

Abstract
Treatment of macrophages with pyridinyl imidazole inhibitors of p38 protein kinases can inhibit lipopolysaccharide-stimulated tumor necrosis factor α secretion. However, bone marrow-derived macrophages from tristetraprolin (TTP)-deficient mice were less sensitive than normal macrophages to this effect of p38 inhibitors, despite evidence for normal p38 activation in response to lipopolysaccharide. TTP is known to cause decreased stability of tumor necrosis factor α and granulocyte-macrophage colony-stimulating factor mRNAs after binding to an AU-rich element in their 3′ -untranslated regions. A recombinant TTP fusion protein could be phosphorylated by a recombinant p38 kinase in cell-free assays and was phosphorylated to the same extent by immunoprecipitated p38 derived from normal and TTP-deficient cells stimulated with lipopolysaccharide; in both cases, the enzyme activity was inhibited by the p38 inhibitors. TTP phosphorylation also was increased in intact macrophages after lipopolysaccharide stimulation, an effect that was blocked by the p38 inhibitors. Finally, TTP in mammalian cell extracts bound less well to an AU-rich element RNA probe than did the same amount of TTP following dephosphorylation. These results suggest that TTP may be a component of the signaling cascade, initiated by inflammatory stimuli and mediated in part by activation of p38, that ultimately leads to enhanced secretion of tumor necrosis factor α.
Lipolysaccharide (LPS)1-induced production of tumor necrosis factor α (TNFα) by monocyte/macrophages is regulated at both transcriptional and post-transcriptional levels. Post-transcriptional regulation of TNFα synthesis occurs in part by modulation of its mRNA stability. This in turn is dependent upon a so-called class II AU-rich element (ARE) found in the 3′-untranslated region of TNFα transcripts (1). This ARE has been implicated in the regulation of both TNFα mRNA stability and its translation (2, 3). Targeted deletion of the TNFα mRNA ARE in mice (△ARE mice) results in the overproduction of TNFα and the development of a systemic inflammatory syndrome (4). A role for the protein serine/threonine kinase p38 has been suggested in ARE-mediated TNFα mRNA processing by numerous studies (5-7), and it was found recently that macrophages from the △ARE mice were relatively insensitive to the p38 inhibitor, SB203580 (4). Conflicting studies suggest that these p38 inhibitors can regulate TNFα synthesis at either the mRNA stability or protein translation level (8-10). Mice lacking the p38 substrate MAPKAPK-2 have been reported to have defective TNFα synthesis following an LPS challenge (11). In this case, the regulation appears not to be due to a decrease in either TNFα mRNA levels or stability but rather to inhibition of translation, suggesting that the effects of the p38 pathway on mRNA stability and translation may be independent and uncoupled.
These and other studies have indicated a role for the p38 signaling pathway in the post-transcriptional regulation of TNFα synthesis through a mechanism involving the ARE. p38 belongs to the growing family of mitogen-activated protein kinases (MAPK). Stress signals, such as LPS, heat shock, and ultraviolet light can initiate a signaling cascade resulting in the activation, by dual tyrosine/threonine phosphorylation, of p38. The activation of p38 results in the phosphorylation of intracellular substrates, among them MAPKAPK-2 and the activating transcription factor 2 (12, 13). There are five known isoforms of p38 (α, β, β2, γ, and δ) in mammals, which differ in expression patterns, activators, inhibitors, and substrate specificity (14).
We have shown previously that the RNA-binding protein tristetraprolin (TTP) promotes TNFα mRNA instability in mouse macrophages through direct interactions with its ARE (15). TTP deficiency in mice results in a severe inflammatory syndrome, characterized by severe polyarticular arthritis, myeloid hyperplasia, autoimmunity, and cachexia (16). This syndrome is largely the result of increased stability of the mRNAs for TNFα and granulocyte-macrophage colony-stimulating factor (GM-CSF) and increased secretion of these cytokines (15, 17, 18). We showed earlier that TTP can be phosphorylated on at least one serine by p42 MAPK (19), and that there are several other consensus phosphorylation sites for mitogen- or stress-activated proline-directed protein kinases in TTP. These observations, together with the characteristics of the inflammatory syndrome exhibited by the TTP-deficient (TTPKO) mice and the fact that TTP expression is induced by several of the same stimuli that activate p38, suggested the possibility that TTP could be part of the signaling cascade through which p38 kinase regulates the stability of certain cytokine mRNAs.
In this paper, we show that bone marrow-derived macrophages (BMMϕ) from TTP-deficient mice are less sensitive than normal macrophages to the p38 kinase inhibitors SB203580 and SB220025, which normally inhibit LPS-stimulated TNFα secretion from these cells. We also show that TTP can be phosphorylated by p38 in a cell-free system and that LPS-stimulated phosphorylation of TTP in macrophages can be inhibited by p38 inhibitors. The absence of TTP did not affect the ability of LPS to activate p38, and p38 derived from TTP-deficient cells was normally sensitive to the p38 inhibitors in a cell-free assay. Finally, we demonstrated that phosphorylated TTP expressed in 293 cells bound less avidly to an ARE RNA probe than did dephosphorylated TTP. These data indicate that TTP may play an important role in mediating the downstream effects of p38 activation and thus may represent a novel therapeutic target for anti-inflammatory drug development.
EXPERIMENTAL PROCEDURES
Mice deficient in TTP, their genotyping, and maintenance of the colony have been described elsewhere (16). Bone marrow-derived macrophages were prepared as described (17) and used after 2 weeks in culture. To measure TNFα secretion, BMMϕ were plated in 96-well plates at a density of 1.2 × 106 cells/ml together with increasing concentrations of the p38 inhibitors SB203580 (7) and SB220025 (20) and the TNFα-converting enzyme (TACE) inhibitor Marimastat (21) (all three compounds were synthesized at AstraZeneca). After a 30-min incubation, LPS (Sigma) was added at 0.1 or 1 μg/ml. Cells were incubated with LPS for 24 h, and supernatants were harvested for the measurement of TNFα accumulation by conventional ELISA techniques (R & D Systems, Minneapolis, MN), as specified by the manufacturer. Duplicate samples were analyzed per data point; each data point represents the average of results from three animals per genotype.
For Western blotting, confluent cultures of BMMϕ were stimulated with LPS (1 μg/ml) for the indicated times and processed as described (22). l00 μg of protein was separated by 12% SDS-polyacrylamide gel electrophoresis (PAGE), transferred to nitrocellulose, and sequentially probed with antibodies against phosphorylated p38 (Cell Signaling, Beverly, MA) or against total p38 (A-12) (Santa Cruz Biotechnology, Inc., Santa Cruz, CA). Detection was performed with the SuperSignal West Pico Chemiluminescent Substrate (Pierce).
For kinase assays, GST-p38 was purchased from Calbiochem and used in a cell-free kinase assay using 5 μg of the maltose-binding protein-human TTP (MBP-hTTP) or maltose-binding protein-mouse TTP (MBP-mTTP ) fusion proteins as substrates. These recombinant proteins were produced in Escherichia coli as amino-terminal fusions of TTP with maltose-binding protein and affinity-purified on amylose resin (New England Biolabs, Beverly, MA) as described previously (23). We also expressed and purified the MBP protein as a control. The details of fusion protein expression and purification will be described elsewhere. The conditions for p38 phosphorylation of the purified fusion proteins were as recommended by the manufacturer. Where indicated, SB203580 or SB220025 was used to inhibit p38 kinase activity at 5 μM. Phosphorylated proteins were resolved by 10% SDS-PAGE, followed by autoradiography. For kinase assays in which endogenous p38 was used, BMMϕ were stimulated with LPS (1 μg/ml) for 15 min, and the cells were harvested as described (22), with the following modifications: sodium deoxycholate and SDS were omitted from the wash buffer, and the kinase reaction was performed at 37 °C for 30 min. MBP-mTTP (2.5 μg) was used as the substrate. Phosphorylated proteins were resolved by 11% SDS-PAGE, followed by autoradiography.
To evaluate TTP phosphorylation in intact cells, confluent 60-mm dishes of BMMϕ were washed with phosphate-free Dulbecco's modified Eagle's medium and incubated for 4 h in the presence of 200 μCi/ml [32P]orthophosphoric acid (PerkinElmer Life Sciences). The p38 inhibitors SB203580 and SB220025 (5 μM) were then added for 30 min followed by 1 μg/ml LPS for another 30 min. Cells were harvested and lysed as described above, and equal amounts of trichloroacetic acid-precipitable counts were immunoprecipitated using a specific anti-serum against TTP (19). Phosphorylated proteins were resolved by 12% SDS-PAGE, followed by autoradiography and quantification using a PhosphorImager Typhoon 8600 and ImageQuant software (Molecular Dynamics, Sunnyvale, CA).
For the 293 cell transfection experiments, we used the TTP expression plasmid CMV.hTTP.tag (24) and plasmid CMV.(his)6N.hTTP. By using the original human TTP cDNA as a template, the latter plasmid was generated by adding oligonucleotides encoding the six histidines using the polymerase chain reaction primer-overlapping mutagenesis technique (25) after the first amino acid of hTTP. Correct sequence of the fusion cDNA was confirmed by dRhodamine Terminator Cycle Sequencing (PerkinElmer Life Sciences). HEK 293 cells (referred to as 293 cells) were maintained, and transient transfection of 1.2 × 106 cells per 100-mm plate with plasmid constructs in calcium-phosphate precipitates was performed, as described (26). Either 5 μg of vector BS+ DNA or 0.5 μg of TTP expression plasmid DNA plus 4.5 μg of vector BS+ DNA was used in each plate. Cytosolic extracts were prepared 24 h after the removal of the transfection mixture as described (26).
To evaluate the effect of TTP dephosphorylation on its binding to RNA, we first performed experiments involving transfected 293 cells and labeling with [32P]orthophosphate. Twenty four h after the removal of the transfection mixture, the culture medium was replaced with 1% (v/v) fetal calf serum in Dulbecco's modified Eagle's medium without phosphate for 2 h at 37 °C. [32P]Orthophosphate (0.2 mCi per ml) was added, and the cells were incubated for an additional 2 h. To prepare cell extracts, the medium was removed, and the cell monolayers were washed three times in ice-cold phosphate-buffered saline. The cells were then lysed in 0.6 ml per 60-mm culture dish of the lysis buffer described previously (24). The extracts were then placed in plastic microcentrifuge tubes and sonicated in an ice water bath in a Heat Systems Ultrasonics, Inc., w-380 Sonicator, setting 8, three times for 10 s each time. The samples were then rotated for 15 min at 4 °C, centrifuged at 10,000 × g for 10 min, and the supernatants removed and frozen at −70 °C until used. Cell extracts prepared from 32P-labeled 293 cells transfected with CMV.(his)6N.hTTP (20 μg of protein) were diluted 1:10 in dephosphorylation buffer (50 mM Tris-HCl (pH 8.5) and 0.1 mM EDTA) and were incubated at 30 °C for 1 h with or without 570 units/ml calf intestinal alkaline phosphatase (CIAP, Stratagene, La Jolla, CA). The CIAP activity was stopped by adding 1 μl of 0.1 M Na2HPO4 (pH 6.5). The reaction mixtures were then rapidly frozen and stored at −20 °C for further analysis. Equal volumes of sample were then loaded onto 12% (w/v) gels and subjected to SDS-PAGE, after which the gels were dried and used for autoradiography.
Cell extracts were prepared in a similar fashion from unlabeled 293 cells transfected with CMV.hTTP.tag or CMV.(his)6N.hTTP. The extracts contained 40 μg of cellular protein in 0.2 ml of 50 mM Tris-HCl (pH 8.5), 0.1 mM EDTA, and 285 units/ml CIAP. Each extract, from six independent but similar transfections, was divided into two tubes and kept on ice. To one tube, 1 M Na2HPO4 (pH 6.5) was added to a final concentration of 100 mM to inactivate the CIAP. Both tubes were then incubated at 30 °C for 2 h, after which the same volume of 1 M Na2HPO4 was added to the other tube. This method was used so that the two tubes from each experiment would contain identical concentrations of all reactants at the conclusion of the experiment. The only difference was that the phosphatase inhibitor was added before the 30 °C incubation in one tube and after the 30 °C incubation in the other tube. At the conclusion of the incubation, the extracts were rapidly frozen and stored at −20 °C for further analysis.
For Western blotting of these extracts, samples containing 5 μg of protein in 0.025 ml of the reaction buffer described above containing CIAP and Na2HPO4 were mixed with 1/5 volume of 5× SDS sample buffer, boiled for 5 min, and then loaded onto 12% SDS-PAGE gels. Western blotting was performed by standard techniques. Membranes were incubated in Tris-buffered saline, 0.3% Tween 20 (TBS/T) with a polyclonal antiserum directed at a recombinant human TTP fusion protein with maltose-binding protein.2
For RNA gel mobility shift assays, the RNA probe was derived from plasmid pGM-CSF-ARE (base pairs 3399–3453 of GenBank™ accession number X03020), containing the AU-rich element in the mouse GM-CSF 3′-untranslated region, 5′-AUUUAUUUAUAUAUUUAUAUUUUUUAAAUAUUUAUUUAUUUAUUUAUUUAUUUUU-3′. This was constructed by inserting double-stranded synthetic oligonucleo tides into the EcoRV-XbaI cloning sites of pSK–. Correct sequence of the plasmid insert was confirmed by dRhodamine Terminator Cycle Sequencing (PerkinElmer Life Sciences). To label the RNA transcript with [α-32P]UTP (800 Ci/mmol), plasmid pGM-CSF-ARE linearized with XbaI was used as a template, and the Promega Riboprobe in vitro Transcription Systems protocol was employed. The resulting radioactive products were separated from the free nucleotides through G-50 columns.
For the gel shift assays, cytosolic extracts prepared from 293 cells transfected with TTP expression constructs were incubated with CIAP, and the phosphatase inhibitor was added either before or after the 30 °C incubation as described above. Various amounts of protein (0.4–1.2 μg) in 2–6 μl of the reaction mixture described above containing CIAP and Na2HPO4 were added to 25 μl of the lysis buffer described above, without protease inhibitors, containing 2 × 105 cpm of RNA probe, and used in mobility shift assays as described (24, 26).
RESULTS
Effect of p38 Inhibitors on TNFα Production by WT and TTPKO Bone Marrow-derived Macrophages—We first evaluated the effect of two p38 inhibitors, SB203580 and SB220025, on the secretion of TNFα by BMMϕ derived from either WT or TTPKO mice. Both p38 inhibitors decreased LPS-stimulated TNFα synthesis in WT BMMϕ, albeit at higher concentrations than required to inhibit TNFα synthesis by LPS-stimulated human monocyte/macrophages. Differences in sensitivity to various p38 inhibitors among animal species have been reported previously (27, 28).3 As shown in Fig. 1A, SB203580 effectively inhibited the LPS-stimulated secretion of TNFα by BMMϕ derived from WT mice (solid circles). The IC50 for this compound was ∼5 μM, similar to the value obtained in previous studies with mouse splenocytes3 and thioglycollate-elicited peritoneal macrophages (4). However, when the same compound was used in parallel experiments in BMMϕ derived from animals deficient in TTP (open circles), there was no inhibition of TNFα production at any concentration of SB203580 used (Fig. 1A).
Fig. 1.
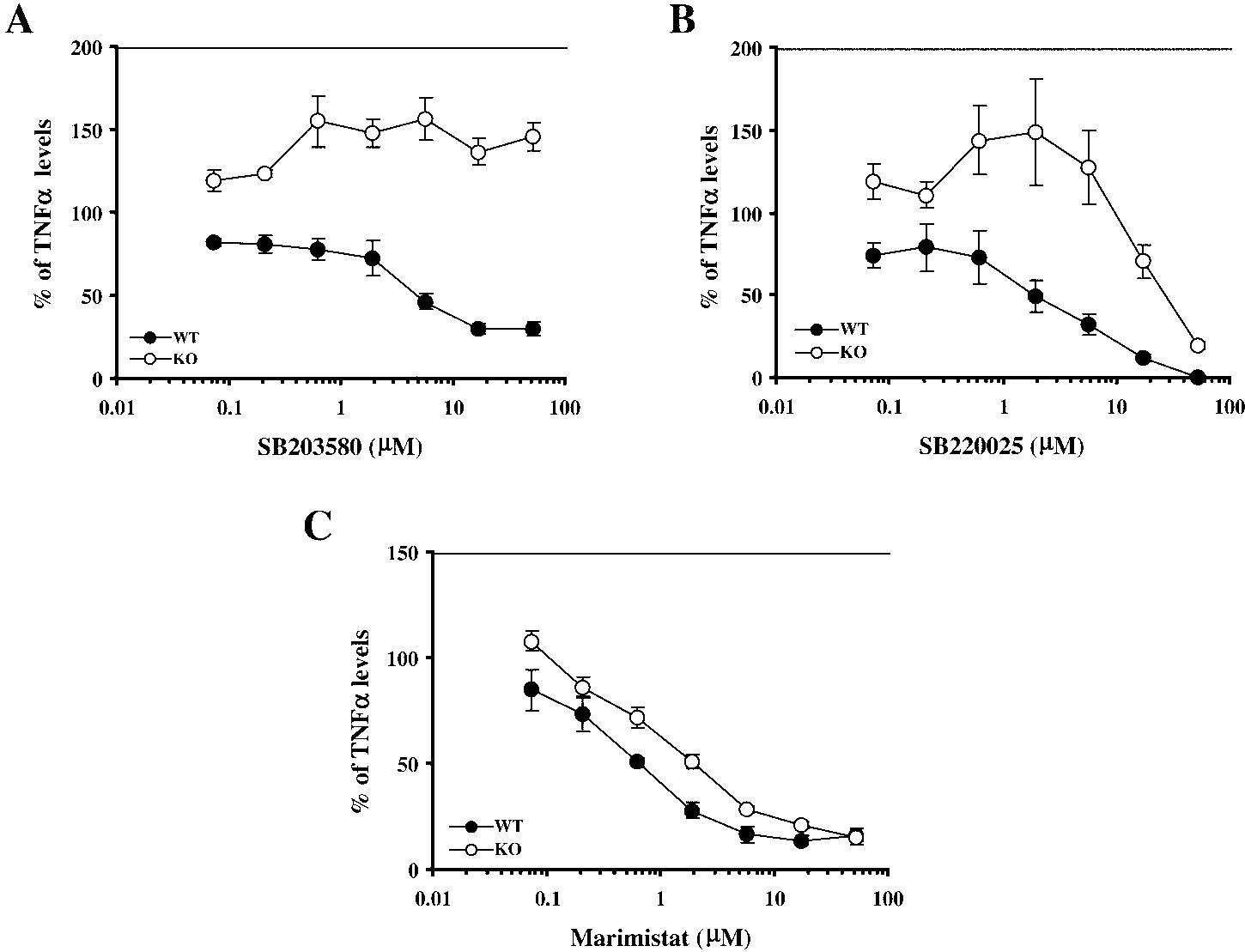
Resistance of TTP-deficient BMMϕ to p38 inhibitors. A,SB203580; B, SB220025; C, Marimastat. BMMϕ derived from WT (solid circles) or TTPKO (open circles) mice were incubated in the presence of increasing concentrations of p38 inhibitors or Marimastat for 30 min, followed by 24 h of incubation with 1 μg/ml LPS. Supernatants were then harvested and TNFα levels were determined by ELISA. Each data point represents the average ± S.E. of duplicate ELISA determinations from two samples per animal from three animals. The values are expressed as the % of LPS-stimulated TNFα levels in the absence of inhibitors. These values for A were 2.5 ± 1.2 ng/ml for the WT and 6.3 ± 1.6 ng/ml for the TTPKO cells; for B, 2.2 ± 1.2 ng/ml for the WT and 8 ± 2 ng/ml for the TTPKO cells; and for C, 1.8 ± 0.9 ng/ml or the WT and 8.8 ± 1.6 ng/ml for the TTPKO cells (mean ± S.E.).
When SB220025 was used, the compound inhibited the production of TNFα by the WT BMMϕ with an IC50 of ∼2 μM (Fig. 1B), similar to the concentrations required previously.3 However, in the TTPKO BMMϕ, the effective inhibitory concentration was shifted markedly to the right, with an IC50 of ∼30 μM (Fig. 1B). Similar results were observed when the cells were stimulated with a lower concentration of LPS, 0.1 μg/ml, and then exposed to the p38 inhibitors (data not shown).
The specificity of this resistance of TTPKO-derived BMMϕ to p38 inhibitors was tested by using a completely unrelated inhibitor of TNFα secretion, the TACE inhibitor Marimastat (21). As seen in Fig. 1C, both WT and TTPKO BMMϕ responded similarly to Marimastat, with the IC50 for inhibition of TNFα secretion being 0.6 μM in the WT and 2 μM in the TTPKO cells, respectively.
Activation of p38 in Bone Marrow-derived Macrophages—To determine whether LPS could activate p38 to the same extent in the WT and TTPKO BMMϕ, the cells were stimulated with LPS (1 μg/ml), followed by immunoblotting for phosphorylated (activated) and total p38. This resulted in the rapid (within 5 min) phosphorylation of p38, which peaked at 15–30 min, and was still apparent after 60 min (Fig. 2). The pattern of phosphorylation was very similar in WT and TTPKO cells (Fig. 2), suggesting that activation of p38 occurred normally, even in the absence of TTP.
Fig. 2.
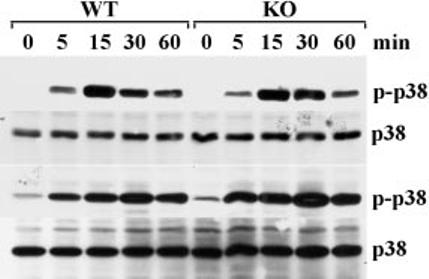
p38 phosphorylation in TTP-deficient and control BMMϕ. BMMϕ were stimulated with LPS (1 μg/ml) for the indicated times. The cells were then washed, harvested, homogenized, and centrifuged as described under “Experimental Procedures,” and then 100 μg of protein from each plate was subjected to 12% SDS-PAGE, and immunoblotted with either an antibody to phosphorylated p38 (p-p38) or to total p38 (p38). Phosphorylated p38 was readily detected within 5 min of LPS stimulation, peaked at 15–30 min, and was still detectable after 60 min. Results from cells derived from two different pairs of age-matched animals are shown in the upper and lower panels. Similar results were obtained with four independent pairs of age-matched animals.
Phosphorylation of a Recombinant MBP-TTP Fusion Protein by Recombinant p38—To determine whether recombinant TTP could serve as a substrate for p38, we performed cell-free kinase assays using commercially available p38, in the form of a GST-p38 fusion protein, and recombinant mouse and human TTP (as fusion proteins with maltose-binding protein or MBP) as substrates. As shown in Fig. 3, p38 was able to phosphorylate both MBP-mTTP (lane 2) and MBP-hTTP (lane 6) (Mr ∼75,000 for both fusion proteins) in this cell-free system. Both SB203580 (5 μM, lanes 3 and 7) and SB220025 (5 μM, lanes 4 and 8) inhibited this phosphorylation. No phosphorylation was observed when the substrate was the same amount (5 μg) of recombinant MBP alone (lane 10) (predicted Mr 41,000). No phosphorylation of either fusion protein or MBP was observed when the p38 kinase was omitted from the reaction (lanes 5, 9, and 11), ruling out the possibility of contaminating kinases in the MBP-TTP preparations. The GST-p38 kinase fusion protein itself appeared to be phosphorylated in these reactions and could be seen as a radioactive band of Mr 68,000 (Fig. 3, lanes 1, 2, 6, and 10). This phosphorylation was also abolished by the p38 inhibitors.
Fig. 3.
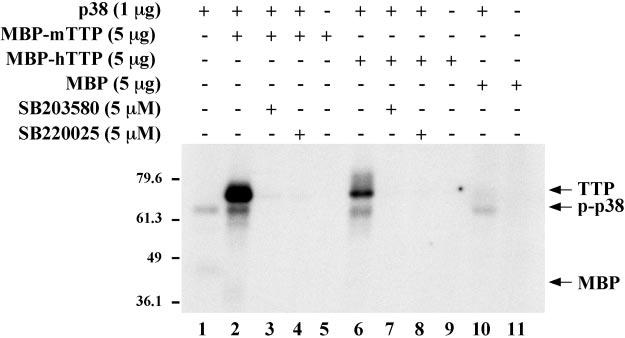
GST-p38 phosphorylation of mouse and human TTP in a cell-free assay. 1 μg of commercial recombinant GST-p38 (p38) was used in cell-free phosphorylation assays using recombinant MBP-mTTP or MBP-hTTP (5 μg) as substrates. GST-p38 could phosphorylate both mouse (lane 2) and human (lane 6) recombinant MBP-TTP; note that the Mr of both fusion proteins is ∼75,000. Background GST-p38 phosphorylation can be seen in lanes 1, 2, 6, and 10 as a radioactive band of Mr ∼68,000 (p-p38). The addition of the p38 inhibitors SB203580 (5 μm) (lanes 3 and 7) or SB220025 (5 μm) (lanes 4 and 9) prevented the phosphorylation of both MBP-TTP fusion proteins and the phosphorylation of p38. No phosphorylation of the MBP-TTP fusion proteins was detected in the absence of p38 (lanes 5 and 9) or when only MBP was present in the reaction (lane 11). The numbers on the left indicate Mr × 10−3. The experiment shown is representative of two independent experiments.
Phosphorylation of TTP in Intact Cells—To determine whether activation of the p38 kinase pathway could lead to TTP phosphorylation in intact cells, we labeled BMMϕ derived from both WT and TTPKO mice with [32P]orthophosphate, and we stimulated them with LPS (1 μg/ml for 30 min) in the presence or absence of the p38 inhibitors (5 μm). Under these conditions (30 min stimulation with LPS), TTP protein levels were not altered.4 As shown in Fig. 4, a specific antiserum against TTP immunoprecipitated a protein of approximately Mr 43,000, whose phosphorylation was increased approximately 3-fold after LPS stimulation (compare lane 2 with lane 1). Preincubation with either SB203580 (lane 3) or SB220025 (lane 4) decreased the level of TTP phosphorylation by ∼50%. The immunoprecipitated, 32P-labeled protein was completely absent in the TTPKO cells (lanes 5–8), confirming its identity as TTP.
Fig. 4.
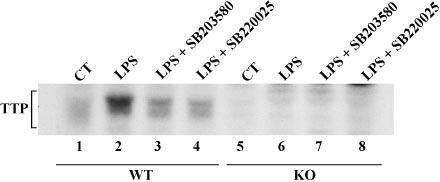
Effect of p38 inhibitors on LPS-stimulated phosphorylation of TTP in BMMϕ. BMMϕ labeled with [32P]orthophosphoric acid were treated as described under “Experimental Procedures,” and cell extracts were immunoprecipitated with a specific anti-TTP antiserum; an autoradiograph of a 12% SDS-PAGE gel containing the immunoprecipitated phosphoprotein is shown. LPS stimulation (1 μg/ml for 30 min) stimulated the phosphorylation of a protein of apparent average Mr 43,000 in the WT cells (lanes 1–4), accompanied by an apparent increase in its average Mr as shown previously (19). Radioactive TTP was absent in the TTPKO cells (lanes 5– 8). The extent of phosphorylation was markedly inhibited by the use of the p38 inhibitors (5 μm), as indicated.
p38 from TTPKO Bone Marrow-derived Macrophages Is Active and Sensitive to p38 Inhibitors—BMMϕ derived from both WT and TTPKO mice were stimulated with LPS (1 μg/ml) for 15 min, and p38 was immunoprecipitated with a specific antibody for total p38. This immunoprecipitate was then used in a cell-free protein kinase assay, using recombinant MBP-mTTP as the substrate, in the presence or absence of 5 μm SB220025. As shown in Fig. 5, p38 immunoprecipitated from either LPS-stimulated WT or TTPKO cells could phosphorylate MBPmTTP to approximately the same extent (lanes 2 and 5), and this phosphorylation could be inhibited by SB220025 (lanes 3 and 6). No phosphorylated proteins were observed in the absence of immunoprecipitated p38 (lane 9) or in the absence of MBP-mTTP (lanes 7 and 8), ruling out the possibility that significant concentrations of nonspecific kinases or substrates were present in the reactions. These results suggested that p38 kinase could be activated by LPS in both the TTPKO and the WT macrophages and that in both cases the kinase was sensitive to the action of p38 inhibitors in the cell-free kinase assay.
Fig. 5.
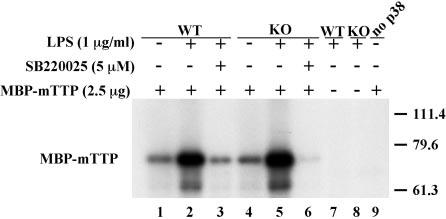
Phosphorylation of recombinant TTP by endogenous BMMϕ p38. BMMϕ were stimulated with LPS (1 μg/ml) for 15 min, and then p38 was immunoprecipitated as described under “Experimental Procedures.” Immune complexes were then used in cell-free kinase assays using MBP-mTTP (2.5 μg) as a substrate. p38 immunoprecipitated from both WT and TTPKO cells was capable of phosphorylating recombinant TTP in an LPS-activated manner (lanes 1, 2, 4, and 5); this LPS-stimulated phosphorylation was sensitive to the p38 inhibitor SB220025 added to the kinase reaction (5 μm) (lanes 3 and 6). Lanes 7 and 8 represent controls in which the substrate (MBP-mTTP) was omitted from the kinase reaction to rule out the presence of a p38 substrate in the immunoprecipitates. Lane 9 represents a control in which immunoprecipitates were omitted (no p38) to control for the presence of contaminating kinases in the MBP-mTTP preparation. The numbers on the right indicate Mr × 10−3.
Effect of Dephosphorylation of Cell-expressed TTP on ARE Binding—We next evaluated the effect of “global” TTP dephosphorylation on its binding to a GM-CSF ARE probe. We first developed experimental conditions that would result in extensive dephosphorylation of TTP expressed in transfected 293 cells labeled with 32P. That these conditions would be adequate for dephosphorylating the phosphorylated protein are demon-strated in Fig. 6A. In this experiment, 32P-labeled TTP was the most prominent phosphoprotein in a crude cell extract; almost all of the 32P-labeling could be removed by the incubation at 30 °C with CIAP, and the decrease in 32P label was also accompanied by a shift to a lower apparent molecular weight in the SDS gel (Fig. 6A).
Fig. 6.
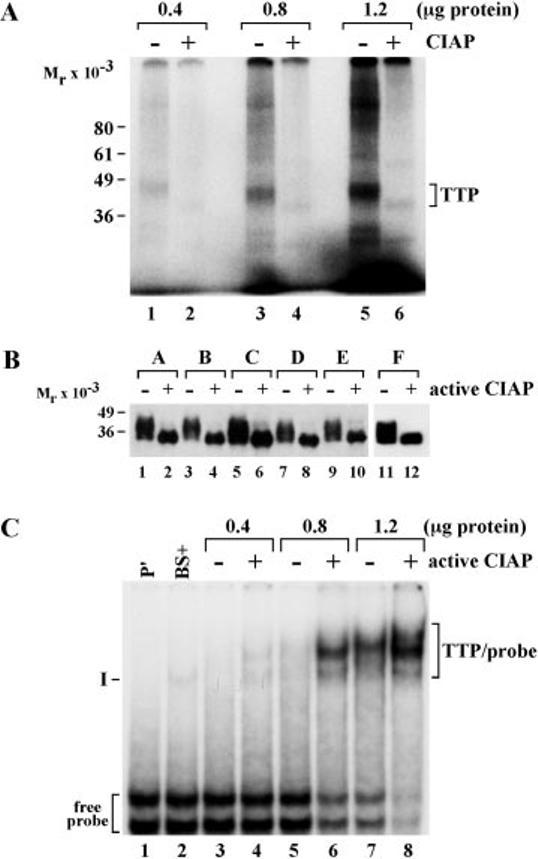
Effect of TTP dephosphorylation on its binding to an ARE probe. A, the indicated amounts of protein from 32P-labeled cellular extracts from 293 cells transfected with CMV.(his)6N.hTTP were incubated with (+) or without (−) active CIAP as indicated for 30 min at 30 °C. An autoradiograph of a gel of these samples is shown here. Both phosphorylated and dephosphorylated TTP are indicated by the labeled bracket on the right; the positions of molecular weight standards are indicated on the left. B shows a Western blot of a similar dephosphorylation experiment involving protein extracts from six independent transfection experiments (labeled A–F at the top of the gel) with the same TTP vector. These extracts were all incubated with active (+) or inactive (−) CIAP for 2 h at 30 °C. A final concentration of the phosphatase inhibitor Na2HPO4 of 0.1 m was present either at the beginning of the incubation (− active CIAP) or added at the end of the incubation (+ active CIAP). The samples were then subjected to SDS-PAGE and Western blotting with a polyclonal antibody directed at human TTP. The bands representing immunoreactive TTP are shown; the positions of molecular weight standards are shown on the left of the gel. C shows an autoradiograph from a gel shift experiment with one of the pairs of extracts shown in B, specifically, the pair of extracts represented by lanes 1 and 2 in B. Different amounts of extract protein were added to each pair of lanes, as indicated, and the legend for active CIAP is the same as for B. P′ indicates the lane (lane 1) containing probe alone, and the positions of the two free probe bands are indicated at the bottom left of the gel. BS+ represents 1 μg of extract protein from 293 cells transfected with the vector BS+ alone; the faint band indicated by I is an endogenous 293 cell protein that shifts this ARE probe. The multiple bands representing TTP-probe complexes are indicated by the bracket labeled TTP/probe to the right of the gel. See the text for further details.
We next performed similar dephosphorylations on non-radioactive 293 cell extracts containing similar amounts of expressed TTP from six independent but similar transfections. Because the CIAP protein migrated close to the Mr of highly phosphorylated TTP in these SDS gels and for other reasons, we took great pains to ensure that the extracts containing phosphorylated and dephosphorylated TTP contained otherwise identical concentrations of reactants after the incubation with CIAP. To do this, we added the phosphatase inhibitor Na2HPO4 (final concentration in the extract, 0.1 mm) to both halves of otherwise identical reaction mixtures containing the cellular extract and CIAP; in one case, the inhibitor was added before the 2-h incubation with CIAP and in the other case after the incubation. In this way, the two halves of the extract contained identical concentrations of all reactants, the only difference being the timing of the addition of the phosphatase inhibitor.
The results of a Western blot of these samples are shown in Fig. 6B, in which TTP was recognized by an antiserum directed against a bacterially expressed human TTP fusion protein with maltose-binding protein.2 In each case, treatment with active CIAP led to extensive dephosphorylation of immunoreactive TTP, resulting in the collapse of the multiple bands or smear representing the normally phosphorylated protein to a single immunoreactive band of approximate Mr 34,000. For convenience, we will refer to these pairs of TTP-containing extracts as phosphorylated and dephosphorylated, respectively.
These same pairs of samples from the six independent cell extracts were then used in RNA mobility shift experiments, using as a probe the ARE from mouse GM-CSF. This was used in preference to a TNFα probe because the single major complex formed in the absence of expressed TTP is usually minor in intensity and does not migrate to the same location on the non-denaturing gels as the TTP-probe complex, in contrast to TNFα ARE probes.4 These gel shift assays were performed with gradually increasing concentrations of cellular protein, so that subtle differences in RNA binding activity could be detected. Each pair of samples was analyzed in parallel on the same gel, and a second identical gel was performed in each case. An example of an autoradiograph from one such electro-phoretic mobility shift experiment is shown in Fig. 6C. The GM-CSF ARE RNA probe migrates as two bands near the bottom of the gel. When an extract (1 μg of protein) from 293 cells transfected with vector alone was used in the gel shift assay, there was almost no detectable shift of the probe, along with the appearance of a minor radioactive band, labeled complex I; this unidentified ARE-binding protein is routinely observed in extracts from untransfected 293 cells4 when this ARE probe is used. When extracts containing progressively increasing concentrations of protein containing the paired phosphorylated and dephosphorylated TTP were evaluated, there was a clear increase in the ability of the dephosphorylated protein to shift the radioactive ARE probe into the gel. This is most obvious when comparing lanes 5 and 6 in Fig. 6C but was apparent at all three protein concentrations used. When evaluated by probe disappearance, the same trend was seen, i.e. more probe was shifted by the dephosphorylated protein in each pair. This is especially apparent when comparing the probe radioactivity in lanes 5 and 6 and in lanes 7 and 8. Similar results were seen in the other 11 gels consisting of the duplicate gel for the one pictured in Fig. 6C as well as the duplicate gels for the remaining five pairs of extracts.
The data from all of these experiments were quantitated by PhosphorImager analysis and subjected to three types of statistical comparisons. First, we treated all 12 sets of PhosphorImager raw data for the TTP-probe complex as independent data points, and we compared the phosphorylated to the dephosphorylated samples by a paired t test. By using this approach, when the data equivalent to lane 3 in Fig. 6C were compared with the data equivalent to lane 4, the dephosphorylated protein bound 2.3-fold more probe than the phosphorylated protein (p = 0.0018). A similar comparison between the samples corresponding to lanes 5 and 6 in Fig. 6C resulted in a 62% average increase in probe binding by the dephosphorylated protein (p = 0.001). At the highest protein concentrations, corresponding to lanes 7 and 8, the difference was not statistically significant (67% increase, p = 0.08).
In a second approach, we expressed each PhosphorImager value of the protein-probe complex as a percentage of the maximum value, in all cases represented by the samples corresponding to lane 8 in Fig. 6C. Results from each pair of gels representing a single sample were averaged, and then the six sets were again averaged. These mean values ± S.E. are illustrated in Fig. 7A. Although these means could not be compared statistically because of the normalization procedure, it is once again apparent that the dephosphorylated protein shifted more probe at each concentration of protein extract than the phosphorylated protein: 2.5-fold more at the 0.4 μg concentration, 2-fold more at the 0.8 μg concentration, and 45% more at the 1.2 μg concentration.
Fig. 7.

Quantitation of the effect of TTP dephosphorylation on ARE binding. Six pairs of control and dephosphorylated TTP-containing protein extracts were used at the indicated protein concentrations to quantitate the effect of dephosphorylation on the amount of probe radioactivity shifted into the gel (A; % means the percent of the maximum value of shifted radioactivity, represented in each case by the samples labeled TTP in lane 8, as illustrated in Fig. 6C) and the amount of radioactive probe remaining in its usual position at the bottom of the gel (B; % means the percent of probe remaining in the probe alone position, exemplified in lane 1 of Fig. 6C). Each pair of bars in the histogram represents the mean values ± S.E. from the control (− active CIAP) and dephosphoryated (+ active CIAP) samples from the six different protein extracts illustrated in Fig. 6B. The results of statistical comparisons of the mean values are listed in the text.
A final analysis was performed on the ability of the protein extracts to retard probe migration to its normal location at the bottom of the gel. In this case, the results of the PhosphorImager values were normalized by expressing them as a percentage of the values obtained from the probe alone lane, corresponding to lane 1 in Fig. 6C. In each case, the values normalized in this way were averaged for each pair of duplicate gels, and then these individual averages were compared by a paired t test. These results are shown in Fig. 7B. At the lowest protein amount of 0.4 μg, the dephosphorylated protein shifted slightly more probe than the phosphorylated protein (89% of probe remaining versus 97%, p = 0.026). At 0.8 μg of protein, the difference was more obvious: 56% of probe remained unshifted by the dephosphorylated protein versus 76% by the phosphorylated protein (p = 0.0027). Finally, at 1.2 μg of protein, 32% of probe remained unshifted by the dephosphorylated protein compared with 53% by the phosphorylated protein (p = 0.0034).
DISCUSSION
One of the major findings in these studies was that macrophages derived from TTPKO mice were less sensitive than normal cells to the effects of p38 inhibitors on LPS-stimulated TNFα release. These data in turn suggest that TTP is part of the p38 signal transduction cascade, initiated by LPS in macrophages, that leads ultimately to stimulated TNFα secretion.
In the TTPKO mouse model, macrophages produce excess amounts of TNFα secondary to stabilization of TNFα mRNA (15, 17). This occurs because of the absence of TTP, which ordinarily binds directly to the ARE in the TNFα mRNA, destabilizing it by a still unknown mechanism (15, 24). p38 has been implicated previously in the regulated synthesis of several inflammatory cytokines; in the case of TNFα, p38 is thought to exert its effects primarily on the regulation of TNFα mRNA stability (5, 6, 29). These and other results have suggested a possible link between p38 and TTP in the regulation of TNFα mRNA stability and thus TNFα secretion. TNFα secretion by macrophages is a process highly sensitive to the use of p38 inhibitors, such as members of the pyridinyl imidazole family; we therefore evaluated the effect of members of this class of p38 inhibitors on the secretion of TNFα by BMMϕ derived from WT and TTPKO mice. Whereas WT cells responded to the inhibitors with a profound decrease in the secretion of TNFα, similar to previous data in a murine macrophage cell line (5), TTPKO cells were markedly less sensitive to these inhibitors. The effect was very dramatic with SB203580, which had no effect at any concentration tested on LPS-stimulated TNFα secretion in the TTPKO cells. However, this p38 inhibitor has been reported to affect other stress-activated kinases, such as the c-Jun aminoterminal kinase (30, 31). At relatively high concentrations of another inhibitor, SB220025, possibly a more specific inhibitor of p38 kinase (20), there was clearly inhibited secretion of TNFα. However, the IC50 for this compound was markedly shifted to the right in the KO cells, with a 15-fold increase over that seen with the WT cells.
These results suggest that TTP may be a component of the cascade by which p38 regulates TNFα production. To exclude the possibility that p38 activation could be impaired in the absence of TTP, we showed that LPS stimulated the phosphorylation of p38 with a very similar time course in both WT and TTPKO cells and that levels of immunoreactive p38 were very similar in the two cell types. Cell-free kinase assays with commercial p38 and recombinant human and mouse MBP-TTP fusion proteins showed that p38 could catalyze the phosphorylation of TTP in a manner inhibited by both SB203580 and SB220025. In addition, TTP phosphorylation in intact macrophages was stimulated when BMMϕ were exposed to LPS, and this stimulated phosphorylation was prevented by the use of the p38 inhibitors. These results strongly suggest that TTP can be phosphorylated in macrophages after activation of p38, even though they do not prove that TTP is directly phosphorylated by p38 in intact cells. Finally, we showed that the activated p38 present in the TTPKO cells after exposure to LPS was able to phosphorylate the MBP-mTTP fusion protein normally in a cell-free kinase assay, a reaction that was again sensitive to the p38 inhibitor SB220025. This is additional evidence against the possibility of abnormal activation or expression of p38 in the absence of TTP.
One important difference between the WT and TTPKO cells is that the TTPKO cells produced about five times more TNFα after LPS stimulation than their WT counterparts, as we have shown previously (17). However, both cell types were similarly responsive to Marimastat, which inhibits TNFα production by inhibiting TACE (21) through a mechanism that is not thought to involve p38 kinases (32). This difference should be kept in mind when evaluating the comparative effects of the inhibitors in these cells.
Another concern is the differential expression of p38 isotypes in macrophages. Human macrophages have been shown to express the p38α and p38δ isotypes (33). These two isotypes differ in their substrate specificity, reactivity with p38 antibodies, mechanisms of activation, and sensitivity to p38 inhibitors (33, 34). The inhibitors used in the present study are thought to be specific for p38α and p38β but to have no effect on p38δ (20, 34). In addition, the antibodies used for both immunoblotting and immunoprecipitation in the present study are thought to recognize only p38α and p38β. We demonstrated that expression of p38, presumably α, was essentially identical in the WT and TTPKO cells used in the present study; however, it was at least theoretically possible that p38δ could be overexpressed in the TTPKO cells, thus providing a mechanism for their resistance to p38 inhibition. However, the expression of p38δ mRNA was at very low levels and was equivalent in the WT and TTPKO cells,5 suggesting that the relevant isotype in our system is p38α.
p38 plays an important role in the regulation of the inflammatory response. It has been shown that p38 activation can increase the stability of mRNAs encoding several pro-inflammatory cytokines (29, 35). p38 activation can also induce the transcription of genes encoding pro-inflammatory factors, and the translation of their mRNAs (36, 37). The use of p38 inhibitors in the treatment of inflammatory diseases has been proposed, and several inhibitors of this type have been used successfully in experimental models of inflammatory diseases (7, 20, 28). However, little is known to date about the downstream components of the signaling pathways that actually regulate this response. The results reported here suggest the possible involvement of TTP downstream from p38. TTP is a phospho-protein that contains several consensus phosphorylation sites for proline-directed, stress-activated kinases (19), and it can be phosphorylated in vivo and in vitro by some of these kinases. Moreover, we found that cell-expressed TTP “globally” dephosphorylated with alkaline phosphatase bound an ARE probe with greater apparent avidity than did the native, phosphorylated protein. TTP appears to be mostly cytosolic in macrophages (15) but can shuttle between the nucleus and cytosol in fibroblasts (38); this is similar to the behavior of p38 itself, which is thought to be transported from the nucleus to the cytoplasm by its phosphorylated substrate MAPKAPK2 (39).
A possible model for a p38-TTP linkage is shown in Fig. 8. According to this proposed model, once TTP was phosphorylated by p38 or another kinase downstream from p38, TTP could change its subcellular localization and/or exhibit decreased binding to the TNFα mRNA ARE, thus increasing the stability of TNFα mRNA and increasing the synthesis and secretion of this cytokine (Fig. 8A1). In contrast, decreased TTP phosphorylation resulting from the use of the p38 inhibitors could promote TTP binding to the TNFα ARE, resulting in the rapid destabilization and degradation of the TNFα mRNA, and consequently, reduction in the levels of secreted protein (Fig. 8A2). The situation in the complete absence of TTP would more closely resemble that seen in Fig. 8A1, i.e. the lack of TTP would promote TNFα mRNA stability and increased TNFα secretion in both the presence and absence of p38 inhibitors (Fig. 8B, 3 and 4). This hypothetical mechanism could explain why the TTPKO cells were relatively or completely resistant to the p38 inhibitors. More studies are necessary to confirm the putative cellular interactions between TTP and p38 (or other downstream kinases), and the cellular compartment(s) in which these interactions take place. However, this apparent placement of TTP in the p38 pathway increases our understanding of the regulation of the inflammatory response by p38 and may provide a new target for the development of anti-inflammatory therapies.
Fig. 8.

Model of p38 inhibition on the regulation of LPS-stimulated TNF secretion in WT and TTPKO macrophages. This model depicts the hypothetical actions of p38 to stimulate the phosphorylation of TTP by p38, either directly or indirectly, and a subsequent effect of this phosphorylation to decrease TTP binding to the TNFα mRNA ARE and increase the stability of the mRNA, leading to increased TNFα secretion (A1). In the presence of p38 inhibitors in the normal cells (A2), dephosphorylated TTP binds more avidly to the TNFα mRNA ARE, promoting destruction of the mRNA and decreased TNFα secretion. In the KO cells, TNFα mRNA is constitutively stabilized in the absence of TTP, and thus activation of p38 has relatively little effect on the mRNA stability (Fig. 8B); TNFα secretion is thus elevated both in the absence (B3) and the presence (B4) of the inhibitors. Both inhibitors are abbreviated SB. + denotes activation of a pathway; – denotes inhibition of a pathway. ↑ and ↓ represent increase and decrease, respectively.
While this paper was under revision, three papers were published that are relevant to our results. In the first (40), the authors demonstrated that TTP could be phosphorylated by p38 kinase, both in a cell-free system and in the Raw 264.7 murine macrophage line. In contrast, Ming et al. (41) evaluated the regulation of IL-3 mRNA stability, which they previously showed could be affected by TTP in a complementation assay (42) and we have shown could be affected by TTP in a cotransfection assay (26). The new study evaluated the roles of the phosphatidylinositol 3-kinase and p38 kinase on the regulation of IL-3 mRNA stability by phorbol esters in transiently transfected NIH-3T3 cells. These authors concluded that TTP was not likely to be a direct substrate of either kinase, because the activation of either pathway failed to overcome the destabilizing effect of TTP on IL-3 mRNA. No experiments on the phosphorylation status of TTP were performed. Finally, Kontoyiannis and colleagues (43) showed that primary macrophages derived from TTP-deficient mice were apparently normally sensitive to the p38 inhibitor SB203580. Although their experimental design differed from ours in several respects, including our use of longer exposure of the cells to the inhibitors, a different source for the inhibitors, and a wider range of inhibitor concentrations, we cannot provide a satisfactory explanation for this difference in results. Considerably more work will be needed to account for some of these disparate data.
Acknowledgments
We thank Chris Heapy for performing the ELISA determinations and Drs. John O'Bryan and Anton Jetten for critical reading of the manuscript.
Footnotes
This work was supported in part by a Cooperative Research and Development Agreement with AstraZeneca Pharmaceuticals. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The abbreviations used are: LPS, lipopolysaccharide; MAPK, mitogen-activated protein kinase; MAPKAPK2, MAPK-activated protein kinase 2; TNFα, tumor necrosis factor α; IL, interleukin; ARE, AU-rich element; TTP, tristetraprolin; GM-CSF, granulocyte-macrophage colony-stimulating factor; BMMϕ bone marrow-derived macrophages; TACE, TNFα-converting enzyme; PAGE, polyacrylamide gel electrophoresis; MBP, maltose-binding protein; CIAP, calf intestinal alkaline phosphatase; ELISA, enzyme-linked immunosorbent assay; WT, wild type.
H. Cao and P.J. Blackshear, unpublished data.
D. Campbell, unpublished data.
W. S. Lai and P. J. Blackshear, unpublished data.
E. Carballo and P. J. Blackshear, unpublished data.
REFERENCES
- 1.Xu N, Chen CY, Shyu AB. Mol. Cell. Biol. 1997;17:4611–4621. doi: 10.1128/mcb.17.8.4611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dean JL, Wait R, Mahtani KR, Sully G, Clark AR, Saklatvala J. Mol. Cell. Biol. 2001;21:721–730. doi: 10.1128/MCB.21.3.721-730.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Piecyk M, Wax S, Beck AR, Kedersha N, Gupta M, Maritim B, Chen S, Gueydan C, Kruys V, Streuli M, Anderson P. EMBO J. 2000;19:4154–4163. doi: 10.1093/emboj/19.15.4154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kontoyiannis D, Pasparakis M, Pizarro TT, Cominelli F, Kollias G. Immunity. 1999;10:387–398. doi: 10.1016/s1074-7613(00)80038-2. [DOI] [PubMed] [Google Scholar]
- 5.Brook M, Sully G, Clark AR, Saklatvala J. FEBS Lett. 2000;483:57–61. doi: 10.1016/s0014-5793(00)02084-6. [DOI] [PubMed] [Google Scholar]
- 6.Wang SW, Pawlowski J, Wathen ST, Kinney SD, Lichenstein HS, Manthey CL. Inflamm. Res. 1999;48:533–538. doi: 10.1007/s000110050499. [DOI] [PubMed] [Google Scholar]
- 7.Badger AM, Bradbeer JN, Votta B, Lee JC, Adams JL, Griswold DE. J. Pharmacol. Exp. Ther. 1996;279:1453–1461. [PubMed] [Google Scholar]
- 8.Prichett W, Hand A, Sheilds J, Dunnington D. J. Inflamm. 1995;45:97–105. [PubMed] [Google Scholar]
- 9.Young P, McDonnell P, Dunnington D, Hand A, Laydon J, Lee J. Agents Actions. 1993;39:C67–C69. doi: 10.1007/BF01972723. [DOI] [PubMed] [Google Scholar]
- 10.Lee JC, Laydon JT, McDonnell PC, Gallagher TF, Kumar S, Green D, McNulty D, Blumenthal MJ, Heys JR, Landvatter SW, Strickler JE, McLaughlin MM, Siemens IR, Fisher SM, Livi GP, White JR, Adams JL, Young PR. Nature. 1994;372:739–746. doi: 10.1038/372739a0. [DOI] [PubMed] [Google Scholar]
- 11.Kotlyarov A, Neininger A, Schubert C, Eckert R, Birchmeier C, Volk HD, Gaestel M. Nat. Cell Biol. 1999;1:94–97. doi: 10.1038/10061. [DOI] [PubMed] [Google Scholar]
- 12.Obata T, Brown GE, Yaffe MB. Crit. Care Med. 2000;28:N67–N77. doi: 10.1097/00003246-200004001-00008. [DOI] [PubMed] [Google Scholar]
- 13.Waskiewicz AJ, Cooper JA. Curr. Opin. Cell Biol. 1995;7:798–805. doi: 10.1016/0955-0674(95)80063-8. [DOI] [PubMed] [Google Scholar]
- 14.Herlaar E, Brown Z. Mol. Med. Today. 1999;5:439–447. doi: 10.1016/s1357-4310(99)01544-0. [DOI] [PubMed] [Google Scholar]
- 15.Carballo E, Lai WS, Blackshear PJ. Science. 1998;281:1001–1005. doi: 10.1126/science.281.5379.1001. [DOI] [PubMed] [Google Scholar]
- 16.Taylor GA, Carballo E, Lee DM, Lai WS, Thompson MJ, Patel DD, Schenkman DI, Gilkeson GS, Broxmeyer HE, Haynes BF, Blackshear PJ. Immunity. 1996;4:445–454. doi: 10.1016/s1074-7613(00)80411-2. [DOI] [PubMed] [Google Scholar]
- 17.Carballo E, Gilkeson GS, Blackshear PJ. J. Clin. Invest. 1997;100:986–995. doi: 10.1172/JCI119649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carballo E, Lai WS, Blackshear PJ. Blood. 2000;95:1891–1899. [PubMed] [Google Scholar]
- 19.Taylor GA, Thompson MJ, Lai WS, Blackshear PJ. J. Biol. Chem. 1995;270:13341–13347. doi: 10.1074/jbc.270.22.13341. [DOI] [PubMed] [Google Scholar]
- 20.Jackson JR, Bolognese B, Hillegass L, Kassis S, Adams J, Griswold DE, Winkler JD. J. Pharmacol. Exp. Ther. 1998;284:687–692. [PubMed] [Google Scholar]
- 21.Barlaam B, Bird TG, Lambert-Van Der Brempt C, Campbell D, Foster SJ, Maciewicz R. J. Med. Chem. 1999;42:4890–4908. doi: 10.1021/jm990377j. [DOI] [PubMed] [Google Scholar]
- 22.Paine E, Palmantier R, Akiyama SK, Olden K, Roberts JD. J. Biol. Chem. 2000;275:11284–11290. doi: 10.1074/jbc.275.15.11284. [DOI] [PubMed] [Google Scholar]
- 23.Maina CV, Riggs PD, Grandea AG, Slatko BE, Moran LS, Tagliamonte JA, McReynolds LA, Guan CD. Gene (Amst.) 1988;74:365–373. doi: 10.1016/0378-1119(88)90170-9. [DOI] [PubMed] [Google Scholar]
- 24.Lai WS, Carballo E, Strum JR, Kennington EA, Phillips RS, Blackshear PJ. Mol. Cell. Biol. 1999;19:4311–4323. doi: 10.1128/mcb.19.6.4311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lai WS, Thompson MJ, Blackshear PJ. J. Biol. Chem. 1998;273:506–517. doi: 10.1074/jbc.273.1.506. [DOI] [PubMed] [Google Scholar]
- 26.Lai WS, Carballo E, Thorn JM, Kennington EA, Blackshear PJ. J. Biol. Chem. 2000;275:17827–17837. doi: 10.1074/jbc.M001696200. [DOI] [PubMed] [Google Scholar]
- 27.van den Blink B, Juffermans NP, ten Hove T, Schultz MJ, van Deventer SJ, van der Poll T, Peppelenbosch MP. J. Immunol. 2001;166:582–587. doi: 10.4049/jimmunol.166.1.582. [DOI] [PubMed] [Google Scholar]
- 28.Wadsworth SA, Cavender DE, Beers SA, Lalan P, Schafer PH, Malloy EA, Wu W, Fahmy B, Olini GC, Davis JE, PellegrinoGensey JL, Wachter MP, Siekierka JJ. J. Pharmacol. Exp. Ther. 1999;291:680–687. [PubMed] [Google Scholar]
- 29.Winzen R, Kracht M, Ritter B, Wilhelm A, Chen CY, Shyu AB, Muller M, Gaestel M, Resch K, Holtmann H. EMBO J. 1999;18:4969–4980. doi: 10.1093/emboj/18.18.4969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Clerk A, Sugden PH. FEBS Lett. 1998;426:93–96. doi: 10.1016/s0014-5793(98)00324-x. [DOI] [PubMed] [Google Scholar]
- 31.Whitmarsh AJ, Yang SH, Su MS, Sharrocks AD, Davis RJ. Mol. Cell. Biol. 1997;17:2360–2371. doi: 10.1128/mcb.17.5.2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Glaser KB, Pease L, Li J, Morgan DW. Biochem. Pharmacol. 1999;57:291–302. doi: 10.1016/s0006-2952(98)00300-1. [DOI] [PubMed] [Google Scholar]
- 33.Hale KK, Trollinger D, Rihanek M, Manthey CL. J. Immunol. 1999;162:4246–4252. [PubMed] [Google Scholar]
- 34.Kumar S, McDonnell PC, Gum RJ, Hand AT, Lee JC, Young PR. Biochem. Biophys. Res. Commun. 1997;235:533–538. doi: 10.1006/bbrc.1997.6849. [DOI] [PubMed] [Google Scholar]
- 35.Holtmann H, Winzen R, Holland P, Eickemeier S, Hoffmann E, Wallach D, Malinin NL, Cooper JA, Resch K, Kracht M. Mol. Cell. Biol. 1999;19:6742–6753. doi: 10.1128/mcb.19.10.6742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Beyaert R, Cuenda A, Vanden Berghe W, Plaisance S, Lee JC, Haegeman G, Cohen P, Fiers W. EMBO J. 1996;15:1914–1923. [PMC free article] [PubMed] [Google Scholar]
- 37.Chin J, Kostura MJ. J. Immunol. 1993;151:5574–5585. [PubMed] [Google Scholar]
- 38.Taylor GA, Thompson MJ, Lai WS, Blackshear PJ. Mol. Endocrinol. 1996;10:140–146. doi: 10.1210/mend.10.2.8825554. [DOI] [PubMed] [Google Scholar]
- 39.Ben-Levy R, Hooper S, Wilson R, Paterson HF, Marshall CJ. Curr. Biol. 1998;8:1049–1057. doi: 10.1016/s0960-9822(98)70442-7. [DOI] [PubMed] [Google Scholar]
- 40.Zhu W, Brauchle MA, Di Padova F, Gram H, New L, Ono K, Downey JS, Han J. Am. J. Physiol. 2001;281:L499–L508. doi: 10.1152/ajplung.2001.281.2.L499. [DOI] [PubMed] [Google Scholar]
- 41.Ming XF, Stoecklin G, Lu M, Looser R, Moroni C. Mol. Cell. Biol. 2001;21:5778–5789. doi: 10.1128/MCB.21.17.5778-5789.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stoecklin G, Ming XF, Looser R, Moroni C. Mol. Cell. Biol. 2000;20:3753–3763. doi: 10.1128/mcb.20.11.3753-3763.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kontoyiannis D, Kotlyarov A, Carballo E, Alexopoulou L, Blackshear PJ, Gaestel M, Davis R, Flavell R, Kollias G. EMBO J. 2001;20:3760–3770. doi: 10.1093/emboj/20.14.3760. [DOI] [PMC free article] [PubMed] [Google Scholar]