

Abstract
We recently found that leukocytes from thrombospondin-1 (TSP1)-deficient mice exhibit significant reductions in cell surface CD44 relative to those from wild type mice. Because TSG-6 modulates CD44-mediated cellular interactions with hyaluronan, we examined the possibility that TSP1 interacts with TSG-6. We show that recombinant full length human TSG-6 (TSG-6Q) and Link module of TSG-6 (Link_TSG6) bind 125I-TSP1 with comparable affinities. Trimeric recombinant constructs containing the N-modules of TSP1 or TSP2 inhibit binding of TSP1 to TSG-6Q and Link_TSG6, but other recombinant regions of TSP1 do not. Therefore, the N-modules of both TSP1 and TSP2 specifically recognize the Link module of TSG-6. Heparin, which binds to these domains of both proteins, strongly inhibits binding of TSP1 to Link_TSG6 and TSG-6Q, but hyaluronan does not. Inhibition by heparin is due to its binding to TSP1, because heparin also inhibits TSP1 binding to Link_TSG6 mutants deficient in heparin binding. Removal of bound Ca2+ from TSP1 reduces its binding to full-length TSG-6. Binding of TSP1 to Link_TSG6, however, is enhanced by chelating divalent cations. In contrast, divalent cations do not influence binding of the N-terminal region of TSP1 to TSG-6Q. This implies that divalent cation-dependence is due to conformational effects of Ca-binding to the C-terminal domains of TSP1. TSP1 enhances covalent modification of inter-α-trypsin inhibitor by TSG-6 and transfer of its heavy chains to hyaluronan, suggesting a physiological function of TSP1 binding to TSG-6 in regulation of hyaluronan metabolism at sites of inflammation.
Components of the extracellular matrix regulate inflammatory responses through their interactions with cell surface receptors on infiltrating immune cells as well as by direct interactions with inflammatory cytokines and chemokines (reviewed in (1–3)). These interactions are important for regulating leukocyte migration and activation at sites of inflammation.
One such extracellular matrix protein that is induced by specific inflammatory signals is thrombospondin-1 (TSP11) (reviewed in (4,5)). TSP1 plays an important role in early phases of wound repair, and its absence prolongs wound repair in the skin (6). TSP1 is specifically induced in the stroma surrounding some tumors (7) and in inflammatory responses associated with rheumatoid arthritis, glomerulonephritis, atopic dermatitis, atherosclerosis, and restenosis (8–13).
Inflammatory responses in the lungs of mice lacking TSP1 implicate TSP1 as a negative modulator of inflammatory responses, in part due to its ability to activate latent TGF-β1 (14,15). Furthermore, TSP1 has direct inhibitory effects on T cell receptor-mediated T cell activation (16) as well as NK cell expansion (17) and dendritic cell activation (18). In contrast, TSP1 primes neutrophils for oxidative burst responses (19) and promotes migration of T cells and other leukocytes (20,21). A lack of monocyte/macrophage recruitment was proposed to explain the delayed wound repair in TSP1 null mice (6). TSP1 also promotes expansion of inflammatory T cells in rheumatoid synovium (22). Therefore, TSP1 may be both a positive and negative modulator of inflammatory responses. These opposing responses to TSP1 may be partially explained by dissecting the opposing signals arising from engaging the several known TSP1 receptors that are expressed on inflammatory cells (8,21,23,24), but direct interactions with secreted inflammatory modulators should also be considered.
We recently found a deficiency in CD44 expression in leukocytes from TSP1 null mice ((25) and manuscript in preparation). CD44 is a cellular receptor for hyaluronan (HA) and also has known functions in regulation of inflammatory responses (26,27) and reviewed in (28). Because to date we have not detected direct interactions between TSP1 and either CD44 or HA, we considered the possibility that TSP1 may interact with other CD44 ligands or HA-binding proteins to influence CD44 expression. HA-binding proteins, also known as hyaladherins, characteristically contain domains related to cartilage link protein (29). TSG-6 (also known as tumor necrosis factor-induced protein-6, TNFIP6 (30)) is a member of this superfamily that attracted our attention based on its known interaction with pentraxin-3 (31), a previous observation that the N-terminal module of TSP1 is evolutionarily related to the pentraxin family (32), and the ability of TSG-6 to modulate CD44 interactions with HA (33). One previous publication reported TSP1 binding to recombinant TSG-6, but this binding was attributed to a nonspecific interaction of TSP1 with the His tag used in the recombinant TSG-6 construct rather than with TSG-6 itself (34).
Here we report that TSP1 is a specific ligand for TSG-6. This interaction is mediated by the N-module of TSP1 and the Link domain of TSG-6. Binding is regulated by divalent cations and is strongly inhibited by the interaction of heparin with the N-module of TSP1 but not by HA binding to the Link domain of TSG-6. One biological activity of TSG-6 is to form covalent complexes with either of the two heavy chains of inter-α-trypsin inhibitor (IαI) (35–37). We further show that TSP1 binding modulates the ability of TSG-6 to covalently modify IαI.
EXPERIMENTAL PROCEDURES
Materials
TSP1 was purified from human platelets obtained from the National Institutes of Health Department of Transfusion Medicine under a protocol approved by the NIH IRB and stored at −70° in 0.02 M Tris, pH 7.6, 0.15 M NaCl, 0.1 mM CaCl2, 20% w/v sucrose (38). Monomeric and trimeric recombinant regions of TSP1 and TSP2 expressed in insect cells were prepared as described (39,40). TSP proteins/domains were labeled with 125I using Iodogen (Pierce, Rockford, IL) as described previously (41). Recombinant full-length TSG-6Q and Link_TSG6 were prepared as described (42–44). Link_TSG6 mutants with either impaired HA-binding (Y12F and Y59F) or heparin-binding (K34A/K54A, K20A/K32A/K41A) activities were constructed and characterized as described in (45,46). Biotinylated-Link_TSG6 was prepared as described in (47).
High molecular weight HA was obtained from BioTechnology General, Rehovot, Israel. The heparin sodium salt from porcine intestinal mucosa (weight average Mr ~ 12,000) was purchased from Eli Lilly, Indianapolis, IN, and heparin coupled to bovine serum albumin was purchased from Sigma, St. Louis, MO.
Solid phase binding assays
Immulon® 2 HB (TermoLabsystems, Franklin, MA) microtiter strips with breakaway wells were coated directly with 50 μl of the indicated concentrations of TSG-6Q or Link_TSG6. For competitive binding studies, 10 μg/ml full-length TSG-6Q or Link_TSG6 by incubating overnight at 4°C in Dulbecco’s PBS without Ca2+, Mg2+. Non-specific sites were blocked with 3% (w/v) BSA in Dulbecco’s PBS (DPBS) at room temperature for 1h. Radioiodinated TSP1 (0.5 μg/ml, 50 μl/well) was added alone or in the presence of increasing concentrations of the indicated unlabelled ligands as competitor in Dulbecco’s PBS, containing 0.5 % (w/v) BSA, 0.1 mM phenylmethylsulfonyl fluoride with or without Ca2+, Mg2+ and incubated at 4°C, 22°C or 37°C for 3 h. The wells were washed with the same cold buffer, and the bound radioactivity was quantified using a gamma counter (Packard BioScience Company, Downers Grove, IL). For some experiments, radioiodinated recombinant NoC1 was added and incubated as above. The effect of pH on the interaction of TSP1 with immobilized TSG-6Q or Link_TSG6 was investigated using a modification of the above assay. Plates were coated, blocked and washed as described for the standard assay. Following this, further dilutions and washes were performed in 100 mM NaCl, 0.5% (w/v) BSA, 0.9 mM CaCl2, 0.5 mM MgCl2, buffered to a specific pH: 50 mM Na-acetate was used from pH 5.5 to 6.0 and 50 mM Na-HEPES (N-[2-hydroxyethyl]piperazine-N’-[2-ethanesulfonic acid]) between pH 6.0 and 7.5.
For some experiments, a reverse assay was employed in which TSP1 was immobilized. TSP1 was immobilized at various concentrations on POLYSORP plates in PBS containing 0.5 mM MgCl2, 0.9 mM CaCl2. Each ELISA was performed in 100 mM NaCl, 50 mM sodium acetate, 0.5 mM MgCl2. 0.9 mM CaCl2, 0.2 % (v/v) Tween-20 pH 6.0 and blocked in 1 % (w/v) BSA in assay buffer prior to incubation with ligand. TSG-6/Link_TSG6 was incubated on the plate for 4 h. TSG-6Q binding was detected by addition of 1:4000 rabbit anti-human polyclonal RAH1 (48) in assay buffer for 45 min, 1:2000 goat anti-rabbit alkaline phosphatase conjugate (Jackson) in assay buffer for 45 min, and 1 mg/ml Na2 p-nitrophenol in development buffer for 20 min.
Pull-down assays
Immobilized antibody recognizing TSP1 (1 μg mouse monoclonal 5G11, Biodesign) was incubated for 3 h at 37° with 1.1 pmol TSP1 and 2 fmol biotinylated Link_TSG6 in 50 μl of DPBS containing Ca2+, Mg2+, 0.5% BSA, and 0.2% Tween-20. After washing, bound Link_TSG6 was solubilized with SDS sample buffer and analyzed by Western blotting with streptavidin-peroxidase. Conversely, a complex of TSP1 with TSG-6Q was pulled down using 1 μg of immobilized TSG-6 antibody Q75 and analyzed by Western blotting using biotinylated TSP1 antibody A6.1 and streptavidin peroxidase.
Analysis of TSP1 effects on the catalytic activity of TSG-6
TSG-6Q (at 5 μg/ml final concentration; 0.17 μM based on a molecular weight of 30 kDa) was pre-incubated for 1h at 37°C with a 4-fold excess of TSP1 (at 100 μg/ml final concentration; 0.67 μM based on a subunit molecular weight of 150 kDa) or mock incubated with the corresponding TSP1 buffer containing sucrose diluted to a final volume of 50 μl in DPBS with divalent cations. TSG-6Q alone, TSG-6Q pre-complexed with TSP1, or mock incubated TSP1 were subsequently incubated with 0.5 μl of IαI-containing fetal bovine serum for 10–60 min. at 37°C. The reaction mixtures were then electrophoresed on 7.5 or 12 % Tris-HCl SDS gels under reducing conditions and transferred onto polyvinylidine difluoride (PVDF) membranes (Bio-Rad, Hercules, CA). The blots were probed with a goat polyclonal antibody against TSG-6 (sc-21828, 1:500; Santa Cruz Biotechnology, Santa Cruz, CA) and rabbit polyclonal antibody against inter-α-trypsin inhibitor (A0301, 1:2000; DAKO A/S, Denmark). Horseradish peroxidase (HRP)-conjugated donkey anti-goat IgG (1:5000; Santa Cruz Biotechnology) and HRP-conjugated goat anti-rabbit IgG (1:5000; Pierce) were used as secondary antibodies. Immunoreactive bands were detected with the ECL™ detection system (Pierce).
Effects of TSP1 on the transfer of IαI heavy chains to HA were assessed essentially as described (37). TSG-6Q (0.2 μg) was preincubated with a 6-fold molar excess of TSP1 (6 μg) for 1 h at 37° prior to addition of purified IαI (8 μg) and a 14-mer HA oligosaccharide (HA14, 2673 Da; 10 μg); i.e., to give final concentrations of 0.27 μM TSG-6, 1.6 μM TSP1, 1.8 μM IαI and 150 μM HA14 in 20 mM Hepes.HCl pH 7.5, 10.2 mM Tris.HCl pH 7.6, 150 mM NaCl, 5 mM MgCl2, 5 mM CaCl2, 10.2 % (w/v) sucrose. After incubating for the indicated times at 37°, the reaction mixtures were analyzed by Tris-tricine SDS gel electrophoresis.
Data analysis
Self-displacement binding experiments were analyzed using Scafit version 2.4 of the LIGAND program (49).
RESULTS
TSP1 binds to the Link module of TSG-6
Human platelet TSP1 was labeled using 125I and tested for binding to human TSG-6 immobilized on plastic (Fig. 1). As shown in Fig.1A, at pH 7.3 in the presence of physiological salt concentrations and divalent cations, TSP1 bound minimally to immobilized full-length TSG-6Q at 4°C. Dose-dependent binding, however, markedly increased with temperature and was highest at 37°C.
Fig.1.

Divalent cation-dependent binding of TSP1 to TSG-6. 125I-TSP1 binding to different amounts of full-length TSG-6Q (panels A, C) or Link_TSG6 (panels B, D) coated onto wells of a microtiter plate was tested by the solid phase assay. Temperature dependence was examined by adding 125I-TSP1 (0.5 μg/ml, 50 μl/well) and incubating at 4°C, 22°C, or 37°C in humidified atmosphere at pH 7.4 for 3h. The assays were performed in the presence of Ca2+ and Mg2+ (Solid lines, panels A, B) or without Ca2+, Mg2+ and in the presence 5 mM EDTA (dotted lines, panels C, D). Background values were subtracted from each data point (66.0 ±15.6, 49.0 ±3.4, 126.1 ±10.9). Results (mean cpm ± S.E.M.) shown are representative of two independent experiments performed in triplicate.
TSG-6 is an approximately 35 kDa secreted protein composed of contiguous Link and CUB modules (30). The TSG-6 Link module binds to HA (50,51), chondroitin 4-sulfate (47), the G1-domain of aggrecan (52), heparin (46,53) and pentraxin-3 (31). The function of the CUB module of TSG-6 remains unknown, although the fact that it is highly conserved between species suggests that it is essential for at least some activities of TSG-6. To determine which domain of TSG-6 mediates binding of TSP1, we examined binding of TSP1 to the HA binding domain of TSG-6 (Link_TSG6). 125I-TSP1 bound to immobilized Link_TSG6 in a similar temperature- and dose-dependent manner as to TSG-6Q (Fig.1B). Therefore, the Link domain of TSG-6 is sufficient for TSP1 binding, but we can not exclude additional interactions of TSP1 with the CUB domain.
To test the influence of calcium ions, which modulate the conformation and some ligand binding properties of TSP1 (54,55), on its binding to TSG-6, 5 mM EDTA was added to chelate free Ca++ (Fig. 1C, D). EDTA decreased binding of TSP1 to TSG-6Q approximately 4-fold at each temperature examined (Fig. 1C). Binding of TSP1 to Link_TSG6, in contrast, was somewhat enhanced in the absence of divalent cations (Fig. 1D).
Interactions between TSP1 and TSG-6 could also be detected when TSP1 was immobilized, and TSG-6 binding was detected by ELISA using an antibody that binds to epitopes distant from the Link domain (i.e., RAH1 (48)). Using this approach, dose-dependent binding of TSG-6 and Link_TSG-6 to immobilized TSP1 was observed (Fig. 2A). TSG-6 antibodies were used to further define the TSP1 binding site. Antibody A38 inhibited TSG-6 binding to TSP1, but antibody Q75 did not (Fig. 2B). Notably, A38 also inhibits HA binding (56), TSG-6·IαI complex formation, and cumulus-oocyte complex expansion (57); Q75 does not affect any of these functions.
Fig. 2.
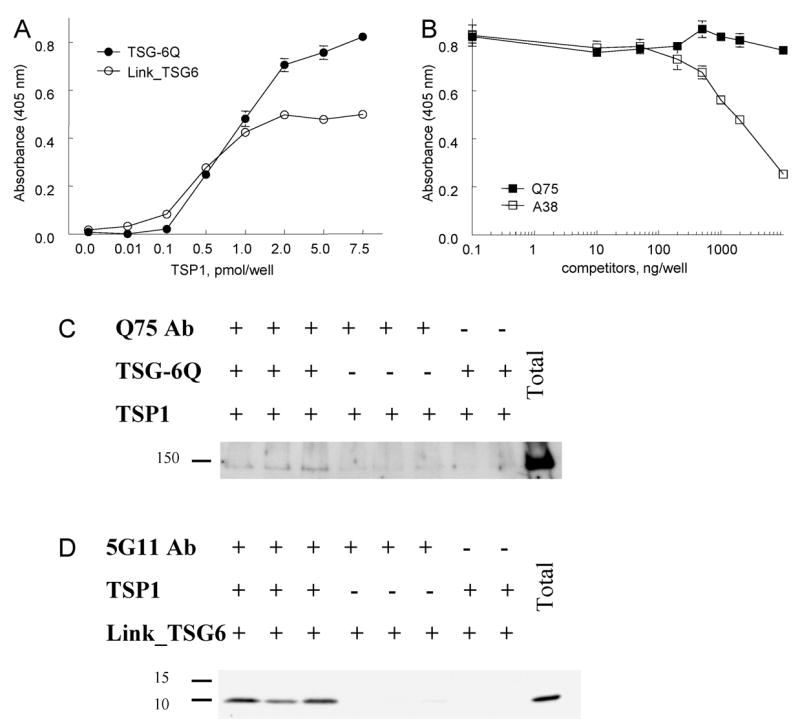
Binding of TSG-6 to immobilized TSP1. Panel A: Binding of 4 pmol solution phase TSG-6Q (filled circles) or 4 pmol Link_TSG6 (open circles) to the indicated amounts of surface bound TSP1 was determined by an ELISA using a rabbit polyclonal anti-TSG-6 antibody. Panel B: Interaction of solution phase TSG-6Q (4 pmol) with surface bound TSP1 (2 pmol) was assessed in the presence of the indicated concentrations of murine monoclonal TSG-6 antibodies Q75 (filled squares) or A38 (open squares). Absorbance at 405 nm is plotted as mean ± SEM, N = 8. Panel C: Binding of TSP1 to TSG-6Q in solution was detected by pull down using immobilized TSG-6 antibody Q75, which did not inhibit binding in B. Bound TSP1 was detected after solubilization in SDS, separation by SDS gel electrophoresis and blotting with biotinylated-TSP1 antibody A6.1, streptavidin-peroxidase, and ECL. Panel D: TSP1-biotinylated-Link_TSG6 complexes were pulled down using immobilized TSP1 antibody 5G11, which recognizes the third properdin repeat of TSP1 (99). Bound Link_TSG6 was detected by electrophoresis on 12% acrylamide Tris/HCl SDS gels and blotting using streptavidin-peroxidase and ECL.
Both of the above assays required surface adsorption of one of the two proteins on polystyrene, which is known in some cases to alter protein conformation and may partially denature the adsorbed protein (58,59). To confirm that TSP1 and TSG-6 can interact in the absence of such potential perturbations, we used pull down assays to detect formation of complexes between TSG-6 and TSP1 in solution. The TSG-6 antibody Q75 was used for this purpose because it did not perturb binding in the solid phase assay (Fig. 2B). Based on detection by Western blotting, TSP1 was pulled down by the Q75 antibody only in the presence of TSG-6Q (Fig. 2C). Conversely, biotinylated Link_TSG6 was pulled down by a TSP1 antibody recognizing an epitope distant from its TSG-6-binding domain (see below) only in the presence of soluble TSP1 (Fig. 2D).
Quantitative analysis of competitive binding experiments using unlabeled TSP1 showed that in the presence of divalent cations at 37°C TSP1 bound with a Kd of 310 nM to immobilized full length TSG-6 (Table 1). In the absence of divalent cations, the affinity of binding decreased approximately 10-fold. In contrast, binding of TSP1 to Link_TSG6 at 37° was approximately 3-fold higher affinity (80 nM) than to the full length protein. The Kd decreased further to 30 nM in the absence of divalent cations. These data indicate an important role of divalent cations in TSP1-TSG-6 interactions, and suggest that the CUB domain of TSG-6 may sterically restrict TSP1 binding.
Table 1.
Divalent cation-dependence for binding of 125I-thrombospondin-1 to immobilized TSG-6Q and Link_TSG6. Apparent dissociation constants ± SD for the specified buffers and temperatures were determined from self displacement data by nonlinear regression using the LIGAND programs.
| Kd (M)
|
|||
|---|---|---|---|
| PBS Ca2+/Mg2+ 37° C | PBS/EDTA 37° C | PBS Ca2+/Mg2+ 22° C | |
| Link_TSG6 | 8.0 ± 3.1 x 10−8 | 3.0 ± 1.2 x 10−8 | 1.13±0.01x10−7 |
| Full-length TSG-6Q | 3.1 ± 0.6 x 10−7 | ~2.4 x 10−6 | 1.2±0.1x10−7 |
The N-module of TSP1 mediates TSG-6 binding
TSP1 is a homotrimeric glycoprotein composed of 150 kDa subunits. Each monomer contains an N-terminal globular module (N), a disulfide-mediated oligomerization site (o), a vWC-like module (C), three properdin/type 1 repeats (P), three EGF-like repeats (E); seven tandem aspartate-rich, Ca2+-binding repeats (Ca); and a C-terminal globular domain (G) (Fig. 3E). Several recombinant regions of TSP1 were examined to define the TSG-6 binding domain (Fig. 3). 125I-TSP1 binding to TSG-6Q (Fig.3A) or Link_TSG6 (Fig.3B) was inhibited by a trimeric N-terminal construct (NoC1) but not by any recombinant TSP1 proteins tested that lacked the N-terminal domain (CP123, P3E123 and E3CaG-1). Lack of inhibition by CP123 suggested that the TSG-6 binding site is in the N-module rather than the C-module, but lack of inhibition by CP123 could also be due to a requirement for trimeric C module for binding to TSG-6.
Fig. 3.
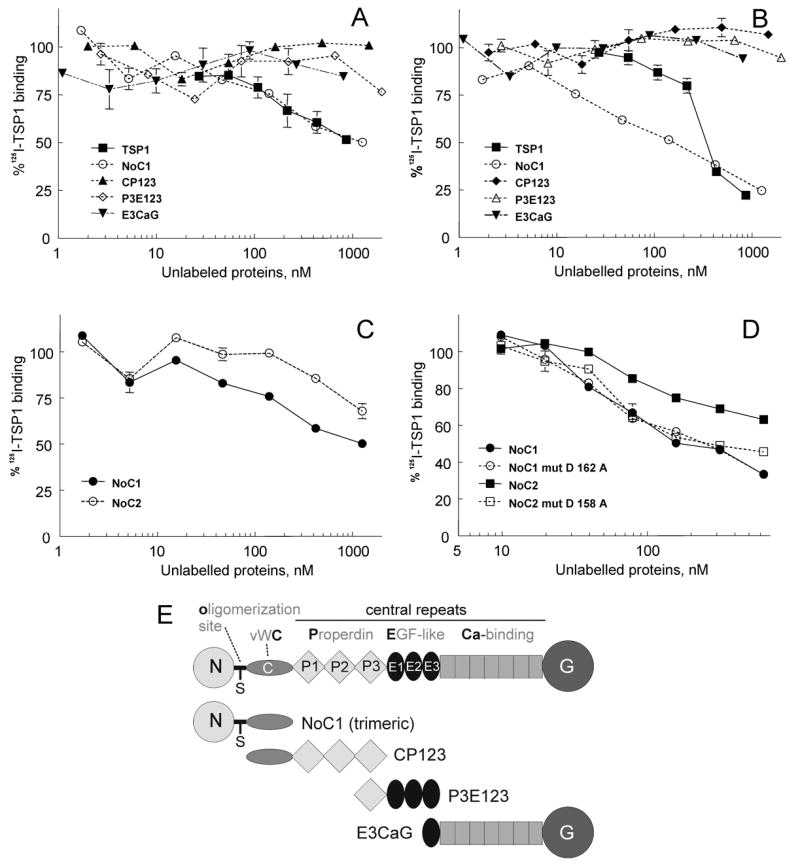
Localization of the TSG-6 binding site in TSP1. Wells were coated using 50μl/well of 10μg/ml TSG-6Q (panel A) or Link_TSG6 (panels B-D), and 125I-TSP1 binding was measured at 37° in the presence of competing concentrations of unlabeled TSP1 or the indicated recombinant regions of TSP1 (as shown in E) or TSP2. Data were normalized and presented as a percent of the specific TSP1 binding in the absence of unlabelled ligands. The assays were performed in the presence of Ca2+, Mg2+.
Within the TSP family, TSP1 and TSP2 are most closely related in their domain organization and primary sequences. TSP1 and TSP2 share the highest degree of sequence identity in their C-terminal regions (60), but the N-modules of human TSP1 and TSP2 are only 36% identical. Despite this divergence, a trimeric recombinant region of TSP2 equivalent to NoC1 (NoC2) also inhibited TSP1 binding to Link_TSG6, albeit with approximately 4-fold lower potency (Fig.3C). The N-module of TSP1 contains binding sites for heparin (61), calreticulin (62), and several β1 integrins (21,63,64). Mutants of NoC1 (D162A) and NoC2 (D158A) were available that disrupt a sequence recognized by α4β1 integrin (21). Both of these mutants inhibited binding of 125I-TSP1 to Link_TSG6 with comparable activities as the native NoC1 (Fig.3D). Therefore, the TSG-6 binding site appears to be distinct from this α4β1 integrin binding site in TSP1 and TSP2.
To confirm specific binding of the N-terminal domains of TSP1 to TSG-6, we examined direct binding of 125I-NoC1 to TSG-6 proteins (Fig. 4). 125I-NoC1 bound with similar dose dependencies to full length TSG-6Q (Fig.4A) and Link_TSG6 (Fig.4B). We also used this assay to further examine the basis for the divalent cation dependence of TSP1-TSG-6 interaction. NoC1 lacks the known Ca2+ binding sites of TSP1, which are located in the type 3 repeats and C-terminal domain (54,65). Unlike full length TSP1 (Fig. 1), NoC1 binding to TSG-6Q was identical in the presence and absence of divalent cations (Fig. 4A). Binding of NoC1 to Link_TSG6 was slightly higher in the absence of cations (Fig. 4B). These results indicate that the divalent cation dependence observed for full length TSP1 binding to TSG-6Q is not due to direct effects of divalent cations on either TSG-6 or its interaction with the N-modules of TSP1; NMR titration studies have shown that there is no specific binding of Ca2+ ions to the TSG-6 Link module2. Rather, binding of Ca2+ to C-terminal regions of TSP1 may indirectly increase the ability of the N-module to interact with the Link module of TSG-6.
Fig. 4.
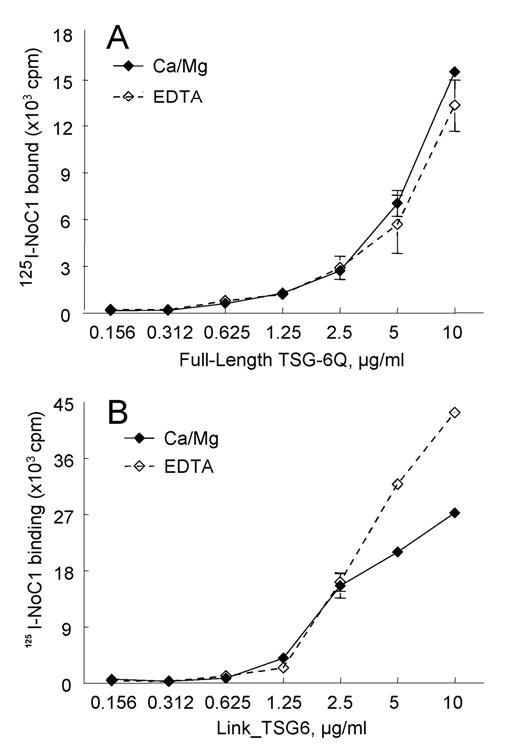
NoC1 binding to TSG-6. 125I-NoC1 binding to different amount of TSG-6Q (A) or Link_TSG6 (B) coated onto wells of a microtiter plate was tested by the solid phase assay. The assays were performed at 37°C in the presence of Ca2+, Mg2+ (solid lines) or in the presence of 5 mM EDTA, (dotted lines). Background values are subtracted from each data point (593 ±43). Results (mean cpm ± S.E.M.) shown are representative of two independent experiments performed in triplicate.
Effects of heparin on TSP1-TSG-6 interactions
TSP1 is a known heparin-binding protein, which is primarily mediated by the N-module (61,66). Heparin, which is very similar in structure to highly sulfated regions of heparan sulfate (HS), is an excellent model for studying HS-protein interactions. The Link module of TSG-6 also interacts specifically with heparin, via a site distinct from its HA-binding surface (30,46,53). Therefore, heparin could potentially inhibit interactions of TSP1 with TSG-6 by binding to either of these proteins.
To further define the molecular basis for the binding of TSP1 to TSG-6, we compared interactions between TSP1 and wild type Link_TSG6 with that of Link_TSG6 mutants that have significantly reduced heparin-binding properties (Fig.5A). Folded mutants K34A/K54A and K20A/K32A/K41A, which have ~60% and ~10% of wild type activity (46), respectively, bound better to TSP1 than did wild type Link_TSG6. This suggested that the TSP1-binding site on TSG-6 is distinct from the heparin-binding site.
Fig. 5.
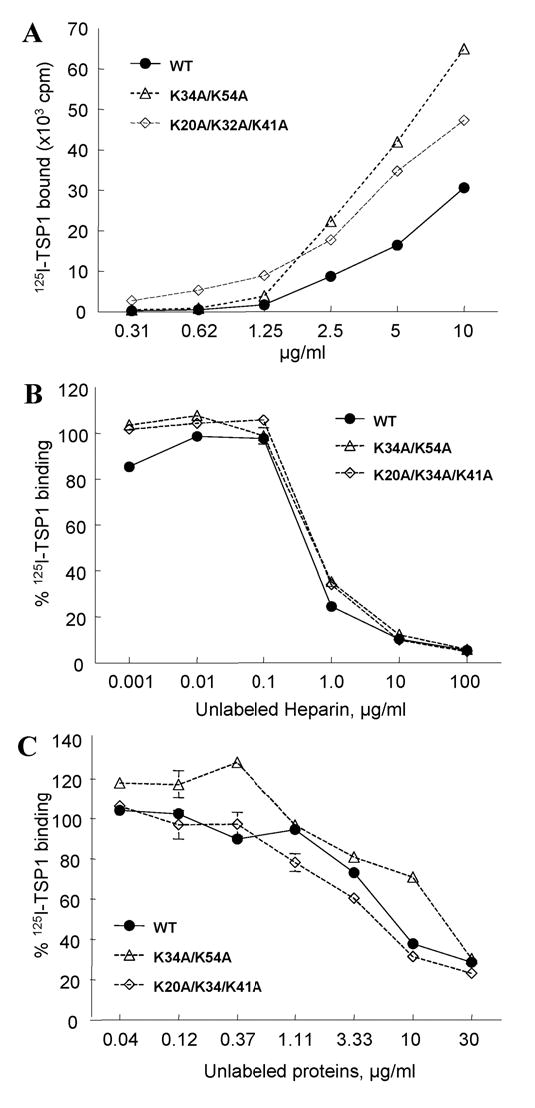
Inhibition of TSP1 binding to Link_TSG6 by heparin. A: Comparison of the TSP1-binding activities of Link module mutants with wild-type Link_TSG6. The binding of 125I-TSP1 to different amounts of wild-type (WT) Link_TSG6 (solid line) or selected mutants (dotted lines) was tested by solid phase assay. Background values are subtracted from each data point (256 ±14). B: The binding of 125I-TSP1 to different amounts of wild-type (WT) Link_TSG6 (solid line) or selected mutants (dotted lines) in the presence of the indicated concentrations of heparin. C: The binding of 125I-TSP1 to wells coated with heparin-BSA was determined in the presence of competing concentrations of unlabeled WT Link_TSG6 (solid line) or mutants (dotted lines). The assays were performed at 37°C, pH 7.3 in the presence of Ca2+, Mg2+. Results (mean cpm ± S.E.M.) shown are representative of two independent experiments performed in triplicate.
In contrast, binding of 125I-TSP1 to immobilized wild type Link_TSG6 was potently inhibited by heparin with an IC50 less than 1 μg/ml (Fig. 5B). However, binding of TSP1 to the Link_TSG6 mutants K34A/K54A or K20A/K32A/K41A was inhibited by heparin with identical IC50 values to that of wild type (Fig.5B). Because the K20A/K32A/K41A mutant has about 90% reduced heparin binding, this result suggested that heparin inhibits primarily via its interaction with TSP1. However, this inhibition could also be explained by co-aggregation of trivalent labeled TSP1 with its multivalent ligand heparin in solution. Thus, inhibition by soluble heparin in the former assay may not be due to direct competition by heparin for a TSG-6 binding site on TSP1. This potential artifact could be circumvented by immobilizing heparin and examining the ability of TSG-6 to inhibit binding of 125I-TSP1. Using this approach, we found that binding of 125I-TSP1 to plates coated with heparin covalently coupled to BSA could be fully inhibited by Link_TSG6 (Fig. 5C). Because the Link_TSG6 mutants with impaired heparin binding (46) exhibited similar dose dependencies as the wild type, we conclude that heparin inhibits TSP1 binding to TSG-6 by interaction with the heparin-binding site in the N-module of TSP1. The heparin and TSG-6 binding sites on TSP1 either overlap or are situated so that heparin binding sterically inhibits binding of TSG-6. Our data also suggests that TSP1 and heparin recognize separate sites on the Link module of TSG-6.
HA and TSP1 bind to distinct sites on Link_TSG6
The HA-binding site of Link_TSG6 has been mapped, and five key residues (i.e., Lys-11, Tyr-12, Tyr-59, Phe-70 and Tyr-78) that contribute to HA-binding have been identified on a single face of the Link module (45,51,67). Two Link_TSG6 mutant constructs that have wild-type folds were selected for comparison. Mutations of Tyr-12 or Tyr-59 (mutants Y12F and Y59F) significantly reduces HA-binding activity (1% and 4% of wild-type binding (45,68). As shown in Fig. 6A, these mutant proteins exhibited the same TSP1 binding efficiency as wild type protein. These findings suggested that TSP1 interacts with an area of the Link module of TSG-6 distinct from the HA-binding site.
Fig. 6.
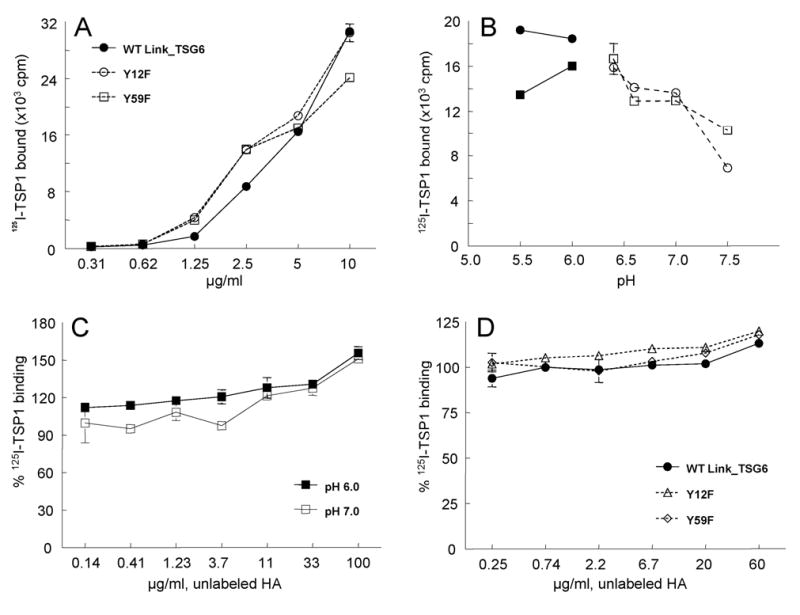
Effect of hyaluronan and pH on TSP1 binding to Link_TSG6. Panel A: Binding of 125I-TSP1 was determined to substrates coated using the indicated concentrations of Link module mutants deficient in hyaluronan binding (○, Y12F; □, Y59F) or wild-type Link_TSG6 (•). Background values are subtracted from each data point (256.3±13.6). Panel B: The pH dependence of 125I-TSP1 binding to immobilized TSG-6Q (•, ○) or Link_TSG6 (▪,□). TSP1 binding was determined at the indicated pH values in two buffer systems (Na-acetate, solid lines with closed symbols; Na-HEPES, dotted lines with open symbols) Panel C: 125I-TSP1 binding to immobilized Link_TSG6 was determined in the presence of the indicated concentrations of HA at a pH optimal for HA binding to TSG-6 (pH 6.0, ▪) and at pH 7.0 (□). Data are presented as a % of the control specific binding determined at each pH in the absence of HA. Panel D: TSP1 binding was determined to immobilized wild type Link_TSG6 (-•-) or the indicated Link_TSG6 mutants with reduced HA-binding activity (dotted lines with open symbols) in the presence of the indicated concentrations of HA. Data are presented as a % of the specific TSP1 binding determined for each Link protein in the absence of HA. For all panels, the results (mean cpm ± S.E.M.) shown are representative of two independent experiments performed in triplicate.
Addition of HA did not inhibit binding of TSP1 to immobilized TSG-6Q at pH 7.3 (results not shown). However, some interactions of TSG-6 with HA are highly pH-dependent, with maximal binding at pH 6 and a dramatic reduction in activity at neutral pH (52), and TSG-6 also binds to aggrecan with a similar pH dependence. Given the reported pH dependency of TSG-6 interactions with HA, these experiments were repeated at pH 6.0. We first examined the effect of pH on the interaction TSP1 with TSG-6 and Link_TSG6 (Fig. 6B). Remarkably, 125I-TSP1 binding to both TSG-6Q and Link_TSG6 coated plates increased with decreasing pH between 7.5 and 5.5. We then explored the possibility that interaction of TSP1 with TSG-6 could be inhibited by HA in a pH-dependent manner. As shown in Fig. 6C, however, binding of 125I-TSP1 to Link_TSG6 slightly increased in the presence of unlabelled HA at both pH 6.0 and pH 7.0. Similar results were obtained for binding of TSP1 to Link_TSG6 mutants defective in HA binding (Fig. 6D). These results support the conclusion that TSP1 and HA recognize separate sites on Link_TSG6. In addition, we have shown that TSP1 does not bind detectably to HA itself (data not shown), suggesting that TSG-6 can mediate interactions between TSP1 and HA by binding simultaneously to both ligands.
TSP1 enhances the interaction between TSG-6 and Inter-α-inhibitor in the absence of HA
To define functional consequences of TSP1 interactions with TSG-6, we examined the effects of this complex formation on the known molecular interactions of TSG-6 with IαI. This interaction has a fundamental role in both normal physiological and pathological processes (36,37).
IαI consists of three polypeptide chains covalently cross-linked by carbohydrate. Two heavy chains, HC1 (76.3 kDa) and HC2 (76.5 kDa) (69), and one light chain named bikunin (30 kDa) are linked together by chondroitin sulfate. TSG-6 influences IαI at two different levels (30). Through the formation of covalent complexes with IαI and catalysis of IαI heavy chain transfer to HA, TSG-6 contributes to the formation of IαI•HA complexes in the extracellular matrix (36,70,71)(37). Second, as a result of non-covalent interactions, TSG-6 enhances the anti-plasmin activity of IαI and thus might modulate the protease network (46,68).
Covalent complexes between TSG-6 and the heavy chains (HC) of IαI are rapidly formed by incubating TSG-6 with purified IαI or serum (37,42) Western blot analysis of IαI-containing fetal bovine serum incubated at 37°C for 10 min. or 60 min. with recombinant human TSG-6 revealed the presence of a TSG-6-immunoreactive band migrating at the known molecular mass of TSG-6·HC (Fig. 7A, lanes 1–3 from three independent experiments). Pre-incubation of TSG-6Q with purified TSP1 substantially enhanced formation of TSG-6·HC at 10 min but less so at 60 min (lanes 4–6 from three independent experiments). Incubation of TSG-6Q with TSP1 in the absence of serum or TSP1 with serum in the absence of TSG-6Q did not form this complex (lanes 8–9). Western blot analysis of the same reaction mixtures with anti-IαI rabbit antibody showed that incubation of serum with TSG-6 led to the expected progressive disappearance of intact IαI (Fig. 7A,B). By contrast, when TSG-6 was pre-incubated with TSP1, more rapid degradation of IαI was observed. This was not a direct effect of TSP1 on IαI because no significant loss of IαI was observed when TSP1 was incubated with IαI in the absence of TSG-6Q. These results indicate that interaction with TSP1 enhances the ability of TSG-6 to mediate the first steps in processing of IαI, i.e. covalent complex formation.
Fig. 7.
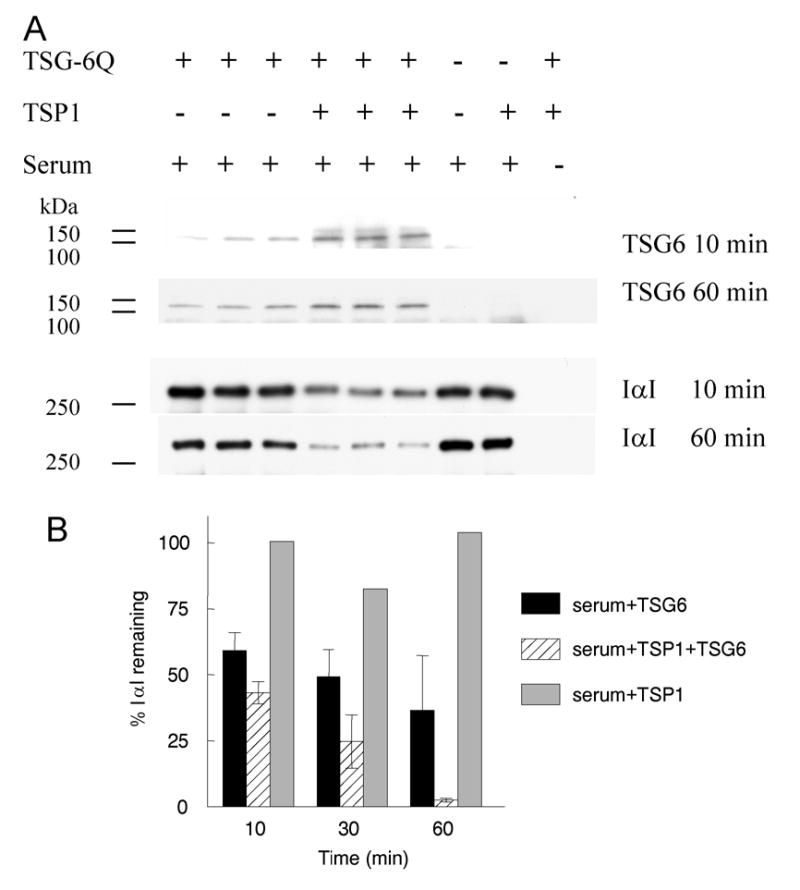
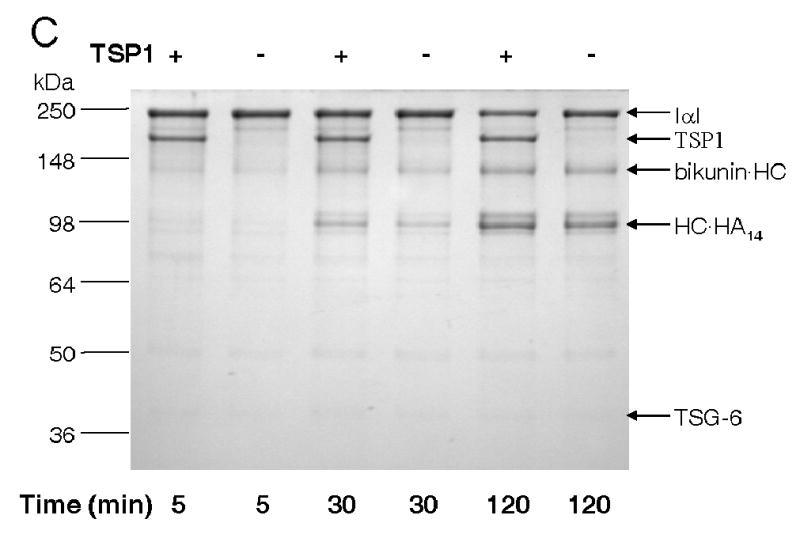
TSP1 enhances TSG-6-mediated IαI degradation (TSG-6·HC complex formation) and transfer of HC onto HA. Panel A: Full-Length recombinant TSG-6Q (at 5 μg/ml final concentration; 0.17 μM based on a molecular weight of 30 kDa) was pre-incubated for 1h at 37°C with TSP1 (at 100 μg/ml final concentration; 0.67 μM based on a subunit molecular weight of 150 kDa) or mock incubated with the corresponding TSP1 buffer diluted to a final volume of 50 μl in DPBS with divalent cations. TSG-6Q alone, TSG-6Q pre-complexed with TSP1, or mock incubated TSP1 was subsequently incubated with 0.5 μl of IαI-containing fetal bovine serum for 10–60 min. at 37°C. Panel A: reaction mixtures were analyzed by SDS gel electrophoresis on 7.5 (IαI) or 12% Tris-HCl acrylamide gels (TSG-6) followed by Western blotting with goat anti-TSG-6 antibody or with rabbit antiserum to IαI. Molecular masses of standards in kDa are indicated at the left border. Panel B: Western blots probed with anti-IαI and exposed at subsaturating densities were analyzed using ImagePro software. Integrated densities are expressed normalized to the mock treated serum signal at each time point (mean ± SD). Panel C: a Coomassie blue-stained SDS-PAGE gel showing enhanced TSG-6·HC complex formation at 30 and 120 min (as evidenced by the increased amount of the bikunin·HC byproduct (37)) and heavy chain transfer onto HA14 (i.e., to form HC·HA14) following pre-incubation with TSP1 for 1h at 37°C.
TSP1 enhances transfer of IαI heavy chains to HA
The TSG-6-HC complex is an intermediate in the covalent transfer of HC to HA (37). By incubating an HA oligosaccharide (HA14) with purified IαI and TSG-6Q that was preincubated in the presence or absence of TSP1, we found that TSP1 also enhances the TSG-6-mediated transfer of IαI heavy chains onto HA (Fig. 7C). Formation of the HC·HA14 product was enhanced at 30 and 120 min in the presence of TSP1, and increased loss of intact IαI was evident in the presence of TSP1 at the latter time point.
DISCUSSION
We have identified TSG-6 as an additional ligand for the N-terminal region of TSP1. This interaction is sensitive to the conformation of TSP1, which is regulated by binding of Ca2+. Depletion of Ca2+ reduces the affinity of TSP1 for TSG-6 approximately 10-fold. One premise for examining this interaction was that the N-module of TSP1 belongs to the pentraxin family, and pentraxin-3 is a known TSG-6 ligand (31). Indeed, we found that the pentraxin module of TSP1 and presumably the same module of TSP2 interact with the Link module of TSG-6. The TSG-6-binding site in pentraxin-3 has not been defined, however, so we can not conclude that pentraxin-3 and TSPs interact with TSG-6 through their paralogous domains. As previously reported for pentraxin-3 (31,36), mutation of residues in TSG-6 required for HA binding do not inhibit TSP1 binding, and we further demonstrate that HA does not inhibit TSP1 interaction with TSG-6. TSP1 binding to TSG-6 is also maintained at pH values permissive for TSG-6 binding to HA (52). Thus, TSG-6 may mediate formation of trimolecular complexes containing HA and TSP1 or TSP2. In contrast, heparin binding to the N-module of TSP1 potently inhibits the same interaction, suggesting that HSPGs may be negative modulators of this interaction. Finally, we demonstrate that TSP1 is an enhancer of the activity of TSG-6 to covalently associate with heavy chains of IαI, and to catalyze the covalent modification of HA by IαI (36,37,70–73).
Insights into other potential functions of the TSP1-TSG-6 interaction may come from examining their patterns of expression. Both proteins have limited expression in normal tissues but are rapidly induced by inflammatory responses. One inflammatory site where these two proteins are clearly co-expressed is in synovial fluid. TSP1 is induced in rheumatoid synovium (74) and accumulates on the surface of fibroblast-like synoviocytes, where it engages T cells through their CD47 receptor (8). Similarly, TSG-6 is elevated in arthritic synovial fluid (75) and joint tissues (76), and is induced by IL-17 on fibroblast-like synoviocytes of rheumatoid arthritis patients (77). TSG-6 shows consistent anti-inflammatory properties in mouse models of inflammation/arthritis (68,78–84), whereas implantation of hydron pellets containing TSP1 in rats during adjuvant-induced arthritis increased inflammation (85). Therefore, TSP1 and TSG-6 may have opposing functions in rheumatoid arthritis.
TSP1 has also been extensively studied in inflammation associated with cardiovascular disease. TSP1 is strongly induced during the intimal hyperplasia associated with atherosclerosis and in mechanical injury, diabetes, and hypercholesterolemia (reviewed in (13)). TSP1 is a positive regulator of vascular smooth muscle cell proliferation and motility. Although less well characterized in cardiovascular disease, TSG-6 is also strongly induced in the neointima following balloon injury of rat iliac arteries, and expression of TSG-6 in vascular smooth muscle cells stimulates their proliferation (86); TSG-6 is one of only a small number of genes upregulated in arterial smooth muscle cells in response to mechanical strain (87). Relevant to the divalent cation dependence we observe for TSP1 binding to TSG-6, a polymorphism in TSP1 associated with familial premature coronary heart disease decreases Ca2+ binding to the protein (88).
TSP1 and TSP2 have well documented suppressive activities in cancer (reviewed in (7,89)), but TSG-6 has not been studied in this inflammatory context. One common link may be the shared regulation of TSP1 and TSG-6 by p53. TSG-6 was identified as a p53-dependent gene expressed following irradiation (90), and TSP1 expression is positively regulated by wild type but not mutant p53 (91).
Phenotypes of TSP and TSG-6 transgenic mice suggest other potential overlapping functions of these proteins. TSP1 null mice have a lung inflammatory phenotype (14), TSP2 null mice have prolonged inflammation in delayed-type hypersensitivity reactions (92), and TSG-6 null mice have several inflammatory abnormalities (83). TSG-6 null mice exhibit female infertility due to a defect on cumulus-oocyte complex expansion (70), as do bikunin null (93,94) and pentraxin-3 null mice (31,95). TSP1 null mice have a mild fertility impairment, but the basis has not been defined (unpublished results). In rats, TSP1 is expressed in early antral phase in the granulosa cells of antral follicles and after ovulation is localized to the developing corpus luteum (96). TSP2 also is expressed in granulosa cells, and both TSP1 and TSP2 are increased in response to LH stimulation. In a growing follicle, TSP is distributed uniformly in the follicular basement membrane and in scattered threadlike masses within the granulosa cell layer (97).
The interactions with TSP1 and TSP2 described here may provide new clues to the mechanism for the potent inhibitory action of TSG-6 in models of acute and chronic inflammation. A large body of evidence supports the conclusion that some anti-inflammatory actions of this protein may involves mechanisms independent of IαI or HA (30,68,83,84,98). Based on the evidence that TSP1 can both inhibit and exacerbate different aspects of specific inflammatory responses, TSG-6 interactions should be considered in efforts to define the molecular bases of these biological activities of TSP1.
Further study is also needed to define the mechanism for TSP1 binding to TSG-6 and to determine whether TSP1 binds to any other HA-binding proteins by recognizing conserved features in their Link modules. Given that pentraxin-3, and related regions of two TSPs recognize the Link module of TSG-6, it will also be of interest to determine whether binding of Link modules is a more widespread property of the pentraxin family.
Acknowledgments
We thank Professor Erik Fries (Uppsala University, Sweden) for kindly providing purified human IαI. AJD acknowledges the support of the Arthritis Research Campaign (grants M0625, 16119 and 16539) and the Medical Research Council.
Footnotes
The abbreviations used are: DPBS, Dulbecco’s phosphate buffered saline; HA, hyaluronan; HSPG, heparan sulfate proteoglycan; IαI, inter-α-trypsin inhibitor; Link_TSG6, residues 36-133 of TSG-6 preprotein; NoC1, trimeric human thrombospondin-1 residues 1-356; TSG-6, tumor necrosis factor-α-stimulated gene-6; TSP, thrombospondin
V. A. Higman & A. J Day, unpublished data
References
- 1.Savino W, Mendes-da-Cruz DA, Silva JS, Dardenne M, Cotta-de-Almeida V. Trends Immunol. 2002;23:305–313. doi: 10.1016/s1471-4906(02)02224-x. [DOI] [PubMed] [Google Scholar]
- 2.Friedl P, Brocker EB. Dev Immunol. 2000;7:249–266. doi: 10.1155/2000/56473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schor H, Vaday GG, Lider O. Dev Immunol. 2000;7:227–238. doi: 10.1155/2000/51902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kuznetsova SA, Roberts DD. Int J Biochem Cell Biol. 2004;36:1126–1134. doi: 10.1016/j.biocel.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 5.Bornstein P, Agah A, Kyriakides TR. Int J Biochem Cell Biol. 2004;36:1115–1125. doi: 10.1016/j.biocel.2004.01.012. [DOI] [PubMed] [Google Scholar]
- 6.Agah A, Kyriakides TR, Lawler J, Bornstein P. Am J Pathol. 2002;161:831–839. doi: 10.1016/S0002-9440(10)64243-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roberts DD. FASEB J. 1996;10:1183–1191. [PubMed] [Google Scholar]
- 8.Vallejo AN, Yang H, Klimiuk PA, Weyand CM, Goronzy JJ. J Immunol. 2003;171:1732–1740. doi: 10.4049/jimmunol.171.4.1732. [DOI] [PubMed] [Google Scholar]
- 9.Gotis-Graham I, Hogg PJ, McNeil HP. Arthritis Rheum. 1997;40:1780–1787. doi: 10.1002/art.1780401009. [DOI] [PubMed] [Google Scholar]
- 10.Riessen R, Kearney M, Lawler J, Isner JM. Am Heart J. 1998;135:357–364. doi: 10.1016/s0002-8703(98)70105-x. [DOI] [PubMed] [Google Scholar]
- 11.Hugo C. Nephrol Dial Transplant. 2003;18:1241–1245. doi: 10.1093/ndt/gfg159. [DOI] [PubMed] [Google Scholar]
- 12.Huang SW, Kao KJ. Allergy Proc. 1993;14:357–361. doi: 10.2500/108854193778774074. [DOI] [PubMed] [Google Scholar]
- 13.Stenina OI, Byzova TV, Adams JC, McCarthy JJ, Topol EJ, Plow EF. Int J Biochem Cell Biol. 2004;36:1013–1030. doi: 10.1016/j.biocel.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 14.Lawler J, Sunday M, Thibert V, Duquette M, George EL, Rayburn H, Hynes RO. J Clin Invest. 1998;101:982–992. doi: 10.1172/JCI1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Crawford SE, Stellmach V, Murphy-Ullrich JE, Ribeiro SMF, Lawler J, Hynes RO, Boivin GP, Bouck N. Cell. 1998;93:1159–1170. doi: 10.1016/s0092-8674(00)81460-9. [DOI] [PubMed] [Google Scholar]
- 16.Li Z, He L, Wilson KE, Roberts DD. J Immunol. 2001;166:2427–2436. doi: 10.4049/jimmunol.166.4.2427. [DOI] [PubMed] [Google Scholar]
- 17.Pierson BA, Gupta K, Hu WS, Miller JS. Blood. 1996;87:180–189. [PubMed] [Google Scholar]
- 18.Doyen V, Rubio M, Braun D, Nakajima T, Abe J, Saito H, Delespesse G, Sarfati M. J Exp Med. 2003;198:1277–1283. doi: 10.1084/jem.20030705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Suchard SJ, Boxer LA, Dixit VM. J Immunol. 1991;147:651–659. [PubMed] [Google Scholar]
- 20.Mansfield PJ, Suchard SJ. J Immunol. 1994;153:4219–4229. [PubMed] [Google Scholar]
- 21.Li Z, Calzada MJ, Sipes JM, Cashel JA, Krutzsch HC, Annis D, Mosher DF, Roberts DD. J Cell Biol. 2002;157:509–519. doi: 10.1083/jcb.200109098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vallejo AN, Mugge LO, Klimiuk PA, Weyand CM, Goronzy JJ. J Immunol. 2000;164:2947–2954. doi: 10.4049/jimmunol.164.6.2947. [DOI] [PubMed] [Google Scholar]
- 23.Barazi HO, Li Z, Cashel JA, Krutzsch HC, Annis DS, Mosher DF, Roberts DD. J Biol Chem. 2002;277:42859–42866. doi: 10.1074/jbc.M206849200. [DOI] [PubMed] [Google Scholar]
- 24.Savill J. Br Med Bull. 1997;53:491–508. doi: 10.1093/oxfordjournals.bmb.a011626. [DOI] [PubMed] [Google Scholar]
- 25.Kuznetsova, S. A., Sharrow, S. O., Lawler, J., and D., R. D. (2004) in Hyaluronan 2003 Conference Proceedings, Matrix Biology Institute
- 26.Wang Q, Teder P, Judd NP, Noble PW, Doerschuk CM. Am J Pathol. 2002;161:2219–2228. doi: 10.1016/S0002-9440(10)64498-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Teder P, Vandivier RW, Jiang D, Liang J, Cohn L, Pure E, Henson PM, Noble PW. Science. 2002;296:155–158. doi: 10.1126/science.1069659. [DOI] [PubMed] [Google Scholar]
- 28.Ponta H, Sherman L, Herrlich PA. Nat Rev Mol Cell Biol. 2003;4:33–45. doi: 10.1038/nrm1004. [DOI] [PubMed] [Google Scholar]
- 29.Day AJ, Prestwich GD. J Biol Chem. 2002;277:4585–4588. doi: 10.1074/jbc.R100036200. [DOI] [PubMed] [Google Scholar]
- 30.Milner CM, Day AJ. J Cell Sci. 2003;116:1863–1873. doi: 10.1242/jcs.00407. [DOI] [PubMed] [Google Scholar]
- 31.Salustri A, Garlanda C, Hirsch E, De Acetis M, Maccagno A, Bottazzi B, Doni A, Bastone A, Mantovani G, Beck Peccoz P, Salvatori G, Mahoney DJ, Day AJ, Siracusa G, Romani L, Mantovani A. Development. 2004;131:1577–1586. doi: 10.1242/dev.01056. [DOI] [PubMed] [Google Scholar]
- 32.Beckmann G, Hanke J, Bork P, Reich JG. J Mol Biol. 1998;275:725–730. doi: 10.1006/jmbi.1997.1510. [DOI] [PubMed] [Google Scholar]
- 33.Lesley J, Gal I, Mahoney DJ, Cordell MR, Rugg MS, Hyman R, Day AJ, Mikecz K. J Biol Chem. 2004;279:25745–25754. doi: 10.1074/jbc.M313319200. [DOI] [PubMed] [Google Scholar]
- 34.Vanguri VK, Wang S, Godyna S, Ranganathan S, Liau G. Biochem J. 2000;347:469–473. doi: 10.1042/0264-6021:3470469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mukhopadhyay D, Hascall VC, Day AJ, Salustri A, Fulop C. Arch Biochem Biophys. 2001;394:173–181. doi: 10.1006/abbi.2001.2552. [DOI] [PubMed] [Google Scholar]
- 36.Day, A., Rugg, M., Mahoney, D., and Milner, C. (2004) in Hyaluronan 2003 Conference Proceedings, Matrix Biology Institute
- 37.Rugg, M. S., Willis, A. C., Mukhopadhyay, D., Hascall, V. C., Fries, E., Fulop, C., Milner, C. M., and Day, A. J. (2005) J Biol Chem [DOI] [PubMed]
- 38.Roberts DD, Cashel J, Guo N. J Tissue Cult Methods. 1994;16:217–222. [Google Scholar]
- 39.Misenheimer TM, Huwiler KG, Annis DS, Mosher DF. J Biol Chem. 2000;275:40938–40945. doi: 10.1074/jbc.M007022200. [DOI] [PubMed] [Google Scholar]
- 40.Anilkumar N, Annis DS, Mosher DF, Adams JC. J Cell Sci. 2002;115:2357–2366. doi: 10.1242/jcs.115.11.2357. [DOI] [PubMed] [Google Scholar]
- 41.Guo NH, Krutzsch HC, Nègre E, Vogel T, Blake DA, Roberts DD. Proc Natl Acad Sci U S A. 1992;89:3040–3044. doi: 10.1073/pnas.89.7.3040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nentwich HA, Mustafa Z, Rugg MS, Marsden BD, Cordell MR, Mahoney DJ, Jenkins SC, Dowling B, Fries E, Milner CM, Loughlin J, Day AJ. J Biol Chem. 2002;277:15354–15362. doi: 10.1074/jbc.M110765200. [DOI] [PubMed] [Google Scholar]
- 43.Day AJ, Aplin RT, Willis AC. Protein Expr Purif. 1996;8:1–16. doi: 10.1006/prep.1996.0068. [DOI] [PubMed] [Google Scholar]
- 44.Kahmann JD, Koruth R, Day AJ. Protein Expr Purif. 1997;9:315–318. doi: 10.1006/prep.1996.0694. [DOI] [PubMed] [Google Scholar]
- 45.Mahoney DJ, Blundell CD, Day AJ. J Biol Chem. 2001;276:22764–22771. doi: 10.1074/jbc.M100666200. [DOI] [PubMed] [Google Scholar]
- 46.Mahoney, D. J., Mulloy, B., Forster, M. J., Blundell, C. D., Fries, E., Milner, C. M., and Day, A. J. (2005) J Biol Chem [DOI] [PubMed]
- 47.Parkar AA, Day AJ. FEBS Lett. 1997;410:413–417. doi: 10.1016/s0014-5793(97)00621-2. [DOI] [PubMed] [Google Scholar]
- 48.Fujimoto T, Savani RC, Watari M, Day AJ, Strauss JF., 3rd Am J Pathol. 2002;160:1495–1502. doi: 10.1016/s0002-9440(10)62575-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Munson PJ, Rodbard D. Anal Biochem. 1980;107:220–239. doi: 10.1016/0003-2697(80)90515-1. [DOI] [PubMed] [Google Scholar]
- 50.Kohda D, Morton CJ, Parkar AA, Hatanaka H, Inagaki FM, Campbell ID, Day AJ. Cell. 1996;86:767–775. doi: 10.1016/s0092-8674(00)80151-8. [DOI] [PubMed] [Google Scholar]
- 51.Blundell CD, Mahoney DJ, Almond A, DeAngelis PL, Kahmann JD, Teriete P, Pickford AR, Campbell ID, Day AJ. J Biol Chem. 2003;278:49261–49270. doi: 10.1074/jbc.M309623200. [DOI] [PubMed] [Google Scholar]
- 52.Parkar AA, Kahmann JD, Howat SL, Bayliss MT, Day AJ. FEBS Lett. 1998;428:171–176. doi: 10.1016/s0014-5793(98)00523-7. [DOI] [PubMed] [Google Scholar]
- 53.Mahoney DJ, Whittle JD, Milner CM, Clark SJ, Mulloy B, Buttle DJ, Jones GC, Day AJ, Short RD. Anal Biochem. 2004;330:123–129. doi: 10.1016/j.ab.2004.03.055. [DOI] [PubMed] [Google Scholar]
- 54.Misenheimer TM, Mosher DF. J Biol Chem. 1995;270:1729–1733. doi: 10.1074/jbc.270.4.1729. [DOI] [PubMed] [Google Scholar]
- 55.Rodrigues RG, Guo N, Zhou L, Sipes JM, Williams SB, Templeton NS, Gralnick HR, Roberts DD. J Biol Chem. 2001;276:27913–27922. doi: 10.1074/jbc.M009518200. [DOI] [PubMed] [Google Scholar]
- 56.Lesley J, English NM, Gal I, Mikecz K, Day AJ, Hyman R. J Biol Chem. 2002;277:26600–26608. doi: 10.1074/jbc.M201068200. [DOI] [PubMed] [Google Scholar]
- 57.Ochsner SA, Day AJ, Rugg MS, Breyer RM, Gomer RH, Richards JS. Endocrinology. 2003;144:4376–4384. doi: 10.1210/en.2003-0487. [DOI] [PubMed] [Google Scholar]
- 58.Hollander Z, Katchalski-Katzir E. Mol Immunol. 1986;23:927–933. doi: 10.1016/0161-5890(86)90122-7. [DOI] [PubMed] [Google Scholar]
- 59.Engel MF, van Mierlo CP, Visser AJ. J Biol Chem. 2002;277:10922–10930. doi: 10.1074/jbc.M106005200. [DOI] [PubMed] [Google Scholar]
- 60.Bornstein P. FASEB J. 1992;6:3290–3299. doi: 10.1096/fasebj.6.14.1426766. [DOI] [PubMed] [Google Scholar]
- 61.Dixit VM, Grant GA, Santoro SA, Frazier WA. J Biol Chem. 1984;259:10100–10105. [PubMed] [Google Scholar]
- 62.Goicoechea S, Orr AW, Pallero MA, Eggleton P, Murphy-Ullrich JE. J Biol Chem. 2000;275:36358–36368. doi: 10.1074/jbc.M005951200. [DOI] [PubMed] [Google Scholar]
- 63.Krutzsch HC, Choe B, Sipes JM, Guo N, Roberts DD. J Biol Chem. 1999;274:24080–24086. doi: 10.1074/jbc.274.34.24080. [DOI] [PubMed] [Google Scholar]
- 64.Calzada MJ, Sipes JM, Krutzsch HC, Yurchenco PD, Annis DS, Mosher DF, Roberts DD. J Biol Chem. 2003;278:40679–40687. doi: 10.1074/jbc.M302014200. [DOI] [PubMed] [Google Scholar]
- 65.Kvansakul M, Adams JC, Hohenester E. Embo J. 2004;23:1223–1233. doi: 10.1038/sj.emboj.7600166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yu H, Tyrrell D, Cashel J, Guo NH, Vogel T, Sipes JM, Lam L, Fillit HM, Hartman J, Mendelovitz S, Panel A, Roberts DD. Arch Biochem Biophys. 2000;374:13–23. doi: 10.1006/abbi.1999.1597. [DOI] [PubMed] [Google Scholar]
- 67.Blundell CD, Almond A, Mahoney DJ, DeAngelis PL, Campbell ID, Day AJ. J Biol Chem. 2005;280:18189–18201. doi: 10.1074/jbc.M414343200. [DOI] [PubMed] [Google Scholar]
- 68.Getting SJ, Mahoney DJ, Cao T, Rugg MS, Fries E, Milner CM, Perretti M, Day AJ. J Biol Chem. 2002;277:51068–51076. doi: 10.1074/jbc.M205121200. [DOI] [PubMed] [Google Scholar]
- 69.Flahaut C, Capon C, Balduyck M, Ricart G, Sautiere P, Mizon J. Biochem J. 1998;333 ( Pt 3):749–756. doi: 10.1042/bj3330749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fulop C, Szanto S, Mukhopadhyay D, Bardos T, Kamath RV, Rugg MS, Day AJ, Salustri A, Hascall VC, Glant TT, Mikecz K. Development. 2003;130:2253–2261. doi: 10.1242/dev.00422. [DOI] [PubMed] [Google Scholar]
- 71.Mukhopadhyay D, Asari A, Rugg MS, Day AJ, Fulop C. J Biol Chem. 2004;279:11119–11128. doi: 10.1074/jbc.M313471200. [DOI] [PubMed] [Google Scholar]
- 72.Jessen TE, Odum L. Reproduction. 2003;125:27–31. doi: 10.1530/rep.0.1250027. [DOI] [PubMed] [Google Scholar]
- 73.Jessen TE, Odum L. Osteoarthritis Cartilage. 2004;12:142–148. doi: 10.1016/j.joca.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 74.Koch AE, Friedman J, Burrows JC, Haines GK, Bouck NP. Pathobiology. 1993;61:1–6. doi: 10.1159/000163752. [DOI] [PubMed] [Google Scholar]
- 75.Wisniewski HG, Maier R, Lotz M, Lee S, Klampfer L, Lee TH, Vilcek J. J Immunol. 1993;151:6593–6601. [PubMed] [Google Scholar]
- 76.Bayliss MT, Howat SL, Dudhia J, Murphy JM, Barry FP, Edwards JC, Day AJ. Osteoarthritis Cartilage. 2001;9:42–48. doi: 10.1053/joca.2000.0348. [DOI] [PubMed] [Google Scholar]
- 77.Kehlen A, Pachnio A, Thiele K, Langner J. Arthritis Res Ther. 2003;5:R186–192. doi: 10.1186/ar762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wisniewski HG, Hua JC, Poppers DM, Naime D, Vilcek J, Cronstein BN. J Immunol. 1996;156:1609–1615. [PubMed] [Google Scholar]
- 79.Bardos T, Kamath RV, Mikecz K, Glant TT. Am J Pathol. 2001;159:1711–1721. doi: 10.1016/s0002-9440(10)63018-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mindrescu C, Thorbecke GJ, Klein MJ, Vilcek J, Wisniewski HG. Arthritis Rheum. 2000;43:2668–2677. doi: 10.1002/1529-0131(200012)43:12<2668::AID-ANR6>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 81.Glant TT, Kamath RV, Bardos T, Gal I, Szanto S, Murad YM, Sandy JD, Mort JS, Roughley PJ, Mikecz K. Arthritis Rheum. 2002;46:2207–2218. doi: 10.1002/art.10555. [DOI] [PubMed] [Google Scholar]
- 82.Mindrescu C, Dias AA, Olszewski RJ, Klein MJ, Reis LF, Wisniewski HG. Arthritis Rheum. 2002;46:2453–2464. doi: 10.1002/art.10503. [DOI] [PubMed] [Google Scholar]
- 83.Szanto S, Bardos T, Gal I, Glant TT, Mikecz K. Arthritis Rheum. 2004;50:3012–3022. doi: 10.1002/art.20655. [DOI] [PubMed] [Google Scholar]
- 84.Cao TV, La M, Getting SJ, Day AJ, Perretti M. Microcirculation. 2004;11:615–624. doi: 10.1080/10739680490503438. [DOI] [PubMed] [Google Scholar]
- 85.Koch AE, Szekanecz Z, Friedman J, Haines GK, Langman CB, Bouck NP. Clin Immunol Immunopathol. 1998;86:199–208. doi: 10.1006/clin.1997.4480. [DOI] [PubMed] [Google Scholar]
- 86.Ye L, Mora R, Akhayani N, Haudenschild CC, Liau G. Circ Res. 1997;81:289–296. doi: 10.1161/01.res.81.3.289. [DOI] [PubMed] [Google Scholar]
- 87.Lee RT, Yamamoto C, Feng Y, Potter-Perigo S, Briggs WH, Landschulz KT, Turi TG, Thompson JF, Libby P, Wight TN. J Biol Chem. 2001;276:13847–13851. doi: 10.1074/jbc.M010556200. [DOI] [PubMed] [Google Scholar]
- 88.Hannah BL, Misenheimer TM, Annis DS, Mosher DF. J Biol Chem. 2003;278:8929–8934. doi: 10.1074/jbc.m211185200. [DOI] [PubMed] [Google Scholar]
- 89.Lawler J, Detmar M. Int J Biochem Cell Biol. 2004;36:1038–1045. doi: 10.1016/j.biocel.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 90.Seidita G, Polizzi D, Costanzo G, Costa S, Di Leonardo A. Carcinogenesis. 2000;21 :2203–2210. doi: 10.1093/carcin/21.12.2203. [DOI] [PubMed] [Google Scholar]
- 91.Dameron KM, Volpert OV, Tainsky MA, Bouck N. Science. 1994;265:1582–1584. doi: 10.1126/science.7521539. [DOI] [PubMed] [Google Scholar]
- 92.Lange-Asschenfeldt B, Weninger W, Velasco P, Kyriakides TR, von Andrian UH, Bornstein P, Detmar M. Blood. 2002;99:538–545. doi: 10.1182/blood.v99.2.538. [DOI] [PubMed] [Google Scholar]
- 93.Sato H, Kajikawa S, Kuroda S, Horisawa Y, Nakamura N, Kaga N, Kakinuma C, Kato K, Morishita H, Niwa H, Miyazaki J. Biochem Biophys Res Commun. 2001;281:1154–1160. doi: 10.1006/bbrc.2001.4475. [DOI] [PubMed] [Google Scholar]
- 94.Zhuo L, Yoneda M, Zhao M, Yingsung W, Yoshida N, Kitagawa Y, Kawamura K, Suzuki T, Kimata K. J Biol Chem. 2001;276:7693–7696. doi: 10.1074/jbc.C000899200. [DOI] [PubMed] [Google Scholar]
- 95.Varani S, Elvin JA, Yan C, DeMayo J, DeMayo FJ, Horton HF, Byrne MC, Matzuk MM. Mol Endocrinol. 2002;16:1154–1167. doi: 10.1210/mend.16.6.0859. [DOI] [PubMed] [Google Scholar]
- 96.Petrik JJ, Gentry PA, Feige JJ, LaMarre J. Biol Reprod. 2002;67:1522–1531. doi: 10.1095/biolreprod.102.007153. [DOI] [PubMed] [Google Scholar]
- 97.Bagavandoss P, Sage EH, Vernon RB. J Histochem Cytochem. 1998;46:1043–1049. doi: 10.1177/002215549804600908. [DOI] [PubMed] [Google Scholar]
- 98.Wisniewski HG, Naime D, Hua JC, Vilcek J, Cronstein BN. Pflugers Arch. 1996;431:R225–226. doi: 10.1007/BF02346350. [DOI] [PubMed] [Google Scholar]
- 99.Calzada MJ, Annis DS, Zeng B, Marcinkiewicz C, Banas B, Lawler J, Mosher DF, Roberts DD. J Biol Chem. 2004;279:41734–41743. doi: 10.1074/jbc.M406267200. [DOI] [PubMed] [Google Scholar]