

Abstract
In addition to providing information on tissue structure, magnetic resonance (MR) technology offers the potential to investigate tissue metabolism and function. MR spectroscopy (MRS) offers a wealth of data on the biochemistry of a selected brain tissue volume, which represent potential surrogate markers for the pathology underlying multiple sclerosis (MS). In particular, the N-acetylaspartate peak in an MR spectrum is a putative marker of neuronal and axonal integrity, and the choline peak appears to reflect cell-membrane metabolism. On this basis, a diminished N-acetylaspartate peak is interpreted to represent neuronal/axonal dysfunction or loss, and an elevated choline peak represents heightened cell-membrane turnover, as seen in demyelination, remyelination, inflammation, or gliosis. Therefore, MRS may provide a unique tool to evaluate the severity of MS, establish a prognosis, follow disease evolution, understand its pathogenesis, and evaluate the efficacy of therapeutic interventions, which complements the information obtained from the various forms of assessment made by conventional MR imaging.
Keywords: Magnetic resonance spectroscopy, multiple sclerosis, N-acetylaspartate, choline, creatine, myo-inositol, metabolite, magnetic resonance imaging, axonal damage, atrophy
Conventional magnetic resonance imaging (MRI) is based on hydrogen nuclei (protons, or 1H) in water molecules,1 which are 1000-fold more abundant than any brain metabolite.2 But when the water-related signals are suppressed, a wealth of additional data emerge, enabling the same technology to offer novel information on the biochemistry of a selected tissue volume.
As early as 1990, it had been suggested that these data—obtained by proton magnetic resonance spectroscopy (MRS)—might hold value in the evaluation of multiple sclerosis (MS).3,4 Today, a substantial body of evidence supports the use of MRS to evaluate the severity of MS, establish a prognosis, follow its progression, understand its pathogenesis, and evaluate the efficacy of therapeutic interventions. However, the details of data interpretation, the nature of underlying pathological changes reflected in MRS findings, and even a standardized protocol for obtaining the data still remain to be firmly established.
Deficiencies of Conventional MRI
The early advocacy for MRS came at a time when conventional MRI evaluation of lesion volume and lesion activity was gaining the approval it continues to hold for assessing MS. Yet conventional MRI findings, such as T2-weighted lesion load, correlate only weakly with clinical disability in cross-sectional studies.5–8 (For a further discussion of this clinical–MRI paradox, please see the accompanying article in this supplement.8) For one thing, MRI-defined lesions often occur without symptoms. Indeed, clinically silent acute lesions are a routine finding, identified 2 to 15 times more often than clinical relapses.9 Conversely, symptoms may occur without MRI-defined lesions. Longitudinal analysis has shown a better predictive value of MRI lesions for long-term disability, yet the strength of association remains moderate at best.8,10
The explanation for such a clinico-pathological paradox11 may lie in the lack of sensitivity and specificity of conventional MRI for detecting important pathophysiological facets of MS.12 Classically, attacks of inflammation, with consequent demyelination and a degree of remyelination, are taken to characterize the early course of relapsing forms of the disease, and neurodegeneration, with accompanying gliosis, marks the later stages (and also the early stages of primary-progressive MS [PPMS]).13,14 Amid this heterogeneity of events, pathological and MRI studies have emphasized the importance of neurodegeneration, and in particular of axonal injury and central nervous system (CNS) atrophy, as identified even in early MS.15 Some studies have used biomolecular markers of damaged or transected axons—amyloid precursor protein16 or nonphosphorylated neurofilaments17—to identify tissue destruction in acute MRI-defined lesions. Others have discovered significant axonal pathology in normal-appearing white matter (NAWM)18 and neuronal pathology in normal-appearing gray matter (NAGM).19,20 For example, a numerical axonal loss of more than 50% has been documented in corpus callosum tissue far from any plaques.21 Consequentially, demyelination is no longer viewed as a prerequisite for axonal pathology, and it is increasingly appreciated that the full impact of MS at every stage of the disease reflects injury both to axons and to myelin sheaths. The evidence also emphasizes axonal pathology and CNS atrophy as major causes of MS-related disability.15,22,23
The lesion contrast at any given time point on either T1- or T2-weighted images is nonspecific for the underlying pathology and may reflect a combination of events including demyelination, remyelination, inflammation, edema, gliosis, axonal loss, and Wallerian degeneration.24–26 The specificity of conventional MRI may be improved by following lesion evolution, such as the transformation of newly appearing lesions into chronic persisting T1 hypointensities.27 (For an in-depth discussion of lesion evolution in MS, please see the companion article in this supplement.27) Despite these refinements, conventional MRI is insensitive to clinically relevant pathology in normal-appearing brain tissue.28 The overall deficiencies of conventional MRI were explored in a recent study of 34 patients with untreated PPMS (Fig 1).29 Using conventional MRI, the patients were classified as having a low (less than 3 cm3) or high T2-weighted lesion load, and brain atrophy and brain metabolite ratios were assessed quantitatively by MRI and MRS in a volume centered on the corpus callosum. In both respects, these 2 groups of patients differed from healthy controls, but no correlations could be identified between lesion load and either atrophy or the N-acetylaspartate (NAA)-creatine (Cr) ratio, supporting the conclusion that in MS, axonal injury and brain atrophy proceed with only a minimal relation to lesion load.
Fig 1.
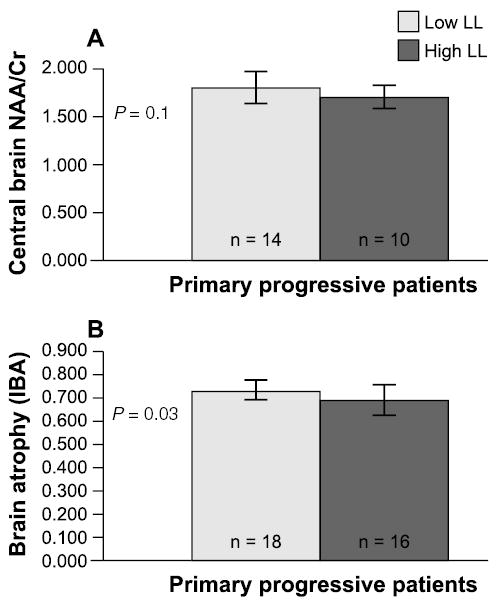
Deficiencies of conventional magnetic resonance imaging as a means of monitoring multiple sclerosis (MS) are illustrated by a study of 34 patients with untreated primary-progressive MS in which subgroups defined by a T2-weighted lesion load (LL) lower (light gray) or higher (dark gray) than 3 cm3 showed no significant difference in central brain N-acetylaspartate (NAA)-creatine (Cr), the ratio of NAA to Cr, a magnetic resonance spectroscopy measure of axonal pathology (A). The subgroups also failed to differ in an index of brain atrophy (B). From Pelletier et al. J Neurol Neurosurg Psychiatry 2003;74:950-952.29 Adapted and reproduced with permission from the BMJ Publishing Group.
MRS Peaks and Their Biological Significance
Unlike the hypo- or hyperintensities seen on conventional MRI,27 the peaks in MRS data quantify specific neurometabolites, in turn potentially representing specific MS-related events, such as demyelination, inflammation, and axonal/neuronal dysfunction. Fundamentally, MRS analyzes a region as small as a single volume element (voxel), a brick-shaped volume typically 2 to 8 cm3, or an array of many voxels (in which case, the data can be displayed as a 2- or 3-dimensional MRS “image”) selected by MRI. By examining one or another species of atomic nucleus—most often 1H or 31P—the spectra can quantitate the concentration of a large number of brain molecules,30 each of which produces a peak, or resonance, at a characteristic position along the frequency scale. The position of the peaks is expressed as parts per million (ppm). The sharpness and height of a given peak are governed not only by a molecule’s identity and concentration but also by its mobility.31 Only freely mobile molecules produce well-defined resonances.
Routine 1H-MRS of CNS tissue, requiring no special upgrade of the MRI technology, exhibits fewer than 15 peaks, but several appear to be highly pertinent to MS. From right to left, the spectrum (Fig 2) may include resonances representing free lipids, lactate, NAA, glutamate/glutamine, Cr/phosphocreatine (the Cr peak), choline (Cho; the Cho peak), and myo-inositol (mI).30 In spectra obtained at long echo times, some of them—including the peaks for NAA, Cr, Cho, and lactic acid—tend to be sharp. By contrast, lipid signals relax rapidly, necessitating short-echo MRS.1 Among the various peaks, the lactate peak should be weak in normal tissue. Representing the biochemical end product of glycolysis, it normally gains prominence only if the selected CNS volume reflects the lactate content of cerebrospinal fluid.30 Lactate does, however, accumulate abnormally during necrosis, inflammation, or other tissue injury.32
Fig 2.
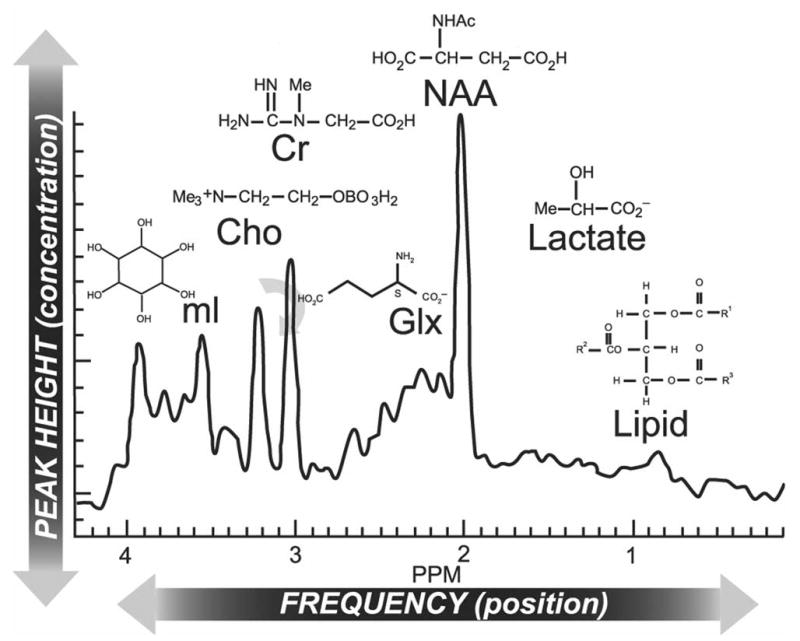
Magnetic resonance spectroscopy (MRS) spectrum of brain along with molecular assignment of resonances. The normal 1H-MRS spectrum is dominated by an N-acetylaspartate (NAA) resonance, flanked at the left by peaks for glutamate/glutamine (Glx), creatine (Cr)/phosphocreatine, choline (Cho)-containing phospholipids, and myo-inositol (mI) and at the right by peaks for lactate and free lipids. In multiple sclerosis, abnormal findings often include a decrease in the NAA peak (indicative of axonal pathology) and an increase in the Cho peak (indicative of demyelination). Adapted with permission from Lin et al. Efficacy of proton magnetic resonance spectroscopy in neurological diagnosis and neurotherapeutic decision making. NeuroRx® 2:197-214.30 Copyright © 2005, American Society for Experimental Neuro Therapeutics. All rights reserved.
The free-lipid peaks should also be weak in normal tissue, in that normal myelin, with its highly compacted structure, affords little mobility in its constituent lipids. In MS, however, the margins of active plaques show myelin breakdown and even myelin debris within macrophages.33 In an early study of the capacity of MRS to identify active MS lesions,3 16 MS patients underwent MRI to identify plaque-containing volumes of interest for 1H-MRS. Among 25 of the resulting spectra, 8 (from 6 patients) differed from the spectra of healthy controls in exhibiting free-lipid peaks. In some patients, their presence was accompanied by clinical or MRI evidence of MS activity, but no conventional MRI findings were predictive of an MRS finding of lipid peaks.
The other peaks can all be expected in 1H-MRS spectra of normal CNS tissue. Among these, the glutamate/glutamine peak represents a mixture of amino acids and bioamines used throughout the CNS as excitatory and inhibitory neurotransmitters, and the mI peak represents a sugar-like molecule thought to be a marker of glial proliferation and now recognized for its importance in osmotic regulation of brain-tissue volume.30 A recent MRS evaluation of glutamate concentrations in 25 MS patients, comprising relapsing-remitting MS (RRMS), secondary-progressive MS (SPMS), and PPMS, found elevated levels of the metabolite in acute, enhancing lesions (P= .02) and in NAWM (P= .03) compared to 16 controls.34 Because of glutamate’s neurotoxic potential when its homeostasis is altered, excess glutamate in active lesions could contribute to axonal damage, brain atrophy, and neurological disability.34
The presence of elevated levels of mI, a possible indicator of glial cell proliferation or gliosis, in patients with clinically isolated syndromes (CIS) and various MS subtypes has also been found.35–37 In a study of 41 patients who had initially presented with CIS 14 years previously, the patients had elevated mI levels compared to 21 controls in NAWM and in lesions, and the mI concentration in NAWM was correlated with Expanded Disability Status Scale (EDSS) score (r= 0.48, P= .005).35 Interestingly, after 14 years, 32 of 41 initial CIS patients had developed clinically definite MS (78%) and 9 remained with CIS (22%); it was the metabolite changes in NAWM and NAGM that separated these 2 groups: MS patients had increased mI in these tissues, and the patients who remained with CIS did not.35 Elevated mI levels in NAWM were also found in an analysis of 96 patients evaluated about 19 weeks after onset of CIS compared with 44 controls (P= .001), but there was no significant correlation of this elevation with the CIS patients’ T2 lesion load.36 Further investigation recently has shown that increased NAWM mI levels did not correlate with disability scores in a study that included 76 patients with RRMS, SPMS, and PPMS; although there was an elevation in mI levels for MS patients compared to the 25 controls, there were no metabolite differences between the disease subtypes.37 This mI increase early in the disease process may hold prognostic promise for future relapses and disability. However, care must be exercised in interpreting the changes in the mI level because of its proximity to the water peak.
The remaining 3 peaks are of special interest in MS.2,32 The Cr/phosphocreatine peak lies at 3.0 ppm. It represents the high-energy biochemical reserves of neurons and even more so of glia.28 Because the concentration of Cr arguably is typically stable and unaffected by the disease process, the other metabolites are expressed as a ratio relative to Cr as a means of normalization. The Cho peak, at 3.2 ppm, encompasses several soluble Cho-containing phospholipid constituents of all cell membranes and, hence, of myelin. Because almost all Cho is normally in insoluble form, a heightened Cho peak may represent abnormal Cho mobility, in turn suggesting inflammation, demyelination, and remyelination.2,30
The NAA peak, at 2.0 ppm, is, perhaps, the most informative spectral feature of all. In normal CNS tissue, it is the highest peak, representing a variety of N-acetyl compounds but chiefly NAA, an amino-acid derivative synthesized almost exclusively in neurons and distributed throughout the cell’s dendrites and axon, along which it travels by anterograde axonal transport.30 NAA, therefore, is thought to constitute a specific marker of neuronal viability or, in white matter, a specific marker of axonal integrity. Decreases in NAA identified in white matter, therefore, are taken to reflect axonal injury or loss.1,30 The presence of NAA in oligodendroglial precursor cells is now deemed highly unlikely to confound the data for mature CNS tissue.2 Meanwhile, an animal study has confirmed NAA’s relationship to axonal damage. In transected adult rat optic nerves, NAA declined to undetectable levels in 24 days.38
As early as 1990, MRS detection of NAA deficiency was proposed for quantifying MS-related axonal dysfunction or loss.3,4 Indeed, the early work correlated NAA deficits with MS-related disability, implicating axonal damage as an important direct cause of chronic functional impairment. Since then, reduced NAA has repeatedly been identified in acute lesions, chronic lesions, NAWM, and NAGM.39–45 The degree of reduction has repeatedly been correlated with the degree of clinical disability,41–44,46 a relationship that emphasizes the importance of axonal pathology in MS.32 In a postmortem study of spinal cord tissue from paralyzed MS patients, diminished axonal abundance and volume correlated with diminished NAA levels, both within lesions and in NAWM, offering direct evidence of relationships between NAA deficiency, axonal pathology, and MS-related disability.22
In MS, an MRS spectrum may exhibit an abnormally strong or weak peak or abnormal patterns in the strengths of several peaks.30 The simplest interpretive strategy is to judge the absolute value of a peak (or the implied concentration of the corresponding brain metabolite) against a baseline or control value or to gauge the peak against the same spectrum’s Cr peak, on the assumption, right or wrong, that the Cr peak is not affected very much by a pathological state.1,47 Although the NAA-Cr ratio is widely used, no standardized method of reporting and interpreting MRS findings has yet been achieved.30
Evaluation of MRS Capabilities
Lesion Evolution
Acute MS lesions, marked by gadolinium enhancement on conventional MRI, have shown a range of MRS abnormalities, including a diminished NAA peak and increased Cho and free-lipid peaks (Fig 3).3,12,47–49 Remarkably, the NAA peak has sometimes been found to regain much or even all of its normal strength.39,41 In one study, 25 MS patients with mild to moderate disability underwent serial MRI, MRS, and lesion-volumetric analyses for as long as 2 years.47 To force an unbiased selection of tissue, the volume of interest was kept within the centrum semiovale without regard to the presence or absence of MRI-defined lesions. Two acute lesions (in different patients) showed transient NAA level declines, which normalized in about 120 and 161 days (Fig 4). Proposed explanations have included an ebbing of lesional edema, remyelination, and reversal of a temporary metabolic disturbance, perhaps in mitochondria.2 By contrast, 25 stable lesions (lacking gadolinium enhancement) showed an enduring NAA deficiency. In 4 patients, strong free-lipid peaks were detected in regions lacking any MRI-defined pathology. In 3 of these patients, an MRI-defined lesion subsequently appeared. (The fourth patient withdrew from the analysis.) When free-lipid peaks occurred in association with lesions, the anatomical source of the lipid signal appeared to extend beyond the MRI-defined lesion.47
Fig 3.
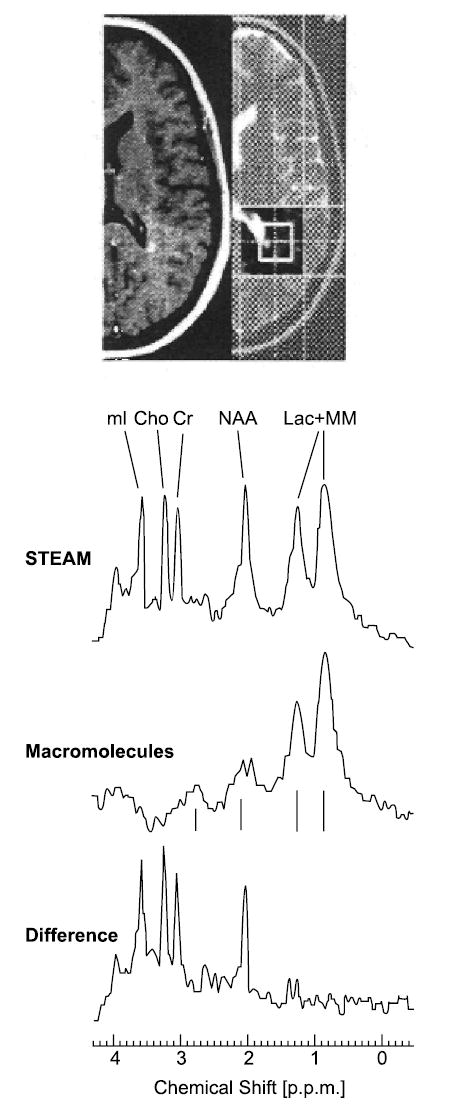
Contrast-enhanced and T1-weighted axial turbo spin-echo images of a small, acute, contrast-enhancing lesion and a large corresponding hyperintense lesion in the periventricular white matter of a patient with multiple sclerosis (MS). The saturation slices and position of the 8-mL volume of interest are noted. The STEAM spectrum shows a reduced N-acetylaspartate level (NAA) and an elevated myo-inositol (mI) level. Cho = choline; Cr = creatine; Lac + MM = lactate + lipids + macromolecules. Reprinted with permission from Mader et al. Proton MR spectroscopy with metabolite-nulling reveals elevated macromolecules in acute multiple sclerosis. Brain 2001;124(Pt 5):953–961,49 by permission of Oxford University Press.
Fig 4.
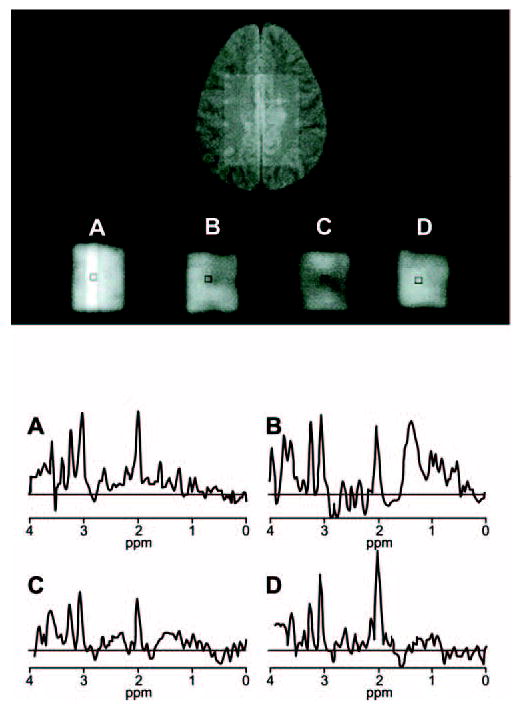
Magnetic resonance spectroscopy of a multiple sclerosis patient’s centrum semiovale exhibits temporary changes in normal-appearing white matter, including the reversal of a depression in N-acetylaspartate (NAA) level. The spectra shown were obtained on days 98 (A), 147 (B), 189 (C), and 259 (D) of a longitudinal study; the second and third in the series show a fall in the NAA peak (at 2.0 ppm), but the fourth spectrum documents a recovery. The second spectrum also shows temporary free-lipid features, toward the right side of the data. The neuroanatomical region subsequently developed a magnetic resonance imaging–defined lesion. Adapted from Narayana et al. Serial proton magnetic resonance spectroscopic imaging, contrast-enhanced magnetic resonance imaging, and quantitative lesion volumetry in multiple sclerosis. Ann Neurol 1998;43:56–71.47Copyright ©1998 Wiley-Liss, Inc., A Wiley Company. Reproduced with permission ofJohn Wiley & Sons, Inc.
Chronic persistent lesions tend not to show similar dynamics.2 They do, however, exhibit persistently diminished NAA levels, particularly if a T1 hypointensity has developed. In one recent study, NAA, Cho, and Cr levels were measured at better than 1-cm3 spatial resolution in nonenhancing lesions and NAWM of 9 RRMS patients and in the white matter of 9 healthy controls.50 Among a total of 66 T2 lesions that had been evident for at least 6 months, 43 could be classified on T1-weighted imaging as hypointense “black holes,” considered to be foci of severe tissue damage, and 23 as isointensities, considered to be prime candidates to become black holes (Fig 5). The mean NAA level was significantly lower in hypointensities than in isointensities in NAWM or in control white matter. Cho and Cr peaks were significantly higher in isointensities and in NAWM than in controls. The overall impression was of abnormal metabolic activity in all MS tissue types.
Fig 5.
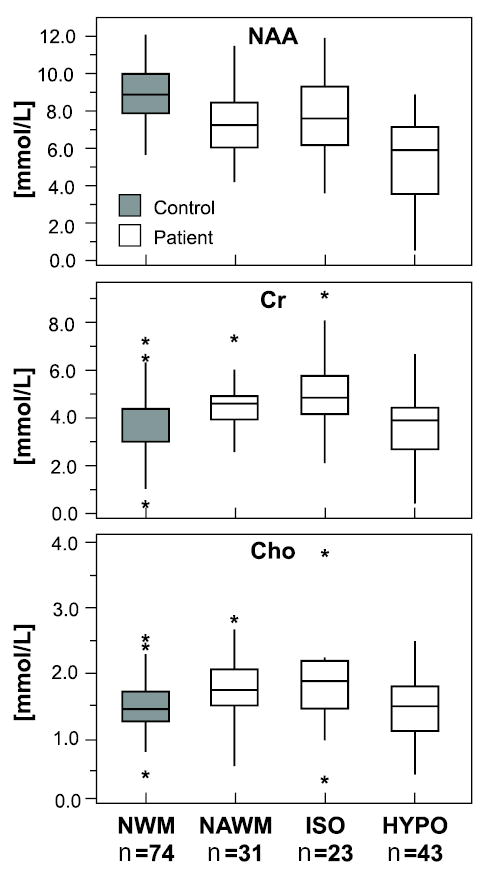
Magnetic resonance spectroscopy findings for a total of 66 T2-weighted lesions that had persisted for at least 6 months in one or another of 9 relapsing-remitting multiple sclerosis (MS) patients showed NAA levels to be lowest in T1-weighted hypointensities, or “black holes” (HYPO), while creatine (Cr) and choline (Cho) are higher in T1-weighted isointensities (ISO) and in normal-appearing white matter (NAWM) than in white matter of normal controls (NWM). In all instances, horizontal lines are medians, boxes show 25% to 75% values, vertical lines span ±95%, and asterisks show outliers. Adapted with permission from He et al. Relapsing-remitting multiple sclerosis: metabolic abnormality in nonenhancing lesions and normal-appearing white matter at MR imaging—initial experience. Radiology 2005;234:211–217.50
Correlations With Disability
Studies to detect correlations between MRI metrics and clinical measures of disability have presented conflicting results, and greater correlation between T1 lesion load and disability than T2 lesion load and disability is observed.8,51,52 MRS measures, however, may prove to correlate better with clinically measured disability.
In a study of 31 patients with clinically definite MS, a significant correlation was found between the NAA-Cr ratio in NAWM and disability as measured by EDSS score (r= −0.38; P< 0.03).53 The study also compared the NAA-Cr ratio in NAWM and white matter lesions to other disability scales, including the Multiple Sclerosis Functional Composite, but no correlations were found. Correlations between EDSS score and NAA-Cr ratio were found in several other studies as well,42,44,46,53 suggesting that MRS measures of brain metabolites are better predictors of clinical disability than are conventional MRI lesion measures.
Evaluating Occult Disease
Several recent reports explored the use of MRS to identify MS-related tissue abnormalities beyond the macroscopic lesions identified by conventional MRI. For a study of occult involvement of the brain’s white matter, 31 RRMS patients underwent MRS and clinical assessment on the same day. The MRS was to evaluate neuronal damage in NAWM, and the clinical assessment was to seek linkage with the patients’ degree of disability.53 The NAA-Cr ratio was lower in patients than in healthy controls, both in NAWM and in MRI-defined lesions. This reduction correlated with EDSS scores but not with MS Functional Composite Scale scores. Another study12 recruited 40 PPMS patients, in whom the NAA-Cr ratio proved to be significantly lower than in healthy controls, with no significant difference between lesion-containing regions and NAWM. In the normal-appearing regions, 24 patients also exhibited strong free-lipid peaks. No MRS findings showed any correlation with conventional MRI measures or with EDSS score.
To evaluate NAWM during clinically silent MS, a third study evaluated 11 RRMS patients, none of whom had experienced a relapse for at least 3 months.28 In normal-appearing callosal white matter, NAA levels were 9% lower, Cr levels 22% higher, and Cho levels 32% higher than in healthy controls (but among these differences, only the Cho level distinguished patients from controls with 100% specificity and better than 90% sensitivity).
For a study of occult involvement of the spinal cord, 9 MS patients underwent MRS of normal-appearing cervical tissue.54 No patient had clinical signs indicative of cervical involvement, and no patient had cervical lesions. Nevertheless, the NAA level was found to be significantly reduced. For Cr, Cho, lipid, lactate, and mI features in the MRS data, the findings for patients differed insignificantly from those for healthy controls.
Gray matter is known to be involved in MS,55,56 but its involvement has been hard to evaluate using conventional MRI because lesion contrast in gray matter is less dramatic than in white matter.57 Intracortical, juxtacortical, and subcortical gray matter lesions have nonetheless been identified, often histopathologically and sometimes by MRI.57–60 For an MRS study of cortical involvement, 52 RRMS patients and 20 normal controls underwent serial MRI (using a method adjusted to heighten lesion contrast) and short-echo MRS over spans of up to 3 years.45 Although few of the identified macroscopic lesions were intracortical, substantial numbers of juxtacortical (positioned amid U-fibers) or subcortical (near cortex but distinct from it) lesions were detected. MRS spectra showed a depressed NAA peak accompanied by elevations in the Cr/phosphocreatine and Cho peaks, but in 13 patients, roughly 10% of the voxels exhibited strong additional free-lipid peaks. These appeared to originate mainly in the gray matter, either affected (with MRI-defined lesions within or nearby) or normal appearing.
The deep gray matter is also a site of significant tissue damage in MS as shown by pathological19 and MRI studies.61,62 MRS has also shown sensitivity for detecting deep gray matter involvement in MS. For a study of 3 such sites—the thalamus, the putamen, and the head of the caudate nucleus—MRS was performed in 15 RRMS patients and 9 controls.63 The 7% decrease in NAA level in these patients was deemed likely to be an effect of distal white matter lesions, and the observed 14% increase in Cho level was judged to be smaller than would be expected for white matter affected by MS, reflecting the lower abundance of myelin in gray matter. The Cr peak showed no significant difference between patients and controls. Overall, findings of gray matter involvement in MS expand the classic view of MS as a white matter disease, perhaps because oligodendrocytes invest axons with myelin sheaths beginning close to the neuronal cell bodies45 or because other direct processes are occurring in gray matter that contribute to the degenerative aspects of the disease.64
Establishing a Prognosis
Because axonal damage may be the dominant cause of irreversible deficits in MS, several research groups have hypothesized that the whole-brain NAA concentration might accurately mirror the extent of a patient’s MS and might also yield a prognosis. In one exploration of this possibility, 42 patients with RRMS had brain atrophy assessed by MRI and also underwent an MRS determination of whole-brain NAA.65 Although atrophy increased and NAA declined linearly with increasing MS disease duration, the rate of decline for NAA was 3.6 times faster than atrophy. The researchers concluded that neuronal dysfunction, as represented by NAA decline, precedes parenchymal loss and therefore might be an earlier, more sensitive marker of disease progression.
In another study, 49 RRMS patients underwent a 1-time MRS assessment of whole-brain NAA.66 To derive a rate of change, each patient’s MS duration was defined as the time since diagnosis of clinically definite MS, and a study of healthy volunteers 16 to 52 years old67 was used to set a mean normal whole-brain NAA (13.2 mmol/L). By this standard, 10 patients (20%) were stable, showing an insignificant decline; 27 patients (55%) showed a moderate rate of decline, at a mean of 2.8% per year; and 12 patients (25%) showed rapid decline, a mean of 27.9% per year. Their median duration of MS was significantly shorter than that of the other subgroups, but no subgroup showed a correlation between whole-brain NAA and patient age, implying that low whole-brain NAA may represent an aggressive MS variant with onset at any age. When MS duration was redefined as time since the first of the 2 episodes required for a clinically definite diagnosis (Fig 6), 5 patients in the rapid group were reclassified into the moderate group, but no patient was reassigned to the stable group.
Fig 6.
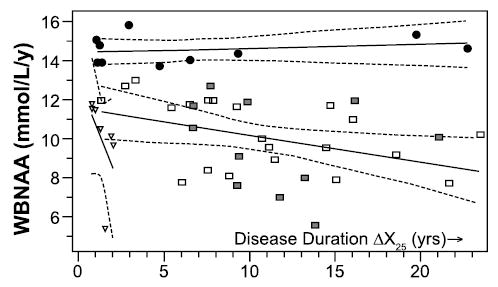
Whole-brain levels of N-acetylaspartate (WBNAA) for 49 patients with relapsing-remitting multiple sclerosis (MS) are plotted against each patient’s duration of MS, beginning with the initial clinical episode. The data define distinct trends for patients with an insignificant rate of NAA decline (black circles), a decline calculated not to exceed 1.7 mmol/L/y (squares, with shaded squares indicating patients who received MS treatment at the time of the NAA measurement), or a rapid decline (triangles). Dashed lines show 95% confidence intervals. Adapted with permission from Gonen et al. Relapsing-remitting multiple sclerosis and whole-brain N-acetylaspartate measurement: evidence for different clinical cohorts—initial observations. Radiology 2002;225:261–268.66
It was proposed that evaluation of whole-brain NAA at the time of a first clinical episode may hold value for identifying a deficiency and for permitting a follow-up measurement at a second episode, in turn permitting a clinically definite diagnosis of MS to be accompanied by accurate assignment to a prognostic category.66 For stable patients, the likely future might be a long period without substantial accrual of MS-related disability. For patients in the rapid group, aggressive intervention might be especially advisable. The intermediate majority of patients might be counseled to expect a “typical” MS course. In research settings, the rapid (or moderate) group might produce the best cohorts in which to test therapies.
Correlations With Fatigue
Defined as a feeling that a given activity requires disproportionate effort (distinct from muscle weakness), fatigue is a prominent MS symptom and often makes major contributions to a patient’s disability.68 MRI studies have shown no consistent relationship between fatigue and lesion burden or atrophy.68,69 To explore the possibility that the symptom might reflect diffuse cerebral axonal damage, data from 73 MS patients who had undergone MRS and had completed the Fatigue Severity Scale (FSS) questionnaire were analyzed.70 In a supracallosal volume chosen to encompass frontal, parietal, and occipital white matter, the NAA-Cr ratio was significantly lower among high-fatigue patients (FSS scores no lower than 5) than among low-fatigue patients (FSS scores no higher than 4). The relationship was independent of EDSS score, T2-weighted lesion volume, patient age, and MS duration, and it persisted after the exclusion of patients on antidepressants, anxiolytics, or fatigue-modifying drugs (such as amantadine) and patients with a diagnosis of depression. Across all patients, the NAA-Cr ratio—but not the T2-weighted lesion volume—showed a correlation with the FSS score.
Correlations With Cognition
MRI studies have generally found no more than a moderate correlation between conventional imaging findings and cognitive function in MS.71 The question is whether identification of diffuse occult damage in tissue that appears normal on conventional MRI might yield a stronger relationship with cognitive function.
One of the most common and disabling cognitive signs in MS is impairment of working memory and attention. One group studied these aspects using a forced-attention dichotic listening task, in which participants were cued by a tone to pay attention to one or the other ear and then report the syllable they heard in that ear.72 RRMS patients had already shown a significant impairment in performing this task, even early in the MS disease course.73 For a pertinent neuroanatomical region, the researchers chose the locus coeruleus, a brainstem cell group prominent as a source of noradrenergic innervation of cerebral cortex, evidently enabling the cell group to modulate cortical attention. Among 19 mildly disabled RRMS patients with no brainstem lesions, the listening task identified attentional dysfunction in 47%. On MRS, the pontine NAA-Cr ratio near the fourth ventricle, in a volume closely matching the location of the locus coeruleus, accounted for 39% of the variance in the cognitive test results (with the lowest levels in patients with the greatest attentional disturbance). When left- and right-side NAA-Cr values were analyzed separately, the right-side value explained 43% of the cognitive variability, a finding consistent with studies associating attentional tasks with higher right-hemisphere activation.74,75 In one such study, the investigators found that atrophy in the central region of the brain accounted for more than 50% of the variance in overall cognitive performance in 37 RRMS and SPMS patients with known cognitive impairment and that this atrophy measure was significantly correlated with NAA-Cr ratios of the right hemisphere.75
Effects of Therapies
Because of MRS’s ability to monitor the cerebral metabolite changes in relatively small volumes of interest, a few studies have now used MRS to evaluate immunomodulatory therapies in RRMS. In a small 6-month study that evaluated lesions in the white matter and NAWM during a first treatment period, 5 untreated RRMS patients were matched with 5 RRMS patients treated with once-weekly intramuscular interferon β-1a (30 μg).76 The untreated patients had insignificant variances in their NAA, Cho, inositol, and Cr peaks over the study time. However, in the treated group, there was a significant increase (P< .001) in the Cho peak area at the 1-month time point compared with baseline, and this elevation was also present at the 6-month time point. Because of the treated group’s Cho increase and the insignificant changes in Cr and NAA peaks, there was a significant increase in the Cho/Cr (P< .02) and Cho/NAA (P< .005) ratios for this group over the 6-month evaluation period compared with baseline values. Although these findings confirmed that cerebral metabolites are markers of MS activity and demonstrated that intramuscular interferon β-1a affects metabolite concentrations in MS lesions, the relatively short time frame of this study could not assess the potential for this treatment to induce remyelination.
An early study of interferon β-1b77 identified a 1-year increase of 5.5% (P= .05) in the NAA-Cr ratio measured in a large, central brain volume among 10 patients with moderately active RRMS, compared with an insignificant decrease in untreated controls. The finding was judged consistent with research showing that declines in NAA levels may reflect axonal dysfunction, which may be partially reversible with interferon therapy’s reduction of inflammation.77 In a longer term study (up to 34 months) that assessed 10 RRMS patients who received interferon β-1b, NAA, total Cr, and Cho-containing compounds were evaluated in unenhanced lesions and contralateral NAWM using MRS.78 The concentrations of Cho and Cr were higher in the MS brain tissue than in brain tissue from 10 healthy controls. In treated patients, there was a lower (insignificant) NAA concentration in lesions than in NAWM, but no other differences were observed. The metabolite concentrations, although exhibiting variability, were stable over time, which the authors speculated may have resulted from the interferon β therapy.
In another study, 11 patients with a median relapse rate of 1.5 per year were monitored during a year of interferon β therapy: 9 patients received subcutaneous interferon β-1a, 22 μg 3 times per week; 1 patient received intramuscular interferon β-1a, 6 MIU once weekly; and 1 patient received interferon β-1b, 8 MIU every other day.79 The patients showed a significantly reduced relapse rate, and the T2-weighted lesion load showed a median 8% decrease. But in a volume centered on the corpus callosum, the NAA-Cr ratio, already 16% lower than in healthy controls, continued to decline, with a mean 6.2% 1-year change. The controls showed no 1-year decline. Small sample sizes in each of these studies limit the generalizability of the results or the ability to draw firm conclusions on the treatment effect of interferon β on MRS metrics.
Similar to the interferon β studies, data on glatiramer acetate are limited to small studies. In a nonrandomized study of glatiramer acetate, MRI and MRS were performed annually for 2 years in 18 RRMS patients who had started to use the drug and in 4 patients who chose to remain untreated.80 In a volume centered on the corpus callosum, the NAA-Cr ratio increased by a mean 10.7% among treated patients and decreased by 8.6% among untreated patients. The treated patients showed decreases in lesion burden and relapse rate, and the untreated patients showed an increase in lesion burden and no change in relapse rate. A decrease in the NAA-Cr ratio of untreated patients is consistent with the findings in an earlier pilot study that compared the NAA-Cr ratios in 15 RRMS patients treated with glatiramer acetate with those of matched, untreated, natural history controls.81 One year after baseline, a significant number of untreated controls had decreased levels of NAA relative to Cr. To more completely understand the effect of disease-modifying therapies on MRS findings in MS, larger prospective studies are needed.
MRS is poised to play a major role in the evaluation of treatment efficacy. The major challenge remains tied to the technical aspects of MRS, which differ from conventional MRI in terms of low signal-to-noise ratio, poor spatial resolution, limited spatial coverage, and the complexity of data-processing paradigms.12 (For a discussion of medical image–processing pipelines, please see the accompanying article in this supplement.82) The routine adoption of MRS imaging to multicenter clinical trials is feasible but cannot happen until it is determined whether there is an imaging-center effect; that is, the MRS imaging results depend on where the images are generated and how the MRS metabolite data on any patient subset compare with preexisting published data for the patient “universe” with the same disease and demographic characteristics. The center effect may be surmountable in multicenter trials if there is strict adherence to protocols, including imaging protocols.12 Metabolite-specific information from patient subsets can be compared to the findings from previous, similar studies, but the power of these findings may be enhanced by calculating absolute concentrations in specific tissues and for selected individual patients and then monitoring the fluctuations over long time periods (3 or more years).12,78
Summary
In seeking to account for the disability caused by MS, the sphere of interest has widened from overt foci of inflammation and demyelination to diffuse axonal and neuronal pathology. MRS is emerging as a valuable tool to demonstrate widespread changes in the brain, such as deficiency of the axonal marker NAA, not only in MRI-defined lesions but also in normal-appearing CNS tissue, even early in MS.1 The MRS findings support pathological studies suggesting widespread tissue destruction extending beyond white matter plaques.31 Within this complexity, nerve fiber loss in NAWM and neuronal loss in NAGM may make a significant contribution to MS-related physical and neuropsychological disabilities.
Research shows a role for NAA decline in MS-related clinical disability—a relationship that may prove to be stronger than those for lesional assessments offered by conventional MRI.1 The value of the NAA peak as a measure of axonal and neuronal integrity is gaining acceptance. The implication is that patients might benefit greatly from neuroprotective measures that specifically address axonal and neuronal dysfunction and damage. The major contribution of MRS, therefore, may prove to be its quantification of tissue biochemistry as a means of not only monitoring MS but also of more fully understanding it in the quest for improved therapies and treatment strategies.
Acknowledgments
This work was partially supported by National Institutes of Health grant R01 EB002095. The author would like to acknowledge Dr Omar Khan for his helpful discussions. This supplement was supported by an educational grant from Teva Neuroscience. BioScience Communications contributed to the editorial refinement of this article and to the production of this supplement. Authors may have accepted honoraria for their supplement contributions.
References
- 1.Wolinsky JS, Narayana PA. Magnetic resonance spectroscopy in multiple sclerosis: window into the diseased brain. Curr Opin Neurol. 2002;15:247–251. doi: 10.1097/00019052-200206000-00004. [DOI] [PubMed] [Google Scholar]
- 2.De Stefano N, Bartolozzi ML, Guidi L, Stromillo ML, Federico A. Magnetic resonance spectroscopy as a measure of brain damage in multiple sclerosis. J Neurol Sci. 2005;233:203–208. doi: 10.1016/j.jns.2005.03.018. [DOI] [PubMed] [Google Scholar]
- 3.Wolinsky JS, Narayana PA, Fenstermacher MJ. Proton magnetic resonance spectroscopy in multiple sclerosis. Neurology. 1990;40:1764–1769. doi: 10.1212/wnl.40.11.1764. [DOI] [PubMed] [Google Scholar]
- 4.Arnold DL, Matthews PM, Francis G, Antel J. Proton magnetic resonance spectroscopy of human brain in vivo in the evaluation of multiple sclerosis: assessment of the load of disease. Magn Reson Med. 1990;14:154–159. doi: 10.1002/mrm.1910140115. [DOI] [PubMed] [Google Scholar]
- 5.Filippi M, Paty DW, Kappos L, et al. Correlations between changes in disability and T2-weighted brain MRI activity in multiple sclerosis: a follow-up study. Neurology. 1995;45:255–260. doi: 10.1212/wnl.45.2.255. [DOI] [PubMed] [Google Scholar]
- 6.Ciccarelli O, Brex PA, Thompson AJ, Miller DH. Disability and lesion load in MS: a reassessment with MS functional composite score and 3D fast FLAIR. J Neurol. 2002;249:18–24. doi: 10.1007/pl00007843. [DOI] [PubMed] [Google Scholar]
- 7.Miki Y, Grossman RI, Udupa JK, et al. Relapsing-remitting multiple sclerosis: longitudinal analysis of MR images—lack of correlation between changes in T2 lesion volume and clinical findings. Radiology. 1999;213:395–399. doi: 10.1148/radiology.213.2.r99oc01395. [DOI] [PubMed] [Google Scholar]
- 8.Zivadinov R, Leist TP. Clinical–magnetic resonance imaging correlations in multiple sclerosis. J Neuroimaging. 2005;15(suppl 1):10S–21S. doi: 10.1177/1051228405283291. [DOI] [PubMed] [Google Scholar]
- 9.Miller DH, Grossman RI, Reingold SC, McFarland HF. The role of magnetic resonance techniques in understanding and managing multiple sclerosis. Brain. 1998;121:3–24. doi: 10.1093/brain/121.1.3. [DOI] [PubMed] [Google Scholar]
- 10.Brex PA, Ciccarelli O, O’Riordan JI, Sailer M, Thompson AJ, Miller DH. A longitudinal study of abnormalities on MRI and disability from multiple sclerosis. N Engl J Med. 2002;346:158–164. doi: 10.1056/NEJMoa011341. [DOI] [PubMed] [Google Scholar]
- 11.Barkhof F. The clinico-pathological paradox in multiple sclerosis revisited. Curr Opin Neurol. 2002;15:239–245. doi: 10.1097/00019052-200206000-00003. [DOI] [PubMed] [Google Scholar]
- 12.Narayana PA, Wolinsky JS, Rao SB, He R, Mehta M PROMiSe Trial MRSI Group. Multicentre proton magnetic resonance spectroscopy imaging of primary progressive multiple sclerosis. Mult Scler. 2004;10(suppl 1):S73–S78. doi: 10.1191/1352458504ms1035oa. [DOI] [PubMed] [Google Scholar]
- 13.Zaffaroni M. Biological indicators of the neurodegenerative phase of multiple sclerosis. Neurol Sci. 2003;24(suppl 5):S279–S282. doi: 10.1007/s10072-003-0174-3. [DOI] [PubMed] [Google Scholar]
- 14.Rammohan KW. Axonal injury in multiple sclerosis. Curr Neurol Neurosci Rep. 2003;3:231–237. doi: 10.1007/s11910-003-0083-0. [DOI] [PubMed] [Google Scholar]
- 15.Bakshi R, Dandamudi VSR, Neema M, De C, Bermel RA. Measurement of brain and spinal cord atrophy by magnetic resonance imaging as a tool to monitor multiple sclerosis. J Neuroimaging. 2005;15(suppl 1):30S–45S. doi: 10.1177/1051228405283901. [DOI] [PubMed] [Google Scholar]
- 16.Ferguson B, Matyszak MK, Esiri MM, Perry VH. Axonal damage in acute multiple sclerosis lesions. Brain. 1997;120:393–399. doi: 10.1093/brain/120.3.393. [DOI] [PubMed] [Google Scholar]
- 17.Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mörk S, Bö L. Axonal transection in the lesions of multiple sclerosis. N Engl J Med. 1998;338:278–285. doi: 10.1056/NEJM199801293380502. [DOI] [PubMed] [Google Scholar]
- 18.Miller DH, Thompson AJ, Filippi M. Magnetic resonance studies of abnormalities in the normal appearing white matter and grey matter in multiple sclerosis. J Neurol. 2003;250:1407–1419. doi: 10.1007/s00415-003-0243-9. [DOI] [PubMed] [Google Scholar]
- 19.Cifelli A, Arridge M, Jezzard P, et al. Thalamic neurodegeneration in multiple sclerosis. Ann Neurol. 2002;52:650–653. doi: 10.1002/ana.10326. [DOI] [PubMed] [Google Scholar]
- 20.Kutzelnigg A, Lassmann H. Cortical lesions and brain atrophy in MS. J Neurol Sci. 2005;233:55–59. doi: 10.1016/j.jns.2005.03.027. [DOI] [PubMed] [Google Scholar]
- 21.Evangelou N, Esiri MM, Smith S, Palace J, Matthews PM. Quantitative pathological evidence for axonal loss in normal appearing white matter in multiple sclerosis. Ann Neurol. 2000;47:391–395. [PubMed] [Google Scholar]
- 22.Bjartmar C, Kidd G, Mork S, Rudick R, Trapp BD. Neurological disability correlates with spinal cord axonal loss and reduced N-acetyl aspartate in chronic multiple sclerosis patients. Ann Neurol. 2000;48:893–901. [PubMed] [Google Scholar]
- 23.Zivadinov R, Bakshi R. Central nervous system atrophy and clinical status in multiple sclerosis. J Neuroimaging. 2004;14(suppl):27S–35S. doi: 10.1177/1051228404266266. [DOI] [PubMed] [Google Scholar]
- 24.Bruck W, Bitsch A, Kolenda H, Bruck Y, Stiefel M, Lassmann H. Inflammatory central nervous system demyelination: correlation of magnetic resonance imaging findings with lesion pathology. Ann Neurol. 1997;42:783–793. doi: 10.1002/ana.410420515. [DOI] [PubMed] [Google Scholar]
- 25.Bitsch A, Kuhlmann T, Stadelmann C, et al. A longitudinal MRI study of histopathologically defined hypointense multiple sclerosis lesions. Ann Neurol. 2001;49:793–796. doi: 10.1002/ana.1053. [DOI] [PubMed] [Google Scholar]
- 26.Bakshi R, Minagar A, Jaisani Z, Wolinsky JS. Imaging of multiple sclerosis: role in neurotherapeutics. NeuroRx. 2005;2:277–303. doi: 10.1602/neurorx.2.2.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Inglese M, Grossman RI, Filippi M. Magnetic resonance imaging monitoring of multiple sclerosis lesion evolution. J Neuroimaging. 2005;15(suppl 1):22S–29S. doi: 10.1177/1051228405282243. [DOI] [PubMed] [Google Scholar]
- 28.Inglese M, Li BS, Rusinek H, Babb JS, Grossman RI, Gonen O. Diffusely elevated cerebral choline and creatine in relapsing-remitting multiple sclerosis. Magn Reson Med. 2003;50:190–195. doi: 10.1002/mrm.10481. [DOI] [PubMed] [Google Scholar]
- 29.Pelletier D, Nelson SJ, Oh J, et al. MRI lesion volume heterogeneity in primary progressive MS in relation with axonal damage and brain atrophy. J Neurol Neurosurg Psychiatry. 2003;74:950–952. doi: 10.1136/jnnp.74.7.950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lin A, Ross BD, Harris K, Wong W. Efficacy of proton magnetic resonance spectroscopy in neurological diagnosis and neurotherapeutic decision making. NeuroRx. 2005;2:197–214. doi: 10.1602/neurorx.2.2.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Matthews PM, De Stefano N, Narayanan S, et al. Putting magnetic resonance spectroscopy studies in context: axonal damage and disability in multiple sclerosis. Semin Neurol. 1998;18:327–336. doi: 10.1055/s-2008-1040884. [DOI] [PubMed] [Google Scholar]
- 32.Arnold DL, Wolinsky JS, Matthews PM, Falini A. The use of magnetic resonance spectroscopy in the evaluation of the natural history of multiple sclerosis. J Neurol Neurosurg Psychiatry. 1998;64(suppl 1):S94–S101. [PubMed] [Google Scholar]
- 33.Adams CW, Poston RN, Buk SJ. Pathology, histochemistry and immunocytochemistry of lesions in acute multiple sclerosis. J Neurol Sci. 1989;92:291–306. doi: 10.1016/0022-510x(89)90144-5. [DOI] [PubMed] [Google Scholar]
- 34.Srinivasan R, Sailasuta N, Hurd R, Nelson S, Pelletier D. Evidence of elevated glutamate in multiple sclerosis using magnetic resonance spectroscopy at 3 T. Brain. 2005;128:1016–1025. doi: 10.1093/brain/awh467. [DOI] [PubMed] [Google Scholar]
- 35.Kapeller P, Brex PA, Chard D, et al. Quantitative 1H MRS imaging 14 years after presenting with a clinically isolated syndrome suggestive of multiple sclerosis. Mult Scler. 2002;8:207–210. doi: 10.1191/1352458502ms822oa. [DOI] [PubMed] [Google Scholar]
- 36.Fernando KTM, McLean MA, Chard DT, et al. Elevated white matter myo-inositol in clinically isolated syndromes suggestive of multiple sclerosis. Brain. 2004;127:1361–1369. doi: 10.1093/brain/awh153. [DOI] [PubMed] [Google Scholar]
- 37.Vrenken H, Barkhof F, Uitdehaag BMJ, Castelijns JA, Polman CH, Pouwels PJW. MR spectroscopic evidence for glial increase but not for neuro-axonal damage in MS normal-appearing white matter. Magn Reson Med. 2005;53:256–266. doi: 10.1002/mrm.20366. [DOI] [PubMed] [Google Scholar]
- 38.Bjartmar C, Battistuta J, Terada N, Dupree E, Trapp BD. N-acetylaspartate is an axon-specific marker of mature white matter in vivo: a biochemical and immunohistochemical study on the rat optic nerve. Ann Neurol. 2002;51:51–58. doi: 10.1002/ana.10052. [DOI] [PubMed] [Google Scholar]
- 39.Davie CA, Hawkins CP, Barker GJ, et al. Serial proton magnetic resonance spectroscopy in acute multiple sclerosis lesions. Brain. 1994;117:49–58. doi: 10.1093/brain/117.1.49. [DOI] [PubMed] [Google Scholar]
- 40.Husted CA, Goodin DS, Hugg JW, et al. Biochemical alterations in multiple sclerosis lesions and normal-appearing white matter detected by in vivo 31P and 1H spectroscopic imaging. Ann Neurol. 1994;36:157–165. doi: 10.1002/ana.410360207. [DOI] [PubMed] [Google Scholar]
- 41.De Stefano N, Matthews PM, Antel JP, Preul M, Francis G, Arnold DL. Chemical pathology of acute demyelinating lesions and its correlation with disability. Ann Neurol. 1995;38:901–909. doi: 10.1002/ana.410380610. [DOI] [PubMed] [Google Scholar]
- 42.Tourbah A, Stievenart JL, Gout O, et al. Localized proton magnetic resonance spectroscopy in relapsing remitting versus secondary progressive multiple sclerosis. Neurology. 1999;53:1091–1097. doi: 10.1212/wnl.53.5.1091. [DOI] [PubMed] [Google Scholar]
- 43.Lee MA, Blamire AM, Pendlebury S, et al. Axonal injury or loss in the internal capsule and motor impairment in multiple sclerosis. Arch Neurol. 2000;57:65–70. doi: 10.1001/archneur.57.1.65. [DOI] [PubMed] [Google Scholar]
- 44.De Stefano N, Narayanan S, Francis GS, et al. Evidence of axonal damage in the early stages of multiple sclerosis and its relevance to disability. Arch Neurol. 2001;58:65–70. doi: 10.1001/archneur.58.1.65. [DOI] [PubMed] [Google Scholar]
- 45.Sharma R, Narayana PA, Wolinsky JS. Grey matter abnormalities in multiple sclerosis: proton magnetic resonance spectroscopic imaging. Mult Scler. 2001;7:221–226. doi: 10.1177/135245850100700402. [DOI] [PubMed] [Google Scholar]
- 46.De Stefano N, Matthews PM, Fu L, et al. Axonal damage correlates with disability in patients with relapsing-remitting multiple sclerosis: results of a longitudinal magnetic resonance spectroscopy study. Brain. 1998;121:1469–1477. doi: 10.1093/brain/121.8.1469. [DOI] [PubMed] [Google Scholar]
- 47.Narayana PA, Doyle TJ, Lai D, Wolinsky JS. Serial proton magnetic resonance spectroscopic imaging, contrast-enhanced magnetic resonance imaging, and quantitative lesion volumetry in multiple sclerosis. Ann Neurol. 1998;43:56–71. doi: 10.1002/ana.410430112. [DOI] [PubMed] [Google Scholar]
- 48.Simone IL, Tortorella C, Federico F, et al. Axonal damage in multiple sclerosis plaques: a combined magnetic resonance imaging and 1H-magnetic resonance spectroscopy study. J Neurol Sci. 2001;182:143–150. doi: 10.1016/s0022-510x(00)00464-0. [DOI] [PubMed] [Google Scholar]
- 49.Mader I, Seeger U, Weissert R, et al. Proton MR spectroscopy with metabolite-nulling reveals elevated macromolecules in acute multiple sclerosis. Brain. 2001;124:953–961. doi: 10.1093/brain/124.5.953. [DOI] [PubMed] [Google Scholar]
- 50.He J, Inglese M, Li BS, Babb JS, Grossman RI, Gonen O. Relapsing-remitting multiple sclerosis: metabolic abnormality in nonenhancing lesions and normal-appearing white matter at MR imaging—initial experience. Radiology. 2005;234:211–217. doi: 10.1148/radiol.2341031895. [DOI] [PubMed] [Google Scholar]
- 51.Simon JH, Lull J, Jacobs LD, et al. A longitudinal study of T1 hypointense lesions in relapsing MS: MSCRG trial of interferon beta-1a. Multiple Sclerosis Collaborative Research Group. Neurology. 2000;55:185–192. doi: 10.1212/wnl.55.2.185. [DOI] [PubMed] [Google Scholar]
- 52.Barkhof F. Assessing treatment effects on axonal loss: evidence from MRI monitored clinical trials. J Neurol. 2004;251(suppl 4):IV6–IV12. doi: 10.1007/s00415-004-1403-2. [DOI] [PubMed] [Google Scholar]
- 53.Ruiz-Pena JL, Pinero P, Sellers G, et al. Magnetic resonance spectroscopy of normal appearing white matter in early relapsing-remitting multiple sclerosis: correlations between disability and spectroscopy. BMC Neurol. 2004;4:8. doi: 10.1186/1471-2377-4-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kendi AT, Tan FU, Kendi M, Huvaj S, Tellioglu S. MR spectroscopy of cervical spinal cord in patients with multiple sclerosis [published correction appears in Neuroradiology 2004;46:789] Neuroradiology. 2004;46:764–769. doi: 10.1007/s00234-004-1231-1. [DOI] [PubMed] [Google Scholar]
- 55.Minagar A. Gray matter involvement in multiple sclerosis: a new window into pathogenesis. J Neuroimaging. 2003;13:291–292. [PubMed] [Google Scholar]
- 56.Sanfilippo MP, Benedict RHB, Sharma J, Weinstock-Guttman B, Bakshi R. The relationship between whole brain volume and disability in multiple sclerosis: a comparison of normalized gray vs. white matter with misclassification correction. Neuroimage. 2005;26:1068–1077. doi: 10.1016/j.neuroimage.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 57.Bakshi R, Ariyaratana S, Benedict RHB, Jacobs L. Fluid-attenuated inversion recovery magnetic resonance imaging detects cortical and juxtacortical multiple sclerosis lesions. Arch Neurol. 2001;58:742–748. doi: 10.1001/archneur.58.5.742. [DOI] [PubMed] [Google Scholar]
- 58.Catalaa I, Fulton JC, Zhang X, et al. MR imaging quantitation of gray matter involvement in multiple sclerosis and its correlation with disability measures and neurocognitive testing. AJNR Am J Neuroradiol. 1999;20:1613–1618. [PMC free article] [PubMed] [Google Scholar]
- 59.Geurts JJ, Bo L, Pouwels PJ, Castelijns JA, Polman CH, Barkhof F. Cortical lesions in multiple sclerosis: combined postmortem MR imaging and histopathology. AJNR Am J Neuroradiol. 2005;26:572–577. [PMC free article] [PubMed] [Google Scholar]
- 60.Geurts JJ, Pouwels PJ, Uitdehaag BM, Polman CH, Barkhof F, Castelijns JA. Intracortical lesions in multiple sclerosis: improved detection with 3D double inversion-recovery MR imaging. Radiology. 2005;236:254–260. doi: 10.1148/radiol.2361040450. [DOI] [PubMed] [Google Scholar]
- 61.Bakshi R, Benedict RHB, Bermel RA, et al. T2 hypointensity in the deep gray matter of patients with multiple sclerosis: a quantitative magnetic resonance imaging study. Arch Neurol. 2002;59:62–68. doi: 10.1001/archneur.59.1.62. [DOI] [PubMed] [Google Scholar]
- 62.Bermel RA, Innus MD, Tjoa CW, Bakshi R. Selective caudate atrophy in multiple sclerosis: a 3D MRI parcellation study. NeuroReport. 2003;14:335–339. doi: 10.1097/00001756-200303030-00008. [DOI] [PubMed] [Google Scholar]
- 63.Inglese M, Liu S, Babb JS, Mannon LJ, Grossman RI, Gonen O. Three-dimensional proton spectroscopy of deep gray matter nuclei in relapsing-remitting MS. Neurology. 2004;63:170–172. doi: 10.1212/01.wnl.0000133133.77952.7c. [DOI] [PubMed] [Google Scholar]
- 64.Bermel RA, Puli SR, Rudick RA, et al. Prediction of longitudinal brain atrophy in multiple sclerosis by gray matter magnetic resonance imaging T2 hypointensity. Arch Neurol. 2005;62:1371–1376. doi: 10.1001/archneur.62.9.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ge Y, Gonen O, Inglese M, Babb JS, Markowitz CE, Grossman RI. Neuronal cell injury precedes brain atrophy in multiple sclerosis. Neurology. 2004;62:624–627. doi: 10.1212/wnl.62.4.624. [DOI] [PubMed] [Google Scholar]
- 66.Gonen O, Moriarty DM, Li BS, et al. Relapsing-remitting multiple sclerosis and whole-brain N-acetylaspartate measurement: evidence for different clinical cohorts—initial observations. Radiology. 2002;225:261–268. doi: 10.1148/radiol.2243011260. [DOI] [PubMed] [Google Scholar]
- 67.Gonen O, Catalaa I, Babb JS, et al. Total brain N-acetylaspartate: a new measure of disease load in MS. Neurology. 2000;54:15–19. doi: 10.1212/wnl.54.1.15. [DOI] [PubMed] [Google Scholar]
- 68.Bakshi R. Fatigue associated with multiple sclerosis: diagnosis, impact and management. Mult Scler. 2003;9:219–227. doi: 10.1191/1352458503ms904oa. [DOI] [PubMed] [Google Scholar]
- 69.Bakshi R, Miletich RS, Henschel K, et al. Fatigue in multiple sclerosis: cross-sectional correlation with brain MRI findings in 71 patients. Neurology. 1999;53:1151–1153. doi: 10.1212/wnl.53.5.1151. [DOI] [PubMed] [Google Scholar]
- 70.Tartaglia MC, Narayanan S, Francis SJ, et al. The relationship between diffuse axonal damage and fatigue in multiple sclerosis. Arch Neurol. 2004;61:201–207. doi: 10.1001/archneur.61.2.201. [DOI] [PubMed] [Google Scholar]
- 71.Benedict RH, Carone DA, Bakshi R. Correlating brain atrophy with cognitive dysfunction, mood disturbances, and personality disorder in multiple sclerosis. J Neuroimaging. 2004;14(suppl):36S–45S. doi: 10.1177/1051228404266267. [DOI] [PubMed] [Google Scholar]
- 72.Gadea M, Martinez-Bisbal MC, Marti-Bonmati L, et al. Spectroscopic axonal damage of the right locus coeruleus relates to selective attention impairment in early stage relapsing-remitting multiple sclerosis. Brain. 2004;127:89–98. doi: 10.1093/brain/awh002. [DOI] [PubMed] [Google Scholar]
- 73.Gadea M, Marti-Bonmati L, Arana E, Espert R, Casanova V, Pascual A. Dichotic listening and corpus callosum magnetic resonance imaging in relapsing-remitting multiple sclerosis with emphasis on sex differences. Neuropsychology. 2002;16:275–281. [PubMed] [Google Scholar]
- 74.Coull JT. Neural correlates of attention and arousal: insights from electrophysiology, functional neuroimaging and psychopharmacology. Prog Neurobiol. 1998;55:343–361. doi: 10.1016/s0301-0082(98)00011-2. [DOI] [PubMed] [Google Scholar]
- 75.Christodoulou C, Krupp LB, Liang Z, et al. Cognitive performance and MR markers of cerebral injury in cognitively impaired MS patients. Neurology. 2003;60:1793–1798. doi: 10.1212/01.wnl.0000072264.75989.b8. [DOI] [PubMed] [Google Scholar]
- 76.Sarchielli P, Presciutti O, Tarducci R, et al. 1H-MRS in patients with multiple sclerosis undergoing treatment with interferon β-1a: results of a preliminary study. J Neurol Neurosurg Psychiatry. 1998;64:204–212. doi: 10.1136/jnnp.64.2.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Narayanan S, De Stefano N, Francis GS, et al. Axonal metabolic recovery in multiple sclerosis patients treated with interferon beta-1b. J Neurol. 2001;248:979–986. doi: 10.1007/s004150170052. [DOI] [PubMed] [Google Scholar]
- 78.Schubert F, Seifert F, Elster C, et al. Serial 1H-MRS in relapsing-remitting multiple sclerosis: effects of interferon-beta therapy on absolute metabolite concentrations. MAGMA. 2002;14:213–222. doi: 10.1007/BF02668215. [DOI] [PubMed] [Google Scholar]
- 79.Parry A, Corkill R, Blamire AM, et al. Beta-interferon treatment does not always slow the progression of axonal injury in multiple sclerosis. J Neurol. 2003;250:171–178. doi: 10.1007/s00415-003-0965-8. [DOI] [PubMed] [Google Scholar]
- 80.Khan O, Shen Y, Caon C, et al. Cerebral axonal recovery in relapsing-remitting multiple sclerosis patients treated with glatiramer acetate. Mult Scler In press.
- 81.Narayanan S, Caramanos Z, Arnold D. The effect of glatiramer acetate treatment on axonal integrity in multiple sclerosis [abstract] Mult Scler. 2004;10(suppl 2):S256. Abstract P633. [Google Scholar]
- 82.Liu L, Meier D, Polgar-Turcsanyi M, Karkocha P, Bakshi R, Guttmann CRG. Multiple sclerosis medical image analysis and information management. J Neuroimaging. 2005;15(suppl 1):103S–117S. doi: 10.1177/1051228405282864. [DOI] [PubMed] [Google Scholar]