

Abstract
Recent findings associate the control of stereochemistry in lipoxygenase (LOX) catalysis with a conserved active site alanine for S configuration hydroperoxide products, or a corresponding glycine for R stereoconfiguration. To further elucidate the mechanistic basis for this stereocontrol we compared the stereoselectivity of the initiating hydrogen abstraction in soybean LOX-1 and an Ala542Gly mutant that converts linoleic acid to both 13S and 9R configuration hydroperoxide products. Using 11R-3H- and 11S-3H-labeled linoleic acid substrates to examine the initial hydrogen abstraction, we found that all the primary hydroperoxide products were formed with an identical and highly stereoselective proS hydrogen abstraction from C-11 of the substrate (97–99% pro-S selective). This strongly suggests that 9R and 13S oxygenations occur with the same binding orientation of substrate in the active site, and as the equivalent 9R and 13S products were formed from a bulky ester derivative (1-palmitoyl-2-linoleoyl-phosphatidylcholine), one can infer that the orientation is tail-first. Both the EPR spectrum and the reaction kinetics were altered by the R product-inducing Ala-Gly mutation, indicating a substantial influence of this Ala-Gly substitution extending to the environment of the active site iron. To examine also the reversed orientation of substrate binding, we studied oxygenation of the 15S-hydroperoxide of arachidonic acid by the Ala542Gly mutant soybean LOX-1. In addition to the usual 5S,15S- and 8S,15S-dihydroperoxides, a new product was formed and identified by HPLC, UV, GC-MS and NMR as 9R , 1 5 S -dihydroperoxy-eicosa-5Z,7E,11Z,13E-tetraenoic acid, the R configuration “partner” of the normal 5S,15S product. This provides evidence that both tail-first and carboxylate end-first binding of substrate can be associated with S or R partnerships in product formation in the same active site.
Polyunsaturated fatty acids are oxygenated to signaling molecules of very specific structure by the dioxygenases lipoxygenase (LOX) and cyclooxygenase, and by the cytochrome P450 class of monooxygenase (1–3). One of the fundamental differences lies in activation of the fatty acid by the dioxygenases compared to oxygen activation by the P450s. In the case of P450s, the oxygen is bound to the heme iron and the control of reaction specificity largely depends on the specific binding of substrate and its presentation to the activated ferryl oxygen (4–6). By contrast, in both classes of dioxygenase the fatty acid substrate is activated to a radical species capable of instantaneous reaction with molecular oxygen (1,2); all elements of reaction specificity thus depend on arranging the meeting of molecular oxygen with the correct site on the substrate radical. All the evidence suggests that the reacting oxygen molecule is untethered and that other factors in the enzyme active site must determine the reaction outcome. In particular, cyclooxygenase enzymes have no residues capable of direct oxygen binding within the oxygenase active site (7,8), while for lipoxygenases there is both spectroscopic and kinetic isotope data to refute the involvement of the active site nonheme iron in oxygen binding (9–11). The possibility of oxygen channeling through the protein is one potential means of achieving the targeting of O2 onto a particular carbon of the reacting fatty acid radical (12–14). The influence of protein residues in shielding some of the reactive carbons must also figure into the control of product regiochemistry and stereochemistry. These two options relate directly to the studies we report here on the specificity of reaction by soybean lipoxygenase.
Soybean LOX-1 is the prototypical enzyme of its class and is the best-studied model of lipoxygenase catalysis and structure. Mechanistic studies are supported by the availability of two high resolution X-ray crystal structures (12,15). Soybean LOX-1 has been used over the years to establish many fundamental concepts of lipoxygenase catalysis. Among these are the high specificity of the reaction and formation of a chiral product (16); the sole product is 13S-hydroperoxyoctadecadienoic acid (13S-HPODE) from linoleic acid, and the corresponding 15S-hydroperoxide (15-HPETE) from arachidonic acid. Early on it was shown that the specific oxygenation is associated with a highly stereoselective initial hydrogen abstraction from the fatty acid and that the two events occur on opposite faces of the reacting pentadiene (an antarafacial relationship) (16,17). Related observations in comparing the 9S-specific corn lipoxygenase with the 13S-specific soybean enzyme led to the important concept of substrate being capable of adopting a reversed orientation of binding in the lipoxygenase active site (18). This reversed orientation hypothesis has remained open to debate in that there is no LOX structure available with bound substrate to directly support or refute the various lines of argument. Indeed, a recent study suggested a head-first substrate orientation for the first oxygenation by soybean LOX-1 (19). In the absence of an X-ray structure with bound substrate, modeling stands as one useful approach to understanding concepts developed by analysis of the stereochemistry of the lipoxygenase reaction and the influence of modifications to the protein structure (14,20).
A recent study described the importance of a conserved lipoxygenase active site residue in controlling the regio- and stereospecificity of arachidonic acid oxygenation (21). The residue is conserved as an Ala in lipoxygenases that form S-specific hydroperoxides and as Gly in the R-hydroperoxide-forming enzymes. The Ala-Gly mutation in the enzymes studied at least partially switched the position and R or S chirality of oxygenation on the substrate such that an enzyme with original 8R-specificity could be mutated to make 12S product, 15S to 11R and so on. These changes correspond to a switch in the position of oxygenation from one end of the fatty acid pentadiene intermediate to the other. The aim of the present study was to investigate some novel aspects of the reaction of wild-type and Ala542Gly mutant soybean LOX-1 including the specificity of the associated hydrogen abstraction and the characteristics of double dioxygenation as they pertain to our understanding of substrate binding and the stereocontrol of oxygenation.
MATERIALS AND METHODS
Materials and Procedures
Mutagenesis primers were synthesized by Integrated DNA Technologies, Inc. (IDT, Coralville, IA). Mutagenesis was performed according to the manufacturer's protocol (Stratagene QuickChange Mutagenesis Kit, La Jolla, CA). Plasmids were purified using QIAprep Spin Miniprep kits (Qiagen, Valencia, CA). Stratagene XL1-Blue super competent cells and BL21-Gold(DE3) cells were transformed for plasmid amplification and protein expression, respectively. BugBuster protein extraction reagent was from Novagen (Madison, WI). It was supplemented with 0.8 mg/ml DNAse and 5.0 mM MgCl2 before use. E. coli spheroplasts were prepared in TSE (0.1M Tris acetate, 0.5 M sucrose, 1mg/ml lysozyme and 0.5 mM EDTA; pH 7.6). Ni-NTA affinity agarose (His·Bind Superflow, Novagen) was prepared in wash buffer (0.01 M Tris-HCl, 0.5M NaCl and 3 mM sodium azide; pH 8.0) to remove ethanol. All buffers from BugBuster through the Ni-NTA buffers contained PMSF and benzamidine, both at 1 mM. Anion exchange chromatography medium was DE-52 (Whatman). The DE-52 column was eluted with a linear gradient of 0-0.5 M sodium chloride in 10 mM Tris-HCl, pH 8.0. HPLC purification was performed using a Dionex (Sunnyvale, CA) HPLC system (GP50 gradient pump) with a size exclusion column (Superdex 200 HR 10/30, Amersham Pharmacia Biotech AB) and a flow rate of 0.4 ml/min. The HPLC buffer was 0.02 M KPi. 0.15 M KCl, 3.0 mM sodium azide, pH 7.3.
Mutagenesis of Soybean LOX-1
Soybean LOX-1 was cloned from whole, mature green pods of plants cultured from organically grown soybeans (22). To insert a N-terminal (His)6 tag, the soybean LOX-1 cDNA was then subcloned into the pET-30Xa/LIC (Novagen, Madison, WI) expression vector following manufacturer's instructions. Mutant plasmids were created using the QuikChange site-directed mutagenesis kit (Stratagene) using overlapping mismatching primers containing the desired mutation following manufacturer’s instructions. Correctly mutated plasmids were identified by sequencing. DNA sequencing was performed at the FSU Biology DNA Sequencing Facility using an Applied Biosystems 3100 Genetic Analyzer.
Cloning and Expression
The plasmid originally constructed from soybean mRNA transcript had three single base mutations that were corrected by mutagenesis to the J02795 coding sequence, and one three-base mutation (AGT to GAG at positions 478–480 encoding a Ser160Glu change) that was retained in the construct for most experiments. Ser160 was introduced for some comparisons, as indicated in the text. The yield of expressed protein was improved by lowering the temperature of incubation after IPTG induction and by lengthening the incubation period, until the final condition of 14 °C for 30 h was determined to be optimal. The isolated protein yield was halved if the cells were harvested after 24 hrs. The mutant proteins examined in this study were Ala542Gly, Ala542Ser, Ala542Thr, and Ala542Val. Yields of the expressed mutant proteins were similar to yields of native lipoxygenase: typically 10 mg lipoxygenase/L of original culture were obtained after purification for wild-type and somewhat less for mutants.
Wild-type and mutant soybean LOX-1 were expressed in E. coli BL21 (DE3) Gold (Stratagene). Overnight cultures in LB (37 °C) were diluted 200-fold into multiple 1 L flasks containing 375 ml TB. Cultures were grown to OD600 > 1.2 (4–5 hrs, 37 °C), cooled to 14 °C, induced with IPTG (0.1 mM), and grown for 30 hrs at 8 °C. Cells were harvested and incubated for 30 min in TSE (0.1M Tris acetate, 0.5 M sucrose, 1 mg/ml lysozyme and 0.5 mM EDTA; pH 7.6). Pelleted spheroplasts were frozen at −70 °C. Thawed pellets were suspended in BugBuster (Novagen, 60 ml/L of original culture). After 20 min at room temperature, the suspension was clarified by centrifugation (18,600 x g in this and subsequent steps) and solid ammonium sulfate was added to the supernatant to 30% of saturation. After 20 min, a pellet was removed by centrifugation, the supernatant was brought to 40% ammonium sulfate saturation, clarified again by centrifugation, and finally the supernatant was brought to 65 %-saturated ammonium sulfate and the suspension was centrifuged immediately. The 65 % ammonium sulfate pellet from 3 L of culture was suspended in wash buffer and immediately cycled several times through a prepared column of Ni-NTA (Novagen, 6 ml from 50% suspension). The Ni-NTA was washed with 100 ml wash buffer, and then His-tagged lipoxygenase was eluted at once with wash buffer containing 250 mM imidazole, and dialyzed overnight (4 °C) in Tris-HCl (10 mM, pH 8). The dialyzed eluate from Ni-NTA was separated by anion exchange chromatography (1 x 15 cm DE-52 Whatman column). Fractions containing soybean LOX-1 activity were brought to 65% of saturation with ammonium sulfate, were centrifuged, and the pellet was suspended in 1–2 ml HPLC buffer and dialyzed in HPLC buffer. Lipoxygenase elutes at ~30 min from the Superdex 200 HR 10/30 HPLC column. Purity was assessed by SDS-PAGE and was >95%.
The amino-terminal His tag in expressed lipoxygenases was removed by incubating 10 units of Factor Xa/mg LOX protein for 4 hrs at room temperature and the reaction was stopped using Xarrest Agarose, according to the manufacurer's protocol (Novagen, Factor Xa Cleavage Capture Kit). Typically, 5–10 mg of protein was digested in one batch and the yield of cleaved LOX protein, after workup, was >50% of the starting uncleaved protein. Completion of cleavage was verified by SDS-PAGE (7.5% gel). Samples were AMS precipitated, brought up to 5–10 mg/ml and dialyzed to remove remaining cleaved peptide.
Samples were prepared for metal analysis by centrifugation and dialysis into 0.01 M Tris, 5 mM sodium azide, pH 7.6. The Tris buffer was passed through a Chelex column before use. Glassware for analytical samples was washed sequentially with nitric acid and water. Metal analyses by Inductively Coupled Plasma Mass Spectrometry (ICP-MS) were performed at the Chemical Analysis Laboratory, University of Georgia (Athens, GA). Percent iron values are based on absorbances at 280 nm for a 1 mg/ml protein solution of 1.28 (with His-tag) and 1.35 (no tag) (23). The active samples analyzed included one of Ser160/Gly542 without His-tag, three of Gly542 with His-tag, one of Ser542 with tag and two of wild type (Glu160) with tag. The samples for metal determination ranged in concentration from 1 to 15 μM LOX protein and iron, nickel, manganese, copper and zinc were analyzed. To determine which metals were correlated with protein concentration, metal analysis and protein concentration data of all samples were graphed using Kaleidagraph (μM metal vs. μM protein). The metal concentrations that correlated with protein concentration (mole ratio metal/protein) were iron (0.62), manganese (0.02) and, weakly correlated, nickel (0.04) (Pearson's R-values of 0.99, 0.96, and 0.69 respectively). Average amounts of zinc (2 μM) and copper (0.2 μM) in the samples were not correlated with protein concentration. Single analyses of iron in Threonine and Valine mutants with His tags indicated iron incorporation similar to that of the active samples. The dialysis buffer at the final sample preparation step for metal analysis had negligible iron, manganese, nickel, copper or zinc content.
Assay system for wild-type and mutant soybean LOX-1
Recombinant wild-type soybean LOX-1 or mutants (1 μg) were incubated with either [1-14C]AA or [1 -14C]LA (100 μM final concentration) in 100 μl 50 mM borate buffer pH 9, 150 mM NaCl for 2 minutes at room temperature. Incubations were stopped by addition of methanol and the pH reduced to ~ 6 by addition of 1N HCl prior to product extraction using the Bligh and Dyer method (24). For experiments studying the second oxygenation reactions, soybean LOX-1 and the Ala542Gly mutant (50 μg) were incubated with 15S-HPETE (10 μg) in 0.5 ml 50 mM pH 9 borate buffer. Reactions were monitored in the UV (200–350 nm) and upon completion products were extracted with dichloromethane (0.5 ml) after acidification to ~pH 6 with 1N HCl. Hydroperoxy derivatives were resuspended in methanol and reduced to hydroxy fatty acids by addition of 5 μl of triphenylphosphine (TPP) (5 mg/ml) prior to HPLC analysis. Wild-type soybean LOX-1 and the Ala542Gly mutant (10 μg) were incubated with either C16/LA-PC or C16/AA-PC (100 μM final concentration) in 0.5 ml 50 mM borate, pH 9, 150 mM NaCl and 10 mM deoxycholate. Reactions were monitored by UV spectrophotometry at 235 nm and stopped by addition of methanol. Products were extracted using the Bligh and Dyer procedure (24), reduced with TPP and transesterified with NaOMe prior to HPLC analysis.
Incubation with stereospecifically labeled linoleic acids
The stereospecifically-labeled 11R-3H and 11S-3H linoleic acids were synthesized previously as described (25). The tritium-labeled linoleates were mixed with [14C]LA, which served as an internal standard for measurement of tritium retention. Incubations were carried out in 1 ml of 50 mM borate pH 9, 150 mM NaCl using 10,000 cpm 3H for incubations with the wild-type enzyme and 20,000 cpm 3H for experiments with the Ala542Gly mutant. The appearance of the conjugated diene chromophore was monitored by UV spectrometry (200–300 nm) and reaction was stopped by addition of methanol at ~80% substrate conversion. The products were acidified and extracted by the Bligh and Dyer procedure (24), reduced with TPP and methylated with diazomethane prior to HPLC analysis. Fractions were collected and counted for 3H and 14C, and the % of 3H retained was estimated by comparison of the 3H/14C ratios of product and substrate.
HPLC analysis
Products of the reaction of LOX enzymes and mutants with [1-14C]LA, [1-14C]AA and 15S-HPETE were analyzed on an Agilent 1100 HPLC equipped with a diode array detector connected online to a Radiomatic FLO-ONE A-100 radioactive detector. Metabolites from [1-14C]LA were analyzed using a Whatman® Partisil 5 silica column (0.46 × 25 cm) eluted at a flow rate of 1 ml/min with hexane/isopropanol/acetic acid (100/2/0.1, by volume) with UV detection at 235 nm. Metabolites from incubation with [1-14C]AA were analyzed on a Waters Symmetry C18 5-μm column (25 x 0.46 cm) using a solvent of methanol/water/acetic acid (80/20/0.01, by volume) eluted at 1 ml/min with UV detection at 235 nm; the unreacted substrate was eluted with methanol. The oxygenated products of 15S-HPETE were eluted with methanol/water/acetic acid (75/25/0.01, by volume) at a flow rate of 1 ml/min.
Methyl esters of products of incubations with either C16/LA-PC or stereospecifically labeled linoleic acids were analyzed on a Whatman® Partisil 5 silica column (0.46 × 25 cm) eluted at a flow rate of 1 ml/min with hexane/isopropanol/acetic acid (100/1/0.1 by volume)
HETE and HODE metabolites collected from the HPLC systems above were converted to methyl esters by treatment with ethereal diazomethane and further purified by SP-HPLC prior to chiral phase HPLC analysis. Chiral analysis of the HETE methyl esters was performed using a Daicel Chiralpak AD (0.46 × 25 cm) eluted at a flow rate of 1 ml/min with hexane/methanol (100/2, by volume), HODE methyl esters were eluted with hexane/methanol (100/5, by volume) with UV detection at 235 nm (26).
Determination of 3H retention in products formed from stereospecifically labeled linoleic acids
HPODE products formed by the wild-type soybean LOX-1 and the Ala542Gly mutant were reduced with TPP, converted to the methyl ester derivative with diazomethane and purified by SP-HPLC and chiral phase HPLC as described under “HPLC analysis”. Fractions of 0.5 ml were collected from the chiral HPLC column directly into scintillation counting vials. The solvent was evaporated under nitrogen, scintillation fluid was added and the samples each counted for 10 min in a Packard 1900 TR liquid scintillation analyzer using the dual channel mode for 3H and 14C. Typically there were 1000–2000 cpm of 14C in the product peaks, and the samples were counted for 30 min to improve precision. The counts in the baseline fractions adjacent to the chromatographic peaks were used as background. The 3H retention was calculated by comparison of the 3H/14C ratio of substrate and product.
Preparation of 9R,15S-diHPETE and 9S,15S-diHPETE
Wild-type soybean LOX-1 (50 μg in 1 ml) was incubated with 9S- or 9R-HPETE (50 μM) prepared by autoxidation of methyl arachidonate, purification by SP-HPLC, resolution as 9R and 9S by chiral chromatography using a Chiralpak AD column (9R elutes before 9S (27)) with a solvent of hexane/methanol 100:2, by volume, and alkaline hydrolysis to give the corresponding free acids (26). To prepare milligram amounts of 9R,15S-diHETE and 9S,15S-diHETE soybean LOX-1 (2 mg, Sigma type V) was incubated with 9R,S-HETE (20 mg) in 100 ml of 50 mM borate buffer pH 9, 150 mM NaCl, products of reaction were extracted in dichloromethane at pH 6, re-dissolved in methanol and reduced with NaBH4 prior to methylation with diazomethane. The analysis of the methyl ester trimethylsilyl ether hydrogenated derivatives of the products was carried out in the positive-ion electron impact mode (70 eV) by using a Hewlett–Packard 5989A mass spectrometer coupled to a Hewlett–Packard 5890 gas chromatograph equipped with an RTX-1701 fused silica capillary column (5 m x 0.25 mm internal diameter). The position of the OH groups was confirmed by the presence of the ions at m/z 259 and 345 (α cleavage at C9) and of the peaks at m/z 173 and 431 (α cleavage at C15).
1H NMR and 2D (H,H-COSY) NMR spectra were recorded on a Bruker DRX 400-MHz spectrometer. 9R,15S-diHETE-Me and 9S,15S-diHETE-Me gave (ppm): δ 0.87 (t, H20), 1.23–1.32 (m, H17–19), 1.53 (H16 under the H2O peak), 1.71 (p, H3), 2.21 (q, H4), 2.31 (t, H2), 2.41–2,50 (m, H10), 3.65 (s, H1), 4.15 (q, H15), 4.24 (q, H9), 5.37–5.49 (m, H5 and H11), 5.67–5.74 (dt, H8 and H14), 6.00 (t, H6), 6.13 (t, H12), 6.44–6.51 (dd, H7 and H13).
Kinetic analysis
Enzymatic activity of purified enzymes was determined by monitoring the increase of the signal at 235 nm in a Perkin Elmer lambda-35 UV-Vis spectrophotometer with LA concentrations ranging from 2 to 100 μM. Rates of reaction were calculated from the initial linear part of the curve. The enzymatic assay for soybean LOX-1 or the Ala542Gly mutant (0.25 μg) was performed in 0.5 ml of 50mM borate pH 9, 150 mM NaCl buffer. 13S-HPODE (1.3 μM) was included in each reaction as hydroperoxide activator. Km and kcat values were calculated with the Michaelis-Menten equation. To determine the effect of different activators on the lag phase, enzymatic assays with soybean LOX-1 (0.05 μg) or the Ala542Gly mutant (0.25 μg) were performed in the presence of 13S-HPODE or 9R-HPODE (1.3 μM). Lag phase was measured from the beginning of the reaction to the time at which a line tangent to the maximum velocity intersects the time axis, as described by Smith and Lands (28).
EPR Spectroscopy
Lipoxygenase samples (1–2 mg/ml in 0.2 M KPi, pH 6.8) were oxidized by addition of one equivalent of previously prepared 13S-HPODE, precipitated immediately by adding saturated ammonium sulfate to produce 65% saturation, and brought up to 4–9 mg/ml in 0.2 M buffer of choice (0.2 M KPi, pH 6.8 in most cases). To remove residual ammonium sulfate and remaining products derived from 13S-HPODE, samples for comparison of EPR spectra were dialyzed overnight against ~10,000 volumes of a buffer. To verify that no significant reduction of the ferric signal had occurred during dialysis, the EPR intensities of individual samples were also compared before and after dialysis. Lineshapes of samples before dialysis were variable because of the presence of residual ammonium sulfate. Samples were also examined in 0.2 M Tris pH 7.5, and in 0.2 M sodium bicarbonate pH 9.0. EPR spectra were examined for at least two separately prepared samples of each mutant. No differences in EPR spectra were found with or without the His-tag present, or when the polymorphism Ser160 replaced Glu160. In further control experiments, spectra of all samples in other buffers showed differences in detail similar to those seen in KPi. Addition of glycerol to 15% increased the proportion of component 2 in all samples, and at 50% gave essentially complete conversion to component 2 with some line width broadening (see RESULTS Figure 8). A Varian E 109 spectrometer equipped with an Oxford Instruments ESR9/10 pumped, liquid helium-flow cryostat was used for EPR spectroscopy. Relevant instrumental parameters are given in the legend to Figure 8. X-band frequencies were measured at the bridge and the magnetic field was calibrated using a sealed quartz tube (0.5 mm ID) of CaO containing a manganous impurity, frozen in a 4 mm X-band quartz EPR tube of sample buffer.
Fig. 8.
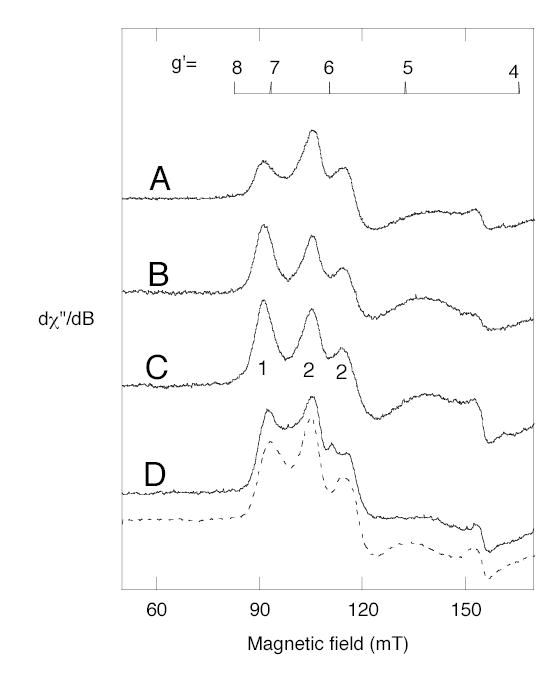
Representative EPR spectra of ferric forms of native soybean LOX-1 and the Ala542 mutants. A, Ala542Gly mutant. B, wild-type enzyme. C, LOX-1 isolated from soybeans. D, Ala542Ser (solid line) and Ala542Thr (dashed line). Peaks arising from two major spectral components, 1 and 2, are labeled on Fig.8C. X-band EPR spectra were recorded at 3.5 K and the instrument parameters were: microwave power, 1 mW; modulation amplitude, 1.0 mT; sweep rate, 13.3 mT/min (except 25 mT/min, spectra D); time constant, 0.5 sec; and gain 12,500.
Computational Docking
Linoleic acid was docked to the soybean LOX-1 structure using the program Autodock 3.03 (29). All the water molecules in the structure, except the iron ligand Wat842, were removed and the iron was assigned +2 oxidation state. The two double bonds and the two C-O bonds in linoleic acid were made non-rotatable and the rest were free to rotate. The search cube was designed to include the major residues that comprise cavity IIa (15), including iron and its ligands. The grid parameter file (GPF) for the Autogrid contained 1,157,625 (1053 in X, Y, Z) grid points, with a spacing of 0.247Å between points. RMSD tolerance was set at 1.5 Å to get convergence. Calculations were run on an AMD Athlon MP 2800+ dual processor Linux workstation with 1GB DDR and the computational time varied from 8–12 hours.
RESULTS
Expression of Soybean LOX-1
Soybean LOX-1 with a N-terminal His-tag and four mutants at position 542 (substitution of wild-type Ala542 for Gly, Ser, Thr, or Val) were expressed in E. coli. The wild-type protein used in these experiments retained Glu160. Some comparisons of Ser160 and Glu160 were made and these experiments are indicated in text. Optimal final conditions for expression were incubation at 14 °C for 30 h. The expressed proteins were purified using a nickel affinity column followed by anion exchange chromatography and a final step of high-resolution gel filtration. Yields after purification were typically 10 mg lipoxygenase/L original culture.
Stereospecificity of hydrogen abstraction from C-11 of linoleic acid
Wild-type soybean LOX-1 produces almost pure 13S-HPODE from linoleic acid, and the Ala542Gly mutation changed this product profile to a 1.25:1 mixture of 13S-HPODE and 9R-HPODE (Fig. 1). The 13S and 9R positions on linoleic acid are at opposite ends of the double bond system, on the same side of the substrate molecule. The corresponding substitution of Ala542 with Ser had little effect on the reaction profile, whereas conversion to either Val or Thr resulted in complete loss of catalytic activity, paralleling the effect on other LOX enzymes (21) (data not shown). As a well-established feature of lipoxygenase catalysis is the initial stereoselective hydrogen abstraction (16,18), it was of great mechanistic interest to compare this reaction in formation of both the 13S- and 9R-hydroperoxy products by the Ala542Gly mutant of soybean LOX-1. Accordingly, wild-type and Ala542Gly soybean LOX-1 were reacted separately with 11S-3H- and 11R-3H-labeled linoleic acid substrates. The 3H-labeled linoleic acids were mixed with [1-14C]linoleic acid as internal standard, allowing measurement of the 3H/14C ratio as an index of the 3H content of substrate and product. The HPODE products were reduced to HODEs, converted to the methyl esters and purified by SP-HPLC. Each sample was then run on chiral phase HPLC using the highly resolving Chiralpak AD column (26), and fractions were collected across the chromatographic peaks and the 3H/14C ratios quantified by scintillation counting. Incubation of wild-type soybean LOX-1 with [11S-3H]linoleic acid substrate resulted in formation of 13S-HPODE with loss of the 3H label (Fig. 2A). The 13S-HPODE and the 9R-HPODE formed by the Ala542Gly mutant also lost the 3H label (Fig. 2B and C). In contrast, when [11R-3H]linoleic acid was used as a substrate the 3H label was retained in the 13S-HPODE formed by the wild-type soybean LOX-1 (Fig. 2D) as well as in the 13S-HPODE and the 9R-HPODE formed from the Ala542Gly mutant (Fig. 2E and F). All results are summarized in Table 1. Clearly, all products formed from the [11S-3H]-LA lost the tritium label (Fig. 3A), whereas the label was almost completely retained in products formed from [11R-3H]-LA (Fig. 3B). These results establish that the reaction to form both 13S-HPODE and 9R-HPODE by the Ala542Gly mutant and 13S-HPODE by the wild-type enzyme proceeds via the same 11-pro-S hydrogen abstraction. Interestingly using linoleic acid substrate, cyclooxygenase behaves similarly to the Ala542Gly mutant by producing a mixture of 13S-HPODE and 9R-HPODE via pro-S hydrogen abstraction (30).
Fig. 1.
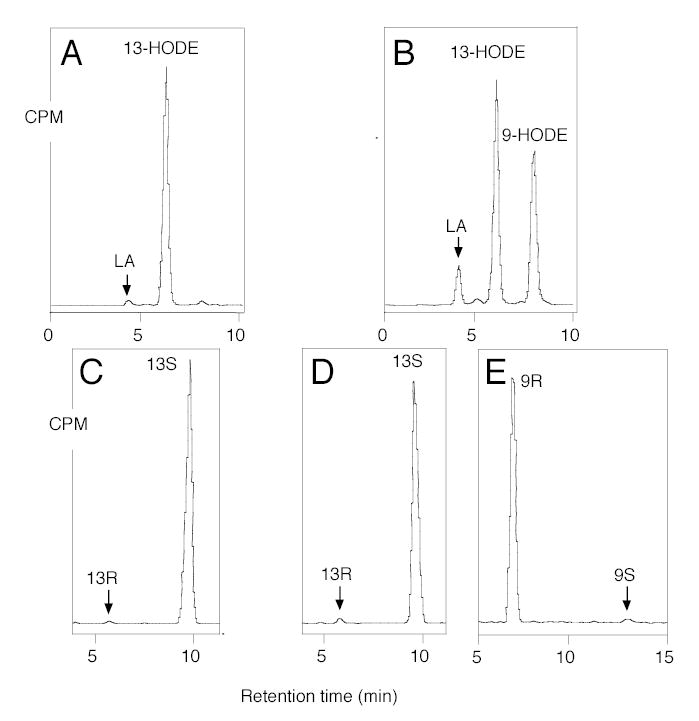
HPLC analysis of products formed from incubation of [1-14C]linoleic acid. A, SP-HPLC analysis, wild-type. B, SP-HPLC analysis, Ala542Gly mutant. C, Chiral HPLC analysis, 13-HODE-Me from wild-type. D, Chiral HPLC analysis 13-HODE-Me from Ala542Gly mutant. E, Chiral HPLC analysis, 9-HODE-Me from Ala542Gly mutant. Me, methyl ester. Products were analyzed using a Whatman® Partisil 5 silica column (0.46 × 25 cm) eluted at a flow rate of 1 ml/min with hexane/isopropanol/acetic acid (100/2/0.1, by volume). Individual metabolites purified by both RP-HPLC and SP-HPLC were analyzed on a Daicel Chiralpak AD (0.46 × 25 cm) eluted at a flow rate of 1 ml/min with hexane/methanol (100/5, by volume) (26).
Fig. 2.
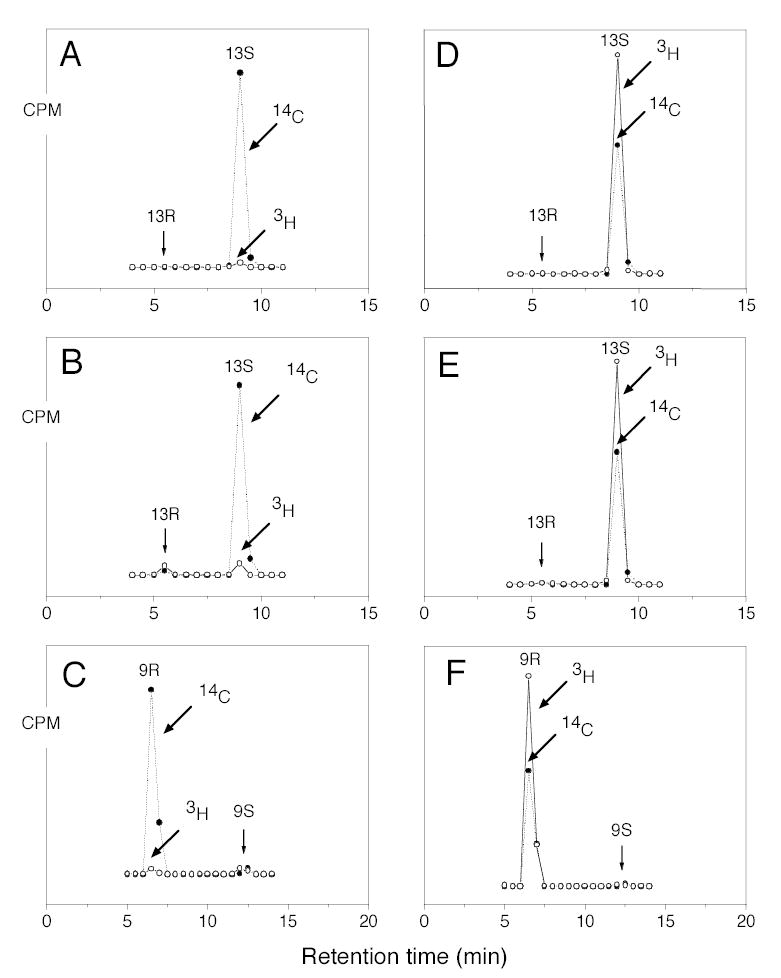
Chiral HPLC analysis of metabolites from incubation of wild-type and mutant soybean LOX-1 with stereospecifically 3H-labeled linoleic acids. A, 13-HODE-Me from incubation of wild-type enzyme with [11S-3H]linoleic acid. B, 13-HODE-Me from incubation of Ala542Gly mutant with [11S-3H]linoleic acid. C, 9-HODE-Me from incubation of Ala542Gly mutant with [11S-3H]linoleic acid. D, 13-HODE-Me from incubation of wild-type enzyme with [11R-3H]-linoleic acid. E, 13-HODE-Me from incubation of Ala542Gly mutant with [11R-3H]linoleic acid. F, 9-HODE-Me from incubation of Ala542Gly mutant with [11R-3H]linoleic acid. Reactions were monitored on a UV spectrophotometer and stopped when 80 % of the substrate had been converted. After reduction with TPP metabolites were converted to their methyl esters with ethereal diazomethane and purified by SP-HPLC prior to chiral HPLC analysis.
Table 1.
Measurement of 3H retention in experiments with stereospecifically labeled linoleic acids.
| Product, enzyme | % 3H retained [11S-3H]-LA | % 3H retained [11R-3H]-LA |
|---|---|---|
| 13S-HODE, wild-type | 1.6 | 99.7 |
| 13S-HODE, Ala542Gly | 3.6 | 98.7 |
| 9R-HODE, Ala542Gly | 1.6 | 97.8 |
Fig. 3.
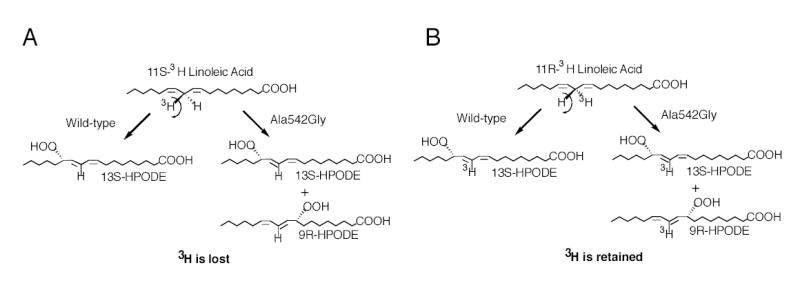
Hydrogen abstraction in the transformation of [11-3H]linoleic acids by wild-type and the Ala542Gly mutant soybean LOX-1.
Investigation on substrate orientation using C16/LA-PC substrate
In view of the recent study that concluded LA and AA bind carboxyl end first in soybean LOX-1 (19), we considered it important to test substrate orientation in wild-type soybean LOX-1 and its Ala542Gly mutant. Given the bulk of the structure of PC and other mechanistic considerations, it is likely that the oxygenation of fatty acids esterified in PC can occur only with a tail-first entry of the substrate into the soybean LOX-1 active site. We used this criterion to investigate the substrate orientation associated with the formation of S and R configuration products by soybean LOX-1. 1-Palmitoyl-2-linoleoyl-phosphatidylcholine (C16/LA-PC) was incubated with the wild-type and mutant enzymes and the reduced products transesterified to give the HODE methyl esters which were analyzed by SP-HPLC and chiral phase HPLC. Wild-type soybean LOX-1 converted C16/LA-PC to a single main product identified by SP-HPLC and chiral HPLC analyses as 13S-HODE (Fig. 4A and C). In accord with the reaction seen with free acid substrate, the Ala542Gly mutant converted C16/LA-PC to two main products in ~2:1 ratio; these were identified by SP-HPLC (after reduction and transesterification) as 13-HODE and 9-HODE methyl esters, respectively (Fig. 4B). Chiral phase HPLC analysis showed that the 13-HODE metabolite had the S stereoconfiguration whereas the 9-HODE had the R stereoconfiguration (Fig. 4D and E). We confirmed also that the arachidonate ester, C16/AA-PC, is oxygenated to the same products as AA free acid. These experiments indicate that the change in stereospecificity caused by the Ala542Gly mutation is not the result of a change in the tail-first substrate binding orientation in the soybean LOX-1 active site.
Fig. 4.
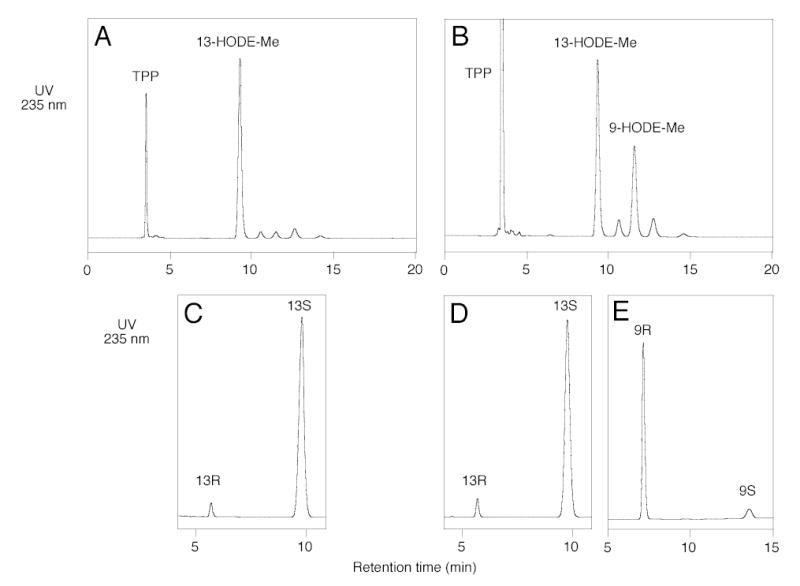
Metabolism of C16/LA-phosphatidylcholine substrate by wild-type and mutant soybean LOX-1. A, SP-HPLC analysis of wild-type products. B, SP-HPLC analysis of Ala542Gly mutant products. C, Chiral HPLC, 13-HODE-Me from wild-type. D, Chiral HPLC, 13-HODE-Me from Ala542Gly mutant. E, Chiral HPLC, 9-HODE-Me from Ala542Gly mutant. Products from the incubation of C16/AA-PC with wild-type and Ala542Gly soybean LOX-1 were reduced with TPP, converted to methyl esters by transesterification, and analyzed on a Whatman® Partisil 5 silica column (0.46 × 25 cm) eluted with hexane/isopropanol/acetic acid (100/1/0.1 by volume) at 1 ml/min with UV detection at 235 nm. Chiral HPLC analysis of the HODE methyl esters was performed as described in Figure 1.
Single and double dioxygenation of arachidonic acid
Similarly to the reactions with linoleic acid, wild-type soybean LOX-1 converted [1-14C] AA to 15S-HPETE as the sole product, whereas the Ala542Gly mutant converted [1-14C] AA to 15S-HPETE and 11R-HPETE in a 2:1 ratio (data not shown). Other mutations at the 542 position also gave comparable results to those seen with linoleic acid (data not shown). To investigate the effect of the Ala542Gly mutation on the well characterized reaction of double dioxygenation catalyzed by soybean LOX-1 (31,32), both the wild type and the mutant enzyme were incubated with 15S-HPETE and the reaction products analyzed by RP-HPLC. As reported, wild-type soybean LOX-1 converted 15S-HPETE to two main metabolites; the earlier-eluting peak exhibited a conjugated triene chromophore with λmax at 269 nm, characteristic of 8S,15S-diHPETE, and the second peak had a distinctive chromophore with λmax at 245 nm, characteristic of 5S ,15S-diHPETE (Fig. 5A) (32). The Ala542Gly mutant incubated with 15S-HPETE gave a different HPLC profile compared to the wild-type enzyme. The two major products were the same, but their ratio was changed in favor of the dioxygenation product at C-8. In addition, a minor but mechanistically significant new product was detectable (Fig. 5B). This product exhibited a distinctive conjugated diene chromophore with uncommonly prominent shoulders and a λmax at 239 nm (Figure 6B, insert). It was reasoned that this additional metabolite could be the “partner” of the C5 oxygenation, i.e 9,15-diHPETE. To test this hypothesis, standards of 9R-HPETE and 9S-HPETE were prepared and reacted separately with soybean LOX-1, giving 9R,15S-diHPETE and 9S,15S-diHPETE products that were characterized by HPLC, UV spectroscopy, GC-MS and 1H-NMR (Experimental Procedures). Each diastereomer exhibited a uv spectrum identical to the distinctive 239nm chromophore of the novel oxygenation product. Comparison of HPLC retention times showed that the 9S,15S standard eluted ahead of the 9R,15S diastereomers (8.4 and 9.5 min respectively), and that the latter co-chromatographed precisely with the novel product. Accordingly, the additional metabolite of the mutant soybean LOX-1 enzyme was identified as 9R,15S-diHPETE. The 5S and 9R positions are on the same side of the fatty acid chain at opposite ends of the reacting pentadiene, and therefore 9R is the counterpart to the 5S dioxygenation product from 15S-HPETE. Notably, the relative abundance of this 9R product compared to the 5S dioxygenation product was far lower than the ratio of the 11R to 15S products found with arachidonic acid as substrate. Furthermore, no R counterpart to the 8S dioxygenation product was found. We examined the RP-HPLC traces carefully for this potential “partner” of 8S,15S-diHPETE (predicted to be a diastereomer of 14,15-diHPETE), but none was detectable. All evidence suggests that this second oxygenation involves a reversal of substrate orientation, with the carboxyl end buried deep in the active site (33). This experiment indicates that the less preferred "head first" mode of substrate binding is possible in the Ala542Gly mutant and that one novel R configuration product, 9R,15S-diHPETE, is formed in the process. According to this model (Fig. 6), it is likely that the counterpart of the 8S dioxygenation product is not formed because there is extended conjugation in the reacting double bond system and the oxygenation should occur at C14, which is four carbons distant from the C10 hydrogen abstraction and out towards the exterior of the active site (Fig. 6).
Fig. 5.
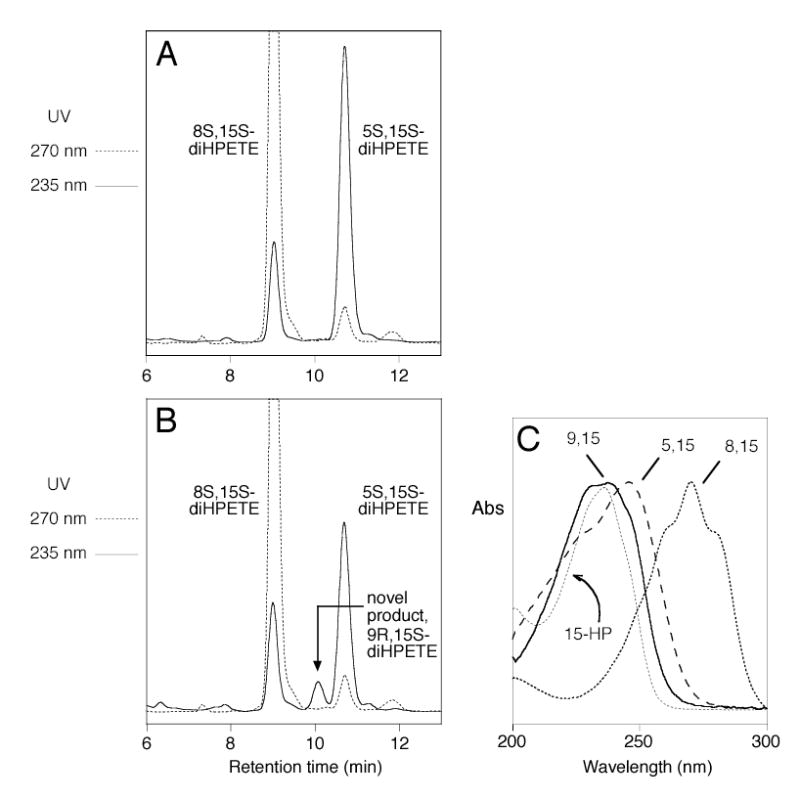
RP-HPLC analysis of products formed from incubation with 15(S)-HPETE. A, wild-type. B, Ala542Gly mutant. Products were analyzed using a Waters Symmetry C18 5-μm column (0.46 × 25 cm) eluted at a flow rate of 1 ml/min with methanol/water/acetic acid (75/25/0.01, by volume). C, Comparison of the uv spectra of 5,15-diHPETE, 8,15-diHPETE, and the novel product, 9,15-diHPETE. The spectra are normalized to λmax and do not convey relative molar extinction values.
Fig. 6.
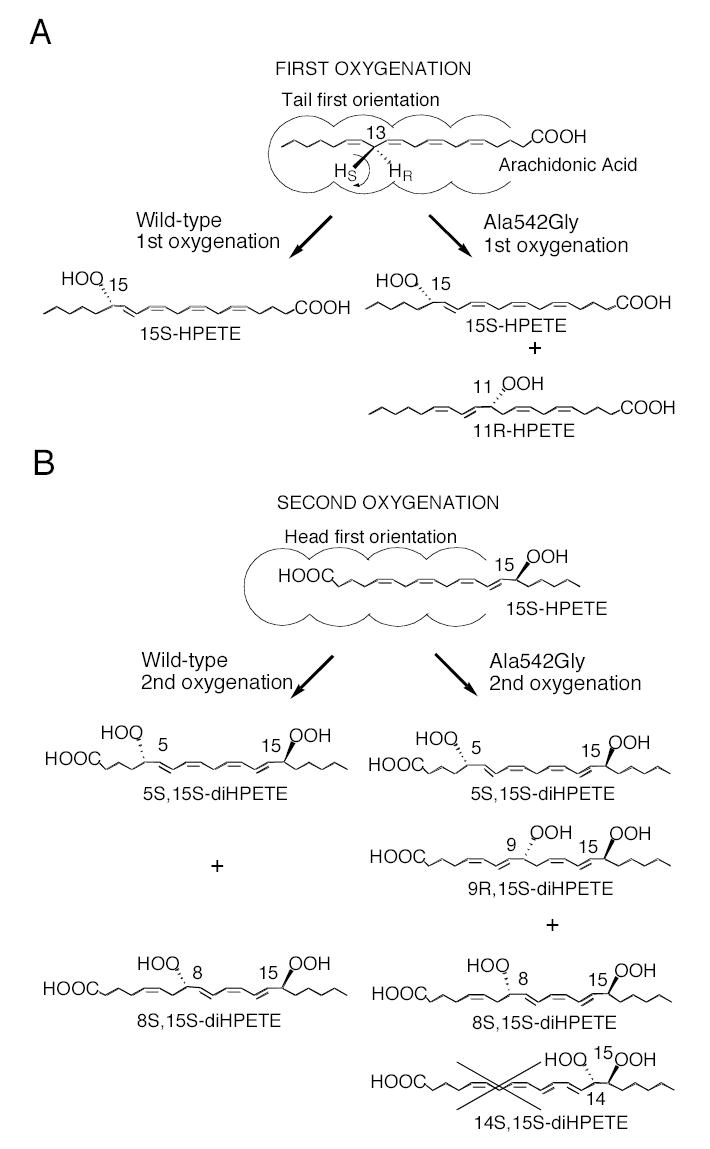
Transformations of arachidonic acid and 15S-HPETE by wild-type soybean LOX-1 and its Ala542Gly mutant.
Enzyme kinetics
In addition to the change in product profile, the Ala542Gly mutation induced a significant change in reaction kinetics compared to the wild-type enzyme, Fig. 7. The maximal rates of reaction calculated as a kcat of 129 sec−1 for the wild-type and 20 sec−1 for the mutant (not corrected for iron content, see below). The respective Km values calculated as 16.7 μM and 6.4 μM. The curve for the Ala542Gly mutant had a tendency to curve downwards at higher concentrations of linoleate, and this substrate inhibition undoubtedly confounds a meaningful estimate of K m, which instead calculates as an artificially low value. It was apparent also that the Ala542Gly mutant reacted with a more pronounced lag phase than evident with the wild-type enzyme (Fig. 7C). The lag could be eliminated by including a low concentration of 13S-HPODE (1.3 μM) in the reaction mixture, but this was effective only at low substrate concentrations (2 – 20 μM) and at higher substrate concentrations the lag reappeared. The exaggerated lag phase with the Ala542Gly mutant enzyme can be explained partly by its lower catalytic activity, resulting in a slower generation of sufficient hydroperoxide activator and lower hydroperoxide/substrate ratios, both of which will accentuate the lag (34). We found also that the product specific to the Ala542Gly mutant, 9R-HPODE, is a very inefficient activator of the soybean LOX-1 (wild-type or mutant) compared to 13S-HPODE (Fig. 7D).
Fig. 7.
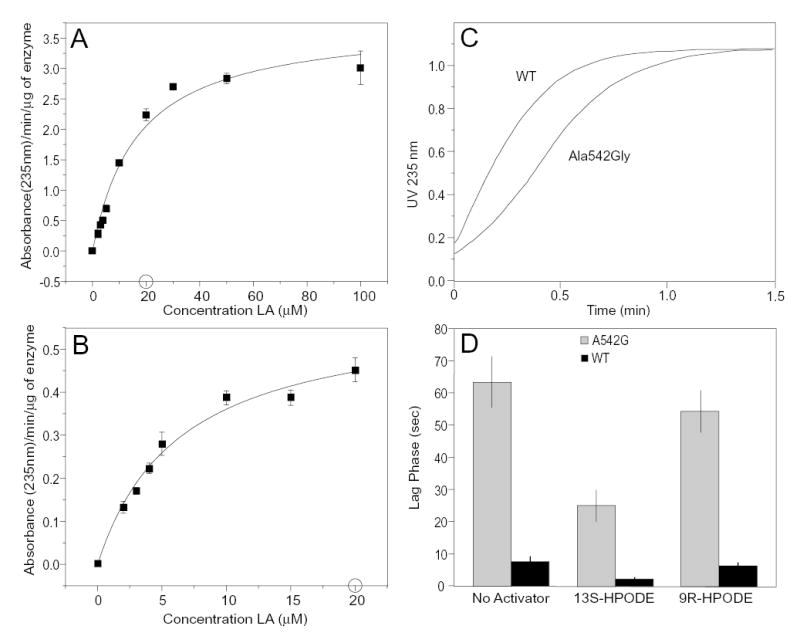
Kinetic analysis of wild-type soybean LOX-1 and the Ala542Gly mutant. A, wild-type enzyme. B, Ala542Gly mutant. Rates are shown for linoleic acid concentrations of 2 – 100 μM for the wild-type enzyme and 2 – 20 μM for the Ala542Gly mutant. C. Typical reaction curve at 235 nm for wild-type and Ala542Gly mutant. D. Lag times compared using 1.3 μM 13S-HPODE, 9R-HPODE or no hydroperoxide activator and 50 μM linoleic acid with wild-type soybean LOX-1 (black bars) and the Ala542Gly mutant (grey bars); mean values +/− S.E., n = 6 are shown. Reactions in panels A and B were performed in 0.5 ml 50 mM borate buffer pH 9, 150 mM NaCl in the presence of 1.3 μM 13S-HPODE. Reaction rates were taken as the steepest slope of the 235 nm absorbance-time curves. Data were fitted using the Michaelis-Menten function on Microcal Origin.
Metal Analysis and EPR Spectroscopy
Expressed, active LOX proteins incorporated iron at a ratio of iron/protein of 0.62, the content of inactive mutants (Thr, Val) was the same or higher, and the iron content of enzyme isolated from soybeans, and prepared for analysis similarly, was 0.74. Therefore it can be concluded that the reduced activity of 542 mutants is not due to lack of incorporation of iron.
Even though inactive mutants incorporated iron, it was possible that they had an altered iron environment, so the activated ferric state of the proteins was examined by EPR spectroscopy. All samples, wild-type and the Ala542Gly, Ala542Ser, Ala542Thr and Ala542Val mutant enzymes, had no ferric iron EPR signals in the form originally isolated and thus were presumed to contain ferrous iron, as expected. Oxidation to ferric iron was achieved for each using one equivalent of 13S-HPODE; thus all had redox-active iron. Because EPR spectra of frozen ferric soybean LOX-1 vary depending on small molecule components of the buffer (35), care was taken to examine samples prepared under identical conditions, of similar concentrations and after thorough dialysis. The spectra shown in Fig. 8 are similar in the sense that there are three prominent peaks in each one, at g'-values of about 7.27, 6.27 and 5.80 (g'-value scale shown at top of Fig. 8). (For reference, if the iron symmetry showed a large change, peaks might be found any place in the magnetic field region shown in Fig. 8.) Most noticeably, the glycine mutant has diminished intensity in the peak labeled 1, possibly reflecting a change in the 5- or 6-coordinate iron liganding (see Discussion). Although all the other EPR spectra have similar intensities in the peaks labeled as components 1 and 2, there is a broad feature, as yet uncharacterized, underlying the peaks in the EPR spectra of serine and threonine mutants that was unchanged after repurification of the samples. The valine mutant was not characterized in detail except to establish that the iron could be oxidized.
DISCUSSION
The results of our study are relevant to the issues of substrate access, substrate orientation, and control of oxygenation in soybean LOX-1. We have established that the initial hydrogen abstraction occurs similarly in the wild-type and Ala542Gly mutant enzymes, with important connotations regarding substrate orientation. Furthermore, our results with single or double oxygenation reactions fit in a consistent picture of tail-first substrate binding in the initial oxygenation and reversed binding (carboxyl end first) in forming the double dioxygenation products of arachidonic acid. In considering how these results all intertwine, firstly it is useful to understand where the Ala542 resides in the structure of soybean LOX-1. The Ala residue is located in helix 11, opposite to the iron, intruding into the available substrate binding pocket and roughly pointing “inwards” back towards the nonheme iron which resides in the center of the catalytic domain (Fig. 9). Other investigators have examined the effect of mutating some of the highly conserved residues in helix 11 of other lipoxygenases and concluded it is clearly an important determinant of kinetic efficiency and regiospecificity (14,36,37). But in none of these other mutants was significantly altered stereospecificity observed.
Fig. 9.
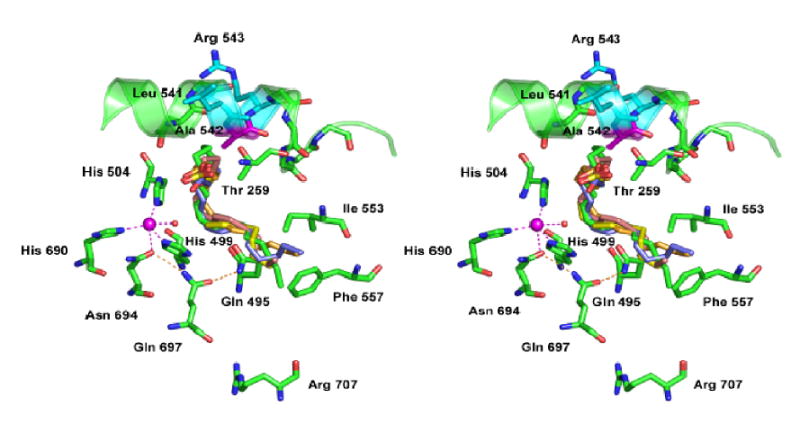
Modeling of linoleic acids in the active site of soybean LOX-1. The site of mutagenesis, Ala 542, is shown in proximity to the catalytic iron (magenta). Helix 11 (Asn535-Gln544) is rendered as a ribbon, with Ala542 (magenta) and the residues flanking it, Leu541 and Arg543 (cyan), highlighted. The ligands to iron shown are His499, His504, His690, Asn694 and water842 (red sphere); the remaining iron ligand, the carboxy terminal O1 of Ile839, is above (toward the viewer) the iron and has been omitted for clarity. A cluster of multiple potential alignments of linoleic acid are shown as computed by Autodock 3.03 (29); Molecular Graphics Laboratory, (http://www.scripps.edu/mb/olson)) The starting coordinates for the ligand linoleic acid (eic.pdb) were acquired from the Hetero Compound Information Center, Uppsala (http://alpha2.bmc.uu.se/hicup). The linoleic acid structures lie with the middle of the carbon chain opposite the iron and water842, with the methyl end deep in the active site near Phe557, and the carboxyl end lying close to the potential access channel for substrate near Leu541 and Thr259 (see main text for further discussion). The figure is based on the coordinates of 1YGE (15) and was drawn using PyMol (http://www.pymol.org).
Substrate access
As there are no X-ray structures available for a LOX with bound fatty acid substrate, (a product-bound structure is available (38)), there has always been debate on the route of substrate access to the soybean LOX-1 catalytic domain and on the mode of substrate binding (12,15). It appears significant to us that Ala542 is situated next to a proposed route for substrate entry (Fig. 9). In fact there is no open route in the X-ray structures of soybean LOX-1, but as noted by Minor et al. “One promising [route] is located at the opposite end of cavity IIa relatively close to the Fe atom. In this case, access to IIa is barred 8–10 Å from the Fe atom by two contacts between side chain atoms: Thr259O1 and Leu541C, 4.6 Å; Lys260C and Leu541C2, 5.3 Å. Sterically unhindered adjustments of the side chains of Thr259 and Leu541 appear to be sufficient to open a channel wide enough (>6 Å) for entry of a fatty acid” (15). Ala542, the determinant of oxygenase specificity, sits close to this point of access (Fig. 9), and taken together with the inferences on substrate orientation, our results are consistent with this being the point of substrate access. This also matches the mode of substrate access and substrate binding inferred from the X-ray structure of the mammalian reticulocyte 15-LOX (which was crystallized with an inhibitor in the active site) (20,39), suggesting many common features in the respective reactions. It might be noted that helices 2 and 21 have side chains in contact with helix 11 and other entries into cavity IIa involving these three helices can also be considered (15).
Substrate orientation
There remain conflicting views on how substrates bind in different lipoxygenases (e.g. (21,33,40–42)). A recent study of soybean LOX-1, the enzyme we studied here, used the results of mutagenesis of two internal amino acids (Trp500 and Arg707) to conclude that the usual orientation of arachidonic acid (as it is converted to 15S-HPETE) is with its carboxyl end buried deep in the protein (19). But this conclusion fails to consider the well established fact that linoleate or arachidonate esterified in phosphatidylcholine are oxygenated by soybean LOX-1 identically to the free fatty acids (43). Here we show that this applies also to reaction of C16/LA-PC and C16/AA-PC with the Ala542Gly mutant, which forms the 9R/13S linoleate products or 11R/15S arachidonate products from the PC ester or the free acids. This is only compatible with a tail-first substrate binding orientation in the soybean LOX-1 active site in all these primary oxygenation reactions. Tail-first binding is further supported by the experiments with stereospecifically labeled linoleic acids showing that the same hydrogen is abstracted to form products with either S or R specificity. Also consistent with the conclusion is the positioning of the Ala542 residue (or 542Gly in the mutant) in the active site, closer to the surface of the enzyme compared to the iron, allowing control of oxygenation at C-9 of linoleate when the fatty acid is bound in the tail-first orientation. Binding in the reversed orientation with the carboxyl end deep in the active site is implicated in some circumstances, notably in the further oxygenation of 15S-HPETE. Here we have provided additional consistent support for this in characterizing oxygenation of 15S-HPETE to 9R,15S-diHPETE by the Ala542Gly mutant enzyme, thus identifying the R configuration “partner” of the usual secondary oxygenation product 5S,15S-diHPETE. These second oxygenation reactions are consistent with substrate binding in the reversed carboxyl end-first orientation.
Our results, therefore, are consistent with the positioning of C9 of linoleic acid in the vicinity of Ala542, and computational docking of the substrate in the active site gave several solutions matching this arrangement (Fig. 9). Many conformations of linoleic acid can be achieved inside the active site because of the high flexibility of the substrate, and several mechanistically relevant possibilities are illustrated in the figure. Linoleic acid is shown with the middle of the carbon chain lying beside the iron and with its methyl end running deep into the active site. The location of Ala542 opposite to the iron is favorable for interference with antarafacial oxygenation on C-9. As discussed above, we hypothesize that the linoleic acid enters the active site through the channel opened by movement of the side chains of Thr259 and the Leu541 residues, but the docking program cannot accurately account for this conformational flexibility of the protein. As a consequence, the linoleic acid appears to be trapped in the active site with its carboxylic end computed to be turning back inwards, rather than exiting the active site.
Oxygen access to the activated radical
Access of oxygen to the pentadienyl radical intermediate of LOX appears to be altered in the Ala542Gly mutant. It has been argued that oxygen approaches the radical intermediate through a side channel near Ile553 and that this channel controls 13S oxygenation reactions of the wild type enzyme (14). Here we observe the appearance of 9R oxygenation in the Ala542Gly mutant in which a methyl group has been removed at position 542. According to our analysis it is unlikely that this could create a new specific channel for 9R oxygenation. And if 9R oxygenation can occur without the need for an oxygen channel, this raises the question whether a channel is the basis of stereocontrol in the usual 13S reaction. On the other hand, it is quite probable that an oxygenpocket (a space to “hold” an O2 molecule next to the substrate) forms part of the structural basis of stereocontrol. This is compatible with the effects of changing Ala542 to Gly and introducing the new 9R oxygenase activity on the carboxy-proximal end of the linoleate molecule. In concert with this, steric factors will shield the reacting pentadiene and thus control positional and stereo specificity. Thus, the most straightforward interpretation of the effects of the Ala542Gly mutation is that it opens up space and allows oxygenation on the side of the activated pentadienyl radical closer to the surface of the enzyme.
Other changes in the active site
EPR spectroscopy and the reaction kinetics suggest that the Ala-542 mutations are also associated with some changes in the way the substrate can interact with the catalytic iron center. General access of fatty acids is clearly not blocked in the inactive mutants because hydroperoxide product oxidizes iron in all Ala542 mutants examined. And since the serine and threonine mutants are active and inactive, respectively, and have similar EPR spectra to wild-type, the data suggest that the inactivity of the threonine mutant results from changes in substrate positioning relative to the catalytic iron rather than from a major change in the properties of the iron center.
The EPR spectrum (Fig. 8A) of the glycine mutant is similar to the wild type spectrum in that there are two major components corresponding to the same degrees of deviation from axial symmetry at iron, but there is considerable variation in their proportions. The signals in the EPR spectra and their intensity are a reflection of the detailed interactions of iron and its ligands, and the two signals may correspond to 5- and 6-coordinate iron liganding. Previously, a network of H-bonds involving iron ligand Asn694 and residues in the iron second coordination sphere has been characterized by mutation, CD, MCD and x-ray structure analyses, with the conclusion that dynamic changes in iron coordination, and especially in the iron-Asn694 bond length, is essential for catalytic function (44,45). It is likely that the helix 11 containing the 542 Ala or Gly is connected through hydrogen bonds of residues and waters in cavity IIa to the network of H-bonds that modulate the iron-ligand bond lengths. We speculate that the volume of the side chain at position 542 in soybean LOX-1 influences the hydrogen bonding details and/or the location of waters throughout cavity IIa. Explaining precisely how these networks are linked awaits characterization of the structure of relevant enzyme intermediates in lipoxygenase catalysis. In addition, the dramatic effects of the Ala-Gly mutation include not only a change in oxygenase specificity, but also the appearance of a profound lag phase in catalysis and a marked reduction in the attainable Vmax(kcat) of the enzyme. This may indicate effects beyond a simple opening up of space, and point to a more profound effect on the structure of the enzyme and/or the conformation of the substrate into the active site. Crystallization of the Ala542Gly mutant would greatly facilitate a deeper understanding of the effects of this mutation.
Finally, it is of interest to note that the formation of hydroperoxides with R stereoconfiguration has already been described for the wild-type of the type-II plant lipoxygenases, which are reasonably close relatives of soybean LOX-1 (46). Although the type II enzymes are usually known for catalysis of non-specific oxygenations, when the native type II enzymes of pea and soybean were incubated with low concentrations of linoleic acid at pH 9, the product profile showed evidence of more specific reaction (46). Under these conditions the products were a 2:1 mixture of 9- and 13-hydroperoxides, and the chirality of the 9-hydroperoxide showed a bias towards 9R (62–78% 9R ) while the 13-hydroperoxide was predominantly 13S (87–91%). Apparently these wild-type type II LOX enzymes with an Ala in the position equivalent to Ala542 of soybean LOX-1 are able to mimic some elements of the reaction promoted by the Ala542Gly mutation in soybean LOX-1, albeit with less control of stereospecificity. As noted, the type II enzymes tend to catalyze stereorandom oxygenation, especially at higher substrate concentrations and at neutral pH (46), implying a comparatively loose fit of the substrate in the active site. This may facilitate their ability to catalyze some 9R oxygenation within the context of the wild-type structure.
Footnotes
This work was supported by NIH grants GM53638 (A.R.B.) and GM65268 (B.J.G.).
We thank Fayi Wu for providing a sample of the Ala542Thr mutant.
The abbreviations used are: LOX, lipoxygenase; AA, arachidonic acid; LA, linoleic acid; H(P)ODE, hydro(pero)xyoctadecadienoic acid; H(P)ETE, hydro(pero)xyeicosatetraenoic acid; diH(P)ETE, dihydro(pero)xyeicosatetraenoic acid; C16/LA-PC, 1-palmitoyl-2-linoleoyl-phosphatidylcholine; C16/AA-PC, 1-palmitoyl-2-arachidonoyl-phosphatidylcholine; PMSF, phenylmethylsulfonyl fluoride; RP-HPLC, reversed-phase high pressure liquid chromatography; SP-HPLC, straight-phase high pressure liquid chromatography; EPR, electron paramagnetic resonance; CPM, counts per minute; TPP, triphenylphosphine.
References
- 1.Brash AR. J Biol Chem. 1999;274:23679–23682. doi: 10.1074/jbc.274.34.23679. [DOI] [PubMed] [Google Scholar]
- 2.Rouzer CA, Marnett LJ. Chem Rev. 2003;103:2239–2304. doi: 10.1021/cr000068x. [DOI] [PubMed] [Google Scholar]
- 3.Capdevila JH, Falck JR. Prostaglandins Other Lipid Mediat. 2002;68–69:325–344. doi: 10.1016/s0090-6980(02)00038-2. [DOI] [PubMed] [Google Scholar]
- 4.Ortiz de Montellano, P. R. (1995) in Cytochrome P450: Structure, mechanism and biochemistry (Ortiz de Montellano, P. R., ed), pp. 245–303, Plenum Press, New York
- 5.Sono M, Roach MP, Coulter ED, Dawson JH. Chem Rev. 1996;96:2841–2887. doi: 10.1021/cr9500500. [DOI] [PubMed] [Google Scholar]
- 6.Ortiz de Montellano PR, De Voss JJ. Nat Prod Rep. 2002;19:477–493. doi: 10.1039/b101297p. [DOI] [PubMed] [Google Scholar]
- 7.Malkowski MG, Ginell SL, Smith WL, Garavito RM. Science. 2000;289:1933–1937. doi: 10.1126/science.289.5486.1933. [DOI] [PubMed] [Google Scholar]
- 8.Kurumbail RG, Kiefer JR, Marnett LJ. Curr Opin Struct Biol. 2001;11:752–760. doi: 10.1016/s0959-440x(01)00277-9. [DOI] [PubMed] [Google Scholar]
- 9.van der Heijdt LM, Schilstra MJ, Feiters MC, Nolting HF, Hermes C, Veldink GA, Vliegenthart JF. Eur J Biochem. 1995;231:186–191. doi: 10.1111/j.1432-1033.1995.tb20685.x. [DOI] [PubMed] [Google Scholar]
- 10.Glickman MH, Klinman JP. Biochemistry. 1996;35:12882–12892. doi: 10.1021/bi960985q. [DOI] [PubMed] [Google Scholar]
- 11.Prigge ST, Boyington JC, Gaffney BJ, Amzel LM. Proteins. 1996;24:275–291. doi: 10.1002/(SICI)1097-0134(199603)24:3<275::AID-PROT1>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 12.Boyington JC, Gaffney BJ, Amzel LM. Science. 1993;260:1482–1486. doi: 10.1126/science.8502991. [DOI] [PubMed] [Google Scholar]
- 13.Knapp MJ, Klinman JP. Biochemistry. 2003;42:11466–11475. doi: 10.1021/bi0300884. [DOI] [PubMed] [Google Scholar]
- 14.Knapp MJ, Seebeck FP, Klinman JP. J Am Chem Soc. 2001;123:2931–2932. doi: 10.1021/ja003855k. [DOI] [PubMed] [Google Scholar]
- 15.Minor W, Steczko J, Stec B, Otwinowski Z, Bolin JT, Walter R, Axelrod B. Biochemistry. 1996;35:10687–10701. doi: 10.1021/bi960576u. [DOI] [PubMed] [Google Scholar]
- 16.Hamberg M, Samuelsson B. J Biol Chem. 1967;242:5329–5335. [PubMed] [Google Scholar]
- 17.Egmond MR, Veldink GA, Vliegenthart JF, Boldingh J. Biochem Biophys Res Commun. 1973;54:1178–1184. doi: 10.1016/0006-291x(73)90816-4. [DOI] [PubMed] [Google Scholar]
- 18.Egmond MR, Vliegenthart JFG, Boldingh J. Biochem Biophys Res Commun. 1972;48:1055–1060. doi: 10.1016/0006-291x(72)90815-7. [DOI] [PubMed] [Google Scholar]
- 19.Ruddat VC, Mogul R, Chorny I, Chen C, Perrin N, Whitman S, Kenyon V, Jacobson MP, Bernasconi CF, Holman TR. Biochemistry. 2004;43:13063–13071. doi: 10.1021/bi0489098. [DOI] [PubMed] [Google Scholar]
- 20.Borngräber S, Browner M, Gillmor S, Gerth C, Anton M, Fletterick R, Kuhn H. J Biol Chem. 1999;274:37345–37350. doi: 10.1074/jbc.274.52.37345. [DOI] [PubMed] [Google Scholar]
- 21.Coffa G, Brash AR. Proc Natl Acad Sci USA. 2004;101:15579–15584. doi: 10.1073/pnas.0406727101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maguire, B. C. (1999) in Ph. D. Thesis, pp. 117, Johns Hopkins University, Baltimore
- 23.Gill SC, von Hippel PH. Anal Biochem. 1989;182:319–326. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- 24.Bligh EG, Dyer WJ. Can J Biochem Physiol. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 25.Schneider C, Boeglin WE, Lai S, Cha JK, Brash AR. Anal Biochem. 2000;284:125–135. doi: 10.1006/abio.2000.4670. [DOI] [PubMed] [Google Scholar]
- 26.Schneider C, Boeglin WE, Brash AR. Anal Biochem. 2000;287:186–189. doi: 10.1006/abio.2000.4847. [DOI] [PubMed] [Google Scholar]
- 27.Milne GL, Porter NA. Lipids. 2001;36:1265–1275. doi: 10.1007/s11745-001-0841-2. [DOI] [PubMed] [Google Scholar]
- 28.Smith WL, Lands WEM. J Biol Chem. 1972;247:1038–1047. [PubMed] [Google Scholar]
- 29.Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ. J Comput Chem. 1998;19:1639–1662. [Google Scholar]
- 30.Hamberg M, Samuelsson B. Biochim Biophys Acta. 1980;617:545–547. doi: 10.1016/0005-2760(80)90022-3. [DOI] [PubMed] [Google Scholar]
- 31.Bild GS, Ramadoss CS, Lim S, Axelrod B. Biochem Biophys Res Commun. 1977;74:949–954. doi: 10.1016/0006-291x(77)91610-2. [DOI] [PubMed] [Google Scholar]
- 32.Van Os CPA, Rijke-Schilder GPM, Van Halbeek H, Verhagen J, Vliegenthart JFG. Biochim Biophys Acta. 1981;663:177–193. doi: 10.1016/0005-2760(81)90204-6. [DOI] [PubMed] [Google Scholar]
- 33.Schwarz K, Borngräber S, Anton M, Kühn H. Biochemistry. 1998;37:15327–15335. doi: 10.1021/bi9816204. [DOI] [PubMed] [Google Scholar]
- 34.Schilstra MJ, Veldink GA, Verhagen J, Vliegenthart JFG. Biochemistry. 1992;31:7692–7799. doi: 10.1021/bi00148a033. [DOI] [PubMed] [Google Scholar]
- 35.Gaffney BJ, Mavrophilipos DV, Doctor KS. Biophys J. 1993;64:773–783. doi: 10.1016/S0006-3495(93)81438-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gan QF, Browner MF, Sloane DL, Sigal E. J Biol Chem. 1996;271:25412–25418. doi: 10.1074/jbc.271.41.25412. [DOI] [PubMed] [Google Scholar]
- 37.Hughes RK, Lawson DM, Hornostaj AR, Fairhurst SA, Casey R. Eur J Biochem. 2001;268:1030–1040. doi: 10.1046/j.1432-1327.2001.01964.x. [DOI] [PubMed] [Google Scholar]
- 38.Skrzypczak-Jankun E, Bross RA, Carroll RT, Dunham WR, Funk MO., Jr J Am Chem Soc. 2001;123:10814–10820. doi: 10.1021/ja011759t. [DOI] [PubMed] [Google Scholar]
- 39.Gilmor SA, Villasenor A, Fletterick R, Sigal E, Browner MF. Nat Struct Biol. 1997;4:1003–1009. doi: 10.1038/nsb1297-1003. [DOI] [PubMed] [Google Scholar]
- 40.van Zadelhoff G, Veldink GA, Vliegenthart JF. Biochem Biophys Res Commun. 1998;248:33–38. doi: 10.1006/bbrc.1998.8910. [DOI] [PubMed] [Google Scholar]
- 41.Butovich IA, Reddy CC. Biochim Biophys Acta. 2001;1546:379–398. doi: 10.1016/s0167-4838(01)00162-5. [DOI] [PubMed] [Google Scholar]
- 42.Walther M, Ivanov I, Myagkova G, Kühn H. Chem Biol. 2001;8:779–790. doi: 10.1016/s1074-5521(01)00050-3. [DOI] [PubMed] [Google Scholar]
- 43.Brash AR, Ingram CD, Harris TM. Biochemistry. 1987;26:5465–5471. doi: 10.1021/bi00391a038. [DOI] [PubMed] [Google Scholar]
- 44.Tomchick DR, Phan P, Cymborowski M, Minor W, Holman TR. Biochemistry. 2001;40:7509–7517. doi: 10.1021/bi002893d. [DOI] [PubMed] [Google Scholar]
- 45.Schenk G, Neidig ML, Zhou J, Holman TR, Solomon EI. Biochemistry. 2003;42:7294–7302. doi: 10.1021/bi027380g. [DOI] [PubMed] [Google Scholar]
- 46.Van Os C, Rijke-Schilder G, Vliegenthart J. Biochim Biophys Acta. 1979;575:479–484. doi: 10.1016/0005-2760(79)90120-6. [DOI] [PubMed] [Google Scholar]