

Abstract
V-ATPases are multisubunit membrane proteins that use ATP binding and hydrolysis to transport protons across membranes against a concentration gradient. Although some cell types express plasma membrane forms of these transporters, all eukaryotes require V-ATPases to maintain an acidic pH in membrane-bound compartments of endocytic and secretory networks to facilitate protein trafficking and processing. Mammalian cells that completely lack V-ATPases are not viable; yet, the abundance of V-ATPases can differ among cell types by an order of magnitude or more, requiring precise control of their expression. We previously showed that mRNA stability appears to play a major role in regulating overall abundance of V-ATPases. In this report, we demonstrate that the stability of V-ATPase mRNA is regulated through AU-rich elements in 3′-untranslated regions. Unlike some mRNAs that are short-lived due to the presence of these elements, V-ATPase mRNAs have half-lives of hours to days. However, during stress induced by ATP depletion, AU-rich elements are necessary to maintain stability of these transcripts and their presence in the cytoplasm. HuR, an RNA-binding protein that interacts with and stabilizes AU-rich mRNAs, shows increased binding to some V-ATPase mRNAs during ATP depletion. siRNA-mediated knockdown of HuR results in diminished V-ATPase expression. These results indicate that AU-rich elements and associated proteins can play a role in regulation of even very stable mRNAs by protecting against loss during cellular stress.
V-ATPases2 are a class of multisubunit transporters that perform numerous functions through their capacity to pump protons across cellular membranes (1). V-ATPases are required for cell survival because of their role in acidification of intracellular compartments, including the endocytic network and components of the secretory pathway. Mammalian cells are not viable in the total absence of V-ATPases (2), and yeast cells lacking these transporters must be maintained in acidic medium for survival (3). In recent years, it has become clear that V-ATPases bind to, and are regulated by, elements of the glycolytic pathway, indicating an intimate association with cellular energetic processes (4-10). Aside from these required functions, isoforms of V-ATPases that reside in plasma membranes of specific cells are involved in multiple physiological functions, including acid-base balance by the kidney (11), bone resorption by osteoclasts (12), and maintenance of pH within the inner ear (13) and the male reproductive tract (14, 15). Gene mutations causing loss of specific V-ATPase isoforms have been shown to result in renal tubular acidosis (13, 16), osteopetrosis (17, 18), and deafness (13).
Mammalian V-ATPases are composed of thirteen different subunits, including a, c, c″, d, and e in the membrane-bound, or V0, sector, and A–H in the cytosolic, or V1, sector. Most subunits can be expressed as tissue-restricted isoforms that are generated from distinct genes. The subunit isoforms encoded by unique genes include A, B1–2, C1–2, D, E1–2, F, G1–3, H, a1–4, d1–2, c, c″, and e. (For a summary of the mammalian V-ATPase gene nomenclature, see Ref. 19). In addition to the variation created by mixing and matching of subunit isoforms, V-ATPases also vary in overall cellular expression levels. Cells that require high levels of these transporters for normal cellular function, such as kidney epithelia, osteoclasts, and macrophages, can express V-ATPases at levels an order of magnitude higher than cells lacking specific requirements for high levels of proton transport. One of our interests has been in the mechanisms by which V-ATPase levels are regulated. We previously showed that macrophages express higher levels of V-ATPase than fibroblasts due to increased mRNA stability (20), and have since extended this conclusion to kidney epithelial cells, which also express high V-ATPase levels. Here we define stability elements in the mRNA transcript of the ubiquitously expressed E1 subunit and find that AU-rich elements are critical for this control.
AU-rich elements (AREs) are the best-studied regulator sequences of mRNA stability. Originally defined in short-lived mRNAs such as those encoding lymphokines, cytokines (21, 22), and proto-oncogenes (23), they consist of U-rich runs, often containing AUUUA cores (24, 25). These sequences are bound by a multiplicity of regulatory proteins that can stabilize or destabilize the transcript, or affect its translatability (26, 27). Among the most widely expressed ARE-binding proteins are the RNA-stabilizing protein HuR (HuA), the destabilizer hnRNP D (AUF-1), and TIAR and TIA-1, which are involved in translational regulation (reviewed in Ref. 26). In addition, other ARE-binding proteins, including several with homology to HuR, have been identified in specific cell types such as neural cells and lymphocytes.
ARE-containing mRNAs are known to be stabilized during various forms of cellular stress, including heat shock (28), UV irradiation (29, 30), hypoxia (31), and nutrient deprivation (32). Because kidney epithelia, particularly proximal tubules cells, are susceptible to damage by ischemic injury (33), and because high V-ATPase levels are critical to kidney cell function, we investigated whether V-ATPase mRNA expression in these cells might be affected by stress. We chose to examine the consequences of ATP depletion in the porcine proximal tubule cell line LLC-PK1, a well studied model that mimics many of the effects of ischemic injury produced in whole animal systems (34-38). We found that AREs in V-ATPase mRNAs are required for maintained stability and cytoplasmic expression of these transcripts under conditions of ATP depletion, and that the RNA-binding protein HuR mediates this stabilization. These results show that even very long-lived mRNAs can possess functional AREs that act primarily not as destabilizing moieties, but as positive regulators of expression during cell stress events.
MATERIALS AND METHODS
Cell Culture—LLC-PK1 cells (American Type Culture Collection, Manassas, VA) were cultured in Dulbecco's modified Eagle's medium containing penicillin/streptomycin supplemented with fetal bovine serum (10%) at 37 °C in 5% CO2. When LLC-PK1 cells were confluent the medium was replenished and incubated overnight. The cells were rinsed twice with phosphate-buffered saline and the culture medium was then replaced with pre-warmed Dulbecco's modified Eagle's medium base supplemented with l-glucose, sodium bicarbonate, and 0.1 μM antimycin A for 0–4 h. Mock treated cells underwent the same procedure, but the culture medium was replaced with normal growth medium. In some experiments, ATP-depleted cells were allowed to recover by replacing the depletion medium with normal growth medium. Using this system, cellular ATP levels dropped to <1% of normal levels within 1 h of culture in ATP depletion medium and remained very low until normal growth medium was added. Four hours of recovery in normal growth medium resulted in ATP levels at ∼75% of original levels.3 This result is consistent with other studies of renal epithelia in which the rebound of ATP levels is incomplete with only a few hours of recovery (34, 38).
Transfection—For transient transfection, human E1 V-ATPase cDNAs in the pcDNA3.1 expression vector (Invitrogen) were transfected singly or pairwise at 70% confluence using Lipofectamine with Plus reagent (Invitrogen). Cells were harvested 48 h post-transfection for analysis. For creation of stably transfected cells, after 48 h of transfection as described above, the medium was replaced with normal growth medium supplemented with G418 (Invitrogen). Colonies were selected and expression confirmed with Northern blot analysis.
For RNA interference studies, three siRNAs against porcine HuR were derived by, and purchased from, Ambion (Austin, TX). All three siRNAs knocked down HuR RNA levels at 50 nM concentrations, but only one was considered for further study, and was used at 10–50 nm. Transfection of siRNA into LLC-PK1 cells was performed using Lipofectamine with Plus reagent (Invitrogen) and transfecting the cells when 50% confluent.
cDNAs and Antibodies—A human E1 V-ATPase cDNA containing 84 bp of 5′-UTR and the entire coding and 3′-untranslated regions was subcloned into expression vector pcDNA3.1 (Invitrogen). Deletions of the 3′-UTR were created by excising specific sequences at convenient restriction sites. Site-directed mutagenesis was performed using the QuikChange site-directed mutagenesis kit (Stratagene). AUUUA core sequences were mutagenized to ACTAGT to create new SpeI sites for convenient screening of mutants. The porcine E1 and HuR sequences were determined by amplifying selected regions from LLC-PK1 cells using RT-PCR and subcloning the products into a TOPO-TA vector from Invitrogen. Reverse transcription was performed using the Superscript First-strand Synthesis System, and PCR was performed using Platinum TaqDNA Polymerase High-Fidelity (both from Invitrogen). All constructs were sequenced by the Plant-Microbe Genomics Facility at The Ohio State University.
For detection of E1, a synthetic peptide corresponding to the mouse E1 C-terminal residues PEVRGALFGANANRKFLD was created and conjugated to keyhole limpet hemocyanin. Peptide synthesis, conjugation to keyhole limpet hemocyanin, and development of rabbit polyclonal antisera were performed by Invitrogen. For detection of HuR, a mouse monoclonal antibody (3A2) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-hnRNP D (T-10) was a goat polyclonal antibody also purchased from Santa Cruz Biotechnology.
RNA and Protein Detection Procedures—RNA blots (Northern blots) were performed using standard conditions. Total RNA was prepared using RNA Bee (Tel-Test Inc., Friendswood, TX), and 10 μg of each sample was analyzed in a formaldehyde agarose gel as previously described (39). The RNA was transferred to a GeneScreen Plus membrane (PerkinElmer Life Sciences) and fixed using a Stratalinker UV Cross-linker (Stratagene). To detect the exogenously expressed human E1 subunit mRNA, an XhoI-KpnI fragment containing 38 bp of 5′-UTR and 335 bp of coding region was labeled using the DECAprime labeling system (Ambion, Austin, TX). This probe did not cross-react with the endogenous porcine form under our conditions. For immunoblots (Westerns), 10 μg of whole cell lysates from LLC-PK1 cells were run in a 12.5% acrylamide gel (Bio-Rad, Hercules, CA) under standard conditions. Proteins were transferred to Hybond P membrane (GE Health-care/Amersham Biosciences) and probed with antisera at a 1:500 (3A2) or 1:1000 (E1) dilution. Primary antibodies were detected using horse-radish peroxidase-conjugated secondary antibodies and an ECL Western blotting detection kit (Amersham Biosciences).
For competitive RT-PCR of the endogenous LLC-PK1 E1 subunit, primers were created to correspond to sequences within the porcine coding region. The sense primer was of the sequence 5′-GCTTGTGCAAACCCAAAGACTG-3′, and the antisense primer was of the sequence 5′-TATGTCTTCAGGCAGGTAGGCC-3′. For an internal standard, a cDNA was created that corresponded to the expected RTPCR product using the primers above but contained an internal deletion of 15%, a T7 promoter element, and a tail of 15 adenosines, as previously described (40). This product was transcribed in vitro using the MAXIscript system (Ambion), and 1 pg of the resulting RNA (the internal standard) was added to 1 μg of LLC-PK1 total RNA prior to reverse transcription and PCR. Reverse transcription was performed using the Superscript First-strand Synthesis System and TaqDNA Polymerase, both from Invitrogen. One-fifth of the PCR products were run in a 2% agarose gel containing ethidium bromide.
Immunohistochemistry and in Situ Hybridization—For immunolocalization of HuR, 3 × 104 LLC-PK1 cells were seeded on glass coverslips in 24-well plates and grown to confluence, at which time the cells were given fresh medium and cultured overnight prior to use. Following the necessary treatments, cells were fixed and permeabilized in 2% formaldehyde in stabilization buffer (41). Cells were then probed with anti-HuR antibody obtained from Santa Cruz Biotechnology and Alexa 488-or Alexa 568-conjugated goat anti-mouse antibodies from Molecular Probes (Eugene, OR). Cells were visualized with a Nikon Eclipse 80i epifluorescent microscope with SPOT software (Diagnostic Instruments, Sterling Heights, MI), or with a Zeiss 510 META laser scanning confocal microscope at the Campus Microscopy and Imaging Facility at The Ohio State University.
For in situ hybridization of exogenously expressed human E1, an antisense probe was transcribed in vitro using the MAXIscript system (Ambion). The XhoI-KpnI fragment used for Northern analysis was subcloned into pBluescript and an antisense probe was made by linearizing the plasmid with BamHI and performing transcription using T3 RNA polymerase. BODIPY-TR-labeled UTP (Molecular Probes) replaced unlabeled UTP in this reaction. A probe corresponding to the porcine E1 subunit was PCR-amplified using the degenerate sense primer (5′-CGSTTGTGCAAACSCAAAG-3′) and antisense primer (5′-CACCAGCTATNTCYTCAGGCAS-3′). This fragment was cloned into the pCRII-TOPO vector (Invitrogen). Linearizing the plasmid with XhoI and performing transcription using T3 RNA polymerase created an antisense probe for endogenous porcine E1. BODIPY-TR-labeled UTP (Molecular Probes) replaced unlabeled UTP in this reaction. A probe against the c subunit was previously described (40). The hybridization procedures were essentially as previously described (40), but the human E1 probe used to detect exogenous V-ATPase expression was hybridized at 42 °C, rather than 37 °C, so it would not bind to the endogenous, porcine sequence.
Protein-RNA Binding Assays—Immunoprecipitation/RT-PCR was performed essentially as described before (42). LLC-PK1 cells were washed 2× in phosphate-buffered saline and lysed in CEB buffer containing Halt Protease Inhibitor (Pierce) and RNaseOUT (Invitrogen) for 40 min. Cells were scraped and centrifuged for 10 min to remove cell debris. Ten micrograms of the HuR antisera (3A2) or anti-hnRNP D (T-10) were added to the supernatant and incubated for 30 min at 4 °C. Five micrograms each of protein A and protein G were added, and the mixture was incubated for 30 min at 4 °C. The reaction was then gently centrifuged, and the supernatant removed. The precipitate was washed five times with CEB buffer, and RNA was extracted using RNA Bee reagent (Tel-Test Inc.).
For detection of the E1 subunit mRNA, the primers were identical to those used for competitive RT-PCR. Because the sequence for the porcine c subunit is unknown, this mRNA was detected by creating degenerate primers that were capable of binding both human and mouse sequences. The sense primer was of the sequence 5′-TCCATCATCCCRGTGGTCATGG-3′, and the antisense primer was of the sequence 5′-ACTTTGTGGAGAGGATRAGGGC-3′.
RESULTS
V-ATPase mRNAs Contain AU-rich Elements—To define sequences within V-ATPase mRNAs that regulate stability, we performed deletion/mutation analyses of the ubiquitously expressed E1 (ATP6V1E1) subunit transcript. The human cDNA was subcloned into expression vector pcDNA3.1, and sequentially larger portions of the 3′-untranslated region (3′-UTR) were deleted, as shown in Fig. 1A. Construct 1 contained the full-length 3′-UTR, whereas constructs 2–4 were shortened at convenient restriction sites as shown. Construct 2 lacked 143 bp of the 3′-UTR, whereas constructs 3 and 4 lacked 328 bp and 456 bp, respectively. To determine relative stabilities of the resulting mRNAs, expression constructs were transfected in equal amounts into LLC-PK1 cells, both singly and pairwise, and levels of the expressed mRNAs were compared by Northern analysis. Lanes 2–5 of Fig. 1B show the mRNA products of cells singly transfected with each construct. Lanes 6–8 of this figure demonstrate expression levels of the products when co-transfected pairwise. Because the co-transfected constructs were transcribed via the same cytomegalovirus promoter, any differences in expression levels between them necessarily were due to differences in transcript stability. In lanes 6–8, the full-length cDNA (construct 1) was co-transfected individually with each deleted construct. These results demonstrate that expression from construct 1 (1. 27 kb) was found to be much lower than expression from constructs 2, 3, or 4. These data, as well as those from similar co-transfection experiments, suggested that the full-length E1 mRNA is less stable than mRNA expressed from the deletion mutants, apparently due to sequences within its 3′ 143 bases. Examination of the nucleotide sequence within this region revealed a highly conserved 50-base AU-rich element, with two canonical AUUUA core sequences, as shown in Fig. 1C. This element resides 9 bases 5′ to the poly-A tail, as indicated in Fig. 1A. To determine whether this ARE was involved in regulation of E1 mRNA stability, the AUUUA core elements were mutated individually in the full-length cDNA and tested by Northern analysis in co-transfection experiments similar to those in Fig. 1B. Fig. 1D shows the results of multiple experiments in which these mutated constructs, as well as the deleted constructs in Fig. 1A, were co-transfected pairwise into LLC-PK1 cells with construct 4, and the relative expression levels (as assayed by Northern blots) were determined by densitometric methods. These data show that constructs 2, 3, and 4 produced mRNAs with stabilities ∼2.5-fold greater than the full-length mRNA. In addition, mutation of the upstream AUUUA core (mutA) resulted in a 2.5-fold stabilization of mRNA, whereas mutation of the downstream AUUUA core (mutB) did not. This lack of effect is not entirely unexpected, because the downstream AUUUA overlaps the polyadenylation signal, and is likely to be involved in that process, rather than binding of ARE-specific RNA-stabilizing proteins. Data from these experiments, as well as other pairwise combinations not shown, strongly suggest that the ARE destabilizes E1 transcripts.
FIGURE 1.
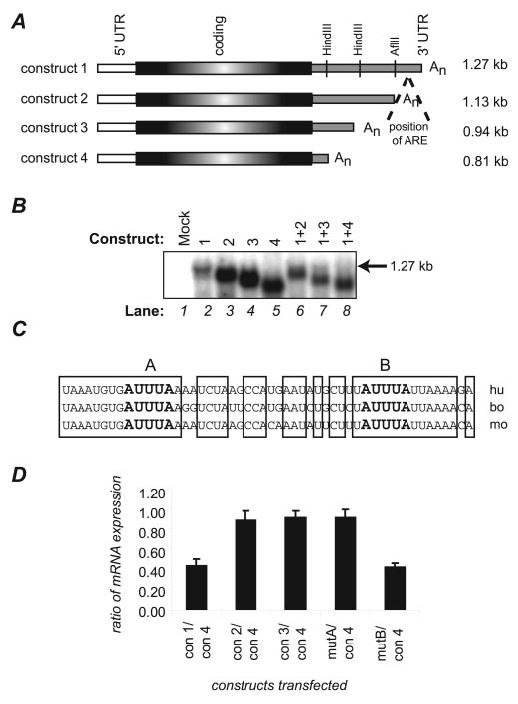
Analysis of sequences involved in V-ATPase mRNA stability. A, deletion constructs used in this experiment were human E1 cDNAs sequentially shortened at the 3′-UTR at convenient restriction sites. All were expressed in the eukaryotic expression vector pcDNA3.1. Construct 1 represents E1 containing the full-length 3′-UTR. B, the deletion constructs shown in panel A were singly or doubly transfected into LLC-PK1 cells and tested for expression levels by Northern analysis. The total amount of plasmid transfected into each sample was kept constant, so that samples in lanes 6–8 were transfected with half the amount of each plasmid as the singly transfected cells in lanes 2–5. Lane 1 represents RNA from mock-transfected cells and demonstrates that the Northern procedures detect only exogenously expressed E1. The arrow denotes the position of mRNA expressed from the full-length construct 1. C, analysis of an AU-rich region of E1 mRNA shows extensive conservation among human (hu), bovine (bo), and mouse (mo) sequences. Bases that are identical among all three species are boxed. AUUUA sequences that often serve as the cores of protein-binding regions are bolded and designated as site A or B. Each site was mutated, and the mutants were tested for expression along with the deletion constructs, as shown in panel D. D, pairwise analyses of mRNA expression from deletion and mutation constructs were performed as in panel B. In this graph, relative expression levels from each construct were compared with that from construct 4. Three to five individual experiments were averaged for each data point; standard errors are denoted.
Because deletion or mutation of this ARE caused the E1 transcript to become more stable, we surmised that the mRNA must be associated with a destabilizing protein under normal growth conditions. A good candidate for this is hnRNP D (or AUF-1), a ubiquitously expressed protein that binds AREs and can destabilize its associated mRNAs. hnRNP D is expressed as four protein isoforms generated from a single mRNA transcript (43). As will be shown below in Fig. 5, we found that E1 mRNA binds to this destabilizing protein under normal growth conditions, thus accounting for the increased stability caused by loss of the ARE.
FIGURE 5.
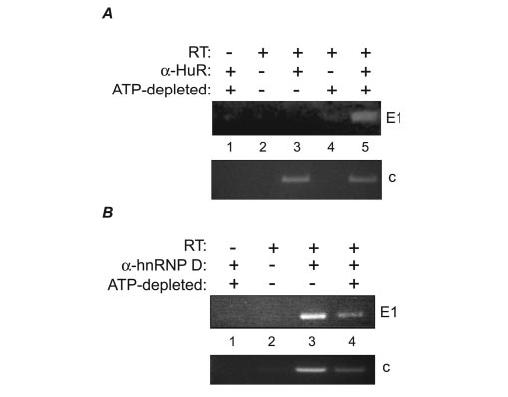
Binding of HuR and hnRNP D (AUF-1) to V-ATPase mRNAs. A, HuR was immunoprecipitated from normal (lane 3) or ATP-depleted (lane 5) LLC-PK1 cells. Bound RNA was harvested and subjected to RT-PCR for the presence of E1 and c transcripts. Negative controls, performed in the absence of either anti-HuR antibody (lanes 2 and 4) or reverse transcriptase (lane 1) are shown. B, hnRNP D was immunoprecipitated from normal (lane 3) or ATP-depleted (lane 4) LLC-PK1 cells, as in panel A, and tested for the presence of bound E1 or c transcripts. Controls lacking reverse transcriptase (lane 1) or anti-hnRNP D antibody (lane 2) also are shown.
AREs Are Required for V-ATPase mRNA Stability during the Stress of ATP Depletion—LLC-PK1 cells are a kidney proximal tubule cell line that have been investigated extensively as models for ischemic injury in native kidney (34-38). The injuries to proximal tubule epithelia resulting from renal ischemia are mirrored by those in LLC-PK1 cells subjected to transient ATP depletion. Because ARE-containing mRNAs can be stabilized under conditions of cellular stress such as heat shock or UV-irradiation, we tested whether E1 is stabilized by the presence of AREs during ATP depletion. Stably transfected cell lines were created that contained some of the individual constructs described in Fig. 1. These lines were subjected to 2h of ATP depletion, and the exogenously expressed E1 mRNAs were assayed by Northern analysis. Fig. 2A (top panel) shows that intact AREs were required for maintenance of normal expression levels during this cellular stress; all mRNAs with either wholesale deletions or mutations of the upstream AUUUA core (constructs 2, 3, and mutA) showed diminished mRNA levels upon energy depletion, whereas the ARE-containing mRNAs (constructs 1 and mutB) did not. Because the relatively low intensity of bands produced by lines expressing constructs 1 and mutB made examination difficult in the top panel, these samples were re-run in a separate experiment and exposed to film for a longer period (Fig. 2A, bottom panel). Further, consistent with the results in Fig. 1B, the mRNAs that lacked ARE sequences were expressed at higher levels than those containing the sequences under normal growth conditions, presumably due to the lack of destabilization by hnRNP D or other proteins. These results are consistent with a role for the ARE in destabilizing E1 mRNA during normal growth, but stabilizing it during cell stress, thus maintaining expression at a consistent level under all conditions.
FIGURE 2.
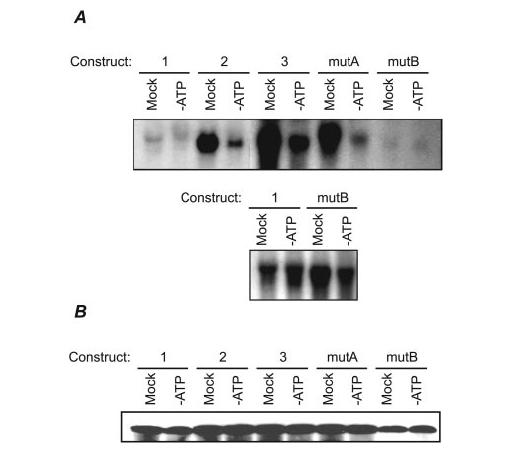
Northern analysis of stable transfectants under normal and ATP-depleted conditions. A, (top panel) LLC-PK1 cells were stably transfected with the constructs used in Fig. 1. Stabilities of the plasmid-encoded mRNAs were tested by culturing these lines either in ATP-depletion medium or fresh growth medium (mock treatment) for 2 h. Ten micrograms of total cellular mRNA from each sample was analyzed. All constructs lacking functional AREs, as defined by the experiments in Fig. 1 (constructs 2, 3, and mutA) were expressed at high levels under normal growth conditions, but were unstable under conditions of ATP depletion. Because of the relatively faint signals produced by constructs 1 and mutB, these samples were run in a separate gel and exposed to film for a longer period for better resolution of the signals (bottom panel). B, E1 protein levels were assayed by Western blotting lysates from cells treated as in panel A. E1 protein levels were slightly elevated in lines lacking functional AREs.
To determine how E1 protein levels might be affected, we performed Western analysis on the same transfected cell lines subjected to energy depletion (Fig. 2B). Two hours of this treatment was not sufficient to caused decreased E1 protein levels (not unexpectedly, because the halflife of E1 in mammalian cells has been estimated at about 6 h (66)). However, E1 levels in the lines containing functional AREs (constructs 1 and mutB) were slightly diminished (by ∼50%). Because the anti-E1 antibody used in this experiment cannot discriminate between the endogenous porcine E1 and the exogenously expressed human form, we are unable to discern what fraction of each band resulted from transcription/translation of the expression constructs. Nonetheless, these results are generally consistent with the notion that AREs in the E1 transcript regulate its expression.
To determine the effects of ATP depletion on endogenous E1 expression, we analyzed the levels of mRNA and protein during this stress event. Western analysis showed that the endogenous E1 protein remained constant during 4 h of ATP depletion (Fig. 3A). Similarly, competitive RT-PCR demonstrated no significant change in E1 mRNA expression during that period (Fig. 3B). These results are consistent with the previous finding that full-length E1 mRNA is not degraded during ATP depletion, and that AREs contribute to this stability. We also tested the expression of E1 protein during recovery from 1h of ATP depletion, and found no significant changes in expression (Fig. 3C). This is consistent with our hypothesis that LLC-PK1 cells regulate E1 mRNA levels to maintain a steady level of protein under normal and stressed conditions.
FIGURE 3.
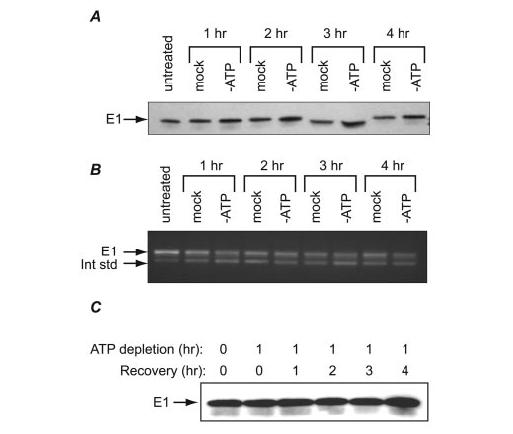
Stability of endogenous E1 under normal and ATP-depleted conditions. A, LLC-PK1 cells were left untreated or cultured in ATP-depletion medium or fresh growth medium (mock treatment) for 1–4 h. Protein was harvested from these cells and assayed by Western analysis for expression of E1. B, total cellular RNA from the same experiment as in A was harvested and analyzed for expression of E1 mRNA using competitive RT-PCR. The internal standard (lower band) was created as described under“Materials and Methods.” C, LLC-PK1 cells were ATP-depleted for 1 h and recovered by culturing in normal growth medium for 1–4 h. Expression of E1 protein was analyzed by Western blotting.
HuR Binds to and Stabilizes the E1 Transcript—Because we found that E1 mRNA is stabilized by AU-rich elements under the stress of ATP depletion, it seemed likely that HuR, a known stabilizer of ARE-containing mRNAs, might be involved. HuR, which shuttles between the nucleus and cytoplasm, is distributed predominantly in the nucleus under normal growth conditions (44). Some forms of cell stress such as heat shock and UV-irradiation cause HuR to become more cytoplasmic in location. To determine whether energy depletion induced a similar re-localization, we depleted LLC-PK1 cells of ATP for up to 4 h and performed immunocytochemistry for HuR. As shown in Fig. 4, HuR in mock treated cells was distributed primarily in nuclei. However, by 2 h in ATP depletion medium, a strong cytoplasmic staining was detected. Recovery in normal medium for an additional 2 h induced a re-localization of HuR back into the nucleus. Thus, ATP depletion, like heat shock or UV irradiation, among other stresses, induced movement of HuR into the cytoplasm in a reversible manner. We have extended these studies with additional time points and found that re-distribution into the cytoplasm continues as long as the cells are cultured in ATP depletion medium, up to 4 h (data not shown). Further, HuR protein levels continually increase over this period and drop back to normal levels when cells are placed in recovery medium.3 These results indicate that energy depletion, like other stresses, alters HuR distribution in kidney epithelia.
FIGURE 4.
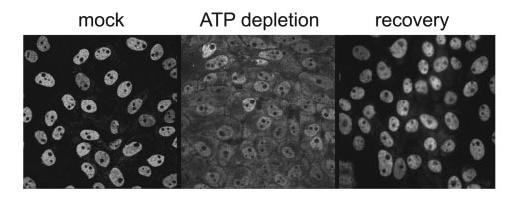
Distribution of HuR under normal, ATP-depleted, and recovered conditions. LLC-PK1 cells were cultured in normal growth medium (left panel) or ATP depletion medium (center panel) for 2 h and prepared for immunocytochemical localization of HuR. Following 2 h of ATP depletion, cells were allowed to recover by incubation in normal growth medium for an additional 2 h (right panel). This figure demonstrates the reversible nucleocytoplasmic redistribution of HuR under ATP-depleted conditions.
To determine whether HuR binds to E1 transcripts under ATP depletion, we performed experiments in which HuR was immunoprecipitated from normal and ATP-depleted cells, and the bound RNA was isolated and subjected to RT-PCR to detect the presence of E1 mRNA. Fig. 5A shows a typical result of these experiments. Although little to no E1 mRNA was bound to HuR under normal growth conditions (lane 3), ATP-depleted cells showed a strong interaction between HuR and E1 transcripts (Fig. 5A, lane 5). In contrast, E1 was readily detectable in hnRNP D immunoprecipitates under both normal (Fig. 5B, lane 3) and ATP-depleted (Fig. 5B, lane 4) conditions, although to a somewhat lesser extent in the stressed cells. We also tested the mRNA for the V-ATPase c subunit in its ability to bind HuR and hnRNP D. The c subunit transcript contains a potential ARE in its 3′-UTR and, recently, was immunoprecipitated in association with HuR from a human colorectal carcinoma cell line (24). The binding profile of c subunit mRNA to hnRNP D was similar to that of E1 (Fig. 5B, lanes 3 and 4), but we routinely detected significant levels of c mRNA binding to HuR under both normal and ATP-depleted conditions (Fig. 5A, lanes 3 and 5). This may be reflective of differences in stability between the E1 and c mRNAs; the c subunit transcript is expressed at higher levels than other V-ATPase mRNAs (40), and the balance of its interactions with HuR and other ARE-binding proteins may regulate its steady-state abundance. The difference in HuR binding of the two mRNAs under different conditions is not unexpected. Although both E1 and c possess class I AREs (containing one to three scattered AUUUA core sequences (25)), the sequences of these AREs differ markedly. In vivo, HuR is capable of discriminating among specific ARE sequences to regulate only particular genes in a given physiological state (45, 46).
To determine how AREs might affect mRNA distribution under normal and stressed conditions, we performed in situ hybridization of both wild-type and mutant V-ATPase transcripts (Fig. 6). Untransfected cells were hybridized with RNA probes against the endogenous porcine E1 following mock treatment or ATP depletion (panels a–d). The endogenous mRNAs were strongly present in both the cytoplasm and the nuclei, and ATP depletion did not significantly alter the distribution of these mRNAs. Similar results were obtained when cells were probed for distribution of the endogenous c subunit mRNA (not shown). We then performed in situ analysis on stably transfected cell lines expressing either the full-length human E1 mRNA (construct 1), a human E1 mRNA deleted of a portion of its 3′UTR containing the ARE (construct 3), or a full-length human mRNA mutated for sequences in its ARE (mutA and mutB). The full-length E1 mRNA (construct 1) showed nuclear and cytosolic distribution in both mock and ATP depleted cells (panels e–h), similar to that of the endogenous mRNA. Nucleolar staining also was present, as in all transfected cell lines; this appears to be a consequence of overexpression of the exogenous mRNAs, but the reasons for this are not clear. In contrast, although mRNA from construct 3 (which lacks an ARE) was nuclear and cytosolic under normal growth conditions (panels i and j), ATP depletion caused this mRNA to be retained primarily in a nuclear or perinuclear distribution (panels k and l). Similar results were obtained using the mutA construct, which is identical to construct 1 except for mutation of a few critical bases of the ARE (panels m–p). As in cells expressing construct 3, ATP depletion of mutA cells caused the mRNA to be distributed predominantly in and around the nucleus. MutB, which maintains a consistent expression level during ATP depletion, was similar to construct 1 in its ability to remain cytosolic during ATP depletion (panels q–t). These results suggest that an ARE is necessary for proper expression of E1 mRNA in the cytosol. An ARE might be required for proper export from the nucleus by HuR, or for stabilization of the mRNA by HuR once in the cytosol (44, 47, 48). Either of these scenarios would require cytoplasmic HuR. To ensure that HuR was still capable of nucleocytoplasmic redistribution in the mutant cells, the lines expressing construct 3 and mutA were ATP depleted for 2 h, and HuR was immunolocalized. HuR was abundant in the cytoplasm of both lines (data not shown); thus, the lack of mutant mRNAs in the cytoplasm cannot be attributed to disrupted HuR re-distribution. However, it is unclear from this experiment whether the ARE is required for nuclear export, as has been previously suggested (49), or is simply required for HuR binding and stability in the cytoplasm.
FIGURE 6.
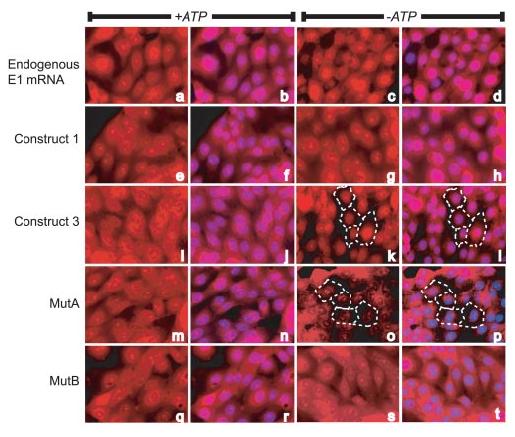
In situ hybridization of V-ATPase mRNAs under normal and ATP-depleted conditions. Wild-type LLC-PK1 cells (panels a–d), or cell lines stably transfected with human E1 expression constructs (panels e–t) were cultured for 2 h in normal or ATP-depletion medium and visualized for mRNA distribution. Locations of nuclei are indicated by 4′,6-diamidino-2-phenylindole staining in the second and fourth columns. The wild-type cells were hybridized with a riboprobe corresponding to the porcine E1 subunit, while a riboprobe corresponding to the human E1 sequence was used to detect expression from the transfected cell lines. In all cases, mock treated cells demonstrated nuclear and cytoplasmic localization of the endogenous or plasmid-encoded mRNAs. However, under ATP depletion, the mRNAs lacking AREs (constructs 3 and mutA) showed decreased expression in the cytoplasm. In panels k and l and o and p, dashed lines indicate the periphery of a few cells, demonstrating that the altered staining patterns are not due to rounding and retraction of these cells.
Finally, we used RNA interference technology to knock down HuR expression and determine its effect on E1 levels. Three siRNAs against porcine HuR were obtained from Ambion; Fig. 7A shows the ability of these inhibitors to affect HuR mRNA expression. Treatment with all three siRNAs resulted in nearly complete inhibition of HuR mRNA expression at 50–100 nm when tested 2 days after transfection. Further experimentation on siRNA 1 showed it to be completely active down to at least 10 nm, and was considered for further study. LLC-PK1 cells were transfected with 10 or 50 nm concentrations of this siRNA, and were examined for HuR protein levels 1–3 days post-transfection. Suppression of HuR protein levels was notable within 1 day of siRNA transfection, and appeared nearly complete within 2–3 days (Fig. 7B). The same samples were tested for expression of the E1 subunit. This protein was notably diminished in expression, although not until 2 days post-transfection, demonstrating that the timing of its loss was subsequent to loss of HuR (Fig. 7C, top panel). To ensure that this diminished E1 expression was due to suppressed mRNA levels, we used competitive RT-PCR to quantify E1 mRNA under the same conditions (Fig. 7C, bottom panel). As expected, cells transfected with siRNAs to HuR showed notable loss of E1 mRNA. These results are consistent with the previous experiments in showing that HuR mediates stabilization and expression of V-ATPase mRNA through ARE sequences.
FIGURE 7.
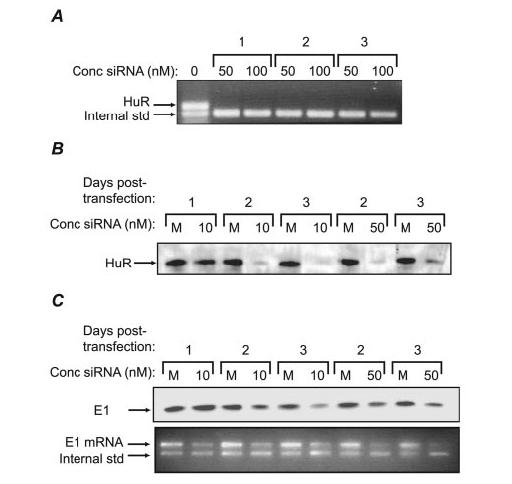
Knock-down of HuR expression in LLC-PK1 cells. A, three siRNAs designed to knock down HuR expression were tested for their capacity to suppress HuR mRNA. Cells were transfected (or mock transfected, first lane) with each of the siRNAs at 50 or 100 nm, and after 2 days, total cellular RNA was harvested and assayed for HuR transcripts by competitive RT-PCR. All three siRNAs were extremely efficient at suppressing HuR expression. B, siRNA 1 from panel A was transfected (or mock transfected, M) into cells at 10 or 50 nm, and after 1–3 days, cells were tested for expression of HuR protein by Western analysis. C, the Western blot in Fig. 7B was stripped and re-probed for E1, demonstrating that HuR suppression diminishes E1 expression (top panel). To confirm that loss of E1 protein was due to diminished E1 mRNA levels, competitive RT-PCR for E1 was performed on cells from the same experiment (bottom panel).
DISCUSSION
AU-rich elements originally were characterized in short-lived transcripts that regulate cell cycle and differentiation, such as c-fos, tumor necrosis factor-α, and granulocyte macrophage-colony stimulating factor (21-23). AREs first were identified as mRNA-destabilizing elements, but more recently, it has become clear that some AREs also can act as translational suppressors. Our identification of a functional ARE in a V-ATPase mRNA initially was somewhat surprising, because these transcripts tend to be very stable, with half-lives of hours to days (20), and V-ATPase proteins are expressed at moderate to very high levels in mammalian cells. Based on our studies, we conclude that the primary role of AREs in V-ATPase transcripts may be as protection against degradation during cell stress and maintenance of a constant expression level, rather than as mediators of short half-lives.
Previous surveys of human mRNA sequences had indicated that several V-ATPase transcripts, including those for A, E1, and F, contained ARE sequences that conformed to a consensus that contained at least one AUUUA core (50-52). However, this criterion is probably conservative, given that a number of studies have shown that U-rich sequences lacking the AUUUA are also capable of being functional AREs (24, 25). Indeed, during the course of our studies, López de Silanes et al. (24) published a report in which they immunoprecipitated HuR from human colorectal carcinoma cells, and performed cDNA array analysis on the bound RNA. They confirmed E1 and c as ligands for HuR, and pulled down mRNAs for additional subunits c″, E2, and G1. Our informal survey of other V-ATPase cDNA sequences suggests to us that most contain potential AREs. Notable exceptions to this are several of the a (ATP6V0A1, ATP6V0A2, TCIRG1, and ATP6V0A4) subunits; however, this is not surprising, because our previous studies showed that high levels of expression of a1 and a3 subunits are mediated at least in part by transcriptional control (20). Another potential exception to regulation by AREs is the B2 subunit, which is widely expressed and is abundant in the proximal tubule. Our previous studies showed that B2 levels are mediated primarily by transcriptional, rather than post-transcriptional regulation (20, 39, 53). Interestingly, however, B2 mRNA is expressed in two alternately polyadenylated forms, one of which contains a potential ARE near its poly(A) tail, whereas the other (shorter) form does not. In macrophages, the ARE-containing transcript is less stable than the short form.4 Thus, it is possible that one pool of B2 subunits is regulated by transcriptional control, while another is regulated by post-transcriptional mechanisms. It is of note that both the B2 and the a subunits appear to be involved in subcellular localization of V-ATPases. By regulating expression of these subunits independently from the core subunits, the cell may achieve more precise control over V-ATPase levels in various membrane compartments. Nonetheless, it appears that AREs provide a mechanism by which the core subunits of V-ATPases can be increased or decreased in concert, as required by the individual cell.
In our renal epithelial cell model, depletion of cellular ATP caused translocation of HuR from the nucleus to the cytosol, while recovery of ATP caused the reverse effect. Our preliminary studies of rat kidneys subjected to ischemia/reperfusion injury also suggest that HuR similarly translocates to the cytoplasm in native energy-depleted kidney epithelia. These results appear to be in conflict with recent studies demonstrating a role for AMP-activated protein kinase (AMPK) in mediating HuR trafficking. Activation of AMPK, a regulator of cellular responses to diminished ATP levels, was shown to increase nuclear localization of HuR in human colorectal carcinoma cells through modification of importin α1, an adaptor protein involved in regulation of nuclear import (54, 55). In another study, addition of extracellular ATP to renal mesangial cells increased cytoplasmic HuR levels, also in contrast with our results (56). Although the reasons for the disparities with our study are unclear, we hypothesize that cell- and tissue-specific differences in AMPK may play a role. The role of AMPK in the kidney is unknown; however, very recent immunohistochemical studies demonstrated that different segments of normal rat kidney nephrons showed markedly different distributions of activated AMPK. Although some nephron segments differed in the subcellular localization of AMPK (e.g. apical versus basolateral staining), proximal tubule cells (the cell type used in this study) were uniformly negative for the activated kinase (57), indicating that regulation of AMPK by renal epithelia differs widely by cell type. Additionally, one hallmark of ischemia-reperfusion injury in proximal tubule cells is a high proliferation rate accompanied by expression of c-fos, a known ligand of HuR (58). This appears to differ from the situation described for colorectal carcinoma cells, in which conditions that activate AMPK (i.e. ATP depletion) down-regulate genes that induce cell proliferation (54). This suggests that alternate mechanisms might be in place to regulate HuR in proximal tubule cells. Indeed, our own studies have indicated that potentially unique genetic regulatory mechanisms are involved in HuR regulation during both energy depletion and recovery in proximal tubule cells.3 Therefore, it appears that proximal tubule cells may utilize distinct pathways to regulate HuR activity.
HuR and hnRNP D (AUF-1) have similar distributions throughout the body, and a given tissue will tend to express similar levels of both proteins (59, 60). Because these proteins work functionally in opposition to each other, it seems likely that their balance is important in regulation of ARE-containing mRNAs. Notably, HuR and hnRNP D are expressed at the lowest levels (59, 60) in tissues where V-ATPase expression is the highest (61, 62), including kidney, brain, heart, lung, and liver. Because hnRNP D is constantly present in the cytoplasm and at minimal levels in these tissues, its low expression may allow V-ATPase mRNAs to remain highly stable and translatable. Maintaining a low HuR level in these tissues would not necessarily negatively affect the stability of these mRNAs, because most HuR resides in the nucleus under normal conditions, and is not likely to contribute as strongly to mRNA stability in this situation. However, transient cell stress such as ischemia would require the maintenance of high V-ATPase levels upon return to normal cellular function, so translocation of HuR to the cytoplasm, where it can stabilize mRNAs, provides this action. In this regard, it is important to note that studies of ARE-containing mRNAs indicate that in the cytoplasm, individual mRNAS are bound either by HuR or hnRNP D (but not both), directing the fates of these transcripts toward either degradation or translation (63).
In our ATP depletion model, we cultured renal epithelial cells in the absence of d-glucose. Over the past few years, it has become clear that glucose plays an important role in regulation of V-ATPase activity. In Saccharomyces cerevisiae, removal of glucose induces a rapid dissociation of V-ATPase into V0 and V1 domains (4), an effect that requires the action of microtubules (64). V-ATPases reassemble when glucose is re-introduced to cells, and their stable assembly requires the presence of the regulator of the ATPase of vacuolar and endosomal membrane complex (8, 65). More recently, kidney V-ATPases were shown to bind glycolytic enzymes: aldolase via the E1 subunit (6, 7), and phosphofructokinase via the a4 subunit (9), suggesting a coupling mechanism with the glucose pathway. Further, as in S. cerevisiae, depleting renal epithelia of glucose induces disassembly into V0 and V1 domains, with reduced acidification of intracellular compartments, and loss of V-ATPases from the plasma membrane. In contrast, stimulation with glucose induced V-ATPase reassembly, activity, and translocation to the plasma membrane (10). Metabolism of glucose through the glycolytic pathway is not required for this stimulation, because 2-dexoyglucose, a glucose analog that readily enters the cytoplasm but is not metabolized further, was sufficient to provide these stimulatory effects. In our ATP depletion system, we deprived the cells of d-glucose but introduced l-glucose to maintain osmolarity. l-Glucose crosses the plasma membrane very slowly through passive diffusion, so it is very likely that V-ATPases did not remain assembled in our system. Nonetheless, the action of HuR maintained a pool of V-ATPase mRNAs so that protein levels remained constant.
In summary, these results show how AU-rich elements can contribute to the maintenance of even long-lived mRNAs under conditions of cell stress. These results also suggest the importance to kidney epithelia and likely, other cells, of maintaining a constant pool of V-ATPases through otherwise traumatic events. Because these transporters are so crucial to cell survival and so closely linked to cellular energetics, their levels are likely to be specifically preserved under a number of adverse conditions that could otherwise alter gene expression profiles throughout the cell.
Acknowledgments
We thank Dr. John Robinson (The Ohio State University) for use of his microscope in the early stages of this work.
Footnotes
This work was supported by Grant DK52131 from the National Institutes of Health (to B. S. L.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The abbreviations used are: V-ATPase, vacuolar H+-translocating ATPase; ARE, adeno-sine-uridine-rich element; UTR, untranslated region; siRNA, small interfering RNA; RT, reverse transcription; hnRNP, heterogeneous nuclear ribonucleoprotein; AMPK, AMP-activated protein kinase.
S. Jeyaraj, D. Dakhlallah, S. R. Hill, and B. S. Lee, manuscript in preparation.
S. Jeyaraj and B. S. Lee, unpublished data.
REFERENCES
- 1.Forgac M. J. Biol. Chem. 1999;274:12951–12954. doi: 10.1074/jbc.274.19.12951. [DOI] [PubMed] [Google Scholar]
- 2.Inoue H, Noumi T, Nagata M, Murakami H, Kanazawa H. Biochim. Biophys. Acta. 1999;1413:130–138. doi: 10.1016/s0005-2728(99)00096-1. [DOI] [PubMed] [Google Scholar]
- 3.Nelson H, Nelson N. Proc. Natl. Acad. Sci. U. S. A. 1990;87:3503–3507. doi: 10.1073/pnas.87.9.3503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kane PM. J. Biol. Chem. 1995;270:17025–17032. [PubMed] [Google Scholar]
- 5.Kane PM, Parra KJ. J. Exp. Biol. 2000;203:81–87. doi: 10.1242/jeb.203.1.81. [DOI] [PubMed] [Google Scholar]
- 6.Lu M, Holliday LS, Zhang L, Dunn WA, Jr., Gluck SL. J. Biol. Chem. 2001;276:30407–30413. doi: 10.1074/jbc.M008768200. [DOI] [PubMed] [Google Scholar]
- 7.Lu M, Sautin YY, Holliday LS, Gluck SL. J. Biol. Chem. 2004;279:8732–8739. doi: 10.1074/jbc.M303871200. [DOI] [PubMed] [Google Scholar]
- 8.Smardon AM, Tarsio M, Kane PM. J. Biol. Chem. 2002;277:13831–13839. doi: 10.1074/jbc.M200682200. [DOI] [PubMed] [Google Scholar]
- 9.Su Y, Zhou A, Al-Lamki RS, Karet FE. J. Biol. Chem. 2003;278:20013–20018. doi: 10.1074/jbc.M210077200. [DOI] [PubMed] [Google Scholar]
- 10.Sautin YY, Lu M, Gaugler A, Zhang L, Gluck SL. Mol. Cell Biol. 2005;25:575–589. doi: 10.1128/MCB.25.2.575-589.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gluck SL, Underhill DM, Iyori M, Holliday LS, Kostrominova TY, Lee BS. Annu. Rev. Physiol. 1996;58:427–445. doi: 10.1146/annurev.ph.58.030196.002235. [DOI] [PubMed] [Google Scholar]
- 12.Blair HC, Teitelbaum SL, Ghiselli R, Gluck S. Science. 1989;245:855–857. doi: 10.1126/science.2528207. [DOI] [PubMed] [Google Scholar]
- 13.Karet FE, Finberg KE, Nelson RD, Nayir A, Mocan H, Sanjad SA, Rodriguez-Soriano J, Santos F, Cremers CW, Di Pietro A, Hoffbrand BI, Winiarski J, Bakkaloglu A, Ozen S, Dusunsel R, Goodyer P, Hulton SA, Wu DK, Skvorak AB, Morton CC, Cunningham MJ, Jha V, Lifton RP. Nat. Genet. 1999;21:84–90. doi: 10.1038/5022. [DOI] [PubMed] [Google Scholar]
- 14.Breton S, Smith PJ, Lui B, Brown D. Nat. Med. 1996;2:470–472. doi: 10.1038/nm0496-470. [DOI] [PubMed] [Google Scholar]
- 15.Breton S, Tyszkowski R, Sabolic I, Brown D. Histochem. Cell Biol. 1999;111:97–105. doi: 10.1007/s004180050339. [DOI] [PubMed] [Google Scholar]
- 16.Smith AN, Skaug J, Choate KA, Nayir A, Bakkaloglu A, Ozen S, Hulton SA, Sanjad SA, Al-Sabban EA, Lifton RP, Scherer SW, Karet FE. Nat. Genet. 2000;26:71–75. doi: 10.1038/79208. [DOI] [PubMed] [Google Scholar]
- 17.Frattini A, Orchard PJ, Sobacchi C, Giliani S, Abinun M, Mattsson JP, Keeling DJ, Andersson AK, Wallbrandt P, Zecca L, Notarangelo LD, Vezzoni P, Villa A. Nat. Genet. 2000;25:343–346. doi: 10.1038/77131. [DOI] [PubMed] [Google Scholar]
- 18.Kornak U, Schulz A, Friedrich W, Uhlhaas S, Kremens B, Voit T, Hasan C, Bode U, Jentsch TJ, Kubisch C. Human Mol. Genet. 2000;9:2059–2063. doi: 10.1093/hmg/9.13.2059. [DOI] [PubMed] [Google Scholar]
- 19.Smith AN, Lovering RC, Futai M, Takeda J, Brown D, Karet FE. Mol. Cell. 2003;12:801–803. doi: 10.1016/s1097-2765(03)00397-6. [DOI] [PubMed] [Google Scholar]
- 20.Wang SP, Krits I, Bai S, Lee BS. J. Biol. Chem. 2002;277:8827–8834. doi: 10.1074/jbc.M111959200. [DOI] [PubMed] [Google Scholar]
- 21.Caput D, Beutler B, Hartog K, Thayer R, Brown-Shimer S, Cerami A. Proc. Natl. Acad. Sci. U. S. A. 1986;83:1670–1674. doi: 10.1073/pnas.83.6.1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shaw G, Kamen R. Cell. 1986;46:659–667. doi: 10.1016/0092-8674(86)90341-7. [DOI] [PubMed] [Google Scholar]
- 23.Wilson T, Treisman R. Nature. 1988;336:396–399. doi: 10.1038/336396a0. [DOI] [PubMed] [Google Scholar]
- 24.Lopez de Silanes I, Zhan M, Lal A, Yang X, Gorospe M. Proc. Natl. Acad. Sci. U. S. A. 2004;101:2987–2992. doi: 10.1073/pnas.0306453101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen CY, Shyu AB. Trends Biochem. Sci. 1995;20:465–470. doi: 10.1016/s0968-0004(00)89102-1. [DOI] [PubMed] [Google Scholar]
- 26.Zhang T, Kruys V, Huez G, Gueydan C. Biochem. Soc. Trans. 2002;30:952–958. doi: 10.1042/bst0300952. [DOI] [PubMed] [Google Scholar]
- 27.Bevilacqua A, Ceriani MC, Capaccioli S, Nicolin A. J. Cell Physiol. 2003;195:356–372. doi: 10.1002/jcp.10272. [DOI] [PubMed] [Google Scholar]
- 28.Andrews GK, Harding MA, Calvet JP, Adamson ED. Mol. Cell Biol. 1987;7:3452–3458. doi: 10.1128/mcb.7.10.3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang W, Furneaux H, Cheng H, Caldwell MC, Hutter D, Liu Y, Holbrook N, Gorospe M. Mol. Cell Biol. 2000;20:760–769. doi: 10.1128/mcb.20.3.760-769.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Westmark CJ, Bartleson VB, Malter JS. Oncogene. 2005;24:502–511. doi: 10.1038/sj.onc.1208224. [DOI] [PubMed] [Google Scholar]
- 31.Levy NS, Chung S, Furneaux H, Levy AP. J. Biol. Chem. 1998;273:6417–6423. doi: 10.1074/jbc.273.11.6417. [DOI] [PubMed] [Google Scholar]
- 32.Yaman I, Fernandez J, Sarkar B, Schneider RJ, Snider MD, Nagy LE, Hatzoglou M. J. Biol. Chem. 2002;277:41539–41546. doi: 10.1074/jbc.M204850200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Padanilam BJ. Am. J. Physiol. 2003;284:F608–F627. doi: 10.1152/ajprenal.00284.2002. [DOI] [PubMed] [Google Scholar]
- 34.Canfield PE, Geerdes AM, Molitoris BA. Am. J. Physiol. 1991;261:F1038–F1045. doi: 10.1152/ajprenal.1991.261.6.F1038. [DOI] [PubMed] [Google Scholar]
- 35.Chen J, Wagner MC. Am. J. Physiol. 2001;280:F619–F627. doi: 10.1152/ajprenal.2001.280.4.F619. [DOI] [PubMed] [Google Scholar]
- 36.Fish EM, Molitoris BA. Am. J. Physiol. 1994;267:F566–F572. doi: 10.1152/ajprenal.1994.267.4.F566. [DOI] [PubMed] [Google Scholar]
- 37.Meldrum KK, Meldrum DR, Sezen SF, Crone JK, Burnett AL. Am. J. Physiol. 2001;281:R359–R364. doi: 10.1152/ajpregu.2001.281.1.R359. [DOI] [PubMed] [Google Scholar]
- 38.van Why SK, Kim S, Geibel J, Seebach FA, Kashgarian M, Siegel NJ. Am. J. Physiol. 1999;277:F227–F234. doi: 10.1152/ajprenal.1999.277.2.F227. [DOI] [PubMed] [Google Scholar]
- 39.Lee BS, Underhill DM, Crane MK, Gluck SL. J. Biol. Chem. 1995;270:7320–7329. doi: 10.1074/jbc.270.13.7320. [DOI] [PubMed] [Google Scholar]
- 40.Lee BS, Holliday LS, Krits I, Gluck SL. J. Bone Miner. Res. 1999;14:2127–2136. doi: 10.1359/jbmr.1999.14.12.2127. [DOI] [PubMed] [Google Scholar]
- 41.Zeng Q, Lagunoff D, Masaracchia R, Goeckeler Z, Cote G, Wysolmerski R. J. Cell Sci. 2000;113:471–482. doi: 10.1242/jcs.113.3.471. [DOI] [PubMed] [Google Scholar]
- 42.Yeap BB, Voon DC, Vivian JP, McCulloch RK, Thomson AM, Giles KM, Czyzyk-Krzeska MF, Furneaux H, Wilce MC, Wilce JA, Leedman PJ. J. Biol. Chem. 2002;277:27183–27192. doi: 10.1074/jbc.M202883200. [DOI] [PubMed] [Google Scholar]
- 43.Wagner BJ, DeMaria CT, Sun Y, Wilson GM, Brewer G. Genomics. 1998;48:195–202. doi: 10.1006/geno.1997.5142. [DOI] [PubMed] [Google Scholar]
- 44.Fan XC, Steitz JA. EMBO J. 1998;17:3448–3460. doi: 10.1093/emboj/17.12.3448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen CY, Xu N, Shyu AB. Mol. Cell Biol. 2002;22:7268–7278. doi: 10.1128/MCB.22.20.7268-7278.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang W, Caldwell MC, Lin S, Furneaux H, Gorospe M. EMBO J. 2000;19:2340–2350. doi: 10.1093/emboj/19.10.2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Atasoy U, Watson J, Patel D, Keene JD. J. Cell Sci. 1998;111:3145–3156. doi: 10.1242/jcs.111.21.3145. [DOI] [PubMed] [Google Scholar]
- 48.Peng SS, Chen CY, Xu N, Shyu AB. EMBO J. 1998;17:3461–3470. doi: 10.1093/emboj/17.12.3461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gallouzi IE, Brennan CM, Steitz JA. RNA (N. Y.) 2001;7:1348–1361. doi: 10.1017/s1355838201016089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bakheet T, Frevel M, Williams BR, Greer W, Khabar KS. Nucleic Acids Res. 2001;29:246–254. doi: 10.1093/nar/29.1.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bakheet T, Williams BR, Khabar KS. Nucleic Acids Res. 2003;31:421–423. doi: 10.1093/nar/gkg023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Frevel MA, Bakheet T, Silva AM, Hissong JG, Khabar KS, Williams BR. Mol. Cell Biol. 2003;23:425–436. doi: 10.1128/MCB.23.2.425-436.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lee BS, Krits I, Crane-Zelkovic MK, Gluck SL. J. Biol. Chem. 1997;272:174–181. [PubMed] [Google Scholar]
- 54.Wang W, Fan J, Yang X, Furer-Galban S, Lopez de Silanes I, von Kobbe C, Guo J, Georas SN, Foufelle F, Hardie DG, Carling D, Gorospe M. Mol. Cell Biol. 2002;22:3425–3436. doi: 10.1128/MCB.22.10.3425-3436.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang W, Yang X, Kawai T, Lopez de Silanes I, Mazan-Mamczarz K, Chen P, Chook YM, Quensel C, Kohler M, Gorospe M. J. Biol. Chem. 2004;279:48376–48388. doi: 10.1074/jbc.M409014200. [DOI] [PubMed] [Google Scholar]
- 56.Huwiler A, Akool E-S, Aschrafi A, Hamada FM, Pfeilschifter J, Eberhardt W. J. Biol. Chem. 2003;278:51758–51769. doi: 10.1074/jbc.M305722200. [DOI] [PubMed] [Google Scholar]
- 57.Fraser S, Mount P, Hill R, Levidiotis V, Katsis F, Stapleton D, Kemp BE, Power DA. Am. J. Physiol. 2005;288:F578–F586. doi: 10.1152/ajprenal.00190.2004. [DOI] [PubMed] [Google Scholar]
- 58.Witzgall R. Exp. Nephrol. 1999;7:15–19. doi: 10.1159/000020579. [DOI] [PubMed] [Google Scholar]
- 59.Gouble A, Morello D. Oncogene. 2000;19:5377–5384. doi: 10.1038/sj.onc.1203910. [DOI] [PubMed] [Google Scholar]
- 60.Lu JY, Schneider RJ. J. Biol. Chem. 2004;279:12974–12979. doi: 10.1074/jbc.M310433200. [DOI] [PubMed] [Google Scholar]
- 61.Nishi T, Forgac M. J. Biol. Chem. 2000;275:6824–6830. doi: 10.1074/jbc.275.10.6824. [DOI] [PubMed] [Google Scholar]
- 62.Sun-Wada GH, Yoshimizu T, Imai-Senga Y, Wada Y, Futai M. Gene (Amst.) 2003;302:147–153. doi: 10.1016/s0378-1119(02)01099-5. [DOI] [PubMed] [Google Scholar]
- 63.Lal A, Mazan-Mamczarz K, Kawai T, Yang X, Martindale JL, Gorospe M. EMBO J. 2004;23:3092–3102. doi: 10.1038/sj.emboj.7600305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Xu T, Forgac M. J. Biol. Chem. 2001;276:24855–24861. doi: 10.1074/jbc.M100637200. [DOI] [PubMed] [Google Scholar]
- 65.Seol JH, Shevchenko A, Shevchenko A, Deshaies RJ. Nature Cell Biol. 2001;3:384–391. doi: 10.1038/35070067. [DOI] [PubMed] [Google Scholar]
- 66.Underhill DM. Assembly and Disassembly of the Mammalian Vascular H+-ATPase. Washington University; St. Louis, MO: 1995. Ph.D. thesis. [Google Scholar]