

The α helix plays a fundamental role in imparting specificity to protein–protein and protein–nucleic acid interactions. Molecules that can predictably and selectively disrupt these interactions would be invaluable as tools in molecular biology and, potentially, as leads in drug discovery.[1] We recently described a new strategy for the synthesis of artificial α helices in which one main-chain i to i + 4 hydrogen bond in the target α helix is replaced with a carbon–carbon bond derived from a ring-closing metathesis reaction (Figure 1).[2] A key feature of this hydrogen-bond surrogate (HBS) approach is that the internal placement of the cross-link affords short helices with minimal perturbations to their molecular recognition surfaces. This method differs significantly from the commonly employed side-chain cross-linking method for helix stabilization. A limitation of the latter approach is that side-chain functionality must be sacrificed to nucleate stable helical conformations. The modified side chains are unavailable for molecular recognition; moreover, the resulting tether blocks at least one face of the putative helix. The HBS approach uniquely allows the synthesis of artificial helices with all side chains available for molecular recognition, and does not place any steric encumbrances on the helix surface. We believe that our artificial α helices have the potential to target protein receptors and regulate protein–protein interactions more successfully than helices with cross-linked side chains.
Figure 1.
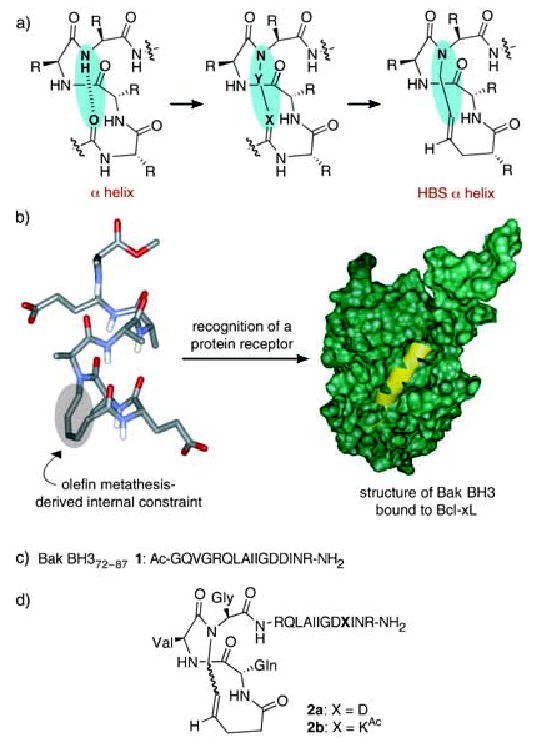
a) Formation of hydrogen-bond surrogate (HBS) derived α helices by replacement of a main-chain hydrogen bond with a carbon–carbon bond. b) HBS α helix and recognition of Bcl-xL (green) with Bak BH3 α helix (yellow); PDB code: 1BXL. c) Sequence of the unconstrained Bak BH3 peptide 1. d) Structures of HBS Bak α helices 2a and 2b.
Our initial studies demonstrated that the HBS approach affords highly stable α helices from alanine-rich peptide sequences. Herein, we show that this metathesis-based method can effectively stabilize α-helical conformations in biologically relevant sequences, that the resulting molecules resist proteolytic degradation as compared with the unconstrained analogues, and that the HBS helices can bind a protein target with high affinity. For these proof-of-principle protein-binding studies with our artificial helices, we chose to target the extensively studied α helix binding protein, Bcl-xL.[3] Bcl-xL is an antiapoptotic protein that regulates cell death by binding the α-helical BH3 domain of a family of proapoptotic proteins (including Bak, Bad, Bid, and Bax).[4–6] NMR spectroscopic studies by Fesik and co-workers have shown that the 16-mer peptide 1 derived from the Bak BH3 domain adopts an α-helical conformation upon binding to Bcl-xL (Figure 1b).[3] Circular dichroism (CD) studies demonstrated that this peptide is unstructured under physiological conditions in the absence of the protein partner and only slightly helical in trifluoroethanol (TFE), a helix-promoting solvent.[7]
Several methods that afford stabilized α helices or helix mimetics have already been used to target Bcl-xL, thus allowing us to directly compare the performance of our internally constrained artificial α helices.[8–10] Significantly, Huang and co-workers recently reported that Bak BH3 α helices stabilized by a lactam-based side-chain cross-linking strategy were unable to bind Bcl-xl.[11] The authors speculated that the lack of binding might be a result of steric clashes between the cross-link and the narrow binding pocket of Bcl-xL. However, Verdine and co-workers found that side-chain-bridged α helices corresponding to the BH3 domain of a different proapoptotic protein, Bid, can target Bcl-xL and suppress the growth of leukemia cells in mice.[1] Judicious placement of the side-chain constraints requires prior knowledge of the protein–ligand complex; otherwise, multiple randomly constrained helices must be prepared and tested. Nevertheless, their report highlights the potential of constrained α helices as tools for the control of protein–protein interactions in vivo. Taken together, these two protein-binding studies from the Huang and Verdine research groups illustrate potential problems with the side-chain bridging strategy. The fundamental advantage of the HBS approach over this strategy for stabilizing helices is that the helix surfaces are not encumbered by the constraining element. The HBS approach should therefore greatly simplify the helix design process. The key question we ask herein is whether the HBS-derived Bak α helix can bind Bcl-xL when the side-chain-constrained (lactam bridge) Bak helix is unable to target this same protein receptor.
The artificial Bak BH3 α helix 2a was synthesized on Rink amide resin by the ring-closing metathesis reaction shown in Scheme 1.[12] HBS α helices can be synthesized from commercially available amino acids or simple amino acid derivatives and do not require preparation of enantiomerically pure amino acid analogues. In the present case, standard solid-phase peptide synthesis using appropriate Fmoc-modified amino acids, dipeptide 4, and pentenoic acid afforded the fully protected resin-bound bis-olefin peptide 3, which was subjected to the Hoveyda–Grubbs ring-closing metathesis catalyst to afford the peptide macrocycles. The metathesized peptide was cleaved from the resin with trifluoroacetic acid to give the constrained peptide 2a as a mixture of the cis- and trans-alkene isomers. We were unable to separate these isomers by HPLC.
Scheme 1.
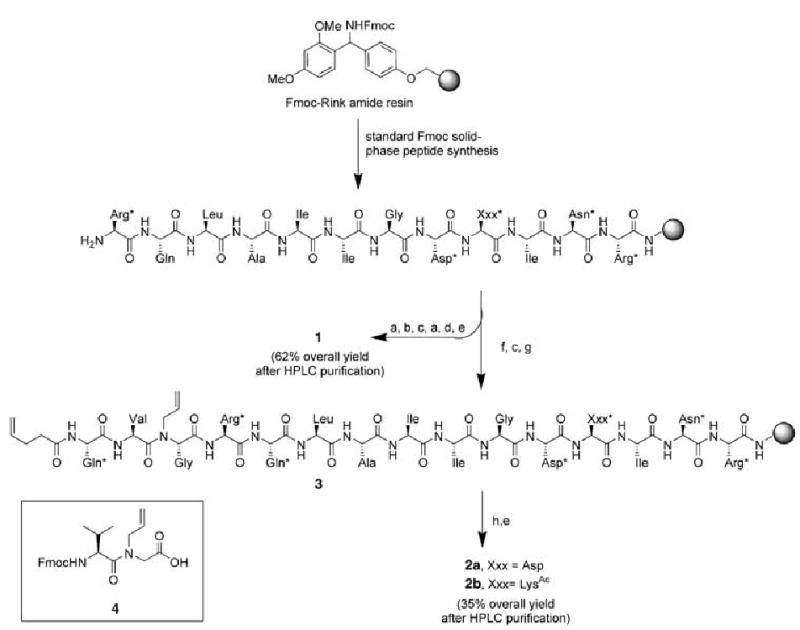
Solid-phase synthesis of HBS α helices 2a and 2b: a) 1. Fmoc Gly-OH (3.2 equiv), HBTU (2.88 equiv), iPr2NEt, NMP; 2. piperidine, NMP; b) 1. Fmoc Val-OH (3.2 equiv), HBTU (2.88 equiv), iPr2NEt, NMP; 2. piperidine, NMP; c) 1. Fmoc Gln*-OH (3.2 equiv), HBTU (2.88 equiv), iPr2NEt, NMP; 2. piperidine, NMP; d) acetic anhydride, iPr2NEt, DMF; e) CF3CO2H/H2O/triisopropylsilane (95:2.5:2.5), 1.5 h; f) 1.4 (3.2 equiv), HBTU (2.88 equiv), iPr2NEt, NMP; 2. piperidine, NMP; g) pentenoic acid (3.2 equiv), HBTU (2.88 equiv), iPr2NEt, NMP; h) Hoveyda–Grubbs catalyst (20 mol%), dichloroethane, 50°C, 24 h. Arg*: Pbf-protected Arg; Asn*: trityl-protected Asn; Asp*: tert-butyl-protected Asp; Gln*: trityl-protected Gln; Xxx*: Asp* or LysAc. Fmoc = 9-fluorenylmethoxycarbonyl; HBTU = O-(benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate; NMP = N-methyl pyrrolidinone; Pbf = 2,2,4,6,7-pentamethyldihydrobenzofuran-5-sulfonyl.
The helical conformation of constrained peptide 2a was investigated by CD. CD studies on 2a and the control peptide 1 were performed in TFE/phosphate-buffered saline (PBS, 1:4) to obtain a quantitative measure of their helical content (Figure 2). The CD results of the artificial α helix 2a display double minima at 206 and 222 nm and a maximum at 189 nm, consistent with those observed for canonical α helices. The HBS α helix 2a is roughly 46% helical as measured by Yang’s method (Supporting Information).[13] In agreement with previous studies,[7] we found that the unconstrained Bak peptide 1 is only weakly helical (≈20%). We hypothesized that the GDD tripeptide residue (residues 82–84 in the Bak BH3 domain) in the middle of the Bak peptide sequence may limit the propagation of the helix and lower the overall helical content of 2a, as glycine is known to be a potent “helix breaker”, and aspartic acid has been implicated as a helix stop signal.[14–16] Fesik and co-workers have previously shown that the residues Gly82 and Asp83 cannot be substituted with alanine without sacrificing binding affinity for the protein; however, Asp84 may be replaced without any deleterious effects.[3] To test the effect of replacing Asp84 on the helicity of Bak peptide, we prepared HBS α helix 2b in which Asp84 is substituted with a side-chain-acetylated lysine (LysAc) (Figure 1). We made this particular substitution because the Bak BH3 peptide with a capped lysine residue was previously shown to bind Bcl-xL with high affinity.[17] We were gratified to find that this single substitution provided a significant boost in α helicity (Figure 2). HBS helix 2b is roughly 65% helical: an increase in helicity of 140% over that of 2a. As expected, this substitution provided a similar increase in helicity for the unconstrained Bak BH3 (Supporting Information). Importantly, this set of experiments shows that the HBS approach can successfully stabilize α-helical conformations in biologically relevant sequences.
Figure 2.
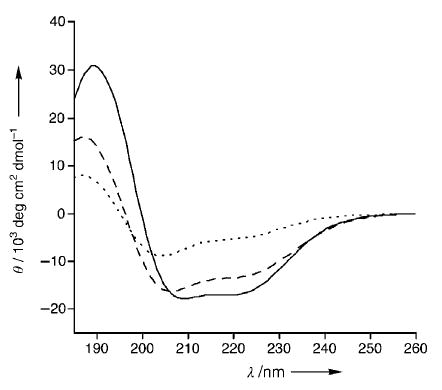
Circular dichroism data of peptides Bak BH3 1 (-----), HBS helix 2 a (– – –), and HBS helix 2 b (—) in TFE/PBS (1:4).
The binding affinities of the unconstrained Bak peptide 1 and artificial Bak α helices 2a and 2b for Bcl-xL were assessed by using a previously described fluorescence polarization assay with a fluorescein-labeled 16-mer Bak peptide (fl-Bak, 5; see Supporting Information). The affinity of fl-Bak for Bcl-xL was determined to be 264 ± 18 nm, which is in agreement with the previously reported values. Competitive inhibition of the fl-Bak–Bcl-xL complex by the Bak peptides (1, 2a, and 2b) is detected by a decrease in fluorescence polarization as the immobilized probe dissociates from the protein. Regression analysis[18] provided a Kd value of 154 ± 23 nm for the unconstrained Bak peptide 1, within range of the previously reported values (Figure 3). Under the same assay conditions, HBS helices 2a and 2b bound Bcl-xL with Kd values of 325 ± 51 and 69 ± 16 nm, respectively. These binding results point to a potential transition-state problem involved in forcing a preformed helix into this pocket, as the unconstrained Bak BH3 peptide 1 targets this protein with twofold higher affinity than the constrained peptide 2a. Nevertheless, the data validate our helix design principle as we find that our internally constrained Bak α helices do indeed access this deep hydrophobic cleft, whereas Bak α helices prepared by the side-chain-bridging method show no affinity for the same target. Moreover, we show that a very high-affinity binder, 2b, for the Bcl-xL can be developed by increasing the helicity of the constrained peptide through rational substitutions. On the basis of these preliminary results, we are now preparing a second generation of HBS Bak α helices designed to be more helical and to bind Bcl-xL with higher affinity than 2b. It remains to be determined if these HBS α helices show selectivity for Bcl-xL over other closely related members of the Bcl-2 family (and over other helix-binding proteins).[8,19,20]
Figure 3.
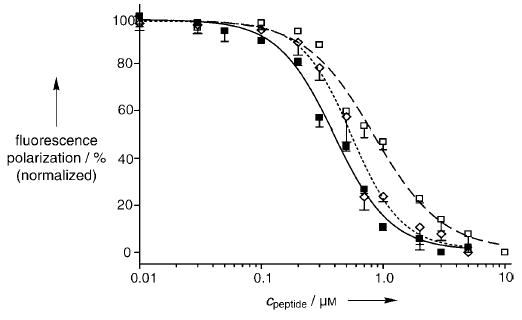
Fluorescence polarization studies indicate that HBS α helices 2a and 2b target Bcl-xL with high affinity. The Kd value for each peptide was determined by competitive inhibition of fluorescein-labeled Bak peptide (15 nm) and Bcl-xL (500 nm) complex. Bak BH3 1 (Kd = 154 ± 23 nm, -----⋄-----), HBS helix 2a (Kd = 325 ± 51 nm, – – –□– – –), and HBS helix 2b (Kd = 69 ± 16 nm, —▪—).
Proteolytic cleavage is one of the principal reasons limiting the in vivo efficacy of peptides as reagents. Proteases are known to bind their substrates in linear or beta-strand conformations,[21] and peptides locked into helical conformations have been shown to resist proteolytic degradation.[22,23] Accordingly, we determined the proteolytic stability of HBS Bak helices 2a and 2b relative to Bak peptide 1 in the presence of trypsin, which is expected to cleave the peptide at the arginine residue (Arg76) positioned two residues away from the macrocycle in helices 2a and 2b. We questioned how this residue that lies outside the constraint, yet is close enough to be in a highly helical conformation, responds to the protease. Comparison of the initial velocities of cleavage by trypsin indicated that the HBS α helix 2a is proteolyzed roughly 30-fold slower than the unconstrained Bak peptide analogue 1 (Figure 4). As expected, an increase in the helicity of the constrained peptide results in a further decrease in the initial velocity of cleavage by trypsin. Thus, the HBS helix 2b is roughly twofold more stable than 2a and 60-fold more stable than 1 against proteolysis by trypsin. The proteolytic stability observed for 2a and 2b is similar to that reported for a side-chain cross-linked α helix.[22]
Figure 4.
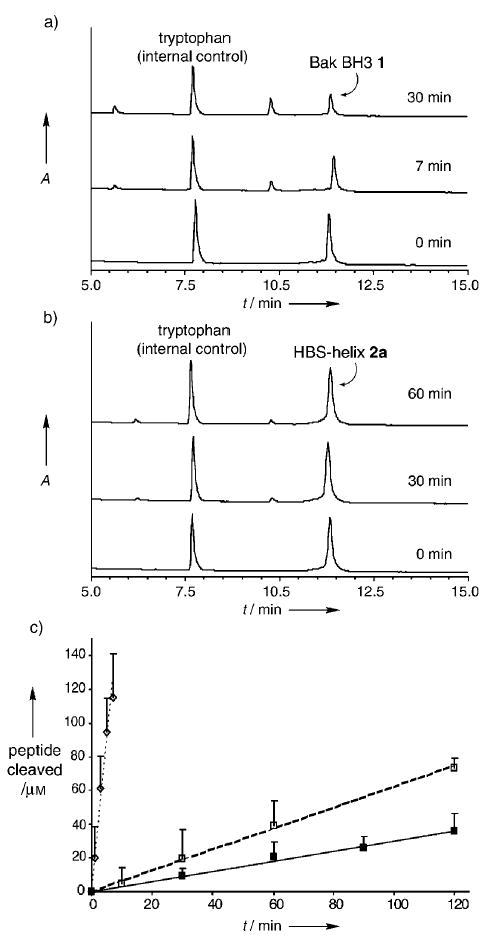
Metabolic stability of HBS α helices. HPLC assay shows rapid proteolysis of a) the unconstrained peptide 1 in the presence of trypsin, whereas b) the HBS α helix 2a degrades at a much slower rate. Tryptophan (500 μm) was used as an internal control for the HPLC studies. c) Comparison of the initial velocities for the proteolysis of Bak BH3 1 (k = 18.0 μm min−1, -----⋄-----), HBS helix 2a (k = 0.60 μm min−1, – – –□– – –), and HBS helix 2b (k = 0.30 μm min−1, —▪—).
In summary, we have demonstrated that artificial α helices prepared by the replacement of a hydrogen bond between the i and i + 4 residues at the N terminus of a short peptide with a carbon–carbon bond can stabilize biologically relevant peptides in helical conformations. These HBS α helices can bind their expected protein receptor with high affinity and resist trypsin-mediated proteolysis. The results are an important step in our efforts to develop rationally designed modulators of protein–protein interactions. These molecules encompass larger surface areas than those displayed by typical small molecules, and may ultimately prove to be more effective in targeting large protein interfaces.
Acknowledgments
[**]We thank Prof. Neville Kallenbach for insightful comments. P.S.A. is grateful for financial support from the NIH (GM073943), the American Chemical Society Petroleum Research Fund, Research Corporation (Cottrell Scholar Award), and the New York State Office of Science, Technology, and Academic Research (James D. Watson investigator). We thank the National Science Foundation for equipment grants for the NMR spectrometer (MRI-0116222) and the capillary LC–ion-trap mass spectrometer (CHE-0234863).
Footnotes
Dedicated to Professor Peter B. Dervan on the occasion of his 60th birthday
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- 1.Walensky LD, Kung AL, Escher I, Malia TJ, Barbuto S, Wright RD, Wagner G, Verdine GL, Korsmeyer SJ. Science. 2004;305:1466–1470. doi: 10.1126/science.1099191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chapman RN, Dimartino G, Arora PS. J Am Chem Soc. 2004;126:12252–12253. doi: 10.1021/ja0466659. [DOI] [PubMed] [Google Scholar]
- 3.Sattler M, Liang H, Nettesheim D, Meadows RP, Harlan JE, Eberstadt M, Yoon HS, Shuker SB, Chang BS, Minn AJ, Thompson CB, Fesik SW. Science. 1997;275:983–986. doi: 10.1126/science.275.5302.983. [DOI] [PubMed] [Google Scholar]
- 4.Cory S, Huang DC, Adams JM. Oncogene. 2003;22:8590–8607. doi: 10.1038/sj.onc.1207102. [DOI] [PubMed] [Google Scholar]
- 5.Letai A, Bassik MC, Walensky LD, Sorcinelli MD, Weiler S, Korsmeyer SJ. Cancer Cell. 2002;2:183–192. doi: 10.1016/s1535-6108(02)00127-7. [DOI] [PubMed] [Google Scholar]
- 6.Rutledge SE, Chin JW, Schepartz A. Curr Opin Chem Biol. 2002;6:479–485. doi: 10.1016/s1367-5931(02)00352-6. [DOI] [PubMed] [Google Scholar]
- 7.Petros AM, Nettesheim DG, Wang Y, Olejniczak ET, Meadows RP, Mack J, Swift K, Matayoshi ED, Zhang H, Thompson CB, Fesik SW. Protein Sci. 2000;9:2528–2534. doi: 10.1110/ps.9.12.2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chin JW, Schepartz A. Angew Chem. 2001;113:3922–3925. Angew. Chem. Int. Ed. 2001, 40, 3806–3809. [Google Scholar]
- 9.Degterev A, Lugovskoy A, Cardone M, Mulley B, Wagner G, Mitchison T, Yuan J. Nat Cell Biol. 2001;3:173–182. doi: 10.1038/35055085. [DOI] [PubMed] [Google Scholar]
- 10.Kutzki O, Park HS, Ernst JT, Orner BP, Yin H, Hamilton AD. J Am Chem Soc. 2002;124:11838–11839. doi: 10.1021/ja026861k. [DOI] [PubMed] [Google Scholar]
- 11.Yang B, Liu D, Huang Z. Bioorg Med Chem Lett. 2004;14:1403–1406. doi: 10.1016/j.bmcl.2003.09.101. [DOI] [PubMed] [Google Scholar]
- 12.Dimartino G, Wang D, Chapman RN, Arora PS. Org Lett. 2005;7:2389–2392. doi: 10.1021/ol0506516. [DOI] [PubMed] [Google Scholar]
- 13.Chen YH, Yang JT, Martinez HM. Biochemistry. 1972;11:4120–4131. doi: 10.1021/bi00772a015. [DOI] [PubMed] [Google Scholar]
- 14.Chakrabartty A, Kortemme T, Baldwin RL. Protein Sci. 1994;3:843–852. doi: 10.1002/pro.5560030514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nelson JW, Kallenbach NR. Biochemistry. 1989;28:5256–5261. doi: 10.1021/bi00438a050. [DOI] [PubMed] [Google Scholar]
- 16.O’Neil KT, DeGrado WF. Science. 1990;250:646–651. doi: 10.1126/science.2237415. [DOI] [PubMed] [Google Scholar]
- 17.Zhang H, Nimmer P, Rosenberg SH, Ng SC, Joseph M. Anal Biochem. 2002;307:70–75. doi: 10.1016/s0003-2697(02)00028-3. [DOI] [PubMed] [Google Scholar]
- 18.Roehrl MH, Wang JY, Wagner G. Biochemistry. 2004;43:16056–16066. doi: 10.1021/bi048233g. [DOI] [PubMed] [Google Scholar]
- 19.Gemperli AC, Rutledge SE, Maranda A, Schepartz A. J Am Chem Soc. 2005;127:1596–1597. doi: 10.1021/ja0441211. [DOI] [PubMed] [Google Scholar]
- 20.Yin H, Lee GI, Park HS, Payne GA, Rodriguez JM, Sebti SM, Hamilton AD. Angew Chem. 2005;117:2764–2767. doi: 10.1002/anie.200462316. Angew. Chem. Int. Ed. 2005, 44, 2704–2707. [DOI] [PubMed] [Google Scholar]
- 21.Tyndall JD, Nall T, Fairlie DP. Chem Rev. 2005;105:973–999. doi: 10.1021/cr040669e. [DOI] [PubMed] [Google Scholar]
- 22.Schafmeister CE, Po J, Verdine GL. J Am Chem Soc. 2000;122:5891–5892. [Google Scholar]
- 23.Shepherd NE, Hoang HN, Abbenante G, Fairlie DP. J Am Chem Soc. 2005;127:2974–2983. doi: 10.1021/ja0456003. [DOI] [PubMed] [Google Scholar]