

Abstract
Two genetically distinct retroviral RNAs can be co-packaged if the RNAs are co-expressed in virion producing cells. For Moloney murine leukemia virus (MLV), co-packaged RNAs are not randomly selected from among all packaging-competent RNAs, but instead primarily associate as homodimers. Here, we tested the hypothesis that the distance between proviral templates might hinder RNA heterodimerization, thus generating the observed preferential homodimerization of co-expressed MLV RNAs. To do this, two genetically distinct RNAs were co-expressed from a single locus and the proportions of hetero- and homodimeric virion RNAs were determined. Unlike RNAs transcribed from two different templates, RNAs transcribed from a single locus dimerized at random. Additionally, in vitro transcription experiments suggested that MLV RNA dimerization can occur more efficiently for longer RNAs during transcription than post-synthesis. Together, these findings show that MLV RNA dimer-partner selection likely occurs either co-transcriptionally or within a pool of transcripts near the proviral template.
Keywords: Retrovirus, RNA packaging, RNA trafficking
Introduction
Two copies of full-length retroviral RNA are co-packaged in a single retroviral particle. Electron microscopy first revealed that these RNAs are linked as a dimer near their 5′ ends in a region termed the dimer linkage site (DLS) (Bender et al., 1978; Bender and Davidson, 1976; Dube et al., 1976; Gonda et al., 1980; Kung et al., 1976; Maisel et al., 1978; Murti et al., 1981). Because the DLS overlaps sequences required for efficient encapsidation of genomic RNA (designated Ψ), studying RNA dimerization in replicating virus is challenging. Therefore, much of what is known about dimerization comes from examining short transcripts in vitro. Under the appropriate thermal and ionic conditions, retrovirus-derived RNAs are able to dimerize spontaneously (Darlix et al., 1990; Marquet et al., 1991; Roy et al., 1990), and such studies have mapped the Moloney murine leukemia virus (MLV) DLS to nucleotides 215 through 420 (Prats et al., 1990).
Although the sequences required for dimerization differ among retroviruses, features of the RNAs’ structures appear to be conserved. Dimerization of MLV genomic RNA is thought to occur via a ‘‘kissing’’ complex formed between palindromic loops within the DLS on either RNA. The loop of one RNA monomer interacts via Watson–Crick base pairing with that of its dimer partner. Formation of this complex induces the melting of local secondary structure and allows additional stabilizing interactions between the RNA strands (Girard et al., 1995).
In vitro studies have addressed the mechanism of dimerization, but the cellular location and timing of viral RNA dimerization are unclear and may differ among retroviruses. Avian retroviruses may initially package two monomeric RNAs (Lear et al., 1995; Ortiz-Conde and Hughes, 1999), but it is generally believed that at least a commitment to RNA dimerization occurs prior to budding for MLV. Isolation of RNA from newly budded MLV virions yields dimers (Fu et al., 1994; Fu and Rein, 1993), and virion-associated genomic RNA is dimeric, even under conditions where packagable RNA levels within infected cells are low and the average RNA copy number per virion is less than one (Hibbert et al., 2004; Levin et al., 1974; Meric and Goff, 1989). Studies aimed at monitoring intracellular dimerization suggest that Ψ+ RNAs associate in a conformation similar to the packaged RNA dimer within the cell prior to encapsidation (Paillart et al., 2004; Pal et al., 1998).
Although co-packaged RNAs are generally identical, genetically distinct RNAs can be co-packaged in virions produced by dually infected cells. This has important ramifications for retroviral recombination, which requires RNA co-packaging (Hu and Temin, 1990), and thus genetic markers should reassort more frequently for viruses that readily co-package two different RNAs than for viruses that tend to encapsidate a single RNA species.
It has been suggested that co-expressed RNAs associate at random, leading to ratios of homo- to heterodimeric RNAs within budding virions that are predictable based on the co-expressed RNAs’ levels of expression (Hu and Temin, 1990). While this assumption appears valid for human immunodeficiency virus (HIV), MLV RNAs have been shown to preferentially homodimerize, resulting in a virus population containing less than half the heterodimers expected from random RNA dimerization (Flynn et al., 2004). Co-expressed MLV RNAs with nearly identical sequences self-associate to the same extent as RNAs that are similar only in their packaging regions, suggesting that homology sensing is not the mechanism of biased RNA homodimerization (Flynn et al., 2004). One possibility that has been raised for nonrandom RNA dimerization is that RNAs generated from separate proviruses might exit the nucleus at separate sites (Hu et al., 1997).
Here, we tested the hypothesis that MLV RNAs commit to dimerization at or near their site of transcription. In contrast to RNAs produced from two separate loci, genetically distinct MLV RNAs dimerized randomly when transcribed from a single template. In vitro transcription experiments suggested that relatively short, incompletely synthesized MLV RNAs may be in a more suitable conformation for dimerization than completed full-length viral RNAs. Together, these data suggest that MLV RNA dimer partner selection occurs early in the replication cycle, near the site of transcription.
Results
Examining heterodimer frequency for two different RNAs transcribed from a single template
In previous experiments that demonstrated preferential homodimerization (Flynn et al., 2004), RNAs were templated either by co-transfected vector expression plasmids or by proviruses integrated at separate sites within the host cell’s chromosomes. One hypothesis that could explain the observed self-association of MLV genomes is that RNAs may commit to dimerization at or near the site of transcription, with transcripts produced from spatially distant loci less capable of forming dimer associations. A prediction of this hypothesis is that RNAs produced from the same template should associate randomly, regardless of their extents of identity outside cis-acting dimerization regions.
To test this prediction, RNA homo- and heterodimer frequencies for two genetically distinct RNAs produced from a single template were compared to frequencies of dimerization when the same two RNAs were each expressed from separate loci. These experiments relied on a specialized MLV-based vector construct, pMΨ Puro-pA-OBD, which contained sequences termed OBD for oligonucleotide binding domain, cloned downstream of the 3′ long terminal repeat (LTR), followed by a strong polyadenylation signal from SV40 (Fig. 1A). Retroviral polyadenylation signals are naturally leaky, and as many as 15% of retroviral transcripts are polyadenylated at sites downstream of the canonical AAUAA found within retroviral LTRs (An and Telesnitsky, 2004; Bohnlein et al., 1989; DeZazzo et al., 1991; Herman and Coffin, 1986; Zaiss et al., 2002). Incomplete processing of RNAs templated by pMΨ Puro-pA-OBD would produce a read-through RNA with the OBD tag near its 3′ end, while processing and polyadenylation at the LTR would result in a shorter, genetically distinct RNA species lacking the OBD (Fig. 1A, SV40 pA RNA and LTR pA RNA, respectively).
Fig. 1.
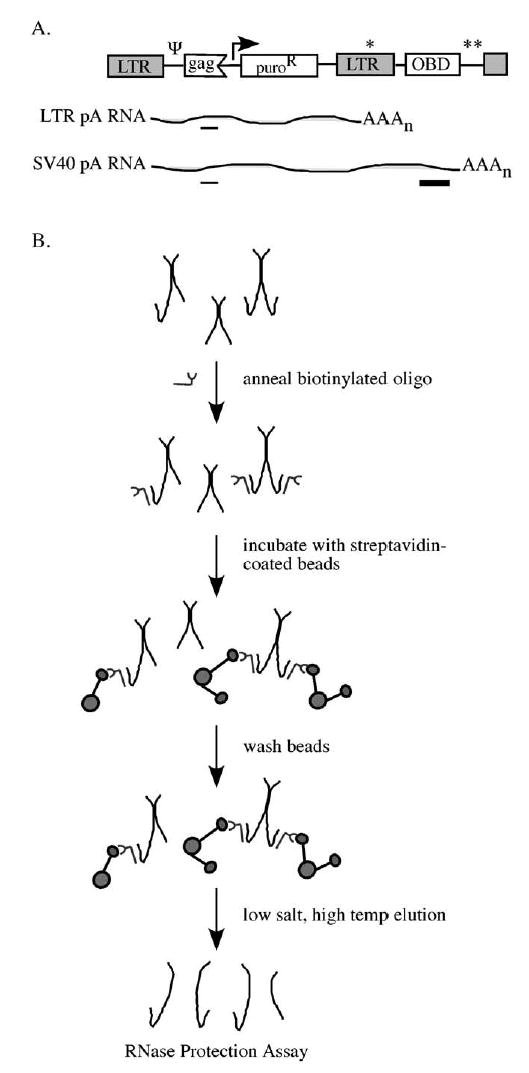
MΨ Puro-pA-OBD RNA capture assay. (A) Schematic representation of the plasmid transfected into 293T-based packaging cells for virus production and RNAs transcribed from this vector (not to scale). Single and double asterisks represent LTR pA and SV40 pA sites, respectively. Thin and thick lines beneath the RNAs depict RNase protection assay gag and OBD protected fragments, respectively. Ψ, packaging signal; OBD, RNA capture assay oligonucleotide binding domain. (B) Schematic overview of the RNA capture assay. Longer hooked RNAs represent OBD-containing read-through RNAs that can bind to the biotinylated oligonucleotide. Shorter RNAs represent LTR polyadenylated RNAs that lack the OBD tag. Oligonucleotide-bound dimers are captured on streptavidin-coated beads and analyzed by RNase protection assay.
Based on the modest levels of polyadenylation signal read-through reported for other retroviruses, the expectation was that among this vector’s RNAs, those polyadenylated at the LTR would be produced and packaged into virions in large excess over OBD-containing read-through RNAs. To test this, 293T-based packaging cells were transfected with pMΨ Puro-pA-OBD, virions were harvested from the media, and virion RNA was isolated under conditions that maintained dimer linkages. RNA content was subsequently analyzed by RNase protection assay using a riboprobe which protected a fragment of gag sequences present on both RNAs transcribed from pMΨ Puro-pA-OBD as well as a longer OBD-specific fragment present only on read-through RNAs (Fig. 1A). Consistent with expectations, the results indicated that the majority of MLV RNAs transcribed from pMΨ Puro-pA-OBD were polyadenylated at the LTR site, yielding the shorter transcripts 94 ± 0.5% of the time (Fig. 2, lane 1 and data not shown). Thus, the shorter RNAs were present at a 16-fold molar excess over the longer OBD-containing RNAs.
Fig. 2.
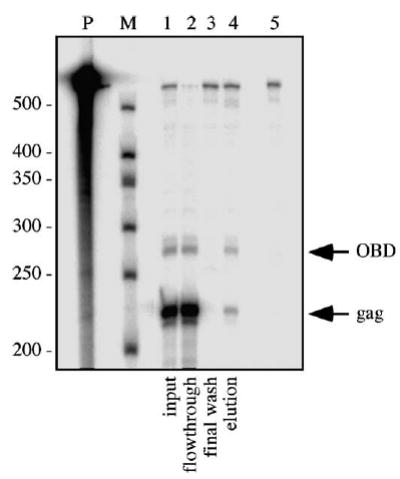
RNase protection assay of single vector RNA capture assay products. Virion RNA isolated from the medium of packaging cells transfected with pMΨ Puro-pA-OBD was analyzed by the RNA capture assay. RNase protection assay of input, flow-through, final wash, and elution (lanes 1– 4, respectively) is illustrated. Lane 5 shows a no sample digested riboprobe control. The migration of OBD and gag-specific protected fragments is indicated at the right. Note that the OBD protected band contains 1.9-fold more radioactive nucleotides; thus, a measured 2:1 gag:OBD molar ratio is represented visually as approximately 1:1. P, undigested riboprobe; M, RNA markers.
Since the OBD-containing RNA represented only a small percent of the total virion RNA population, random dimerization as modeled by the Hardy–Weinberg equation (Hu and Temin, 1990) predicts that almost all of the OBD-containing RNAs will be linked in heterodimers with LTR polyadenylated RNAs. Thus, if dimerization was random, the two RNAs would be present in nearly equal molar proportions in the RNA capture elution fraction. Because both RNA species contain gag sequences but the OBD is found only on read-through RNAs, molar equivalents of LTR polyadenylated and read-through RNAs would yield an expected gag:OBD ratio of 2:1. Alternately, an excess of OBD-containing RNA in the elution would be indicative of preferential homodimerization.
To examine whether or not two RNAs from a single transcription unit dimerized at random, the heterodimer frequency of the two RNA species transcribed from pMΨ Puro-pA-OBD was analyzed using an RNA capture assay (Flynn et al., 2004) (Fig. 1B). First, a 3′-biotinylated oligonucleotide complementary to sequences within the OBD tag was incubated with viral RNA dimers. The oligonucleotide-bound RNAs, as well as their dimer partners, were then captured on streptavidin-coated beads and analyzed by RNase protection assay (Fig. 2). The input sample (lane 1) indicated the proportion of LTR polyadenylated and OBD-containing RNAs in the viral RNA population. The flow-through sample (lane 2) represented RNA not captured by the streptavidin beads, and the elution sample (lane 4) consisted of those RNAs retained by the interaction between streptavidin and the biotinylated oligonucleotide. The absence of RNA in the final wash (lane 3) demonstrated that all detectable nonspecifically bound RNAs had been removed before elution of retained RNAs.
The ratio of the smaller gag band to the larger OBD-specific band in the elution fraction was 2.3:1 in the sample assay here (Fig. 2, lane 4), consistent with values predicted for random dimerization. In several repetitions of this experiment, the average gag:OBD ratio was 2.1:1.
Examining heterodimer frequency of RNAs transcribed from two templates
RNA capture experiments were also performed to test whether or not the two RNA transcripts generated in the above experiment preferentially self-associated when co-expressed from two distinct vectors, as predicted by previous findings (Flynn et al., 2004). The plasmid used to express an RNA identical to the LTR polyadenylated RNA of pMΨ Puro-pA-OBD was pMΨ Puro, which lacks the OBD segment and SV40 pA (Fig. 3A). To generate constitutively expressed OBD-tagged RNA, the pMΨ Puro-ΔpA-OBD vector was constructed (Fig. 3A), which contained two mutations in the canonical LTR AAUAA, but was otherwise identical to pMΨ Puro-pA-OBD.
Fig. 3.
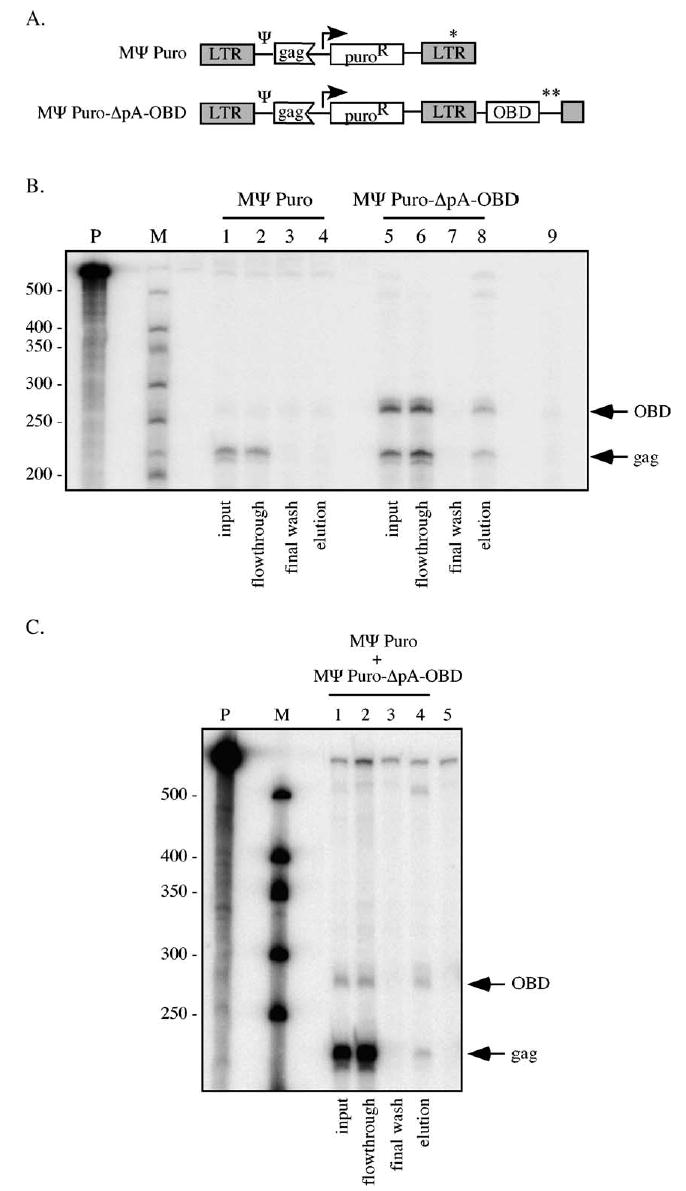
RNA capture assay of RNAs transcribed from two vectors. (A) Schematic representation of vectors used to transfect packaging cells for virus production. pMΨ Puro templates transcription of a 2.7 kb RNA identical to the LTR polyadenylated transcript of MΨ Puro-pA-OBD, and lacks sequences complimentary to the biotinylated oligonucleotide used in the RNA capture assays. pMΨ Puro-ΔpA-OBD contains two base substitutions in the 3′ LTR which decreases use of the viral pA site, and causes most RNAs transcribed from this vector to incorporate sequences complimentary to the biotinylated oligonucleotide. Single and double asterisks represent LTR pA and SV40 pA sites, respectively. Ψ, packaging signal; OBD, RNA capture assay oligonucleotide binding domain. (B) RNA isolated from virions produced by packaging cells transfected with pMΨ Puro (lanes 1 –4) or pMΨ Puro-ΔpA-OBD (lanes 5–8) was analyzed by RNA capture assay. Products of input samples (lanes 1 and 5), flow-through samples (lanes 2 and 6), final washes (lanes 3 and 7), and elutions (lanes 4 and 8) are shown. Lane 9 represents a digested riboprobe control. The migration of OBD and gag-specific protected fragments is indicated at the right. P, undigested riboprobe; M, RNA marker. (C) MΨ Puro/MΨ Puro-ΔpA-OBD RNA capture assay. Viral RNA isolated from the media of cells transfected with a 1:20 ratio of pMΨ Puro:pMΨ Puro-ΔpA-OBD was subjected to the RNA capture assay. RNase protection assay illustrates input, flow-through, final wash, and elution samples (lanes 1–4, respectively). Lane 5 is a digested riboprobe alone control. The migration of OBD and gag-specific protected fragments are indicated at the right. P, undigested riboprobe; M, RNA marker.
As a control to ensure that LTR polyadenylated RNA would not be retained via nonspecific binding to the streptavidin-coated beads in the absence of OBD RNA, LTR polyadenylated RNA was expressed alone in packaging cells from the pMΨ Puro vector. RNA purified from these virions was subjected to the RNA capture assay. In the absence of OBD-containing RNA, LTR polyadenylated RNA was undetectable in the elution fraction (Fig. 3B, lane 4).
To confirm that the polyadenylation site mutations in pMΨ Puro-ΔpA-OBD resulted in increased read-through RNA, viral RNA harvested from pMΨ Puro-ΔpA-OBD transfected cell media was analyzed by RNase protection assay (Fig. 3B). The results indicated that LTR polyadenylation was largely disrupted, with an average of 79 ± 1% of the packaged RNAs from this template reading through the LTR and containing the OBD tag (Fig. 3B, lane 5 and data not shown).
Virion RNA from packaging cells transfected with pMΨ Puro-ΔpA-OBD alone was subjected to the RNA capture assay (Fig. 3B, lanes 5–8). Since LTR-terminated RNAs were a small minority of the total virion RNA and only OBD-containing RNAs should be captured, most RNAs should be OBD-containing homodimers. Thus, the expected ratio of gag and OBD protected fragments in the elution was approximately 1:1. Consistent with this expectation, the gag:OBD elution ratio averaged 1.0:1 (Fig. 3B, lane 8).
The dimerization of these two RNAs co-expressed from separate plasmids was then examined. 293T-derived packaging cells were co-transfected with pMΨ Puro-ΔpA-OBD (which templates the longer OBD-containing RNA) and pMΨ Puro (which lacks the OBD and generates LTR polyadenylated RNAs) at a 1:20 molar ratio to mimic the levels of OBD-containing and LTR polyadenylated RNAs produced by cells transfected with pMΨ Puro-pA-OBD alone. Virus was harvested from the transfected cell media and viral RNA dimers were analyzed by RNA capture (Fig. 3C).
As represented in the autoradiogram shown in Fig. 3C, 3% of the total packaged RNA that resulted from this plasmid co-transfection was the longer OBD-containing RNA (Fig. 3C, lane 1). In contrast to what was observed in the single vector experiments, the gag:OBD elution ratio for the two vector experiments was 1.5:1 (Fig. 3, lane 4), indicative of biased homodimerization. This value was consistent with previous findings for RNAs co-expressed from two distinct templates (Flynn et al., 2004), and indicated that the packaged RNA population contained less than half the heterodimers expected from random dimerization.
The data from single vector and two vector experiments are summarized in Fig. 4. When RNAs were transcribed from a single locus, the captured RNA ratio averaged 2.1:1 (Fig. 4, MΨ Puro-pA-OBD). However, when the same RNAs were transcribed from distinct vectors, the average elution ratio fell to 1.5:1 (Fig. 4, MΨ Puro-ΔpA-OBD/MΨ Puro). This difference was statistically significant, with a P value of less than 0.01, and suggested that generating MLV transcripts from a single locus facilitated random dimerization. When OBD-containing RNA was generated in the absence of the MΨ Puro RNA, the elution ratio was 1.0:1. This value was significantly different from both the MΨ Puro-pA-OBD ( P value < 0.01) and the MΨ Puro-ΔpA-OBD/MΨ Puro ( P value < 0.05) elution ratios and confirmed the assay sensitivity.
Fig. 4.
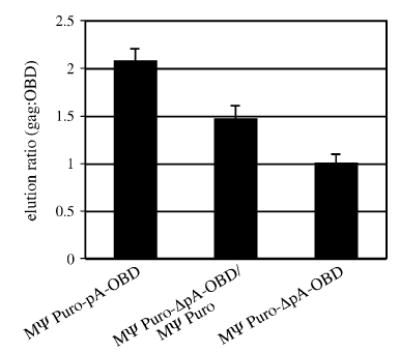
Summary of dimerization frequencies determined by RNA capture assay data. Bars depict the measured ratio of gag to OBD-specific fragments in RNA capture assay elution lanes. As explained in the text, 2 is the value predicted by random dimerization while 1.5 suggests biased homodimerization of the magnitude previously reported for MLV (Flynn et al., 2004). Standard errors are indicated. MΨ Puro-pA-OBD data represent nine experimental repetitions. Data for MΨ Puro-ΔpA-OBD/MΨ Puro are from five experiments, and data for MΨ Puro-ΔpA-OBD are from 3 experiments.
Visualizing RNA dimers from in vitro transcription reactions
Dimerization of short in vitro transcribed retroviral RNAs has been well characterized. However, in vitro dimerization of full-length genomic RNAs reportedly is not achievable (Pal et al., 1998). Conceivably, the most stable fold of a full-length viral RNA may not contain the DLS in a conformation conducive to initiating dimerization whereas the fold of a transcription intermediate, or possibly an RNA remodeled by the nucleocapsid protein, could dimerize. To address the hypothesis that viral RNAs may be able to dimerize before they are completely transcribed, we examined the extent to which RNAs of varying lengths, generated by T7 phage RNA polymerase in in vitro transcription reactions, formed dimer pairs. Templates for transcripts containing authentic viral 5′ ends and various 3′ ends were generated using PCR to fuse a T7 promoter to 400, 800, 1000, or 1200 nucleotides of MLV sequence. RNAs were radiolabeled during the transcription reactions. Aliquots of newly synthesized RNA were either left untreated, denatured by heating to 95 °C, or first denatured and then subjected to conditions previously determined to promote dimerization in vitro (Laughrea and Jette, 1997 and data not shown). The RNA samples were separated on native agarose gels, and the extent of dimerization was quantified by phosphorimager analysis. To maintain weaker dimer linkages, electrophoresis buffer included MgCl2. Parallel gels run in buffers that lacked divalent cations showed similar trends, although overall extents of dimerization were decreased (data not shown).
A representative gel is illustrated in Fig. 5A, and quantification of RNA dimerization for the various length transcripts is presented in Fig. 5B. As seen in lanes 1, 4, 7, and 10 of Fig. 5A, reduced mobility RNA dimers were readily detectable in the untreated samples. The 800, 1000, and 1200 nt RNAs generated during transcription dimerized to similar extents with approximately 25% of the initial transcripts migrating as dimers (Fig. 5B). In contrast, more than 60% of the untreated 400 nt transcripts were dimerized (Fig. 5A, lane 1 and Fig. 5B). As expected, dimers dissociated to monomers upon incubation at 95 °C (Fig. 5A, lanes 2, 5, 8, and 11). To confirm that band shifts represented MLV-specific monomer to dimer transitions, transcripts of the same lengths but derived from lacZ were generated and run in parallel. Unlike the MLV RNAs, these lacZ RNAs did not produce the secondary bands that were designated MLV RNA dimers (data not shown).
Fig. 5.
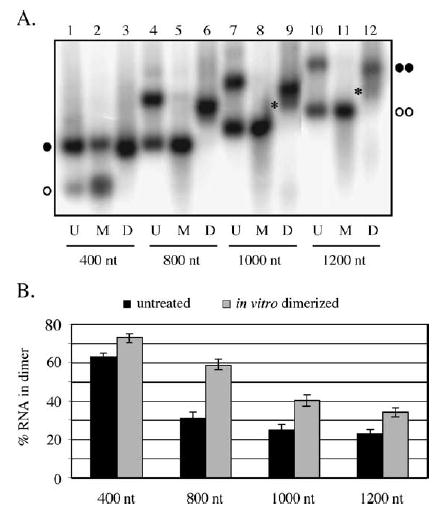
In vitro transcription of MLV RNAs (A) T7 RNA polymerase-synthesized MLV RNA transcripts. The various length transcripts all contained the authentic viral 5′ end but differed in 3′ truncation site. Lanes designated U contain untreated transcription reaction products, those indicated M display samples that were incubated at 95 °C to generate monomeric RNAs. D lanes show the products of in vitro dimerization reactions, performed as described in Materials and methods. Migration of 400 and 1200 nt monomers is indicated by single and double open circles, respectively, and migration of 400 and 1200 nt dimers is indicated by single and double filled circles, respectively. Asterisks represent migration of RNAs with intermediate mobilities. (B) Summary of in vitro transcription product dimerization from two independent experiments. Black bars, untreated samples; gray bars, in vitro dimerized samples. Error bars depict standard error.
In contrast to the fairly uniform extent of dimerization observed for most of the untreated transcription reaction samples (black bars in Fig. 5B), the extent of dimerization observed after denatured RNAs was subjected to in vitro dimerization conditions appeared to decrease as the length of the transcript increased (grey bars in Fig. 5B). While 70% of the 400 nt transcripts were induced to dimerize after denaturation, only approximately 30% of the 1200 nt RNAs migrated more slowly than the monomer after exposure to dimerization conditions (compare Fig. 5A, lanes 3 and 12 and data in Fig. 5B). Interestingly, a significant portion of the slower migrating 1000 and 1200 nt RNAs was of an intermediate mobility (Fig. 5A, lanes 9 and 12 indicated by asterisks) suggesting that the RNAs may have adopted alternate conformations or folded via RNA–RNA tethering interactions outside the dimer linkage region (Hibbert and Rein, 2005).
The data above suggest that the longer RNAs, once unfolded, were less able to adopt the tertiary structures required for stable dimer formation, and thus, may be restricted in their ability to dimerize post-synthesis. To test this hypothesis, the ability of two different-length RNAs to associate in heterodimers either co-transcriptionally or after synthesis was examined. For these experiments, 800 and 1200 nt RNAs were either generated in separate reactions as above then mixed or were co-transcribed by including the templates for the 800 and 1200 nt RNAs in a single transcription reaction mixture. When the RNAs were co-transcribed and separated on native agarose gels, as seen in lane 4 of Fig. 6A, a distinct RNA species was visible. This RNA, which migrated between the 800 and 1200 nt dimers, was presumed to be an RNA–RNA heterodimer. In addition to associating as heterodimers, a portion of the co-transcribed RNAs was monomeric, while the remaining RNAs appeared to homodimerize. Note that in the representative experiment illustrated in Fig. 6A the template ratios were skewed such that an excess of the 400 nt RNA was generated in relation to the 1200 nt RNA, and thus, the 1200 nt RNA dimer was not readily detectable. However, when the 1200 nt RNA dimer was produced in excess of the 400 nt RNA, 1200 nt RNA homodimers were visible (data not shown).
Fig. 6.

Examining RNA heterodimer formation during in vitro transcription. (A) Heterodimers are generated in two template transcription reactions. Templates for the production of 800 and 1200 nt RNAs were mixed prior to transcription and products were separated on native agarose (lane 4). The migration of homodimers, heterodimers, and monomers is indicated. Lanes designated U contain untreated transcription reaction products, those indicated M display samples that were incubated at 95 °C to generate monomeric RNAs. D lanes show the products of in vitro dimerization reactions, performed as described in Materials and methods. (B) MLV RNA heterodimer formation is limited post-synthesis. 800 and 1200 nt RNAs were generated in vitro, mixed, and left untreated (lane 2), heated to 95 °C to denature (lane 3), or incubated at 37 °C in transcription buffer (lane 1). The migration of homo- and heterodimers is indicated.
To examine the ability of these RNAs to form heterodimers post-transcription, full-length 800 and 1200 nt RNAs were mixed after synthesis and incubated together under transcription reaction conditions. In contrast to the co-transcribed RNAs that more readily associated as heterodimers, only a small proportion of the mixed RNAs migrated between the 800 and 1200 nt RNA dimers (Fig. 6B, lane 1). This suggests that heterodimer formation after the completion of transcription, although not completely inhibited, was limited.
Discussion
Here, homo- and heterodimer frequencies for two genetically distinct MLV RNAs, co-expressed either from a single vector or from two distinct templates, were compared using RNA capture assays. When templated by a single DNA, MLV RNAs dimerized in random proportions. In contrast, and consistent with previous findings (Flynn et al., 2004), the same two RNAs were observed to homodimerize preferentially when produced from separate loci, generating less than half the heterodimers expected if dimerization was random. This nonrandom dimerization of RNAs expressed from separate locations, which reflects the situation during natural co-infection, contributes to the marked differences in genetic marker segregation observed for HIV-1 and MLV (Onafuwa et al., 2003).
Dimerization was also examined for 3′ truncated MLV RNAs that were generated in vitro by T7 phage RNA polymerase. RNA dimers were readily detectable in aliquots taken directly from in vitro transcription reaction mixtures. Approximately 60% of the shorter 400 nt RNAs associated in dimers, and all transcripts between 800 and 1200 nt dimerized to a similar extent with roughly 25% of the RNA migrating as a dimer. When these same RNAs were denatured and then subjected to standard in vitro dimerization conditions, longer RNAs reassociated less well than shorter transcripts, and the percentage of RNA that migrated as dimers decreased as the template length increased. This may suggest that the longer RNAs, once unfolded, were less able to adopt the tertiary structures required for stable dimer association. Consistent with this model, when full-length RNAs were incubated together post-synthesis, only a small amount of heterodimers was generated. In contrast, the increased level of dimerization observed for the 400 nt transcripts may imply that these RNAs could dimerize co-transcriptionally and also maintained a dimerization-competent conformation post-synthesis. Together, these data are consistent with a model in which an alternate RNA structure that promotes dimerization is prevalent or more stable in shorter RNAs or RNA intermediates that exist before transcription is completed than in longer MLV RNAs.
In additional in vitro work presented here, two different-length RNAs were co-transcribed from separate templates in a single reaction. Under these conditions, RNA heterodimers were detectable in transcription reaction products. This suggests that there may be no intrinsic block to the heterodimerization of RNAs generated from two distinct templates but that the proximity of the template DNAs during transcription may promote heterodimer formation. These data support the hypothesis that MLV RNA dimer partners are determined at or near the site of transcription, and that RNAs produced in separated locations are less available to form random associations.
The work here analyzed RNAs directly, and complements genetic studies that Kharytonchyk et al. (2005) published while this work was in progress. In the Kharytonchyk study, RNA co-packaging was inferred by monitoring MLV recombination rates, and it was concluded that tandemly integrated DNAs yielded co-packaged MLV RNAs more frequently than did spatially distant templates. The observed magnitudes of effects in our cell-based assays and in the Kharytonchyk studies were similar, and thus the findings here suggest that reported effects of transcription template proximities on recombination frequencies were mediated by differences in RNA dimerization.
Functional consequences for alternate RNA structures, such as those postulated above, have been documented for several retroviruses (D’Souza and Summers, 2004; Dirac et al., 2002; Huthoff and Berkhout, 2001). In vitro experiments suggest that HIV-1 transcripts alternate between two conformers, one which is more thermodynamically stable but incapable of dimer formation and a second which exposes motifs important for initiation of dimerization (Huthoff and Berkhout, 2001). This change in RNA structure appears to be mediated by the nucleocapsid domain of the Gag polyprotein and may serve as a regulatory switch between the RNA’s mRNA and genome roles (Huthoff and Berkhout, 2001). A similar mechanism appears to regulate HIV-2 dimerization (Dirac et al., 2002). For MLV, on the other hand, changes in RNA structure induced upon dimerization expose nucleocapsid domain binding sites which are absent in the RNA monomers and thus may promote specific packaging of genome dimers (D’Souza and Summers, 2004; Rein, 2004).
Whether or not the dimer associations these results describe occur during transcription or immediately following is not clear; however, the results presented here suggest that some portion of MLV RNA is likely transported from the nucleus to sites of assembly as a dimer. The possibility of RNA dimerization prior to nuclear exit has important implications for retroviral trafficking and assembly. Full-length viral RNAs serve both as genomes and as the mRNAs for Gag and Gag-Pol polyproteins. Although MLV genomes and mRNAs are identical in sequence, they appear to reside in separate, nonequilibrating, pools within the cytoplasm of infected cells (Dorman and Lever, 2000; Levin et al., 1974; Levin and Rosenak, 1976; Messer et al., 1981). How full-length RNAs are sorted into one functional pool or the other is unknown. A possible model, based on the suggestion that RNAs may dimerize in the nucleus, is that full-length RNA monomers and dimers may associate with different host factors, thereby shuttling them to their fate as either an mRNA or genomic RNA, respectively.
Materials and methods
Plasmids
pMΨ Puro was an MLV-based retroviral vector containing cis-acting sequences required for vector RNA dimerization and packaging, as well as a puromycin resistance gene driven by a simian virus 40 (SV40) early promoter (Kulpa et al., 1997). pMΨ Puro-pA-OBD, a derivative of pMΨ Puro, contained 290 bp of MLV pol sequence (MLV RNA nucleotides 2683 to 2973) followed by an SV40 pA site cloned downstream of the pMΨ Puro 3′ long terminal repeat (LTR). The inserted 290 bp of MLV sequences, designated OBD for oligonucleotide binding domain, annealed to the biotinylated oligonucleotide used in the RNA capture assay described below. pMΨ Puro-ΔpA-OBD was identical to pMΨ Puro-pA-OBD except for two nucleotide substitutions in the 3′ LTR pA signal (AAUAAA to AAGGAA, nucleotide substitutions are in bold type). Cloning details are available upon request.
To construct the riboprobe template used for the RNA capture assay, PCR was used to fuse 230 nt of MLV gag sequence (MLV RNA nucleotides 784 to 1013) to 284 nt of MLV pol sequences contained in the OBD fragment (MLV RNA nucleotides 2689 to 2972). The amplified sequences were then blunt-end cloned into the pBluescript II SK(+) (Stratagene) EcoRV site to create pJF 1404-1.
Virus production
For virus production, 80% confluent φNXE cells (a 293T-derived MLV-based packaging cell line) (Pear et al., 1993) were transiently transfected or co-transfected with the vector plasmids described for each experiment. Transfections were performed in 6-cm dishes using a total of 4 μg of plasmid DNA and 6 μl of FuGENE 6 reagent (Roche) per manufacturer’s instructions. Culture medium (Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum (Gemini)), was replaced 24 h post-transfection, and a total of 9 ml of virus-containing medium per 6-cm dish was harvested at 36, 48, and 60 h post-transfection. Residual cells were removed by filtration through 0.2 μm filters, and virus-containing medium was stored at −70 °C prior to use.
Isolation of dimeric viral RNA
Approximately 90 ml of virus-containing medium was centrifuged at 115,000 × g for 2 h at 4 °C. Pelleted virus was suspended in 400 μl of lysis buffer (50 mM Tris (pH 7.5), 10 mM EDTA, 1% sodium dodecyl sulfate (SDS), 100 mM NaCl, 50 μg of tRNA/ml, 100 μg of proteinase K/ml) and incubated at 37 °C for 30 min, extracted with an equal volume of phenol–chloroform–isoamyl alcohol (25:24:1 (vol/vol/vol)), and ethanol precipitated. Precipitated RNA was then suspended in 100 μl of DNase mix (25 mM Tris–Cl (pH 8), 50 mM MgCl2, 5 mM dithiothreitol, 100 U of RNase-free DNase (Roche)/ml, 400 U of RNasin (Promega)/ml) and incubated at 37 °C for 15 min. DNase treatment was stopped by addition of 25 μl of 50 mM EDTA, 1.5 M sodium acetate (pH 5.2), 1% SDS. Samples were phenol–chloroform–isoamyl alcohol (25:24:1 (vol/vol/vol)) extracted, ethanol precipitated, and resuspended in 15 μl of 10 mM Tris, 1 mM EDTA, 100 mM NaCl, 1% SDS.
RNA capture assay
The RNA capture assay protocol was adapted from that described previously (Flynn et al., 2004). Viral RNA isolated from 30 ml of transfected cell medium was combined with 200 pmol of a 3′-biotinylated anti-MLV pol oligonucleotide (5′-CTCTGTATGTGGGGCTTG-3′), incubated in 2 × SSC at 42 °C for 15 min (100 μl total volume), and cooled to 25 °C over 30 min. Cooled oligonucleotide-bound RNA was added to streptavidin-coated beads (Promega; suspended in 100 μl of 0.5 × SSC) and mixed gently for 10 min at room temperature. Beads were captured using a magnetic stand and the supernatant (flow-through) was transferred to a new tube. Beads were washed four times with 300 μl of 0.5 × SSC to remove residual unbound RNA, and supernatant from the final wash was retained (final wash) for analysis. To elute bound RNA, beads were resuspended in 100 μl of RNase-free water and incubated at 85 °C for 5 min. This eluate was combined with that from a second 150 μl elution (elution). To generate the input sample, a separate aliquot of viral RNA isolated from approximately 10 ml of transfected cell medium was diluted to a final volume of 30 μl in RNase-free water. All samples –input, flow-through, final wash, and elution–were ethanol precipitated with 15 μg of carrier tRNA and resuspended with a >10-fold molar excess of riboprobe.
To generate the riboprobe, pJF 1404-1 was linearized with EcoRI and transcribed using T7 RNA polymerase (Promega) to create a 580 nt runoff transcript. The riboprobe was radio-labeled by including (α-32P) CTP (Perkin-Elmer) in the reaction mixture and was resuspended in 400 μl of hybridization buffer (80% formamide, 40 mM piperazine-N,N′-bis(2-ethanesulfonic acid) (pH 6.4), 400 mM NaCl, 1 mM EDTA).
For RNase protection assays, samples were incubated in 10 μl of probe-containing hybridization buffer, digested with RNases A and T1, and separated on 5% acrylamide-8 M urea gels by standard techniques. Dried gels were exposed to phosphorimager screens (Molecular Dynamics) for quantitative analysis using ImageQuant software. To deduce molar ratios, phosphorimager values were normalized for the number of radiolabeled C residues in each protected fragment. A two-sample t test assuming unequal variances was used for statistical analyses presented in Fig. 4.
In vitro RNA transcription and dimerization
DNA templates for T7 RNA polymerase-directed in vitro transcription of various length MLV- and lacZ-derived RNAs were generated by PCR using the wild-type MLV proviral clone pNCA (Colicelli and Goff, 1988) or the MLV vector pLacPuro (Pfeiffer et al., 1999), respectively, as the template. RNAs transcribed from these templates initiated at the same nucleotide as native viral transcripts but differed in 3′ truncation site.
T7NCA (5′-GAA TTC GGC CAG TGA ATT GTA ATA CGA CTC ACT ATA GGC GCG CCA GTC CTC CGA-3′) was the sense primer for all pNCA templated PCRs, and T7LacZ (5′-GAA TTC GGC CAG TGA ATT GTA ATA CGA CTC ACT ATA GGC GCA GCC TGA ATG GCG-3′) was the sense primer for all pLacPuro templated reactions. Approximate template lengths and corresponding antisense primers were as follows:
JF67: 5′-CCACAAAAACGGCCCC-3′(400 bp, MLV)
JF68: 5′-GAGGTCTCGGTTAAAGGTGC-3′ (800 bp, MLV)
JF69: 5′-GAGGCGGGGTCGAACG-3′ (1000 bp, MLV)
JF70: 5′-TTGGGGAGGGGTCCG-3′ (1200 bp, MLV)
AntiLac2: 5′ -GCGAGGCGGTTTTCTCC-3′ (400 bp, lacZ)
AntiLac3: 5′ -GCAGTTCAACCACCGCAC-3′ (800 bp, lacZ)
AntiLac4: 5′ -AAAGTTGTTCTGCTTCATCAGC-3′ (1000 bp, lacZ)
AntiLac5: 5′ -GGGTGATTACGATCGCGC-3′ (1200 bp, lacZ).
RNAs were produced by incubating template DNA or DNAs at 37 °C for 1 h with 1 mM ATP, GTP, UTP, and CTP, 10 mM DTT, 40 U of RNasin (Promega), 3.3 pmol (α-32P) CTP (Perkin-Elmer), and 20 U of T7 RNA polymerase (Promega) in 1 × transcription buffer (Promega). To degrade the DNA template, 5 U of RQ1 DNase (Promega) was added, and samples were incubated for an additional 15 min at 37 °C. Samples were phenol–chloroform–isoamyl alcohol (25:24:1 (vol/vol/vol)) extracted twice, chloroform–isoamyl alcohol (24:1 (vol/vol)) extracted, and ethanol precipitated. Finally, RNA pellets were suspended in 100 μl of H2O.
To generate both RNA monomers and dimers, 16 μl of in vitro transcribed RNA in H2O was first denatured at 95 °C for 3 min, then placed on ice for 2 min before addition of 4 μl of 5 × monomer buffer (250 mM sodium cacodylate (pH 7.5), 200 mM KCl, 0.5 mM MgCl2) or 5 × dimer buffer (250 mM sodium cacodylate (pH 7.5), 1.5 M KCl, 25 mM MgCl2), respectively. Monomer samples were kept on ice until the gel was loaded. Dimer samples were incubated at 65 °C for 15 min, then cooled to room temperature over 30 min.
In vitro transcribed RNA was either monomerized or dimerized as described above, or loaded directly after extraction, precipitation, and suspension in water after transcription reactions, onto a 1.5% agarose gel. Gels were run in 1 × TBM at 115 V for 3 to 5 h at 4 °C or 1 × TBE at 70 V for 3 h at room temperature, dried under vacuum, and exposed to phosphorimager screens (Molecular Dynamics) for analysis with ImageQuant software. For this assay, the percentage of RNA in dimers was determined by dividing the amount of radioactivity migrating between the top and bottom of the major dimer band by the total radioactive signal located between the top of the dimer and the bottom of the major monomer band. Note that the RNAs with intermediate mobility, indicated with asterisks in Fig. 5A, were not included as dimers in these calculations.
To examine post-synthesis heterodimer formation, untreated 800 and 1000 nt RNAs were mixed in 1 × transcription buffer (Promega) and incubated at 37 °C for approximately 40 min.
Acknowledgments
We thank Nicole Robson-Dixon for performing the initial in vitro dimerization experiments as well as David Miller, Steven King, and Adewunmi Nuga for critical reading of the manuscript. This work was supported by NIH grant CA69300 to A.T. J.A.F. was supported in part by NIH training grant T32-GM08353.
References
- An W, Telesnitsky A. Human immunodeficiency virus type 1 transductive recombination can occur frequently and in proportion to polyadenylation signal readthrough. J Virol. 2004;78 (7):3419–3428. doi: 10.1128/JVI.78.7.3419-3428.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender W, Davidson N. Mapping of poly(A) sequences in the electron microscope reveals unusual structure of type C oncornavirus RNA molecules. Cell. 1976;7 (4):595–607. doi: 10.1016/0092-8674(76)90210-5. [DOI] [PubMed] [Google Scholar]
- Bender W, Chien YH, Chattopadhyay S, Vogt PK, Gardner MB, Davidson N. High-molecular-weight RNAs of AKR, NZB, and wild mouse viruses and avian reticuloendotheliosis virus all have similar dimer structures. J Virol. 1978;25 (3):888–896. doi: 10.1128/jvi.25.3.888-896.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohnlein S, Hauber J, Cullen BR. Identification of a U5-specific sequence required for efficient polyadenylation within the human immunodeficiency virus long terminal repeat. J Virol. 1989;63 (1):421–424. doi: 10.1128/jvi.63.1.421-424.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colicelli J, Goff SP. Sequence and spacing requirements of a retrovirus integration site. J Mol Biol. 1988;199 (1):47–59. doi: 10.1016/0022-2836(88)90378-6. [DOI] [PubMed] [Google Scholar]
- D’Souza V, Summers MF. Structural basis for packaging the dimeric genome of Moloney murine leukaemia virus. Nature. 2004;431 (7008):586–590. doi: 10.1038/nature02944. [DOI] [PubMed] [Google Scholar]
- Darlix JL, Gabus C, Nugeyre MT, Clavel F, Barre-Sinoussi F. Cis elements and trans-acting factors involved in the RNA dimerization of the human immunodeficiency virus HIV-1. J Mol Biol. 1990;216 (3):689–699. doi: 10.1016/0022-2836(90)90392-Y. [DOI] [PubMed] [Google Scholar]
- DeZazzo JD, Kilpatrick JE, Imperiale MJ. Involvement of long terminal repeat U3 sequences overlapping the transcription control region in human immunodeficiency virus type 1 mRNA 3′ end formation. Mol Cell Biol. 1991;11 (3):1624–1630. doi: 10.1128/mcb.11.3.1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirac AM, Huthoff H, Kjems J, Berkhout B. Regulated HIV-2 RNA dimerization by means of alternative RNA conformations. Nucleic Acids Res. 2002;30 (12):2647–2655. doi: 10.1093/nar/gkf381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorman N, Lever A. Comparison of viral genomic RNA sorting mechanisms in human immunodeficiency virus type 1 (HIV-1), HIV-2, and Moloney murine leukemia virus. J Virol. 2000;74 (23):11413–11417. doi: 10.1128/jvi.74.23.11413-11417.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dube S, Kung HJ, Bender W, Davidson N, Ostertag W. Size, subunit composition, and secondary structure of the Friend virus genome. J Virol. 1976;20 (1):264–272. doi: 10.1128/jvi.20.1.264-272.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn JA, An W, King SR, Telesnitsky A. Nonrandom dimerization of murine leukemia virus genomic RNAs. J Virol. 2004;78 (22):12129–12139. doi: 10.1128/JVI.78.22.12129-12139.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu W, Rein A. Maturation of dimeric viral RNA of Moloney murine leukemia virus. J Virol. 1993;67 (9):5443–5449. doi: 10.1128/jvi.67.9.5443-5449.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu W, Gorelick RJ, Rein A. Characterization of human immunodeficiency virus type 1 dimeric RNA from wild-type and protease-defective virions. J Virol. 1994;68 (8):5013–5018. doi: 10.1128/jvi.68.8.5013-5018.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girard PM, Bonnet-Mathoniere B, Muriaux D, Paoletti J. A short autocomplementary sequence in the 5′ leader region is responsible for dimerization of MoMuLV genomic RNA. Biochemistry. 1995;34 (30):9785–9794. doi: 10.1021/bi00030a016. [DOI] [PubMed] [Google Scholar]
- Gonda MA, Rice NR, Gilden RV. Avian reticuloendotheliosis virus: characterization of the high-molecular-weight viral RNA in transforming and helper virus populations. J Virol. 1980;34 (3):743–751. doi: 10.1128/jvi.34.3.743-751.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman SA, Coffin JM. Differential transcription from the long terminal repeats of integrated avian leukosis virus DNA. J Virol. 1986;60 (2):497–505. doi: 10.1128/jvi.60.2.497-505.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibbert CS, Rein A. Preliminary physical mapping of RNA–RNA linkages in the genomic RNA of moloney murine leukemia virus. J Virol. 2005;79 (13):8142–8148. doi: 10.1128/JVI.79.13.8142-8148.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibbert CS, Mirro J, Rein A. mRNA molecules containing murine leukemia virus packaging signals are encapsidated as dimers. J Virol. 2004;78 (20):10927–10938. doi: 10.1128/JVI.78.20.10927-10938.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu WS, Temin HM. Genetic consequences of packaging two RNA genomes in one retroviral particle: pseudodiploidy and high rate of genetic recombination. Proc Natl Acad Sci USA. 1990;87 (4):1556–1560. doi: 10.1073/pnas.87.4.1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu WS, Bowman EH, Delviks KA, Pathak VK. Homologous recombination occurs in a distinct retroviral subpopulation and exhibits high negative interference. J Virol. 1997;71 (8):6028–6036. doi: 10.1128/jvi.71.8.6028-6036.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huthoff H, Berkhout B. Two alternating structures of the HIV-1 leader RNA. RNA. 2001;7 (1):143–157. doi: 10.1017/s1355838201001881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharytonchyk SA, Kireyeva AI, Osipovich AB, Fomin IK. Evidence for preferential copackaging of Moloney murine leukemia virus genomic RNAs transcribed in the same chromosomal site. Retrovirology. 2005;2 (1):3. doi: 10.1186/1742-4690-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulpa D, Topping R, Telesnitsky A. Determination of the site of first strand transfer during Moloney murine leukemia virus reverse transcription and identification of strand transfer-associated reverse transcriptase errors. EMBO J. 1997;16 (4):856–865. doi: 10.1093/emboj/16.4.856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kung HJ, Hu S, Bender W, Bailey JM, Davidson N, Nicolson MO, McAllister RM. RD-114, baboon, and woolly monkey viral RNA’s compared in size and structure. Cell. 1976;7 (4):609–620. doi: 10.1016/0092-8674(76)90211-7. [DOI] [PubMed] [Google Scholar]
- Laughrea M, Jette L. HIV-1 genome dimerization: kissing-loop hairpin dictates whether nucleotides downstream of the 5′ splice junction contribute to loose and tight dimerization of human immunodeficiency virus RNA. Biochemistry. 1997;36 (31):9501–9508. doi: 10.1021/bi970862l. [DOI] [PubMed] [Google Scholar]
- Lear AL, Haddrick M, Heaphy S. A study of the dimerization of Rous sarcoma virus RNA in vitro and in vivo. Virology. 1995;212 (1):47–57. doi: 10.1006/viro.1995.1452. [DOI] [PubMed] [Google Scholar]
- Levin JG, Rosenak MJ. Synthesis of murine leukemia virus proteins associated with virions assembled in actinomycin D-treated cells: evidence for persistence of viral messenger RNA. Proc Natl Acad Sci USA. 1976;73 (4):1154–1158. doi: 10.1073/pnas.73.4.1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin JG, Grimley PM, Ramseur JM, Berezesky IK. Deficiency of 60 to 70S RNA in murine leukemia virus particles assembled in cells treated with actinomycin D. J Virol. 1974;14(1):152–161. doi: 10.1128/jvi.14.1.152-161.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maisel J, Bender W, Hu S, Duesberg PH, Davidson N. Structure of 50 to 70S RNA from Moloney sarcoma viruses. J Virol. 1978;25 (1):384–394. doi: 10.1128/jvi.25.1.384-394.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquet R, Baudin F, Gabus C, Darlix JL, Mougel M, Ehresmann C, Ehresmann B. Dimerization of human immunodeficiency virus (type 1) RNA: stimulation by cations and possible mechanism. Nucleic Acids Res. 1991;19 (9):2349–2357. doi: 10.1093/nar/19.9.2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meric C, Goff SP. Characterization of Moloney murine leukemia virus mutants with single-amino-acid substitutions in the Cys– His box of the nucleocapsid protein. J Virol. 1989;63 (4):1558–1568. doi: 10.1128/jvi.63.4.1558-1568.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messer LI, Levin JG, Chattopadhyay SK. Metabolism of viral RNA in murine leukemia virus-infected cells; evidence for differential stability of viral message and virion precursor RNA. J Virol. 1981;40 (3):683–690. doi: 10.1128/jvi.40.3.683-690.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murti KG, Bondurant M, Tereba A. Secondary structural features in the 70S RNAs of Moloney murine leukemia and Rous sarcoma viruses as observed by electron microscopy. J Virol. 1981;37 (1):411–419. doi: 10.1128/jvi.37.1.411-419.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onafuwa A, An W, Robson ND, Telesnitsky A. Human immunodeficiency virus type 1 genetic recombination is more frequent than that of Moloney murine leukemia virus despite similar template switching rates. J Virol. 2003;77 (8):4577–4587. doi: 10.1128/JVI.77.8.4577-4587.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz-Conde BA, Hughes SH. Studies of the genomic RNA of leukosis viruses: implications for RNA dimerization. J Virol. 1999;73 (9):7165–7174. doi: 10.1128/jvi.73.9.7165-7174.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paillart JC, Dettenhofer M, Yu XF, Ehresmann C, Ehresmann B, Marquet R. First snapshots of the HIV-1 RNA structure in infected cells and in virions. J Biol Chem. 2004;279 (46):48397–48403. doi: 10.1074/jbc.M408294200. [DOI] [PubMed] [Google Scholar]
- Pal BK, Scherer L, Zelby L, Bertrand E, Rossi JJ. Monitoring retroviral RNA dimerization in vivo via hammerhead ribozyme cleavage. J Virol. 1998;72 (10):8349–8353. doi: 10.1128/jvi.72.10.8349-8353.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pear WS, Nolan GP, Scott ML, Baltimore D. Production of high-titer helper-free retroviruses by transient transfection. Proc Natl Acad Sci USA. 1993;90 (18):8392–8396. doi: 10.1073/pnas.90.18.8392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer JK, Topping RS, Shin NH, Telesnitsky A. Altering the intracellular environment increases the frequency of tandem repeat deletion during Moloney murine leukemia virus reverse transcription. J Virol. 1999;73 (10):8441–8447. doi: 10.1128/jvi.73.10.8441-8447.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prats AC, Roy C, Wang PA, Erard M, Housset V, Gabus C, Paoletti C, Darlix JL. cis elements and transacting factors involved in dimer formation of murine leukemia virus RNA. J Virol. 1990;64 (2):774–783. doi: 10.1128/jvi.64.2.774-783.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rein A. Take two. Nat Struct Mol Biol. 2004;11 (11):1034–1035. doi: 10.1038/nsmb1104-1034. [DOI] [PubMed] [Google Scholar]
- Roy C, Tounekti N, Mougel M, Darlix JL, Paoletti C, Ehresmann C, Ehresmann B, Paoletti J. An analytical study of the dimerization of in vitro generated RNA of Moloney murine leukemia virus MoMuLV. Nucleic Acids Res. 1990;18 (24):7287–7292. doi: 10.1093/nar/18.24.7287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaiss AK, Son S, Chang LJ. RNA 3′ readthrough of oncoretrovirus and lentivirus: implications for vector safety and efficacy. J Virol. 2002;76 (14):7209–7219. doi: 10.1128/JVI.76.14.7209-7219.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]