mice (Chierzi et al., 1999; Lodovichi et al., 2001). Thus, the question arises whether factors other than Bcl-2 are required to support the intrinsic growth mechanisms of RGC axons or whether the CNS environment develops inhibitory mechanisms that block regeneration in Bcl-2tg mice after P5. If it is the latter, Bcl-2tg mice, whose neurons retain their intrinsic capacity to regenerate axons, would provide an excellent model for defining inhibitory mechanisms in the mature CNS environment. In this study, we used the optic nerve model to assess the effect of neuronal Bcl-2 expression and reactive gliosis on the regeneration of RGC axons in vivo.
Materials and Methods
Animals
Wildtype (wt) and Bcl-2tg mice were obtained from crossing wt C57BL/6J females with Bcl-2tg males in which transgene expression is controlled by the neuron-specific enolase promoter (line 73a) (Martinou et al., 1994). Wildtype littermates of Bcl-2tg mice served as controls. Mice deficient in glial fibrillary acidic protein and vimentin (GFAP−/−Vim−/−) were generated as described (Colluci-Guyon et al., 1994; Pekny et al., 1995; Pekny et al., 1999). Bcl-2tgGFAP−/−Vim−/− mice were generated by crossing GFAP+/−Vim+/− females with Bcl-2tgGFAP+/−Vim+/− males, which were generated from crossing GFAP−/−Vim−/− females with Bcl-2tg males.
Optic nerve surgery and anterograde labeling of axons
Mouse pups younger than P6 were anesthetized by hypothermia; older mice were anesthetized with a mixture of ketamine (12.5 mg/ml, Sigma) and xylazine (2.5 mg/ml, Sigma) (5 μl/g). In each mouse, one optic nerve was exposed intraorbitally and crushed with fine surgical forceps for 5 seconds about 0.5–1 mm from the posterior pole of the eyeball. In some experiments the crush site was labeled by sutures or by dipping the tips of forceps in a solution containing 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI) (Molecular Probes, Eugene, OR) (10% in dimethyl formamide) before the crush. In sham-operated controls, the optic nerve was exposed but not crushed. An anterograde tracer, cholera toxin B subunit conjugated with rhodamine (CTB-R) or fluorescein (CTB-F) (List Biological Lab, Campbell, CA) (2.5 μg/μl), was injected into the vitreous cavity immediately after the crush to allow visualization of axons 1–4 days later. To study the role of mature astrocytes in nerve regeneration, immediately after optic nerve crush, gelfoam soaked in PBS containing 10 mg/ml L-α-aminoadipate or PBS alone (control) was placed against the crush site and replaced every 3 days.
Histology and immunofluorescence labeling
Mice were anesthetized, and the eyecups, entire optic nerve, and brains were dissected and sectioned (12–40 μm). Cresyl violet and immunofluorescence staining were carried out on adjacent sections. Primary antibodies against growth-associated protein 43 (GAP-43) and neurofilament protein of medium molecular weight (NF-M) (Chemicon, Temecula, CA) were used to reveal axonal morphology. The crush site was identified by staining with isolectin B4 (Sigma). The sections were examined with a Nikon TE300 microscope equipped with fluorescence illumination and with a Leica confocal microscope.
Retrograde labeling of RGCs
Immediately after the optic nerve surgery, a midline incision was made in the scalp above the superior colliculus (SC). A 1 mm3 piece of gelfoam (Upjohn, Kalamazoo, MI) was soaked in 4% FluoroGold solution (Fluorochrome, Denver, CO) and inserted over the SC. Eleven days later, retinas were dissected, flat mounted, and examined by fluorescence microscopy. Eight standard rectangular regions (0.09 mm2), radially distributed at 1–2 mm from the optic nerve head, were photographed at 40× magnification, and all FluoroGold-labeled cells with RCG-like morphologies were counted.
Retina-brain slice co-culture
Retina-midbrain slice co-cultures were prepared as described (Chen et al., 1995). Briefly, mouse pups were coded, their brains and retinas were dissected, and coronal brain slices containing the SC were prepared. Each retinal explant was placed to abut a brain slice on a six-well cell culture insert and cultured in neurobasal medium supplemented with B27 (Gibco, Grand Island, NY). After 4 days, the cultures were fixed, and DiI crystals were placed on each retinal explant. The dye was allowed to diffuse for 1 week, and retinal axons that had invaded the brain slices were counted by fluorescence microscopy.
Western blot
Midbrains containing the SCs of E14-P14 mice were dissected, and extracts were prepared. Protein concentration was measured with a Micro BCA Protein Assay Reagent Kit (Pierce, Rockford, IL), and 100 μg of protein from each sample was loaded onto a gel and transferred to nitrocellulose membranes. The blots were reacted with primary antibodies against GFAP (Sigma, 1:100), vimentin (Sigma, 1:100), myelin basic protein (MBP) (Chemicon, 1:100), myelinassociated glycoprotein (MAG) (Chemicon, 1:100) and actin (Chemicon, 1:1000) as a control in a solution containing 0.05% Tween-20, 1% bovine serum albumin, and 4% nonfat dry milk at 4°C overnight. The membrane was incubated with the corresponding peroxidase- or horseradish-peroxidase-conjugated secondary antibody (1:10,000) for 1 hour at room temperature. The enhanced chemiluminescence system (Pierce) was used for antibody detection. To compare the responses of astrocytes to injury at different developmental ages, some P0 and P5 pups underwent transection at the midbrain (SC) level. Two days later (P2 and P7), brains from injured and uninjured mice were collected, and the proteins were blotted with anti-GFAP antibody.
Reverse transcription (RT)-PCR
RT-PCR was performed as described (Huang et al., 2003). After extraction with Trizol (Gibco, Rockville, MD), total mid-brain RNA (1 μg) was subjected to RT. For relative quantification, each PCR reaction contained equivalent amounts of cDNA. The relative amounts of target genes in different samples were compared with the amount of the internal standard control gene, glyceraldehyde-3-phosphate dehydrogenase (G3PDH). The PCR primers used to detect astrocyteand myelin-related mouse genes are listed in Table 1. Amplified PCR products were resolved by electrophoresis with 2% agarose gels and photographed with a Kodak DC120 digital camera (Eastman Kodak, Rochester, NY).
Table 1.
Primer sequences used for RT-PCR
| Gene | PCR primers |
|---|---|
| G3PDH | 5′-AGAACATCATCCCTGCATCC
5′-AGCCGTATTCATTGTCATACC |
| GFAP | 5′-AAAACCGCATCACCATTCC
5′-CCTCTTCCCTTCCAATTCTAAC |
| MAG | 5′-ACCCCATCCTTACCATCTTC
5′-TACACAAACTCCCTCTCCG |
| MBP | 5′-ATAACCATTCCCTGCCTCC
5′-TCAACCATCACCTGCCTTC |
| Nogo-A | 5′-ACAAAGCTCTCCACTGAGCC
5′-TTCATTAGCATCACAGCCTCC |
| Vimentin | 5′-CAGCAGTATGAAAGCGTGG
5′-GGAAGAAAAGGTTGGCAGAG |
Transmission electron microscopy
In mice used for ultrastructural studies, the crush site was labeled with a suture. Mice were sacrificed by cervical dislocation, and the eyeballs were dissected, fixed with Karnovsky’s fixative (pH 7.4) for 24 hours at 4°C, and embedded in Epon-Araldite. Semithin sections (1–2 μm) of the optic nerves at 500 μm posterior to the crush site were prepared and stained with Toluidine Blue; ultrathin sections (60–90 nm) were stained with uranyl acetate and lead citrate. The sections were examined and photographed with a transmission electron microscope (EM410; Philips; Eindhoven, The Netherlands). Nine standard rectangular regions (234 μm2) randomly selected from each optic nerve section were photographed at 4400× magnification. All axons in the photomicrograph were counted.
Results
Overexpression of Bcl-2 leads to optic nerve regeneration in P3 mice
To ascertain whether Bcl-2 supports the intrinsic growth capacity of CNS axons, we sought to determine whether overexpression of Bcl-2 alone promotes optic nerve regeneration in mice aged P3 or younger, when the CNS environment is likely to be more permissive for growth. Optic nerve crush was performed in Bcl-2tg (Martinou et al., 1994) and wt littermate mouse pups. Transgenic mouse pups appeared normal and were phenotypically indistinguishable from their wt littermates. Regeneration was assessed by three labeling methods: anterograde axon tracing, immunofluorescent staining for regenerating axons, and retrograde labeling of RGCs that regenerated their axons. An anterograde tracer, CTB-F or CTB-R, was injected into the eye immediately after the nerve was crushed. In sham-operated mice (n=6) and mice with incomplete optic nerve crush (n=4), CTB was transported along the undamaged axons to the lateral geniculate nuclei (LGN) within 24 hours and reached its final target, the SC, within 48 hours.
We first assessed optic nerve regeneration in P3 wt and Bcl-2tg mouse pups at 24–48 hours after surgery (day 1–2) (Fig. 1). No regenerative response was observed in wt mice (n=11) (Fig. 1A,B). CTB-labeled axons stopped proximal to the crush site, and morphological analysis showed ‘bulb-like’ structures, signifying rapid, ongoing axonal degeneration (Fig. 1A). Similar results were obtained by immunofluorescence staining for GAP-43 (not shown) and NF-M (Fig. 1B), markers of regenerating/live axons.
Fig. 1.
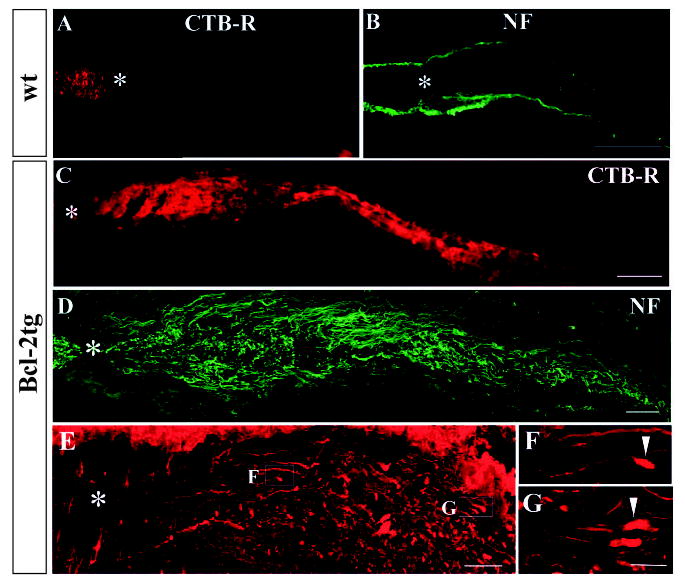
Robust and rapid optic nerve regeneration in P3 Bcl-2tg mice. Photomicrograph montages of adjacent longitudinal optic nerve sections and confocal epifluorescence photomicrographs show axonal morphology of wt (A,B) and Bcl-2tg (C-G) mice at 24 (A-D) and 48 hours (E-G) after optic nerve crush. The sections revealed CTB-R labeling (A,C) or were stained with antibodies against NF-M (B,D-G). F and G show highermagnification views of axon and growth cone morphologies (100×). Asterisks indicates the crush site; arrowheads indicate growth-cone-like structures. Bar, 100 μm (A-C); 40 μm (D,E); 10 μm (F,G).
Remarkably, in all Bcl-2tg mice (n=8), large numbers of CTB-labeled axons extended beyond the lesion site on day 1 (Fig. 1C), indicating robust axonal regeneration. The crush site was clearly identified by a traumatized zone containing degenerated cells and tissue debris or by staining for isolectin, a marker of activated microglia (Ohmi et al., 2003) (data not shown). At day 1 after crush, CTB-labeled axons stopped 500–1000 μm caudal to the lesion. No labeling was seen beyond this point or in the brain sections, suggesting that these were not fibers spared from crush injury, which would have passed through the optic chiasm into the brain at this time point. Since CTB labeling does not reveal fine morphology of axons, we assessed the extent of axon regeneration in Bcl-2tg mice by using immunohistochemistry. Similar patterns of axonal regeneration in Bcl-2tg mice were observed by immunofluorescence labeling of GAP-43 (not shown) and NFM (Fig. 1D).
On day 2, axonal degeneration was further confirmed in all optic nerve sections of wt mice (n=19), which contained only degenerating fibers. In the Bcl-2tg mice, however, labeled axons extended further and stopped 1000–2000 μm caudal to the lesion (n=5), as shown by CTB and immunofluorescence labeling. Many axons terminated in bullet-shaped structures closely resembling growth cones (Fig. 1E-G), consistent with active growth rather than mere survival of axons after injury.
Target recognition and innervation by regenerating axons in Bcl-2tg mice
By day 4, regenerating axons had entered the brain in Bcl-2tg mice (n=12) (Fig. 2A–F). Therefore, we examined CTBlabeled axon trajectories reconstructed in three dimensions in coronal brain sections from days 1–4. As expected, no CTB labeling was found in the brains of wt mice after nerve injury or in Bcl-2tg mice on day 1 and 2 after injury. However, extensive CTB labeling appeared in the brain sections of Bcl-2tg mice on day 4 (n=6). Most labeled axons traveled along optic tract pathways and reached their midbrain targets, including the LGN, pretectal nuclei, and SC (Fig. 2A–F). Aberrant projections to areas outside of these pathways or targets were noticed. Surprisingly, in all cases examined, regenerating axons predominantly innervated their ipsilateral brain targets (Fig. 2A–F); contralateral labeling of regenerating axons was also detected but was much less dense than the ipsilateral. This projection pattern mirrored that in shamoperated controls (Fig. 2G,H), where more than 90% of RGC axons innervated the contralateral brain targets. These results further demonstrate axonal regeneration, rather than mere survival, after nerve injury in Bcl-2tg mice and suggest that regenerating axons follow guidance cues along the optic pathways and find the appropriate visual targets in the midbrain, although they reached the ipsilateral side. Overall, from days 1–4, axons failed to grow and rapidly degenerated in 33 of 34 wt mice, whereas robust optic nerve regeneration, with many axons passing beyond the lesion sites and/or innervating the brain target, was seen in 24 (96%) of 25 Bcl-2tg mice.
Fig. 2.
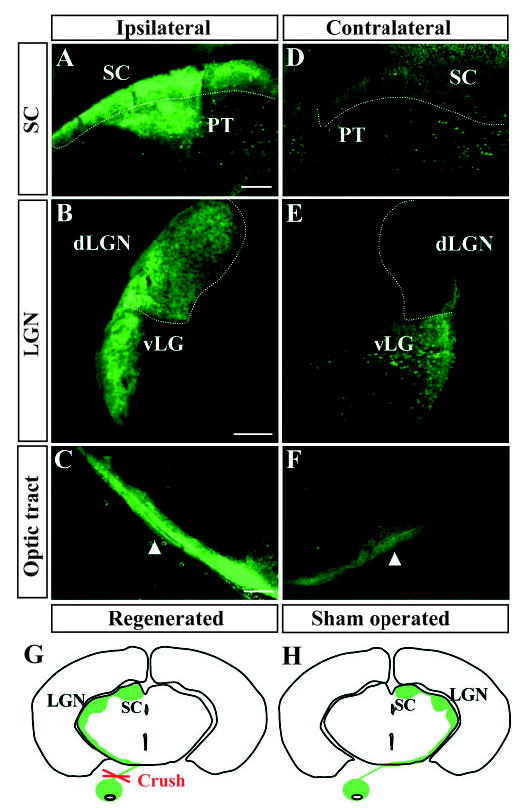
The majority of RGC axons in Bcl-2tg mice regenerate and reach the ipsilateral brain targets within 4 days. (A-F) Epifluorescence photomicrographs of coronal brain sections from a Bcl-2tg mouse on day 4. Note green fluorescence (CTB) in the ipsilateral SC, pretectal nuclei (PT) (A), dorsal (dLGN) and ventral LGN (vLGN) (B), and the optic tract (C). Weak fluorescence is present in the corresponding contralateral targets (D-F). Dotted lines outline the SC and dLGN. Arrows indicate regenerating axons showing positive fluorescence in the optic tract. Bar, 200 μm. (G,H) Schematic drawings of CTB-labeled retinofugal projections in coronal sections of mouse brain. (G) Retinofugal projection formed by regenerating axons in Bcl-2tg mice. (H) Retinofugal projections in normal wt and Bcl-2tg mice. No apparent abnormality of retinal axon projection is noted in uninjured Bcl-2tg mice compared with wt controls.
To determine the percentage of RGCs that regenerated their axons, we placed a retrograde tracer, FluoroGold, bilaterally into the SC (the most distal brain target of the optic nerve) after nerve crush or sham operation and counted labeled RGCs (Fig. 3A-D). In uninjured mice, FluoroGold labeled numerous round or oval RGCs, as well as spiny microglia, which were usually found in different optical planes with RGCs. Since Bcl-2 overexpression prevents the developmental loss of RGCs, these cells were more abundant in uninjured Bcl-2tg mice than in wt mice (Bonfanti et al., 1996) (Fig. 3A,B,E). Eleven days after injury, very few labeled cells were detected in the retinas of wt mice, consistent with their lack of axon regeneration (Fig. 3C,E). By contrast, numerous labeled RGCs, equivalent to approximately 70% of those from the uninjured eye, were present in the retinas of Bcl-2tg mice (n=6) (Fig. 3D,E). Thus, the majority of RGC axons in Bcl-2tg mice regenerated over long distances and reached the SC, while none of those from wt mice did so under similar conditions.
Fig. 3.
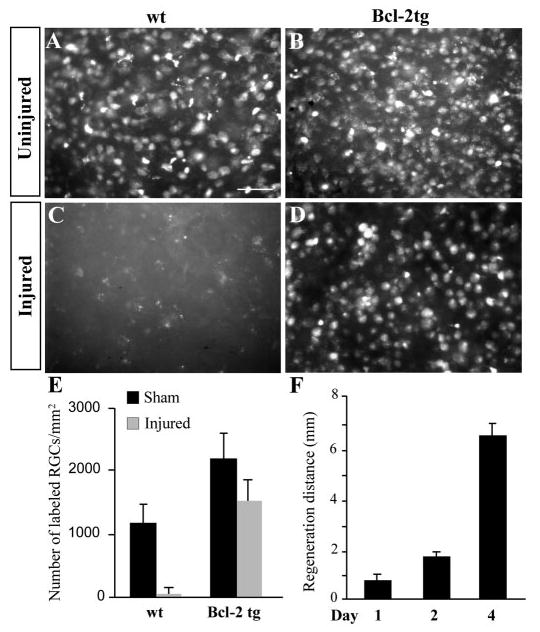
Quantitative assessment of axonal regeneration. (A-D) Photomicrographs of FluoroGold-labeled RGCs in wholemount retinas from injured and uninjured wt and Bcl-2tg mice on day 11 after optic nerve crush. Bar, 50 μm. (E) Number of retrogradely labeled RGCs in the retinal whole-mounts of wt and Bcl-2tg mice that underwent optic nerve crush (injured) or a sham procedure. (F) Distance of axonal regeneration on days 1–4. Values are mean±s.e.m.
Bcl-2 supports the intrinsic growth capacity of mature RGC axons
To further assess the ability of Bcl-2 to maintain the intrinsic growth potential of RGC axons, we determined the speed of axon regeneration in Bcl-2tg mice (Fig. 3F). By subtracting the distance of axon regeneration measured on day 2 (1.0–2.0 mm) from that on day 1 (0.5–1.0 mm), we obtained a rate of 0.5–1.5 mm/day for the fastest growing axons. Using an alternative method, we measured the full length of the optic nerve and added it to the length of the optic tract from the optic chiasm to the posterior border of the SC in mice on day 4, when regenerating axons normally reached the SC. The total distance was 6–8 mm, yielding an average rate of 1.5–2.0 mm/day. Thus, Bcl-2 induced optic nerve regeneration at 0.5–2 mm/day for the fastest growing axons, a rate similar to that reported during embryonic stages (Goldberg et al., 2002; Jhaveri et al., 1991). Therefore, overexpression of Bcl-2 appears to prevent the loss of the intrinsic regenerative potential of neonatal RGC axons.
Next, we asked whether overexpression of Bcl-2 supports the intrinsic growth capacity of RGC axons up to adulthood. Using retina-midbrain slice co-cultures, we previously showed that retinas of adult Bcl-2tg mice grow axons into a permissive brain environment (e.g. a brain slice obtained at E14) (Chen et al., 1997), which suggests that their RGCs maintain their intrinsic growth potential. We confirmed this in P14 retinamidbrain co-cultures, where the retina is populated with mature RGCs. A retinal explant from a P14 mouse was placed against a midbrain slice from an E14 or a P14 mouse (before and after glial maturation and myelination). Consistent with their lack of intrinsic growth capacity, P14 wt retinal explants exhibited poor axonal growth regardless of the age of the brain slice (data not shown). By contrast, P14 Bcl-2tg explants extended few axons into P14 brain slices, but grew robustly and innervated E14 brain slices, confirming that mature retinas can grow axons a permissive environment (Fig. 4A).
Fig. 4.

Onset of optic nerve regenerative failure in P5 Bcl-2tg mice coincides with astrocyte maturation. (A,B) Quantification of retinal axon regrowth in retina-midbrain slice co-cultures. Values are mean±s.e.m. (**P<0.01; ***P<0.001). (C) Photomicrograph montages of adjacent longitudinal optic nerve sections at day 4 after optic nerve crush in a P5 wt mouse and a Bcl-2tg mouse. The asterisk indicates the crush site. Bar, 250 μm. (D,E) Western blot analysis (D) and RT-PCR (E) reveal developmental expression patterns of myelin/oligodendrocyte-associated proteins and astrocyte markers in E14-P14 mouse midbrains. (F) Western blot analysis of GFAP expression in P0 and P5 normal (−) and injured (+) midbrain tissues. (G) Western blot analysis confirms the absence of MBP and MAG in the midbrains of jimpy mice. (H) Quantification of retinal axon regrowth into midbrain slices of P14 wt and jimpy mice in retina-midbrain slice co-cultures. Values are mean±s.e.m. (P=0.6).
Onset of optic nerve regenerative failure in Bcl-2tg mice after P5
To confirm that optic nerve regeneration fails in Bcl-2tg mice after P5 and to identify the mechanism that causes this failure, we performed optic nerve crush on P5 wt and Bcl-2tg mouse pups. In contrast to the robust regeneration in Bcl-2tg mice injured on P3, surviving fibers stopped anterior to the crush site and failed to grow in most Bcl-2tg mice injured on P5 (n=16). By day 4, in a few cases (n=2), labeled axons extended about 200 μm posterior to the crush site but not beyond (Fig. 4C). In all wt mice (n=15), few axons in optic nerve sections were identified by CTB or anti-GAP-43 labeling, even anterior to the crush site, indicating massive axonal degeneration (Fig. 4C). None of the mouse brain sections, regardless of genotype, was positive for CTB. These findings are consistent with previous reports that overexpression of Bcl-2 fails to support optic nerve regeneration after P5 (Chierzi et al., 1999; Lodovichi et al., 2001).
To provide further evidence that the regenerative failure in P5 Bcl-2tg mice reflects a maturational change in the CNS environment, we set up a series of co-culture experiments. Retinal explants of P14 Bcl-2tg mice were placed against E14-P14 midbrain slices containing the RGC target (the SC). Again, P14 retinal explants of Bcl-2tg mice growth robustly into E14 brain slices (Fig. 4A,B). The growth inhibition presented by the midbrain slices increased with age and was maximal at P4; few axons from the P14 retinas of Bcl-2tg mice grew into the SC of P4 mice (Fig. 4B). This growth inhibition appeared to remain unchanged in midbrain slices after P4; similar numbers of axons grew from P14 retinal explants into the brain slices of P4 or older mice. Thus, the growth inhibitory mechanisms in the brain environment appears to reach its adult level at around P4, which coincides with the onset of optic nerve regenerative failure in Bcl-2tg mice.
Next, we looked for correlations between the onset of growth inhibition and developmental events in the midbrain SC, focusing on CNS myelin formation and on the maturation of astrocytes or their ability to form glial scars, both of which have been proposed to contribute critically to growth inhibitory mechanisms (McGee and Strittmatter, 2003; Silver, 1994). Expression of the myelin markers MBP, MAG and proteolipid protein was not detectable in the SC at E14-P8, long after the observed onset of midbrain growth inhibition (Fig. 4D,E). Expression of Nogo-A, an oligodendrocyte/myelin-associated protein, however, was detected in the SC as early as E14, when the midbrain environment is highly permissive for axon extension (Fig. 4E). These results are similar to those reported by others (Hunt et al., 2002; Jhaveri et al., 1992; Josephson et al., 2002) and suggest that the expression of myelin/oligodendrocyte-associated proteins does not correlate with the onset of CNS growth inhibition in Bcl-2tg mice.
To further determine whether CNS myelin affects the ability of retinal axons to regenerate, we cultured retinal explants of P14 Bcl-2tg mice with brain slices of wt and jimpy mice (Vela al., 1998). The absence of CNS myelin and associated proteins in the midbrains of jimpy mice was confirmed by western blot analysis (Fig. 4G). However, there was no significant difference in axonal invasion into wt and jimpy brain slices (n=3/group) (Fig. 4H), suggesting that CNS myelin does not prevent optic nerve regeneration in Bcl-2tg mice.
To assess the maturation of astrocytes in the midbrain, we examined the expression of vimentin (highly expressed in immature astrocytes) and GFAP (a marker of mature astrocytes) (Dahl et al., 1981; Lazarides, 1982). GFAP expression increased in parallel with growth inhibition, being absent at E14, when the environment is permissive for growth, and rising steadily to a plateau at about P4 (Fig. 4D,E). Conversely, vimentin expression in the SC was high at E14 and decreased thereafter (Fig. 4D,E). GFAP expression was also much higher in the midbrains of P5 mice than P0 mice after injury, indicating a greater ability of astrocytes to become reactive after injury at P5 than at P0 (Fig. 4F). Thus, growth inhibition appears to increase as astrocytes mature and acquire the ability to become reactive but is not associated with the presence of CNS myelin.
Inducing optic nerve regeneration by Bcl-2 overexpression and local elimination of astrocytes
We hypothesized that if mature astrocytes are key barriers to optic nerve regeneration in adult mice, elimination of these cells would reverse the growth failure of the severed optic nerves in adult Bcl-2tg mice. To test this, we treated adult (2–3-month-old) wt and Bcl-2tg mice with astrotoxin (L-α-aminoadipate), which selectively kills astrocytes but has minimal effects on surrounding neurons and myelin (Khurgel et al., 1996; McGraw et al., 2001; Takada and Hattori, 1986). Immediately after optic nerve crush, gelfoam soaked in L-alphaaminoadipate solution (10 mg/ml in PBS, Sigma) or in PBS was placed against the crush site. As shown by immunofluorescence staining, astrotoxin created an astrocyte-free, GFAP-negative area in the optic nerve (Fig. 5A,B) but had little effect on myelin and neuronal processes (Fig. 5A-H). In the absence of astrotoxin, no sign of axonal regeneration was seen in adult wt or Bcl-2tg mice. Immunofluorescence labeling of GAP-43 showed that nerve fibers retracted in wt mice, but survived and failed to grow in Bcl-2tg mice (n=3). After astrotoxin treatment in wt mice, a few axons sprouted past the lesion site and grew into the GFAP-negative area (n=5) (Fig. 5A,C). These axons appeared to grow slowly and stopped not far beyond the crush site. By contrast, robust regeneration was seen in all astrotoxin-treated Bcl-2tg mice (n=5). Regenerating axons extended exclusively into and normally reached the end of the GFAP-negative area, but then stopped abruptly where GFAP staining appeared (Fig. 5B,D). Unlike GFAP expression, intense MBP staining colocalized with regenerating axons in most optic nerve sections (Fig. 5E,F), providing further evidence that CNS myelin does not necessarily obstruct axonal regrowth.
Fig. 5.
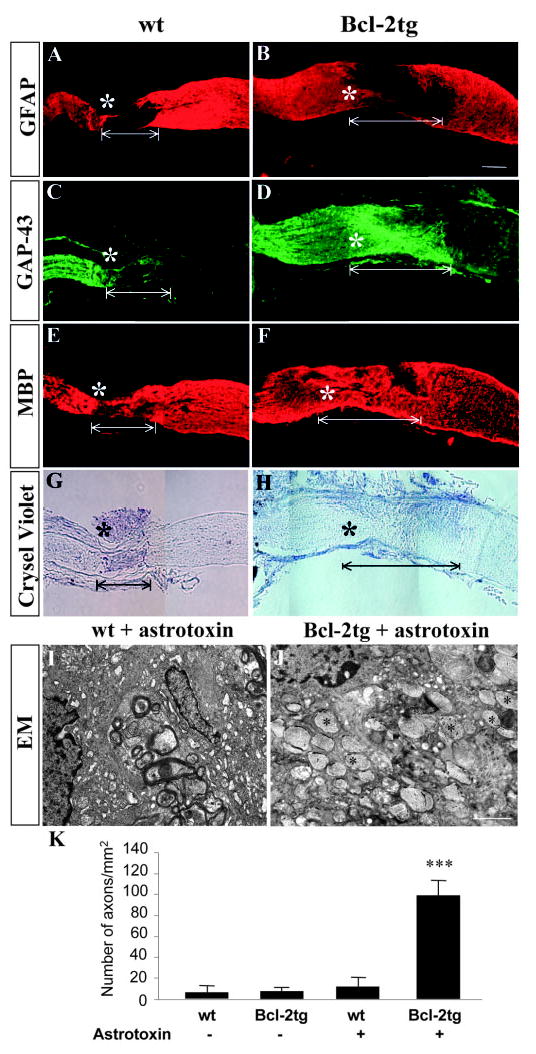
Robust optic nerve regeneration in adult Bcl-2tg mice after treatment with astrotoxin. (A-H) Photomicrograph montages of adjacent longitudinal optic nerve sections on day 8 after optic nerve crush in adult wt and Bcl-2tg mice. The asterisk indicates the crush site. Bar, 250 μm. (I,J) Electron micrographs (EM) of optic nerve sections collected at 0.5 mm posterior to the crush from wt (I) and Bcl-2tg (J) mice treated with astrotoxin. Asterisks indicate crush sites; white lines mark GFAP-negative area. Bar, 1 μm. (K) Quantification of regenerating axons from optic nerve sections. Values are mean±s.e.m. (***P<0.001).
To quantify axonal regeneration, we used electron microscopy to examine optic nerve sections of adult wt and Bcl-2tg mice treated with astrotoxin or PBS. In all groups, degenerating myelin and tissue debris were found in sections collected 0.5 mm posterior to the crush site (Fig. 5I,J). In astrotoxin-treated Bcl-2tg mice (n=5), whose optic nerve sections were devoid of astrocytes, many regenerating axons were consistently observed (Fig. 5J). Most axons were unmyelinated, suggesting they were newly regenerated. In the other groups (n=3/group), however, few axons were present in optic nerve sections collected 0.5 mm posterior to the crush site. Regenerating axons were 20-fold more abundant in astrotoxin-treated Bcl-2tg mice than in PBS-treated wt mice (P<0.001) (Fig. 5K). Thus, elimination of mature astrocytes promoted robust optic nerve regeneration in adult Bcl-2tg mice.
Optic nerve regeneration in Bcl-2tgGFAP−/−Vim−/− mice
To further assess the role of reactive astrocytes in preventing optic nerve regeneration in vivo, we studied GFAP−/−Vim−/− mice, a model characterized by the lack of intermediate filaments, decreased ability to form compact glial scars (Pekny et al., 1999), limited hypertrophy of astrocytic processes after injury, and improved post-traumatic synaptic regeneration (Wilhelmsson et al., 2004) (reviewed by Pekny and Pekna, 2004). Retinal development, axonal projections, and optic nerve myelination in these mice appear to be normal (Kinouchi et al., 2003; Lundkvist et al., 2004).
Optic nerve crush was performed in wt, Bcl-2tg, GFAP−/−Vim−/− and Bcl-2tgGFAP−/−Vim−/− mice on P5 or P14, after astrocytes have become more mature and growth inhibition in the midbrain has reached its peak. In wt mice, most axons degenerated and rapidly retracted after injury (P14, n=4). In Bcl-2tg mice, many severed axons survived but failed to regenerate and stopped anterior to the lesion site (P5, n=6; P14, n=3). In GFAP−/−Vim−/− mice (P5, n=6; P14, n=4), as in the astrotoxin-treated group, a few labeled axons grew past the lesion and extended 100–200 μm posterior to the crush site by day 4 (Fig. 6A,B). The estimated rate of axon extension was 25–50 μm/day, similar to that of axonal sprouting from mature RGC axons (<60 μm/day). By contrast, severed axons regenerated rapidly and extensively in all Bcl-2tgGFAP−/− Vim−/− mice (P5, n=4; P14, n=4) (Fig. 6C-G). Many regenerating axons had structures resembling growth cones at their tips (Fig. 6E-G). By day 4 after injury at both ages examined, labeled axons had extended the entire length of the optic nerve and innervated their brain targets – the LGN and SC – on the ipsilateral side (Fig. 6H,I). Aberrant projections to areas outside of the visual targets were more numerous in Bcl-2tgGFAP−/−Vim−/− mice than in Bcl2tg mice injured on P3. Although the P14 optic nerves and brains of Bcl-2tgGFAP−/−Vim−/− mice were myelin-enriched compared with those at P5, the extent of axon regeneration appeared similar at both ages. However, although Bcl-2tgGFAP−/−Vim−/− mice exhibited a prolonged window of optic nerve regeneration, regeneration did not occur in adult mutant mice (>2 months; n=4). Thus, eliminating GFAP and vimentin appears to delay or attenuate, rather than abolish, growth inhibition by mature reactive astrocytes.
Fig. 6.
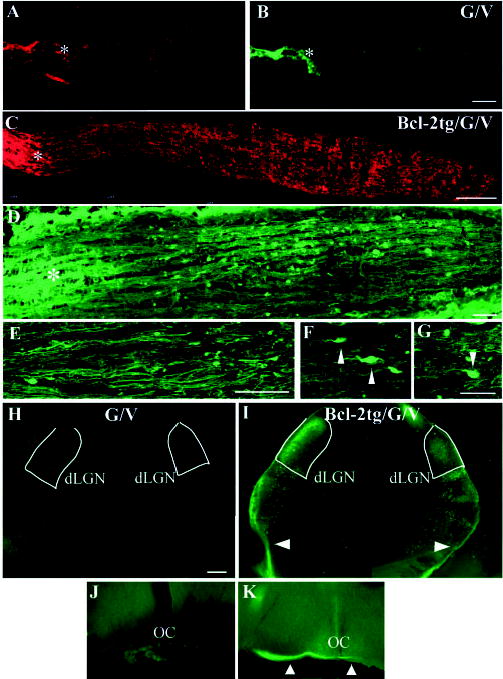
Robust optic nerve regeneration and target innervation in Bcl-2tgGFAP−/−Vim−/− mice after P5. (A-D) Photomicrograph montages of adjacent longitudinal optic nerve sections on day 4 after optic nerve crush in P5 GFAP−/−Vim−/− (G/V) (A,B) and Bcl-2tgGFAP−/−Vim−/− (Bcl-2tg/G/V) (C,D) mice. The asterisk indicates the crush site. Bar, 100 μm. (E-G) Higher-power views of axon and growth cone morphology (40×) revealed by anti-NF staining. Bars, 40 μm (E); 20 μm (F,G); 5 μm (H-K). Epifluorescence photomicrographs of coronal brain sections from GFAP−/−Vim−/− (H,J) and Bcl-2tgGFAP−/−Vim−/− (I,K) mice subjected to optic nerve crush on P14 and examined 4 days later. Note the positive labeling in the ipsilateral optic chiasm (K), dLGN and vLGN, and the optic tract (arrowheads) (I) of Bcl-2tgGFAP−/−Vim−/− brain sections. Dotted lines outline the dLGN. Bar, 200 μm.
To quantify regeneration we placed FluoroGold in the SCs of wt, Bcl-2tg, GFAP−/−Vim−/− and Bcl-2tgGFAP−/−Vim−/− mice at P5 immediately after optic nerve crush to label RGCs whose axons connected or regenerated to the SC (Fig. 7). Since Bcl-2tg and Bcl-2tgGFAP−/−Vim−/− mice normally have more RGCs in the retina than wt and GFAP−/−Vim−/− mice, the number of RGCs with regenerated axons was expressed as a percentage of RGCs in the uninjured retina (Fig. 7D). Despite the increase in aberrant axonal projections after regeneration, approximately 30% of the severed axons reached the SC in Bcl-2tgGFAP−/−Vim−/− mice (n=3). In the three other groups (n=3/group), only a few cells were labeled. Thus, through their ability to become reactive and form scars after injury, mature astrocytes represent a key barrier to optic nerve regeneration in older Bcl-2tg mice. Attenuation of glial scarring after injury is sufficient to allow rapid regeneration of the severed optic nerve over long distances and target innervation in mature Bcl-2tg mice. Our results show that induction of Bcl-2 expression and manipulation of reactive astrocytes promotes CNS regeneration in postnatal mice.
Fig. 7.

Target innervation by regenerating axons of Bcl-2tgGFAP−/−Vim−/− mice after P5. Photomicrographs of whole-mount retinas from Bcl-2tg (A), GFAP−/−Vim−/− (B), and Bcl-2tgGFAP−/−Vim−/− (C) mice on day 11 after optic nerve injury, showing FluoroGold-labeled RGCs. Bar, 50 μm. (D) Number of retrogradely labeled RGCs in retinal whole-mounts. Values are mean±s.e.m. (***P<0.001).
Discussion
These studies demonstrate two parallel mechanisms that affect optic nerve regeneration: an intrinsic developmental program for axon elongation supported by Bcl-2 and inhibition of axonal growth mediated by reactive astrocytes. By increasing Bcl-2 expression in neurons and simultaneously attenuating reactive gliosis, we succeeded to restore the regenerative potential of mature RGC axons up to P14 and achieved a rapid regeneration of severed optic nerves over long distances, culminating in target innervation.
Mouse RGCs lose Bcl-2 expression and the intrinsic mechanisms that support axonal growth around E18 (Chen et al., 1997). Our findings showed that overexpression of Bcl-2 was sufficient to maintain the regenerative potential of RGC axons in the postnatal stage and allowed robust regeneration of severed CNS axons in vivo in a permissive brain environment. However, Bcl-2 did not affect the development or maturation of RGCs. The patterns of expression of RGC markers and proteins associated with neuronal differentiation, such as Brn-3b, β-III tubulin, microtubule-associated protein 2, NF-M, p53, and p21WAF1, were identical in Bcl-2tg mice and wt mice (data not shown). Expression of Bcl-2 restored the growth rate of regenerating axons of postnatal RGCs to values characteristic of embryonic life, suggesting a novel function of Bcl-2 in addition to its regulation of apoptosis. Although the exact molecular mechanism is unclear, enhanced intracellular calcium signaling has been implicated (Jiao et al., 2005).
Our findings also indicate that reactive astrocytes contribute critically to the blockade of axonal regeneration. Although glial scarring has long been thought to have inhibitory effects, treatments aiming at suppressing or eliminating those effects produced limited regrowth, which usually stopped before the axons reached their targets. Thus, it was unclear whether components of glial scarring are essentially involved in CNS regenerative failure or whether other factors, such as myelin-associated growthinhibitory proteins, are more important in blocking CNS regeneration.
It was previously suggested that the failure of neurite outgrowth from transplanted neurons in vivo is associated with reactive glial cells (Davies et al., 1997; Davies et al., 1999). By taking advantage of the fact that mature RGC axons of Bcl-2tg mice have improved their intrinsic capacity to regenerate, we demonstrated that induction of reactive astrocytes after injury is a key barrier to axonal regeneration. Genetic suppression of glial scar formation (Pekny et al., 1999) or application of astrotoxin to destroy astrocytes without disrupting CNS myelin (Khurgel et al., 1996; McGraw et al., 2001) eliminated the growth barrier presented by the CNS environment and supported vigorous optic nerve regeneration in Bcl-2tg mice aged P4 to P14. Interestingly, eliminating GFAP and vimentin failed to support optic nerve regrowth in adult Bcl-2tgGFAP−/−Vim−/− mice. The absence of intermediate filaments in astrocytes did not abolish the production of chondroitin sulfate proteoglycan (data not shown), but might have delayed the onset of astrocyte maturation and prolonged the window of optic nerve regeneration in GFAP−/−Vim−/− mice. These data support the hypothesis that mature astrocytes present a key barrier to CNS axon regeneration in vivo.
Elongation of mature RGC axons is distinct from axonal plasticity – slow sprouting and collateral formation of severed CNS axons. Several recent studies indicated that axonal plasticity does exist in the adult CNS of mammals and that it can contribute to functional improvement after injury under certain circumstances (Fouad et al., 2001; Menet et al., 2003; Wilhelmsson et al., 2004). Our findings support and extend those observations: mature RGCs showed limited growth ability and a small number of their axons extended slowly past the lesion site in a permissive environment (in astrotoxintreated or GFAP−/−Vim−/− mice). By contrast, mature RGC axons in Bcl-2tgGFAP−/−Vim−/− mice regenerated robustly and rapidly, at the speed of embryonic axon elongation. Indeed, many severed CNS axons in postnatal mice regenerated for long distances and reached the appropriate targets in vivo.
Our results cannot be explained by incomplete injury of the optic nerve. First, in every case, the crush site in the optic nerve was identified both by a traumatized zone containing degenerated cells and tissue debris, and by immunofluorescence labeling of microglial markers in optic nerve sections. Second, numerous growth cones were observed at the growing tip of the axons, strongly indicating active fiber regeneration that would not be observed with spared axons. Third, labeled axons extended for various distances caudal to the lesion at 1–4 days after injury, strongly suggesting that these were regenerated axons, since labeling of uncut, spared axons would be seen in the entire optic nerve and the brain within 24 hr after injury. Finally, labeled axons reached ipsilateral brain targets after day 4, consistent with regeneration, since the large majority (>90%) of uncut, spared axons would be found in the contralateral midbrain targets.
Recently, expression of EphB1 by a subset of RGCs was reported to contribute critically to the formation of ipsilateral retinal projections through its repulsive interaction with ephrin-B2 in glial cells of the optic chiasm (Williams et al., 2003). EphB1-null mice exhibit a highly reduced ipsilateral projection. During development, EphB1 is found exclusively in regions of the retina that give rise to the ipsilateral projection. After E17, when the retinal ipsilateral projection has formed, EphB1 expression is no longer confined to a small population of RGCs but is upregulated in other parts of the retina. A likely hypothesis is that wide expression of EphB1 in postnatal RGCs directs the majority of regenerating axons to the ipsilateral brain targets in Bcl-2tg and Bcl-2tg/GFAP−/−Vim−/− mice.
We conclude that Bcl-2 plays a key role in supporting the intrinsic growth mechanisms for CNS axon elongation in vivo, whereas reactive astrocytes, acting as another major player in the environment, prevent axon regeneration. Thus, activating Bcl-2-supported growth mechanisms in CNS neurons and manipulating astrocyte function are two promising avenues for promoting regeneration in the adult mammalian CNS.
Acknowledgments
We thank William D. Snider for comments and Patricia Pearson for electron microscopy. This study was supported by grants to D.F.C. from the National Institutes of Health/National Eye Institute, the Massachusetts Lion’s Eye Research Fund, the Department of the Army and the Minda de GunzBurg Research Center for Retinal Transplantation at the Schepens Eye Research Institute, and by grants to M.P. from the Swedish Cancer Foundation, the Swedish Research Council (11548), the Swedish Society for Medicine, the Swedish Society for Medical Research, the King Gustaf V Foundation, Volvo Assar Gabrielsson Fund and the Swedish Stroke Foundation.
References
- Bonfanti L, Strettoi E, Chierzi S, Cenni MC, Liu XH, Martinou JC, Maffei L, Rabacchi SA. Protection of retinal ganglion cells from natural and axotomy-induced cell death in neonatal transgenic mice overexpressing bcl-2. J Neurosci. 1996;16:4186–4194. doi: 10.1523/JNEUROSCI.16-13-04186.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bregman BS. Regeneration in the spinal cord. Curr Opin Neurobiol. 1998;8:800–807. doi: 10.1016/s0959-4388(98)80124-4. [DOI] [PubMed] [Google Scholar]
- Chen DF, Tonegawa S. Why do mature CNS neurons of mammals fail to re-establish connections following injury-functions of bcl-2. Cell Death Differ. 1998;5:816–822. doi: 10.1038/sj.cdd.4400431. [DOI] [PubMed] [Google Scholar]
- Chen DF, Jhaveri S, Schneider GE. Intrinsic changes in developing retinal neurons result in regenerative failure of their axons. Proc Natl Acad Sci USA. 1995;92:7287–7291. doi: 10.1073/pnas.92.16.7287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen DF, Schneider GE, Martinou JC, Tonegawa S. Bcl-2 promotes regeneration of severed axons in mammalian CNS. Nature. 1997;385:434–439. doi: 10.1038/385434a0. [DOI] [PubMed] [Google Scholar]
- Chierzi S, Strettoi E, Cenni MC, Maffei L. Optic nerve crush: axonal responses in wild-type and bcl-2 transgenic mice. J Neurosci. 1999;19:8367–8376. doi: 10.1523/JNEUROSCI.19-19-08367.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colucci-Guyon E, Portier MM, Dunia I, Paulin D, Pournin S, Babinet C. Mice lacking vimentin develop and reproduce without an obvious phenotype. Cell. 1994;79:679–694. doi: 10.1016/0092-8674(94)90553-3. [DOI] [PubMed] [Google Scholar]
- Cristino L, Pica A, Della Corte F, Bentivoglio M. Coinduction of nitric oxide synthase, bcl-2 and growth-associated protein-43 in spinal motoneurons during axon regeneration in the lizard tail. Neuroscience. 2000;101:451–458. doi: 10.1016/s0306-4522(00)00393-6. [DOI] [PubMed] [Google Scholar]
- Dahl D, Rueger DC, Bignami A, Weber K, Osborn M. Vimentin, the 57 000 molecular weight protein of fibroblast filaments, is the major cytoskeletal component in immature glia. Eur J Cell Biol. 1981;24:191– 196. [PubMed] [Google Scholar]
- Davies SJ, Fitch MT, Memberg SP, Hall AK, Raisman G, Silver J. Regeneration of adult axons in white matter tracts of the central nervous system. Nature. 1997;390:680–683. doi: 10.1038/37776. [DOI] [PubMed] [Google Scholar]
- Davies SJ, Goucher DR, Doller C, Silver J. Robust regeneration of adult sensory axons in degenerating white matter of the adult rat spinal cord. J Neurosci. 1999;19:5810–5822. doi: 10.1523/JNEUROSCI.19-14-05810.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fouad K, Dietz V, Schwab ME. Improving axonal growth and functional recovery after experimental spinal cord injury by neutralizing myelin associated inhibitors. Brain Res Brain Res Rev. 2001;36:204–212. doi: 10.1016/s0165-0173(01)00096-0. [DOI] [PubMed] [Google Scholar]
- Fournier AE, Strittmatter SM. Repulsive factors and axon regeneration in the CNS. Curr Opin Neurobiol. 2001;11:89–94. doi: 10.1016/s0959-4388(00)00178-1. [DOI] [PubMed] [Google Scholar]
- Goldberg JL, Barres BA. The relationship between neuronal survival and regeneration. Annu Rev Neurosci. 2000;23:579–612. doi: 10.1146/annurev.neuro.23.1.579. [DOI] [PubMed] [Google Scholar]
- Goldberg JL, Klassen MP, Hua Y, Barres BA. Amacrine-signaled loss of intrinsic axon growth ability by retinal ganglion cells. Science. 2002;296:1860–1864. doi: 10.1126/science.1068428. [DOI] [PubMed] [Google Scholar]
- Hilton M, Middleton G, Davies AM. Bcl-2 influences axonal growth rate in embryonic sensory neurons. Curr Biol. 1997;7:798–800. doi: 10.1016/s0960-9822(06)00339-3. [DOI] [PubMed] [Google Scholar]
- Horner PJ, Gage FH. Regenerating the damaged central nervous system. Nature. 2000;407:963–970. doi: 10.1038/35039559. [DOI] [PubMed] [Google Scholar]
- Huang X, Wu DY, Chen G, Manji H, Chen DF. Support of retinal ganglion cell survival and axon regeneration by lithium through a Bcl-2-dependent mechanism. Invest Ophthalmol Vis Sci. 2003;44:347–354. doi: 10.1167/iovs.02-0198. [DOI] [PubMed] [Google Scholar]
- Hunt D, Coffin RS, Anderson PN. The Nogo receptor, its ligands and axonal regeneration in the spinal cord; a review. J Neurocytol. 2002;31:93–120. doi: 10.1023/a:1023941421781. [DOI] [PubMed] [Google Scholar]
- Jhaveri S, Edwards MA, Schneider GE. Initial stages of retinofugal axon development in the hamster: evidence for two distinct modes of growth. Exp Brain Res. 1991;87:371–382. doi: 10.1007/BF00231854. [DOI] [PubMed] [Google Scholar]
- Jhaveri S, Erzurumlu RS, Friedman B, Schneider GE. Oligodendrocytes and myelin formation along the optic tract of the developing hamster: an immunohistochemical study using the Rip antibody. Glia. 1992;6:138–148. doi: 10.1002/glia.440060208. [DOI] [PubMed] [Google Scholar]
- Jiao, J. W., Huang, X. A., Feit-Leithman, R. A., Neve, R. L., Snider, W., Dartt, D. A. and Chen, D. F. (2005). Bcl-2 enhances Ca2+ signaling to support the intrinsic regenerative capacity of CNS axons. EMBO J. (in press). [DOI] [PMC free article] [PubMed]
- Josephson A, Trifunovski A, Widmer HR, Widenfalk J, Olson L, Spenger C. Nogo-receptor gene activity: cellular localization and developmental regulation of mRNA in mice and humans. J Comp Neurol. 2002;453:292–304. doi: 10.1002/cne.10408. [DOI] [PubMed] [Google Scholar]
- Khurgel M, Koo AC, Ivy GO. Selective ablation of astrocytes by intracerebral injections of alpha-aminoadipate. Glia. 1996;16:351– 358. doi: 10.1002/(SICI)1098-1136(199604)16:4<351::AID-GLIA7>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Kinouchi R, Takeda M, Yang L, Wilhelmsson U, Lundkvist A, Pekny M, Chen DF. Robust neural integration from retinal transplants in mice deficient in GFAP and vimentin. Nat Neurosci. 2003;6:863– 868. doi: 10.1038/nn1088. [DOI] [PubMed] [Google Scholar]
- Lazarides E. Intermediate filaments: a chemically heterogeneous, developmentally regulated class of proteins. Annu Rev Biochem. 1982;51:219– 250. doi: 10.1146/annurev.bi.51.070182.001251. [DOI] [PubMed] [Google Scholar]
- Li Y, Sauve Y, Li D, Lund RD, Raisman G. Transplanted olfactory ensheathing cells promote regeneration of cut adult rat optic nerve axons. J Neurosci. 2003;23:7783–7788. doi: 10.1523/JNEUROSCI.23-21-07783.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodovichi C, di Cristo G, Cenni MC, Maffei L. Bcl-2 overexpression per se does not promote regeneration of neonatal crushed optic fibers. Eur J Neurosci. 2001;13:833–838. doi: 10.1046/j.1460-9568.2001.01440.x. [DOI] [PubMed] [Google Scholar]
- Lundkvist A, Reichenbach A, Betsholtz C, Carmeliet P, Wolburg H, Pekny M. Under stress, the absence of intermediate filaments from Müller cells in the retina has structural and functional consequences. J Cell Sci. 2004;117:3481–3488. doi: 10.1242/jcs.01221. [DOI] [PubMed] [Google Scholar]
- Martinou JC, Dubois-Dauphin M, Staple JK, Rodriguez I, Frankowski H, Missotten M, Albertini P, Talabot D, Catsicas S, Pietra C, et al. Overexpression of BCL-2 in transgenic mice protects neurons from naturally occurring cell death and experimental ischemia. Neuron. 1994;13:1017–1030. doi: 10.1016/0896-6273(94)90266-6. [DOI] [PubMed] [Google Scholar]
- McGee AW, Strittmatter SM. The Nogo-66 receptor: focusing myelin inhibition of axon regeneration. Trends Neurosci. 2003;26:193– 198. doi: 10.1016/S0166-2236(03)00062-6. [DOI] [PubMed] [Google Scholar]
- McGraw J, Hiebert GW, Steeves JD. Modulating astrogliosis after neurotrauma. J Neurosci Res. 2001;63:109–115. doi: 10.1002/1097-4547(20010115)63:2<109::AID-JNR1002>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Menet V, Prieto M, Privat A, Gimenez y Ribotta M. Axonal plasticity and functional recovery after spinal cord injury in mice deficient in both glial fibrillary acidic protein and vimentin genes. Proc Natl Acad Sci USA. 2003;100:8999–9004. doi: 10.1073/pnas.1533187100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merry DE, Korsmeyer SJ. Bcl-2 gene family in the nervous system. Annu Rev Neurosci. 1997;20:245–267. doi: 10.1146/annurev.neuro.20.1.245. [DOI] [PubMed] [Google Scholar]
- Merry DE, Veis DJ, Hickey WF, Korsmeyer SJ. bcl-2 protein expression is widespread in the developing nervous system and retained in the adult PNS. Development. 1994;120:301–311. doi: 10.1242/dev.120.2.301. [DOI] [PubMed] [Google Scholar]
- Morgenstern DA, Asher RA, Fawcett JW. Chondroitin sulphate proteoglycans in the CNS injury response. Prog Brain Res. 2002;137:313–332. doi: 10.1016/s0079-6123(02)37024-9. [DOI] [PubMed] [Google Scholar]
- Oh YJ, Swarzenski BC, O’Malley KL. Overexpression of Bcl-2 in a murine dopaminergic neuronal cell line leads to neurite outgrowth. Neurosci Lett. 1996;202:161–164. doi: 10.1016/0304-3940(95)12235-4. [DOI] [PubMed] [Google Scholar]
- Ohmi K, Greenberg DS, Rajavel KS, Ryazantsev S, Li HH, Neufeld EF. Activated microglia in cortex of mouse models of mucopolysaccharidoses I and IIIB. Proc Natl Acad Sci USA. 2003;100:1902– 1907. doi: 10.1073/pnas.252784899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekny M, Pekna M. Astrocyte intermediate filaments in CNS pathologies and regeneration. J Pathol. 2004;204:428–437. doi: 10.1002/path.1645. [DOI] [PubMed] [Google Scholar]
- Pekny M, Leveen P, Pekna M, Eliasson C, Berthold CH, Westermark B, Betsholtz C. Mice lacking glial fibrillary acidic protein display astrocytes devoid of intermediate filaments but develop and reproduce normally. EMBO J. 1995;14:1590–1598. doi: 10.1002/j.1460-2075.1995.tb07147.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekny M, Johansson CB, Eliasson C, Stakeberg J, Wallen A, Perlmann T, Lendahl U, Betsholtz C, Berthold CH, Frisen J. Abnormal reaction to central nervous system injury in mice lacking glial fibrillary acidic protein and vimentin. J Cell Biol. 1999;145:503–514. doi: 10.1083/jcb.145.3.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver J. Inhibitory molecules in development and regeneration. J Neurol. 1994;242:S22–S24. doi: 10.1007/BF00939236. [DOI] [PubMed] [Google Scholar]
- Takada M, Hattori T. Fine structural changes in the rat brain after local injections of gliotoxin, alpha-aminoadipic acid. Histol Histopathol. 1986;1:271–275. [PubMed] [Google Scholar]
- Vela JM, Gonzalez B, Castellano B. Understanding glial abnormalities associated with myelin deficiency in the jimpy mutant mouse. Brain Res Brain Res Rev. 1998;26:29–42. doi: 10.1016/s0165-0173(97)00055-6. [DOI] [PubMed] [Google Scholar]
- Wilhelmsson U, Li L, Pekna M, Berthold CH, Blom S, Eliasson C, Renner O, Bushong E, Ellisman M, Morgan TE, et al. Absence of glial fibrillary acidic protein and vimentin prevents hypertrophy of astrocytic processes and improves post-traumatic regeneration. J Neurosci. 2004;24:5016–5021. doi: 10.1523/JNEUROSCI.0820-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams SE, Mann F, Erskine L, Sakurai T, Wei S, Rossi DJ, Gale NW, Holt CE, Mason CA, Henkemeyer M. Ephrin-B2 and EphB1 mediate retinal axon divergence at the optic chiasm. Neuron. 2003;39:919–935. doi: 10.1016/j.neuron.2003.08.017. [DOI] [PubMed] [Google Scholar]
- Yin Y, Cui Q, Li Y, Irwin N, Fischer D, Harvey AR, Benowitz LI. Macrophage-derived factors stimulate optic nerve regeneration. J Neurosci. 2003;23:2284–2293. doi: 10.1523/JNEUROSCI.23-06-02284.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang KZ, Westberg JA, Holtta E, Andersson LC. BCL2 regulates neural differentiation. Proc Natl Acad Sci USA. 1996;93:4504– 4508. doi: 10.1073/pnas.93.9.4504. [DOI] [PMC free article] [PubMed] [Google Scholar]