

Abstract
Tristetraprolin (TTP) is a hyperphosphorylated protein that destabilizes mRNA by binding to an AU-rich element (ARE). Mice deficient in TTP develop a severe inflammatory syndrome. The biochemical properties of TTP have not been adequately characterized, due to the difficulties in protein purification and lack of a high-titer antiserum. Full-length human TTP was expressed in human HEK293 cells and purified to at least 70% homogeneity. The purified protein was free of endogenous ARE binding activity, and was used for investigating its size, zinc dependency, and binding kinetics for tumor necrosis factor α mRNA ARE. A high-titer rabbit antiserum was raised against the MBP–hTTP fusion protein expressed in Escherichia coli. Cellular localization studies of the transfected cells indicated that approximately 80% of the expressed TTP was in the cytosol, with 20% in the nuclei. TTP from both locations bound to the ARE and formed similar complexes. The purified TTP was shown to be intact by N-terminal His-tag purification, C-terminal peptide sequencing, and mass spectrometry analysis. Results from size exclusion chromatography are consistent with the predominant form of active TTP being a tetramer. TTP's ARE binding activity was increased by 10 μM Zn2+. The half-maximal binding of TTP from HEK293 cells was approximately 30 nM in assays containing 10 nM ARE. This value was about twice that of TTP from E. coli. TTP from HEK293 cells was highly phosphorylated, and its electrophoretic mobility was increased by alkaline phosphatase treatment and somewhat by T271A mutation, but not by PNGase F or S186A mutation. The gel mobility of TTP from E. coli was decreased by in vitro phosphorylation with p42/ERK2 and p38 mitogen-activated protein kinases. These results suggest that TTP's zinc-dependent ARE binding affinity is reduced by half by posttranslational modifications, mainly by phosphorylation but not by glycosylation, in mammalian cells. The results support a model in which each subunit of the TTP tetramer binds to one of the five overlapping UUAUUUAUU sequences of the ARE, resulting in a stable TTP–ARE complex.
Tristetraprolin (TTP)1 (also known as ZFP36, Nup475, TIS11, and G0S24) is a prototypical member of a small family of mammalian proteins with tandem CCCH (CX8CX5CX3H) zinc finger motifs separated by 18 amino acid residues (1). Similar zinc finger sequences are found in at least 19 species in the GenBank database, including human, cow, mouse, rat, sheep, chimpanzee, dog, horse, pig, Xenopus, carp, zebrafish, Drosophila, Caenorhabditis elegans, baker's yeast, fission yeast, oyster, rice, and Arabidopsis. TTP mRNA and protein are detected in a number of tissues, including spleen, thymus, lymph node, lung, liver, and intestine, consistent with its proposed antiinflammatory role (2, 3). The expression levels of TTP mRNA and protein in mammalian cells are increased in response to several kinds of stimuli, including insulin and other growth factors, and stimulators of innate immunity, such as the endotoxin lipopolysaccharide (LPS) (2, 3).
TTP is an mRNA-binding protein with high binding specificity for the so-called class II AU-rich elements (AREs) within the 3′-untranslated mRNAs (4-11). TTP and its related proteins can bind to the 3′-untranslated AREs of certain clinically important mRNAs, such as those encoding tumor necrosis factor α (TNF) (5, 7, 9), granulocyte-macrophage colony-stimulating factor (GM-CSF) (6), inter-leukin 3 (8), cyclooxygenase 2 (12, 13), and plasminogen activator inhibitor type 2 (14). This specific binding of TTP to the AREs results in the destabilization of the mRNAs in transfected human embryonic kidney (HEK) 293 cells (5, 7). Mice deficient in TTP develop a severe inflammatory syndrome, including polyarticular arthritis, myeloid hyperplasia, autoimmunity, and cachexia (15, 16). This syndrome is largely due to the increased stability of mRNAs for TNF and GM-CSF, and the resulting enhanced secretion of these proinflammatory cytokines (5, 6).
Some properties of TTP have been described recently using recombinant TTP purified from overexpressed Escherichia coli cells (9, 11) and the tandem zinc finger peptide (10). TTP fusion protein and its tandem zinc finger peptide each bind zinc with an affinity similar to those of other wellcharacterized zinc finger peptides (17-19). Zinc binding is required for the TNF mRNA ARE binding activity of TTP (9). The active form of the MBP–hTTP fusion protein binding to TNF mRNA ARE appears to be in the form of an oligomer rather than a monomer (9). TTP purified from E. coli can be phosphorylated by multiple mitogen-activated protein (MAP) kinases in vitro (9, 20-24) with kinetics similar to those of other protein substrates (9). Finally, the tandem zinc finger peptide of TTP binds ARE sequences with high affinity (10). The binding between the TTP fusion protein and ARE is quite stable over a wide range of pH and salt concentrations (11), and is resistant to 0.5% Nonidet P-40 (NP-40, a nonionic detergent) and 0.04% sodium dodecyl sulfate (SDS, an ionic detergent) in the assay buffers (9).
The biochemical properties of TTP from mammalian sources have not been adequately characterized, largely due to the unavailability of high-titer antibodies and a lack of highly purified TTP from mammalian sources. It is wellknown that TTP is a highly phosphorylated protein in mammalian cells (3, 20-23, 25). Phosphorylation of TTP in mammalian cells partially inhibits its binding to GM-CSF mRNA ARE and enhances its binding to the 14-3-3 protein (20, 25). Given the dramatic differences in posttranslational modifications between E. coli proteins and mammalian proteins, as well as the differences between the full-length TTP and its peptide domains, it is therefore important to characterize the full-length TTP purified from mammalian cells.
In this study, some of the biochemical properties of human TTP (hTTP) were investigated using a high-titer rabbit antiserum and TTP purified from transfected HEK293 cells and from overexpressed E. coli cells. A reliable procedure was developed for purifying active TTP, free of endogenous ARE binding activity, from mammalian cells. These techniques were used to localize active TTP in the transfected cells, to estimate the size of the active protein, to analyze the effect of zinc on its ARE binding activity, to characterize its binding kinetics for TNF mRNA ARE, and to investigate the nature of its posttranslational modifications. The results suggest that TTP's zinc-dependent ARE binding affinity is reduced by posttranslational phosphorylation in the transfected mammalian cells, and support a model in which each subunit of TTP tetramer binds to one of the five overlapping UUAUUUAUU sequences of the ARE, resulting in a stable TTP–ARE complex.
EXPERIMENTAL PROCEDURES
Protein Expression Plasmids. Plasmids pMBP-hTTP and pMBP-mTTP were used to express full-length human TTP (GenBank accession no. NP_003398) and mouse TTP (GenBank accession no. NP_035886) as maltose binding protein (MBP) fusion proteins (MBP–hTTP and MBP–mTTP, respectively) in E. coli as described previously (9). Plasmid pHis-hTTP was used to express full-length hTTP as a His-tagged protein in human HEK293 cells. This plasmid described previously (20) as CMV·(His)6N·hTTP contains DNA sequences encoding the six consecutive histidine residues inserted between the first methionine and the second aspartic acid residues of full-length hTTP. Plasmid pEF-XEH was used as a negative control for expressing a His-tagged Xenopus Eps15 homology (EH) domain of intersectin (26) in plasmid vector pEF6/V5-His-TOPO (Invitrogen, Gaithersburg, MD).
Site-Directed Mutagenesis. The wild-type pHis-hTTP was used as a template for site-directed mutagenesis (S186A and T271A) by two rounds of PCR with Platinum pfx DNA polymerase using the primer overlapping technique. The first round of PCRs for the S186A mutation included PCR 1 [pHis-hTTP, primer 1 (5′-CATTAGGGAGACCCAAGCTTGGTACC-3′), primer 2 (5′-GGAGAATGCGATGCTCTG-3′); the HindIII site and S186A mutation are underlined] and PCR 2 [pHis-hTTP, primer 3 (5′-CAGAGCATCGCATTCTCC-3′), primer 4 (5′-CCTCTAGATGCATGCTCGAGTC-3′); the S186A mutation and XbaI site are underlined]. The first round of PCRs for the T271A mutation included PCR 3 [pHis-hTTP, primer 1, primer 5 (5′-GTACAGAGGGTGCCCGAACCAGG-3′); the T271A mutation is underlined] and PCR 4 [pHis-hTTP, primer 4, primer 6 (5′-CCTGGTTCGGGCACCCTCTGTAC-3′); the T271A mutation is underlined]. The second round of PCRs included PCR 5 (for the S186A mutation, primers 1 and 4, and the products of PCRs 1 and 2) and PCR 6 (for the T271A mutation, primers 1 and 4, and the products of PCRs 3 and 4). The products of PCRs 5 and 6 were digested with HindIII and XbaI, and the resultant mutated DNA fragments were used to replace the DNA fragment from the wild-type pHis-hTTP. The mutations were confirmed with DNA sequencing using the dRhodamine Terminator Cycle Sequencing kit (Perkin-Elmer Life Sciences, Gaithersburg, MD).
Expression of TTP in Transfected Human Cells. Full-length TTP was expressed as a His-tagged protein (His-hTTP) in HEK293 cells. This cell line does not have detectable endogenous TTP mRNA (7). Cells were transiently transfected with pHis-hTTP plasmids encoding the wild-type, S186A, and T271A proteins, using the calcium phosphate precipitation method as described previously (7). HEK293 cells (0.5–1 million cells/10 cm dish) were grown overnight at 37 °C with 5% CO2 in modified Eagle's medium (Invitrogen) supplemented with 10% (v/v) heat-inactivated fetal calf serum (FCS), 100 units/mL penicillin, 100 μg/mL streptomycin, and 2 mM L-glutamine. The cells were transfected with 0.5 μg of pHis-hTTP and cotransfected with 4.5 μg of pBS+. The transfection mixture per 10 cm dish was prepared by mixing 0.5 mL of a DNA/calcium solution (0.5 μg of pHis-hTTP plasmid, 4.5 μg of pBS+ carrier plasmid, and 250 mM CaCl2) with 0.5 mL of a HEPES/phosphate solution [50 mM HEPES, 280 mM NaCl, 2 mM NaH2PO4, and 4mM Na2HPO4 (pH 7.1)]. The DNA/calcium solution was added dropwise to the HEPES/phosphate solution which was being bubbled with a stream of nitrogen gas. The transfection mixture was incubated for 20 min at room temperature before being added to the dish (1 mL/10 cm dish). Cells were washed the next morning, incubated under the same conditions, and collected 24 h later. The transfected cells were washed with phosphate-buffered saline (PBS) and lysed on ice for 30 min in lysis buffer [10 mM HEPES (pH 7.6), 3 mM MgCl2, 40 mM KCl, 0.5% NP-40, 8 μg/mL leupeptin, and 0.5 mM phenylmethanesulfonyl fluoride (PMSF)]. The cell lysate was centrifuged at 1000g for 5 min, resulting in the 1000g supernatant and the 1000g pellet. The 1000g supernatant was then centrifuged at 10000g for 10 min, resulting in the 10000g supernatant and the 10000g pellet. HEK293 cells were also transfected with two control plasmids, pBS+ and pEF-XEH. The supernatants were mixed with glycerol to 20% (v/v), frozen in liquid nitrogen, and stored at − 70 °C.
Purification of TTP from Transfected Human Cells. His-hTTP was purified from the transfected HEK293 cells using nickel-nitrilotriacetic agarose (Ni–NTA) beads (Qiagen, Valencia, CA). For batch purification, the 10000g supernatant (1.5 mL, ∼3 mg) was diluted 2-fold with dilution buffer (556 mM NaCl, 20 mM imidazole, and 100 mM NaH2PO4). Ni–NTA slurry {50 μL of a 50% slurry in 10 mM imidazole in purification buffer [50 mM NaH2PO4, 300 mM NaCl, and 0.05% Tween 20 (pH 8.0)]} was added to the tube and the mixture incubated for 3 h at 4 °C with gentle rotation. Following centrifugation at 1000g for 5 min, the beads were transferred to a Cyto-spin column (Qiagen) and washed four times with 0.5 mL each time of 20 mM imidazole in purification buffer. The bound proteins were eluted with 100 μL of elution buffer (50–250 mM imidazole in purification buffer). The nuclear extract (NE) was extracted from P1000g with 1 volume of a high-salt buffer [0.45 M KCl, 20 mM Tris-HCl (pH 8), 1.5 mM MgCl2, 20% glycerol, 50 mM NaF, 0.2 mM EDTA, 0.2 mM PMSF, 1 mM dithiothreitol (DTT), 10 μg/μL leupeptin, and 1 μg/μL pepstain] following a previously described procedure (3, 27). Protein samples were stored in 20% glycerol as described above.
For purification with Ni–NTA affinity chromatography, a 2 mL Ni–NTA column was prepared using a Bio-Rad mini column (0.7 cm × 15 cm). The column was washed with 20 mL of wash buffer (10 mM imidazole in purification buffer). The 10000g supernatant in 10% glycerol (140 mg of protein) was applied to the Ni–NTA column and washed with 10 mL of wash buffer. Proteins were eluted with 10 mL of elution buffer (250 mM imidazole in purification buffer). Aliquots of the fractions were used for the protein assay, SDS–PAGE, and immunoblotting using anti-MBP-hTTP antibodies.
Purification of MBP–hTTP, MBP–mTTP, and TTP from Overexpressed E. coli Cells. MBP–hTTP and MBP–mTTP were expressed in E. coli and purified as described previously (9), using an AKTAFPLC system (Amersham Pharmacia Biotech, Uppsala, Sweden) with amylose resin affinity, Superose 6 size exclusion, and Mono Q anion exchange columns. For the purification of nonfusion full-length TTP, MBP–hTTP purified by amylose resin affinity column was digested with PreScission protease (Amersham). TTP free from the MBP fusion partner was further purified by continuous-elution gel electrophoresis using the model 491 Prep Cell (Bio-Rad Laboratories, Hercules, CA) as described previously (9). In some experiments, expressed MBP was purified by amylose affinity chromatography as described previously (9).
Anti-MBP–hTTP Antibody Production in Rabbits. MBP–hTTP was concentrated with a Centricon-10 concentrator (Amicon, Beverly, MA) to a protein concentration of 1 mg/ mL for antibody production. Anti-MBP–hTTP serum was produced in rabbits immunized with the purified MBP–hTTP fusion protein according to standard procedures (Covance Research Products, Denver, PA). Briefly, 250 μg of the purified antigen was diluted into 0.5 mL in PBS, mixed with 0.5 mL of Freund's complete adjuvant, and injected into a female New Zealand white rabbit. Three boosts of 125 μg each of the purified antigen in Freund's incomplete adjuvant were performed every 4 weeks following the primary injection. The antiserum that contained the highest-titer anti-TTP activity was centrifuged at 10000g for 10 min; sodium azide was added to a final concentration of 0.02% (w/v), and the serum was stored at −70 °C.
Determination of Protein Concentrations. Protein concentrations were determined using the Protein Assay Dye Reagent Concentrate (Bio-Rad) following treatment of the samples with NaOH as described previously (28). Briefly, protein samples were treated with 0.5 M NaOH according to the method of Stoscheck (29) in 60 μL for 10 min at room temperature. Then 1 mL of the diluted dye reagent (1:5 in water) was added to each sample. Following incubation for 20 min at room temperature, the absorbance at 595 nm was measured using a DU 640 spectrophotometer (Beckman Instruments, Inc., Fullerton, CA). Bovine serum albumin (BSA) from Bio-Rad was used as the protein standard. The relative quantity of TTP in the Ni–NTA-purified fraction was determined by densitometry and analyzed by NIH Image software essentially as described previously (28).
SDS–PAGE and Immunoblotting. Sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) and immunoblotting followed previously described procedures (9). Protein gels were stained with Coomassie blue (30) or silver reagent (31) from Bio-Rad. Proteins on the immunoblots were detected using SuperSignal West Pico Chemilumines-cent Substrate (Pierce, Rockford, IL) as described previously (3). The primary antibodies were the anti-MBP–hTTP serum as described above and the anti-MBP serum (New England Biolabs, Beverly, MA). The secondary antibodies were affinity-purified goat anti-rabbit IgG (H+L) horseradish peroxidase-conjugated antibodies with human IgG absorbed (GAR–HRP, Bio-Rad).
Immunocytochemistry and Confocal Microscopy. HEK293 cells were grown overnight on glass coverslips in a 24-well tissue culture plate (Becton Dickinson and Co., Lincoln Park, NJ). The cells were transfected with pHis-hTTP (50 ng of DNA per milliliter per well) and incubated overnight as described above. After another 24 h incubation, the cells were subjected to immunocytochemistry using a similar procedure as described previously (3) with the following modifications. (1) The primary antiserum was the anti-MBP–hTTP serum as described above and was used at 1:5000 dilution. (2) The goat anti-rabbit Alexa Fluor 488 was used at 1:1000 dilution and was incubated for 1 h. (3) PBS was used instead of TBS. The slides were examined and imaged with an LSM510 UV confocal microscope (Zeiss, Thornwood, NY).
Trypsin Digestion and HPLC Separation. His-hTTP was purified with Ni–NTA beads as described above. Five micrograms of the eluted protein was digested for 24 h at 37 °C with 2 μg of modified trypsin treated with L-(tosylamido-2-phenyl) ethyl chloromethyl ketone (TPCK) (Sigma, St. Louis, MO) in 50 μL of elution buffer containing 200 mM imidazole. The peptide fragments were purified by reverse phase chromatography using an AKTAbasic system and a Sephasil Peptide C18 5 μm ST4.6/100 column (Amersham). The peptides were eluted using a linear gradient from 100% buffer A [0.065% trifluoroacetic acid (TFA) in 2% acetonitrile] to 100% buffer B (0.05% TFA in 100% acetonitrile). Fractions 38 and 39 were dried by Speed Vacuum, suspended in 100 μL of 0.1% TFA, vortexed, incubated for 1 h at room temperature, and used for protein sequencing.
Protein Sequencing. The suspended peptides from fractions 38 and 39 were supplemented with 5 μL of the Biobrene working solution (100 μg of Biobrene plus, 70% methanol, and 0.01% TFA) before being loaded onto a ProSorb Cartridge. The N-terminal sequences of peptides in fractions 38 and 39 were sequenced by the Edman sequencing with the amino-terminal protein sequencer (Procise Model 49X cLC Protein Sequencing System, Applied Biosystems, Foster City, CA), and the results were analyzed with ABI 610A 2.1 data analysis software and FASTA programs as described previously (9).
MALDI-TOF Mass Spectrometry Analysis. For matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF MS) analysis, proteins purified with Ni–NTA affinity beads were separated by 10% SDS–PAGE. After the gel was stained with Coomassie blue, protein bands were excised from the gel, processed by an in-gel digestion with trypsin using the Investigator ProGest Protein Digestion Station (Genomic Solutions, Ann Arbor, MI), and analyzed with a Voyager-DE-STR MALDI/TOF mass spectrometer (Perseptive Biosystems, Framingham, MA) as described previously (32).
Size Exclusion Chromatography. The size of His-hTTP was estimated by size exclusion chromatography using Superose 6 and Superose 12 HR 10/30 columns (Amersham) using a procedure similar to that described previously (9, 33). Proteins in the 10000g supernatant from the transfected cells were separated with the size columns using a buffer [50 mM NaH2PO4, 300 mM NaCl, 20 mM imidazole, and 0.05% Tween 20 (pH 8.0)]. Fractions were analyzed by immunoblotting using anti-MBP–hTTP serum and by ARE binding activity assay. In some experiments, TTP was first purified from the transfected HEK293T cells using a Ni–NTA affinity column. The TTP peak fractions were pooled together and centrifuged at 10000g for 10 min before being further separated with the Superose 12 column as described above. The molecular mass of TTP was estimated using a standard curve generated from known protein standards on the same columns. The protein standards (Amersham) are described in the figure legend.
RNA ARE Binding Activity Assay. TNF mRNA ARE binding activity was measured with the RNA gel mobility shift assay (GMSA) procedure as described previously (5, 9), using a TNF mRNA ARE probe (∼100000–200000 cpm/ reaction) with the following sequence: 5′-CUCUAUUUAUAUUUG|CACUUAUUAUUUAUUAUUUAUUUAUUAUUUAUUUAUUUG|CUUAUG|AAUG|UAUUUA-3′ (the three UUAUUUAUU repeats are underlined, and the two overlapping repeats are in bold letters; the vertical bar represents the RNase T1 recognition site). The ARE probe was transcribed from the mouse TNF mRNA ARE region (nucleotides 1281–1350 of GenBank accession no. X02611) (34), using [α-32P]UTP (NEN Life Sciences, Boston, MA) and T7 RNA polymerase with the Promega's RiboProbe In Vitro Transcription System (Promega Corp., Madison, WI). For the quantitative assay of TNF mRNA ARE binding activity, all of the TTP proteins were diluted with 100 mM imidazole elution buffer to 8.5 μL before being assayed in a 15 μL reaction mixture by GMSA. The binding buffer contained 10 μM ZnCl2 and 10 nM radiolabeled ARE probe with or without 300 nM unlabeled ARE probe. The concentration of the ARE probe was calculated from the specific activity of [α-32P]UTP, the radioactivity of the probe, and the number of U bases in the probe (this ARE probe has 43 U bases). The binding reaction mixtures were incubated for 30 min at room temperature before digestion with 100 units of RNase T1 (Epicentre, Madison, WI) for 15 min at 30 °C. The coprecipitation step with 50 μg of heparin and 1.2 μg of yeast tRNA before RNase T1 digestion (5, 9) was determined to be unnecessary and was therefore omitted in the binding kinetics analysis (data not shown). The TTP–probe complexes and free probes were separated by 6% native PAGE, detected by autoradiography on X-ray film (Eastman Kodak, Rochester, NY), and quantified by a phosphorimager (Molecular Dynamics, Sunnyvale, CA). The extent of half-maximal binding was determined as the protein concentration at which 50% of the TTP–probe complexes formed or 50% of the free probes remained in the assay mixtures (35).
Dephosphorylation Assay. The dephosphorylation assay was similar to that described previously (3, 20). The 10000g supernatant (20 μg of protein) of the transfected HEK293 cells was mixed with 0.5 μL (35 units) of calf intestine alkaline phosphatase (CIAP, Stratagene) in 30 μL of reaction buffer [50 mM Tris-HCl (pH 8.5) and 1 mM EDTA] for 60 min at 30 °C. The reactions were terminated with SDS–PAGE sample buffer followed by SDS–PAGE and immunoblotting analyses.
Deglycosylation Assay. Each deglycosylation assay contained between 20 and 40 μg of the 10000g supernatant. Fetuin (40 μg, Sigma) was used as a positive control for the deglycosylation reaction. Samples were mixed in 1× glycoprotein denaturing buffer (0.5% SDS and 1% β-mercap-toethanol) and incubated for 10 min at 100 °C. Following the addition of 10% (v/v) 10× G7 buffer (0.5 M sodium phosphate, pH 7.5 at 25 °C) and 10% (v/v) NP-40, proteins were deglycosylated with 1000 units of PNGase F (peptide: N-glycosidase F, New England Biolabs) for 60 min at 37 °C. The reactions were terminated with SDS–PAGE sample buffer followed by SDS–PAGE and immunoblotting analyses.
In Vivo Phosphate Labeling and Immunoprecipitation. HEK293 cells were transfected with pHis-hTTP as described above and incubated in the cell culture medium containing [32P]orthophosphate following a previous procedure (20). The cells were incubated with phosphate for 1.5 h and lysed directly in the Petri dishes. TTP was purified from the 10000g supernatant with Ni–NTA beads as described above, or by immunoprecipitation using the anti-MBP–hTTP serum following a procedure similar to that described previously (36). The 10000g supernatant from the labeled HEK293 cells (100 μL) was mixed with 20 μL of the antiserum. After incubation for 90 min at 4 °C with gentle rotation, 50 μL of the 50% slurry of Protein A–Sepharose CL-4B (Amersham Pharmacia Biotech) was added. This mixture was incubated for 30 min at 4 °C and centrifuged at 2000g for 5 min. The beads were washed three times and suspended in 25 μL of the SDS–PAGE sample buffer. The Ni–NTA bead-purified and immunoprecipitated samples were separated by 10% SDS–PAGE. The radiolabeled TTP was detected by autoradiography.
In Vitro Phosphorylation Assay. MBP–hTTP and MBP–mTTP were purified from E. coli by amylose resin columns as described previously (9). The protein samples were phosphorylated by recombinant His-tagged rat p42/ERK2 MAP kinase, GST-tagged mouse p38 MAP kinase, or calmodulin binding peptide (24 amino acid residues)-tagged rat JNK purified from E. coli (Calbiochem, La Jolla, CA) as detailed previously (9).
RESULTS
Production and Characterization of Anti-MBP–hTTP Serum. The N-terminal MBP-tagged hTTP was used to generate a high-titer antiserum against the full-length hTTP. MBP–hTTP was overexpressed in E. coli and purified to near homogeneity by three chromatographic steps, including amylose resin affinity, Superose 6 size exclusion, and Mono Q anion exchange columns. MBP–hTTP was detected with Coomassie blue staining (Figure 1A) and identified by immunoblotting with anti-MBP antibodies (Figure 1B). The Mono Q fractions with the highest purity of MBP–hTTP (Figure 1A, lane 6) were concentrated and used for immunizations.
Figure 1.
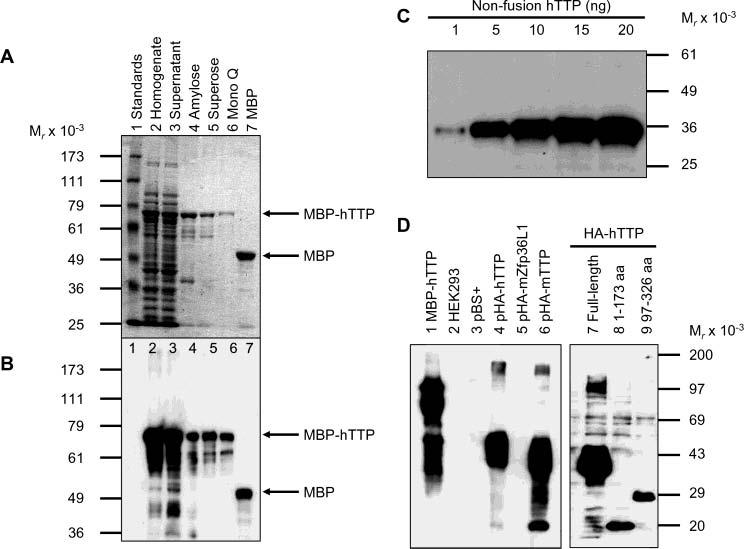
Purification of MBP–hTTP from E. coli and characterization of anti-MBP–hTTP antibodies. (A) Purification of MBP–hTTP stained with Coomassie blue: lane 1, protein size standards; lane 2, homogenate (50 μg of protein); lane 3, supernatant (50 μg); lane 4, amylose resin fraction (5 μg); lane 5, Superose 12 fraction (2 μg); lane 6, Mono Q fraction (1 μg); and lane 7, MBP eluted from the amylose resin column (5 μg). The positions of MBP–hTTP and MBP are indicated. (B) Detection of MBP–hTTP with anti-MBP serum. The samples were identical to those in panel A except that ∼10% of the amount of protein was used in each lane. (C) Detection limit of anti-MBP–hTTP serum by Western blotting. Nonfusion hTTP purified from E. coli as shown in Figure 7A (lane 2) (1, 5, 10, 15, and 20 ng) as indicated was probed with the anti-MBP–hTTP serum (1:10000) for 1 h and with GAR–HRP (1:10000) for 30 min and exposed to X-ray film for 1 min. (D) Characterization of the anti-MBP–hTTP serum by Western blotting. Protein samples were probed with the anti-MBP–hTTP serum (1:10000) for 1 h and with GAR–HRP (1:10000) for 1 h and exposed to X-ray film for 1 min: lane 1, purified MBP–hTTP (50 ng); lane 2, untransfected HEK293 cell extract (50 μg); lane 3, pBS+ transfection extract (50 μg); lane 4, pHA-hTTP transfection extract (50 μg); lane 5, pHA-mZfp36L1/mTIS11b transfection extract (50 μg); lane 6, pHA-mTTP transfection extract (50 μg); lane 7, transfection extract with full-length hTTP (50 μg); lane 8, transfection extract with amino-terminal residues 1–173 of hTTP (50 μg); and lane 9, transfection extract with carboxyl-terminal residues 97–326 of hTTP (50 μg).
The anti-MBP–hTTP serum at a 1:10000 dilution was able to detect as little as 1 ng of the purified nonfusion hTTP (Figure 1C) and MBP–hTTP (Figure 1D). The antiserum was also tested using extracts from HEK293 cells expressing the full-length and truncated hTTP. The antiserum at a 1:10000 dilution was able to detect full-length hTTP in the 10000g supernatant from the transfected cells when micrograms of proteins were loaded into the gel (Figure 1D). Anti-MBP–hTTP serum also recognized mTTP in the transfected cell extracts, but it did not recognize mouse Zfp36L1/TIS11b in the transfected cell extracts under these immunoblotting conditions (Figure 1D). Finally, this antiserum recognized hTTP fragments with amino-terminal residues 1–173 and carboxyl-terminal residues 97–326 (Figure 1D). Endogenous TTP could not be detected by immunoblotting using the anti-MBP–hTTP antibodies in the untransfected or pBS+-transfected HEK293 cells (Figure 1C). Preliminary results indicated that the anti-MBP–hTTP serum was able to detect endogenous hTTP in A549 cells (a human non-small-cell lung cancer cell line) after stimulation with 10% FCS for 3 h, and that this antiserum was also able to detect endogenous mTTP in mouse RAW264.7 cells after stimulation with 0.1 μg/mL LPS for 1–3.5 h (data not shown).
Expression and Localization of Active TTP in Transfected Human Cells. Endogenous TTP could not be detected in HEK293 cells, since little immunofluorescence was detected in the cells transfected with pBS+ (a control plasmid) with a confocal microscope following immunostaining with the anti-MBP-hTTP serum (Figure 2A). In contrast, TTP was overexpressed in HEK293 cells following transfection with pHis-hTTP, because bright immunofluorescence was detected by immunocytochemistry as a vesicular pattern in the outer region of the cell (Figure 2B). For the optimum expression of TTP, several factors were important: initial cell density, phosphate buffer pH, and harvest time. Poor expression was observed when too many cells were used or the phosphate buffer used in the transfection was not close to pH 7.1. Much less protein was recovered when cells were allowed to grow for an overly long period after transfection, at least partly due to many cells being detached from the surface of the dishes. However, the question of whether the transfected cells became apoptotic, as reported previously using NIH3T3 cells, was not examined critically (37).
Figure 2.
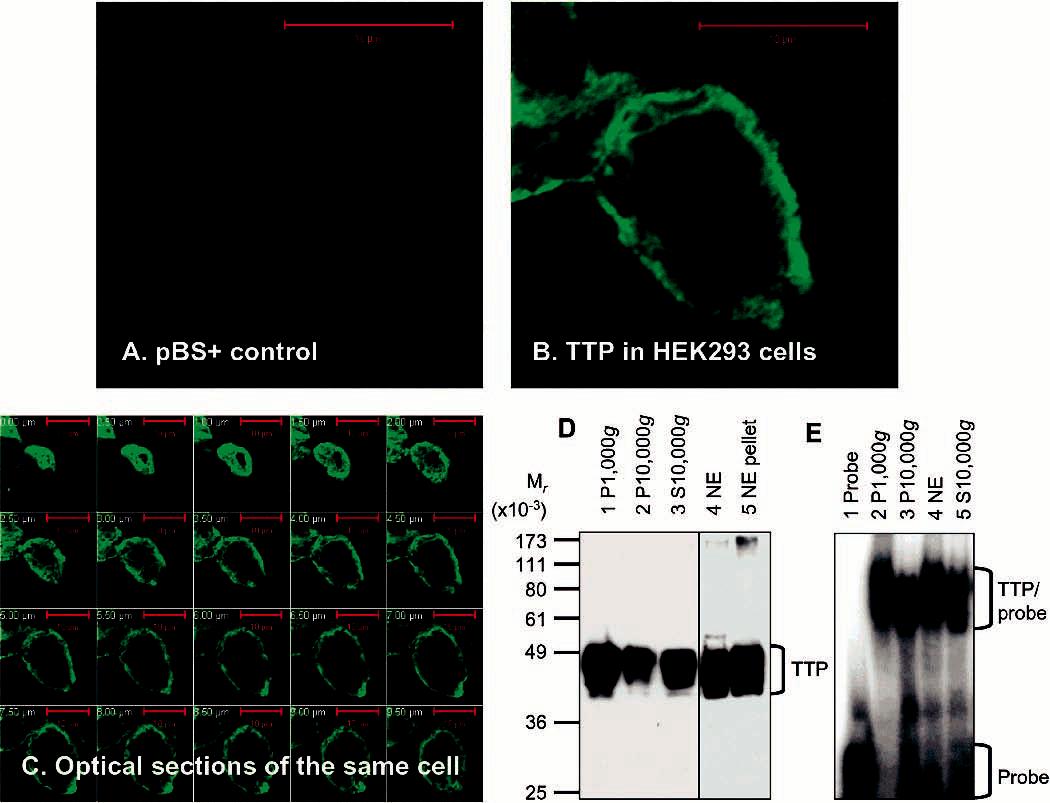
Expression and localization of active TTP in transfected human cells. (A–C) Expression and localization of TTP by immunocytochemistry. HEK293 cells were transfected with pBS+ control plasmid (A) and pHis-hTTP plasmid (B and C). The cells were immunostained with the anti-MBP-hTTP serum (1:5000 dilution) and labeled with goat anti-rabbit Alexa Fluor 488 (1:1000 dilution). Immunofluorescence was recorded with a confocal microscope as either a single image (A and B) or serial images of optical sections of a single cell with a 0.5 μm interval (C). (D) Expression and localization of TTP by immunoblotting. HEK293 cells were transfected with pHis-hTTP. The lysate was centrifuged at 1000g, resulting in S1000g and P1000g. S1000g was further centrifuged at 10000g, resulting in S10000g and P10000g. P1000g was extracted with a buffer containing 0.45 M KCl and centrifuged at 10000g, resulting in the 10000g supernatant (nucleic extract, NE) and the 10000g pellet (NE pellet). Proteins (8 μL) were separated by 10% SDS–PAGE, transferred onto a nitrocellulose membrane, and detected with the anti-MBP–hTTP serum. The position of TTP is indicated. The S10000g lane was loaded with proteins extracted from 10% of the cells used for other samples. (E) ARE binding activity by GMSA. The ARE binding activity was assayed by using proteins prepared as described for panel D. Each assay used 1 μL of each fraction. The positions of TTP–probe complexes and free probes are indicated.
Cellular localization of TTP was examined by immunocytochemistry and immunoblotting using transfected HEK293 cells. Confocal microscopic images were taken from serial optical sections of a single transfected cell following immunostaining with the anti-MBP–hTTP serum (Figure 2C). The great majority of the bright immunofluorescence was detected in the outer regions of all of the sections, suggesting that the expressed TTP was mainly localized in the cytosol of the transfected cell. The expressed TTP was also detected by immunoblotting using the anti-MBP–hTTP serum with apparent molecular masses ranging from 40 to 50 kDa in the 1000g pellet, the 10000g pellet, the 10000g supernatant (S10000g), the nuclear extract (NE), and the residue (NE pellet) (Figure 2D). The observed size of His-hTTP was much greater than the calculated size (Mr = 34 826), suggesting extensive posttranslational modifications of TTP in the transfected cells. It was estimated that TTP levels in lane 4 (NE) and lane 5 (NE pellet) were approximately 3-fold greater than the level in lane 3 (S10000g) (Figure 2D). Since the S10000g lane was loaded with proteins from transfected cells equivalent to 10% of the cells used for the NE and NE pellet lanes, it was estimated that approximately 80% of the expressed TTP was localized in the cytosol and that approximately 20% of TTP was localized in the nuclei of the transfected cells.
Previous studies have shown that cytosolic TTP functions as an mRNA ARE-binding protein (5). However, it was not clear if TTP in the nucleus could also bind to ARE. The nuclear proteins were extracted with 0.45 M KCl from P1000g from the transfected cells following several washes. ARE binding activity was analyzed by GMSA using a 32P-labeled TNF mRNA ARE probe. TTP–ARE complexes were detected using proteins from both the nuclear extract, the 10000g cytosolic fraction, and proteins from P1000g and P10000g (Figure 2E).
Figure 5.
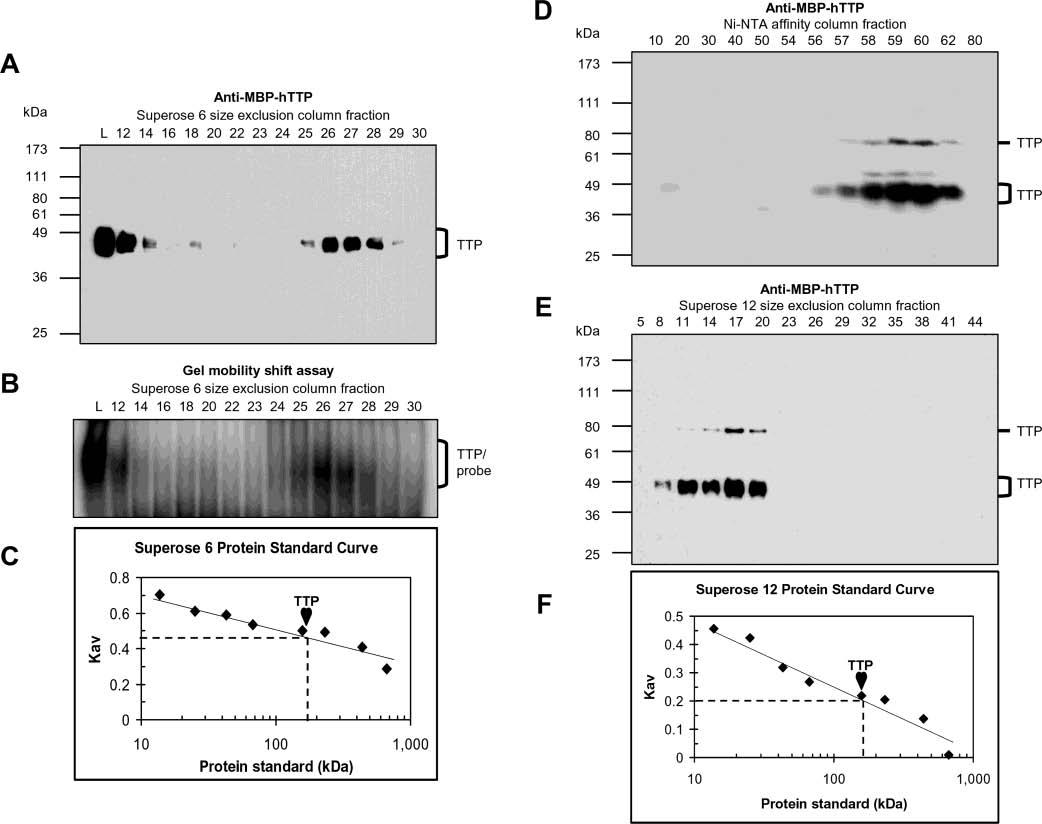
Size of active TTP from transfected human cells. The size of TTP was estimated by size exclusion chromatography using the Superose 6 column and the 10000g supernatant from HEK293 cells transfected with pHis-hTTP (A–C) and using the Superose 12 column and TTP purified by a Ni–NTA affinity column from HEK293T cells transfected with pHis-hTTP (D–F). Fractions were analyzed for the presence of TTP by immunoblotting with anti-MBP–hTTP antibodies (A, D, and E) and analyzed for ARE binging activity by GMSA (B). Lane L in panels A and B represents the initial proteins loaded in the size exclusion columns. The positions of TTP and TTP–probe complexes are indicated. The size of TTP was estimated using a standard curve generated with protein standards run on the same column under identical conditions (C and F), in which Kav = (Ve − Vo)/(Vt − Vo), where Ve, Vo, and Vt are the elution volume of the protein determined by the experiment, the void volume determined with blue dextran (2000 kDa), and the bed volume of the column provided by the manufacturer (Amersham), respectively. The protein standards that were used were provided by the manufacturer (Amersham): bovine pancreas ribonuclease A (13.7 kDa), bovine pancreas chymotrypsinogen (25 kDa), hen egg ovalbumin (43 kDa), bovine serum albumin (67 kDa), rabbit muscle aldolase (158 kDa), bovine liver catalase (232 kDa), horse spleen ferritin (440 kDa), and bovine thyroid thyroglobulin (669 kDa). Similar results were obtained using the Superose 12 column and the same extracts as in panels A–C (data not shown).
Purification of Active TTP from Transfected Human Cells. TTP was purified from the 10000g supernatant of the transfected HEK293 cells using Ni–NTA affinity beads. His-hTTP bound to Ni–NTA beads by the batch purification method was eluted with successively increased imidazole concentrations ranging from 50 to 250 mM (Figure 3A). The predominant protein bands between 40 and 50 kDa on the silver staining gel (Figure 3A) were identified as TTP by immunoblotting using anti-MBP–hTTP antibodies (Figure 3B), and by MALDI-TOF MS (data not shown). However, ∼20% of the bound TTP was still associated with the beads even after multiple imidazole elutions (data not shown). Densitometry analysis estimated that TTP was 70% of the total proteins in the 100 mM imidazole elution (Figure 3A). The integrity of the carboxyl terminus of TTP was confirmed by direct protein sequencing following trypsin digestion and HPLC separation. HPLC fractions 38 and 39 contained the same peptide with LPIFNR sequences, which corresponded to hTTP residues 316–321 (GenBank accession no. NP_003398).
Figure 3.
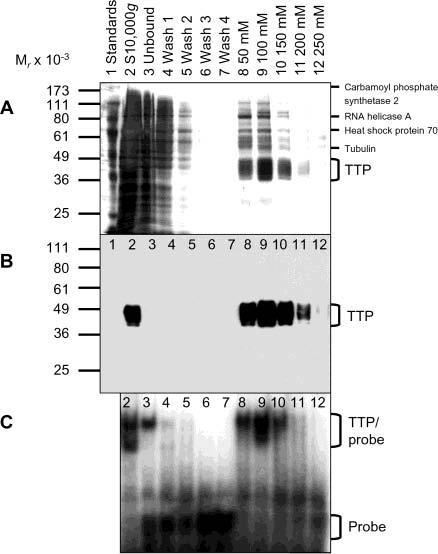
Purification of active TTP from transfected human cells. The 10000g supernatant (lane 2) was mixed with Ni–NTA beads with gentle rotation. The mixtures were then centrifuged at 1000g, resulting in the 1000g supernatant (unbound, lane 3) and the 1000g pellet (beads). The 1000g pellet was washed four times with 20 mM imidazole buffer, resulting in the 1000g supernatants as washes 1–4 (lanes 4–7). Proteins bound to the washed beads were eluted successively with 50, 100, 150, 200, and 250 mM imidazole buffer (lanes 8–12, respectively). (A) Silver staining. Proteins (20 μL) were separated by 10% SDS–PAGE and detected by silver staining. TTP and some of the minor contaminated proteins were also identified by MALDI-TOF MS as indicated. (B) Immunoblotting. Proteins (4 μL/lane) were separated by 10% SDS–PAGE, and TTP was detected with the anti-MBP–hTTP serum. The position of TTP is indicated. (C) Gel mobility shift assay. Each assay used 1 μL of each fraction generated in the Ni–NTA purification process. The positions of TTP–probe complexes and free probes are indicated.
Silver staining detected multiple minor protein bands in the purified protein samples that did not react with the antibodies (Figure 3A,B). The contaminated proteins were analyzed by MALDI-TOF MS. MS analysis identified several other proteins present in the TTP samples (Figure 3A), including RNA helicase A, heat-shock protein 70, carbamoyl phosphate synthetase 2, and tubulin. Attempts to purify TTP further by other chromatographic procedures failed, at least partly due to a precipitation problem when TTP was present at a high concentration (data not shown).
The purified TTP was demonstrated to be active by GMSA. TTP–ARE complexes were detected using proteins eluted with 50, 100, 150, and 200 mM imidazole, but not in the washes or in the 250 mM imidazole fractions (Figure 3C). However, significant ARE binding activity was detected in the unbound fraction (Figure 3C). Since TTP was barely detected in the unbound fraction (Figure 3B), other endogenous ARE-binding protein(s) in HEK293 cells might contribute to the ARE binding activity observed in this fraction (see below). Nonetheless, the results described above are consistent with TTP purified from the transfected cells being intact and capable of binding TNF mRNA ARE.
Identification of Purified TTP Free of Endogenous ARE Binding Activity. To rule out the potential contribution of ARE binding activity by the contaminated proteins in TTP preparations, proteins purified from HEK293 cells transfected with pHis-hTTP were compared to those with buffer alone, pBS+, and pEF-XEH (Figure 4). Silver staining detected multiple minor bands in proteins eluted with 100 mM imidazole buffer from all of the samples, including the negative controls (Figure 4A), but they did not react with the TTP antibodies (Figure 4B). These results suggest that the minor contaminated proteins might be due to nonspecific binding to the beads rather than their associations with TTP.
Figure 4.
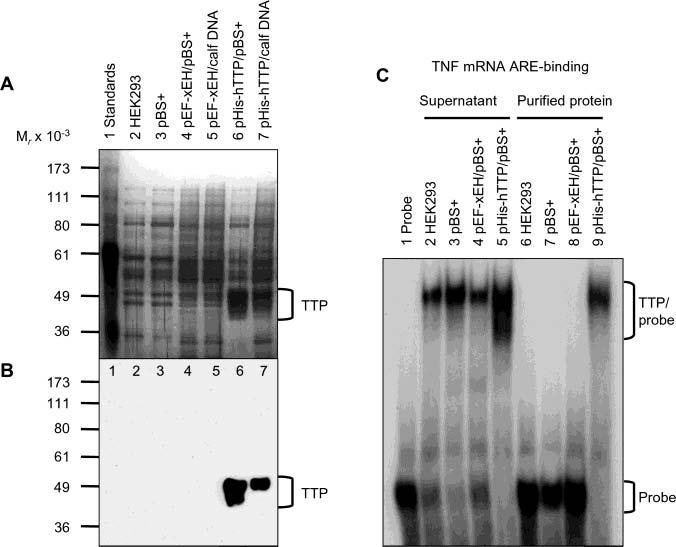
Identification of purified TTP free of endogenous ARE binding activity. HEK293 cells were transiently transfected with pHis-hTTP (0.5 μg) and cotransfected with 4.5 μg of pBS+ (lane 6) or calf thymus DNA (lane 7). As negative controls, cells were also transfected with buffers only (lane 2), pBS+ only (5 μg) (lane 3), or pEF-XEH encoding a His-tagged Xenopus EH domain of intersectin (0.5 μg) and cotransfected with 4.5 μg of pBS+ (lane 4) or calf thymus DNA (lane 5). Proteins were purified with Ni–NTA beads. (A) Silver staining. Each lane was loaded with 5 μL of the purified proteins eluted with 100 mM imidazole buffer. (B) Immunoblotting. Each lane was loaded with 5 μL of the 10000g supernatant and detected with the anti-MBP–hTTP antibodies. (C) Gel mobility shift assay. Each assay used 4 μL of the 10000g supernatant (lanes 2–5) or 4 μL of the purified proteins in 100 mM imidazole elution buffer (lanes 6–9). The positions of TTP, TTP–probe complexes, and free probes are indicated.
GMSA showed that all of the 10000g supernatants from HEK293 cells transfected with pHis-hTTP and the controls were capable of binding to the same ARE probe, but the supernatant from pHis-hTTP-transfected cells showed the highest ARE binding activity (Figure 4C). ARE binding activity was detected only in the protein purified from cells transfected with pHis-hTTP, but not from the negative controls (Figure 4C). The results described above suggested that ARE binding activity in the purified fraction was due to TTP. These results indicated that TTP purified from the transfected cells was probably folded correctly and free of other ARE binding activity.
Size of Active TTP in Transfected Human Cells. To investigate the size of active TTP, proteins in the 10000g supernatant of the transfected cells were separated using a Superose 6 column (Figure 5A-C). Anti-MBP–hTTP antibodies detected TTP in a number of fractions and peaked at fractions 26 and 27 (Figure 5A). Some TTP was also detected in fraction 12 with the elution volume immediately after the void volume (Figure 5A). All fractions containing TTP showed TNF mRNA ARE binding activity (Figure 5B). The elution volume of the majority of TTP was close to that of rabbit muscle aldolase (158 kDa). The size of TTP was estimated against the protein standards to be ∼160 kDa, consistent with active TTP forming a tetramer (Figure 5C). The size of TTP in fraction 12 corresponded to a much larger complex, and probably resulted from oligomerization, precipitation of TTP, and/or binding complexes with other partners.
To confirm the results described above from the Superose 6 column, the size of TTP was estimated using a Superose 12 column. A similar size of active TTP was observed using the same extract (data not shown). To rule out potential interference by other cellular proteins in the supernatant, the size of TTP was determined with a Superose 12 column using TTP purified by a Ni–NTA column (Figure 5D-F). TTP was detected in fractions 56–62 from the Ni–NTA column (Figure 5D) and fractions 8–20 from the Superose 12 column (Figure 5E). The size of the TTP peak in fraction 17 was estimated against protein standards to be ∼160 kDa, a value similar to that from the Superose 6 column (Figure 5F). TTP in fraction 11 corresponded to oligomers much larger than tetramers. Immunoblots showed that the size of some TTP was similar to that of a dimer (Figure 5D,E), probably due to covalent cross-linking of the monomers during the purification process.
Effect of Zinc, EDTA, and SDS on ARE Binding Activity of TTP. Previous studies using TTP purified from over-expressed E. coli cells indicate that TTP's ARE binding activity is dependent on zinc (9). Since proteins expressed in mammalian cells can be dramatically different from proteins expressed in E. coli in terms of posttranslational modifications, the effect of zinc on the ARE binding activity of TTP purified from the transfected HEK293 cells was therefore investigated. The ARE binding activity of TTP was stimulated with 10 μM ZnCl2 but decreased with higher concentrations (Figure 6A). The ARE binding activity was decreased with EDTA in the assay buffers (Figure 6A). The inhibitory effect of 0.1 mM zinc chloride on ARE binding activity was partially neutralized by 0.5 mM EDTA in the assay buffer (Figure 6A).
Figure 6.
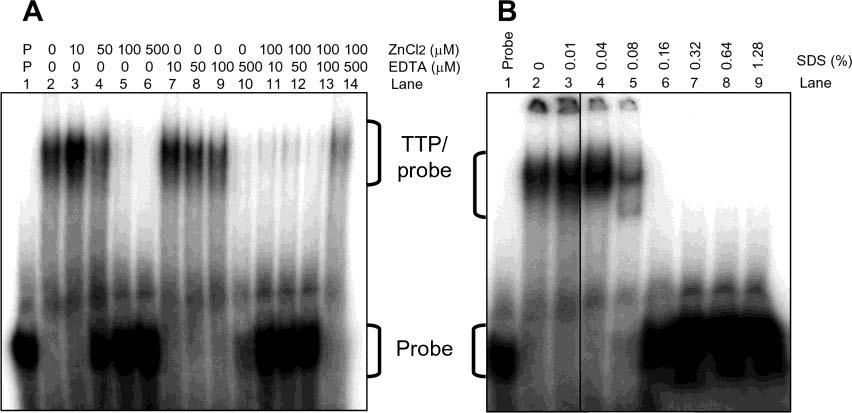
Effect of zinc, EDTA, and SDS on the ARE binding activity of TTP. ARE binding activity was assayed by GMSA using TTP (1 μL) purified by Ni–NTA beads and eluted with 100 mM imidazole buffer as described in the legend of Figure 4A (lane 6). The positions of TTP–probe complexes and free probes are indicated. (A) Effect of zinc and EDTA. GMSA buffers contained various concentrations of ZnCl2, EDTA, or mixtures of ZnCl2 and EDTA as indicated. (B) Effect of SDS. GMSA buffers contained various concentrations of SDS as indicated.
The solubility of TTP is a major problem for the purification of active TTP (9). A previous study using hTTP purified after overexpression in E. coli cells showed that a low concentration of SDS in the assay buffer helped solubilize some precipitated TTP and resulted in a higher ARE binding activity (9). Low concentrations of SDS were found to have the same effect on the ARE binding activity of TTP purified from mammalian cells. The ARE binding activity of TTP was quite stable in assay mixtures containing up to 0.04% SDS, but was completely inhibited by high concentrations of SDS (Figure 6B). The radioactivity detected in the wells of the gel was gradually decreased following addition of increased concentrations of SDS in the assay buffers (Figure 6B), suggesting that low concentrations of SDS in the assays (0.01–0.04%) might help in solubilization of TTP purified from the transfected cells.
Kinetics of ARE Binding Activity of TTP for TNF mRNA ARE. The results described above indicate that TTP expressed in HEK293 cells was similar to TTP expressed in E. coli cells in several aspects, including the size of the active protein and the responsiveness of ARE binding activity to zinc, EDTA, and SDS in the assay buffers. The difference in binding kinetics between TTP from transfected HEK293 cells and TTP from overexpressed E. coli cells (Figure 7A) was explored using two concentrations of TNF mRNA ARE probes in the assay buffers. Zinc chloride (10 μM) was added to the assay mixtures since this concentration of zinc was shown to increase TTP's ARE binding activity (Figure 6). The coprecipitation step with heparin and tRNA was determined to be unnecessary and was omitted in the binding kinetics analysis (data not shown). GMSA revealed only one major TTP–ARE complex near the origin of the gel when purified TTP was used in the assays (Figures 6 and 7), suggesting that TTP and ARE form a single binary complex (35).
Figure 7.
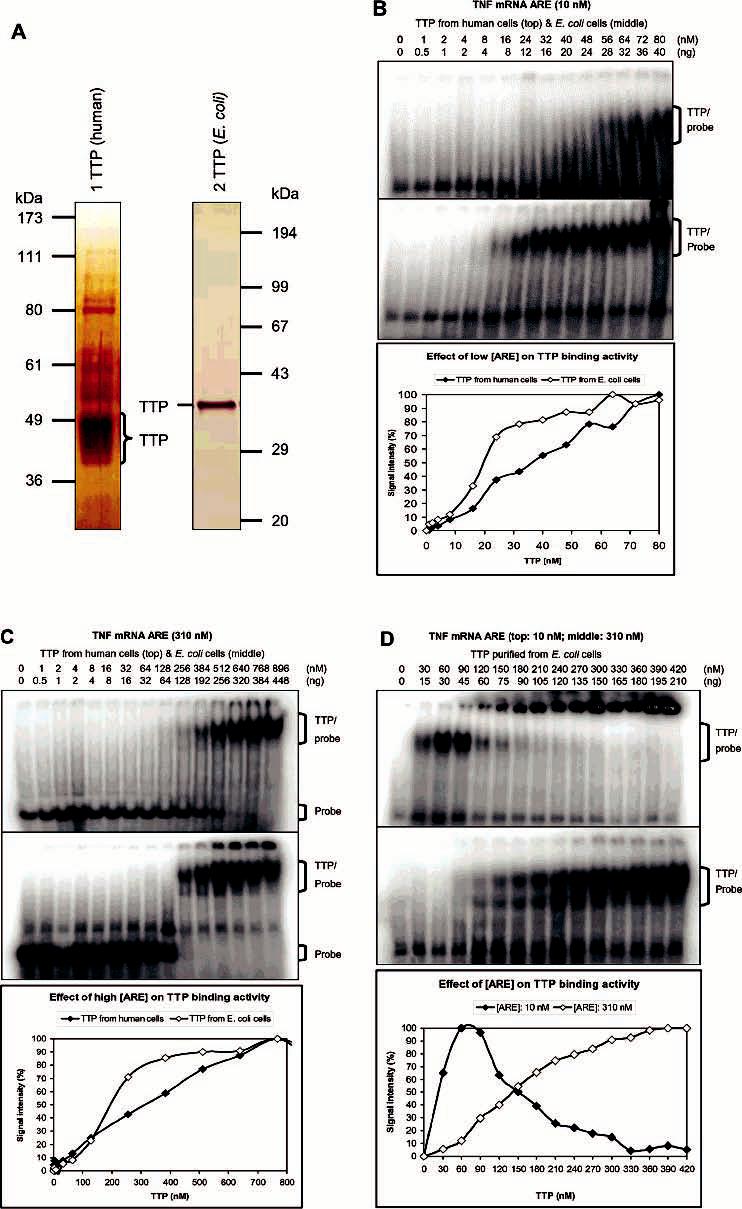
Binding kinetics of TTP for TNF mRNA ARE. Proteins were diluted with 100 mM imidazole buffer to 8.5 μL before being assayed for ARE binding activity in a 15 μL reaction mixture by GMSA. Gels were exposed to phosphorimager screens, and the signal intensity of the TTP–probe complexes and free probes was analyzed with ImageQuant 5.1. The results represented the means of two to four duplications. The positions of TTP–probe complexes and free probes are indicated. (A) Silver staining of TTP used for the binding kinetic studies. TTP was purified from transfected HEK293 cells and separated by 10% SDS–PAGE (lane 1) [72 ng/μL of a 70% pure sample, 50 ng (1.5 pmoL)−1 (μL of TTP)−1] or purified from overexpressed E. coli cells and separated by 12% SDS–PAGE (lane 2) [600 ng (18 pmoL)−1 μL−1, 100% purity]. (B) Effect of low ARE concentration (10 nM labeled ARE probe) on ARE binding kinetics: (top) TTP from HEK293 cells, (middle) TTP from E. coli cells, and (bottom) plot of the signal intensity of TTP-probe complexes vs the concentration of TTP. (C) Effect of high ARE concentration (10 nM labeled and 300 nM unlabeled ARE probe) on ARE binding kinetics: (top) TTP from HEK293 cells, (middle) TTP from E. coli cells, and (bottom) plot of the signal intensity of TTP–probe complexes vs the concentration of TTP. (D) Effect of ARE concentrations on the binding kinetics of TTP from E. coli cells: (top) 10 nM labeled ARE probe, (middle) 10 nM labeled and 300 nM unlabeled ARE probe, and (bottom) plot of the signal intensity of TTP–probe complexes vs the concentration of TTP.
ARE binding activity was first determined by GMSA using a low concentration of the ARE probe in the assay buffers, as recommended previously (35). When 10 nM labeled TNF mRNA ARE probes were used in the assays, the level of TTP–probe complexes increased gradually (Figure 7B), while the level of free ARE probes decreased gradually (data not shown), over a range of TTP concentrations. Phosphor-imager analysis was used to quantify the signal intensity of the binding complexes (Figure 7B) and the free ARE probes (data not shown). The values of half-maximal binding for TTP from HEK293 cells were 36 nM (based on the binding complexes) and 28 nM (based on the free probes). However, the half-maximal binding values for TTP from E. coli cells were approximately half of the values for TTP from HEK293 cells: 18 nM (based on the binding complexes) and 13 nM (based on the free probes) under identical assay conditions.
To confirm the significant difference of the half-maximal binding values between TTP from human cells and TTP from E. coli cells, GMSA was performed using assay conditions with 10 nM labeled and 300 nM unlabeled TNF mRNA ARE probes, such a high concentration of ARE probes not being recommended for binding kinetic assays (35). A whole gel was shown to demonstrate that only a single TTP–ARE complex was detected at a normal position even under this unusually high concentration of ARE probes (Figure 7C). The formation of detectable binding complexes required much higher concentrations of TTP when using this high concentration but low specific radioactivity of the ARE probes (Figure 7C). The concentrations of TTP required for half-maximal binding were 300 and 180 nM for TTP from HEK 293 cells and from E. coli cells, respectively (Figure 7C). A significant amount of radioactivity was detected in the wells when using high concentrations of TTP in the assays, and much more radioactivity accumulated in the wells using TTP from E. coli cells than using TTP from HEK293 cells (Figure 7C).
The effect of ARE concentrations on TTP precipitation was further investigated. When 10 nM ARE probes were used in the assays, TTP–ARE complexes were detected at normal positions using low TTP concentrations, but they were detected in the wells using high TTP concentrations (Figure 7D). On the other hand, when 30-fold unlabeled ARE was added to the assay mixtures, much less TTP–ARE complex was detected in the wells and the binding complexes gradually accumulated at normal positions (Figure 7D). The concentration of TTP required for half-maximal binding at normal positions was increased from 15 nM TTP with 10 nM ARE probes to 150 nM TTP with addition of a 30-fold molar excess of unlabeled ARE probes in these assays (Figure 7D).
Phosphorylation of TTP in Vivo and in Vitro. Immunoblotting results showed that the size of TTP from human cells ranged from 40 to 50 kDa (calculated His-hTTP Mr) = 34 826), much higher than that for TTP purified from overexpressed E. coli cells (calculated hTTP Mr) = 33 872) (Figure 8A). To test if the increased size of TTP expressed in HEK293 cells was due to phosphorylation and/or glycosylation, the 10000g supernatant of the transfected cell extract was treated with CIAP and PNGase F. Immunoblotting showed that CIAP treatment resulted in a dramatic reduction of TTP size, which was close to the size of TTP purified from E. coli cells (Figure 8A). On the other hand, PNGase F treatment did not change the gel mobility of TTP (Figure 8A). As a positive control, the gel mobility of fetuin was dramatically increased following deglycosylation with PNGase F (Figure 8A). Phosphorylation of TTP in the transfected HEK293 cells was further supported by in vivo labeling of proteins with radioactive phosphate, followed by Ni–NTA purification, or by immunoprecipitation with the anti-MBP–hTTP serum (Figure 8A). However, the T271A mutation but not the S186A mutation in hTTP, corresponding to two of the MK2-phosphorylated sites at S264 and S178 of mTTP (23), modestly altered the gel mobility of the mutant protein (Figure 8A).
Figure 8.
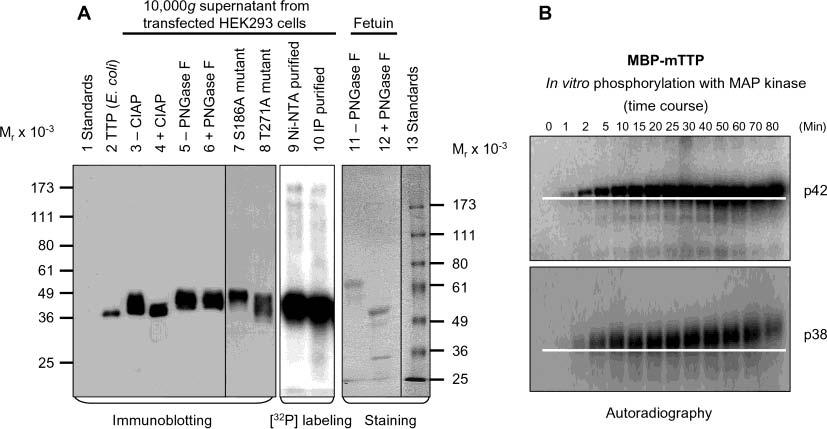
Phosphorylation of TTP in vivo and in vitro. (A) The 10000g supernatant from HEK293 cells transfected with wild-type pHish-TTP was treated with or without CIAP (lanes 3 and 4) or with or without PNGase F (lanes 5 and 6). Nonfusion hTTP purified from E. coli cells as shown in Figure 7A (lane 2) was used as a size standard for estimating the extent of the dephosphorylation (lane 2). HEK293 cells transfected with the wild-type plasmid were also labeled with [32P]orthophosphate in vivo. TTP was then purified from the 10000g supernatant of the labeled cells by affinity purification using Ni–NTA beads (lane 9) or by immunoprecipitation using the anti-MBP–hTTP serum (lane 10). The 10000g supernatants from HEK293 cells transfected with two mutant plasmids (S186A and T271A) are also shown (lanes 7 and 8). Proteins were separated by 10% SDS–PAGE, and TTP was detected with anti-MBP–hTTP antibodies (lanes 2–8). The labeled protein was detected by autoradiography (lanes 9 and 10). As a positive control for deglycosylation, fetuin was treated with or without PNGase F under identical conditions and detected with Coomassie blue staining (lanes 11 and 12). (B) Phosphorylation of MBP–mTTP by MAP kinases in vitro. MBP–mTTP (1 μM) purified from E. coli by amylose affinity resin was treated with MAP kinases, including p42/ERK2 (top) and p38 (bottom). Aliquots of the reaction mixtures were taken at various times as indicated and separated by 10% SDS–PAGE. The gels were exposed to X-ray film. A white line on the image highlights the gel mobility shift of the phosphorylated protein. Similar results were obtained with JNK, and with MBP–hTTP as the substrate (data not shown).
To provide additional evidence that supports the observation that phosphorylation caused the slower mobility of TTP in mammalian cells (Figure 8A), the relationship between gel retardation and phosphorylation was further investigated using an in vitro phosphorylation assay (Figure 8B). The sizes of MBP–mTTP purified from overexpressed E. coli cells were increased in a time-dependent manner by in vitro phosphorylation with p42, p38, and JNK MAP kinases (Figure 8B and data not shown). Similar results were obtained using MBP–hTTP in the phosphorylation assays (data not shown). Both in vivo and in vitro phosphorylation results supported the conclusion that the retarded gel mobility of TTP from transfected HEK293 cells was primarily due to phosphorylation, but not glycosylation, in the transfected cells.
DISCUSSION
TTP and related mammalian proteins, ZFP36L1 and ZFP36L2, are highly phosphorylated ARE-binding proteins (1). The properties of this family of proteins and their interactions with AREs have not been well characterized, largely due to the difficulties of obtaining high-titer antibodies and purified full-length proteins of this family from any sources. This study reports a rabbit serum capable of recognizing 1 ng of TTP on immunoblots and described a reliable procedure for purifying active TTP free of endogenous ARE binding activity normally present in HEK293 cells. The purified TTP was intact and functionally active. This was supported by several lines of evidence, including N-terminal His-tag purification, C-terminal peptide sequencing, mass spectrometry analysis, and ARE binding activity assay.
Immunocytochemistry and subcellular fractionation studies showed that the great majority of TTP was localized in the cytosol and that a small amount of TTP was localized in the nuclei of transfected cells. Nuclear TTP was able to bind TNF mRNA ARE in a manner similar to that of cytosolic TTP. Nuclear localization of TTP in normal and stimulated cells is very minimal, with a high abundance in dividing cells (3). The localization of TTP primarily in the cytosol is in agreement with previous studies using THP-1 myelomonocytic cells (5, 38), mouse tissues and cells (3), and GFP-tagged TTP in transfected cells (7). It is known that in response to mitogen stimulation, TTP is able to shuttle rapidly from the nuclei to the cytosol in NIH/3T3 mouse fibroblasts (39), and that the extent of cytoplasmic localization of TTP is also increased by association with 14-3-3 protein (25). This shuttle activity of TTP is dependent upon the primary sequence of TTP (40, 41), and partially affected by phosphorylation at S178 but not at S220 of mouse TTP (25, 39). However, there is no prior conclusive evidence for any function of TTP in the nuclei. Here we show that nuclear TTP is capable of binding to TNF mRNA ARE, resulting in the formation of TTP–ARE complexes similar to those of the cytosolic TTP. This finding raises an important question about the physiological function of TTP in the nucleus.
Gel filtration chromatography showed that active TTP was predominantly in the form of a tetramer in the transfected cells. Some of the TTP was in the form of a much larger oligomer. The formation of a large oligomer of TTP was also supported by GMSA, in which active TTP–ARE complexes were found in the wells of the autoradiograph at high TTP concentrations. Immunoblotting showed that some TTP was in a size of dimer even under SDS denaturation (Figure 5), in a manner similar to those following cross-linking with disuccinimidyl suberate (42) and the E. coli glycerol facilitator on SDS–PAGE (43). The major form of active TTP from transfected mammalian cells being a tetramer is in agreement with previous results using the MBP–hTTP fusion protein purified from overexpressed E. coli cells (9). Recent NMR results showed that the tandem zinc finger motif of the TTP-like protein (TIS11d) binds to only one copy of the 5′-UUAUUUAUU-3′ sequence (44). Since the TNF mRNA ARE probe used in this study has five overlapping UUAUUUAUU sequences and TTP–ARE complexes form a single binding complex, it is possible that one TTP tetramer binds to a single ARE probe with each TTP molecule binding one 5′-UUAUUUAUU-3′ sequence. The size of active TTP from HEK293 cells reported here and MBP–hTTP from E. coli (9) as a tetramer is different from the monomer structure of mTTP in LPS-stimulated mouse RAW 264.7 cells (3). However, it was not known which size of mTTP is capable of binding ARE in that study (3). Further experiments will be needed to determine the size of functional mTTP in LPS-stimulated RAW cells.
Gel mobility shift assays indicated that the ARE binding activity of TTP purified from HEK293 cells was dependent on zinc in the assay mixtures. ARE binding activity of the purified TTP was increased by low concentrations of zinc, suggesting that the tandem zinc finger domains of the purified TTP were not fully occupied by zinc. The zinc dependency of its ARE binding activity was further supported by the inhibitory effect of EDTA, which could extract zinc from the protein. The effect of zinc on TTP's ARE binding activity was also observed using recombinant TTP fusion proteins purified from expressed E. coli cells (9, 11). Previous studies showed that zinc in recombinant TTP purified from E. coli is essential for its ARE binding activity (9). EDTA treatment of the purified protein could destroy its ARE binding activity, but addition of zinc to the EDTA-treated TTP results in only partial recovery of its ARE binding activity. We proposed earlier that zinc is required for maintaining the structural integrity of TTP (9). This was supported by recent studies in which they showed that zinc is essential for ARE binding activity of the TTP zinc finger domain (45), and that zinc is required for the folding of the zinc finger domain of TIS11d, a TTP-like protein (44). These results suggest that TTP is a zinc-dependent mRNA ARE-binding protein, and both proteins from mammalian and E. coli cells may have similar core molecular structures involved in the interaction with the AREs.
Kinetic studies indicated that posttranslational modifications of TTP in mammalian cells significantly reduced the affinity of TTP for TNF mRNA ARE. This conclusion was based on comparative studies using TTP purified from transfected human cells and from overexpressed E. coli cells. TTP bound to TNF mRNA ARE with high affinity (46). The values for the half-maximal binding were determined to be 28–36 nM for TTP from HEK293 cells and 13–18 nM for TTP from E. coli cells, when 10 nM ARE was used in the GMSA mixtures. The difference in binding affinity between the two TTP preparations was further supported using a much higher concentration of ARE probes in GMSA mixtures. The half-maximal binding values of full-length TTP using 10 nM TNF mRNA ARE in the assays are similar to recently reported values of the TTP tandem zinc finger peptide using 0.2 nM ARE in their assays (10, 45), but significantly lower than those of a TTP single-zinc finger peptide at micromolar levels (47). The half-maximal binding values of TTP for TNF mRNA ARE reported in this study are within the ranges of other ARE-binding proteins, such as His6-AUF1 for c-fos and c-myc proto-oncogene mRNA AREs (48), GST–HuR for human β-adrenergic receptor 160-mer ARE (49), and Xenopus transcription factor IIIA for 5S rRNA (35). These results suggest that the affinity of TTP for TNF mRNA ARE is reduced by approximately half by posttranslational modification machinery in mammalian cells, since proteins from E. coli cells are barely modified posttranslationally (50).
Protein labeling experiments showed that TTP was highly phosphorylated in intact cells. TTP from transfected HEK293 cells appeared as a broad band with a molecular mass much higher than that of the predicated unmodified TTP, while TTP purified from overexpressed E. coli cells appeared as a sharp band on immunoblots. Dephosphorylation, but not deglycosylation, of TTP from HEK293 cells collapsed the broad band into a sharp band on immunoblots. Conversely, the size of MBP–TTP from E. coli was increased following in vitro phosphorylation with MAP kinases. These results suggest that phosphorylation, but not glycosylation, contributes to the gel retardation of TTP from mammalian cells. Previous studies compared ARE binding activity between CIAP-treated and CIAP-untreated crude preparations (the 10000g supernatant from transfected HEK293 cells). The relative ARE binding activity of the CIAP-treated preparations was higher than that of the untreated samples (20). The conclusions from the current studies using purified TTP and from the earlier studies using crude extracts are similar; i.e., unphosphorylated TTP from E. coli and dephosphorylated TTP from HEK293 cells have higher binding affinity and relative binding activity, respectively. There are similar cases in which RNA binding activities are decreased by protein phosphorylation, including potato virus A coat protein (51) and RNA-dependent protein kinase (52). It is possible that the difference in ARE binding affinity between TTP from mammalian cells and E. coli is due to phosphorylation but not glycosylation. In addition to the effect of phosphorylation on TTP's mRNA ARE binding activity reported here and previously (20), phosphorylation may also affect other potential function(s) of TTP, such as subcellular localization (25), apoptosis (37), association with exosome (53), stress granule (54), and other proteins (55-57), as well as TTP's stability (3) and autoregulation (58, 59). We are currently pursuing this investigation further and have identified multiple phosphorylation sites in hTTP by mass spectrometry, protein sequencing, and site-directed mutagenesis (H. Cao and P. J. Blackshear, unpublished results).
In summary, the results reported here suggest that active TTP forms a tetramer and its zinc-dependent ARE binding affinity is reduced by posttranslational phosphorylation in the transfected mammalian cells. The results support a model in which each subunit of the TTP tetramer binds to one of the five overlapping UUAUUUAUU sequences of the ARE, resulting in a stable TTP–ARE complex. The technologies described here, including producing the high-titer anti-MBP–hTTP serum and developing the reliable protocol for purifying full-length TTP free of endogenous ARE binding activity, should facilitate further studies in developing a better understanding of the structure–function relationships of TTP.
ACKNOWLEDGMENT
I greatly appreciate Dr. Perry J. Blackshear for encouragement and insights and Rui Lin for technical assistance. I also thank Dr. Wi S. Lai for CMV·(His)6·N·hTTP and TNF DNA and HA-tagged 293 cell extracts, Elizabeth A. Kennington for advice on protein purification, Jane S. Tuttle for advice on mammalian cell culture, Dr. Deborah J. Stumpo for plasmid pBS+, Dr. John O'Bryan for plasmid pEF-XEH, Dr. Leesa J. Deterding for fetuin, Josh Dubin in Dr. Kenneth B. Tomer's group for MALDI-TOF MS analysis, and Drs. Frank Kari, Robert M. Petrovich, and Mohamed Trebak for critical reading of the manuscript.
Footnotes
This work was supported in part by NIH Intramural Research Training Award TAXP003505.
A preliminary report of this study was presented at the 2nd Human Proteomics Organization and the 19th International Union of Biochemistry and Molecular Biology Joint World Congress in Montreal, Canada, on October 8–11, 2003.
Abbreviations: TTP, tristetraprolin; hTTP, human TTP; mTTP, mouse TTP; HA-hTTP, hemagglutinin epitope-tagged hTTP; His-hTTP, six-histidine-tagged hTTP; MBP–hTTP, maltose binding protein–hTTP fusion protein; MBP–mTTP, maltose binding protein–mTTP fusion protein; ARE, AU-rich element; BSA, bovine serum albumin; CIAP, calf intestine alkaline phosphatase; DTT, dithiothreitol; FCS, fetal calf serum; GAR–HRP, goat anti-rabbit IgG (H+L)–horseradish peroxidase conjugate; GM-CSF, granulocyte-macrophage colony-stimulating factor; GMSA, gel mobility shift assay; HEK, human embryonic kidney; LPS, lipopolysaccharide; MALDI-TOF MS, matrix-assisted laser desorption ionization time-of-flight mass spectrometry; MAP, mitogen-activated protein; MBP, maltose binding protein; NE, nuclear extract; Ni–NTA, nickel–nitrilotriacetic agarose; NP-40, Nonidet P-40; PBS, phosphate-buffered saline; PMSF, phenylmethanesulfonyl fluoride; SDS–PAGE, sodium dodecyl sulfate–polyacrylamide gel electrophoresis; TFA, trifluoroacetic acid; TNF, tumor necrosis factor α.
REFERENCES
- 1.Blackshear PJ. Tristetraprolin and other CCCH tandem zinc-finger proteins in the regulation of mRNA turnover. Biochem. Soc. Trans. 2002;30:945–952. doi: 10.1042/bst0300945. [DOI] [PubMed] [Google Scholar]
- 2.Lai WS, Stumpo DJ, Blackshear PJ. Rapid insulin-stimulated accumulation of an mRNA encoding a proline-rich protein. J. Biol. Chem. 1990;265:16556–16563. [PubMed] [Google Scholar]
- 3.Cao H, Tuttle JS, Blackshear PJ. Immunological characterization of tristetraprolin as a low abundance, inducible, stable cytosolic protein. J. Biol. Chem. 2004;279:21489–21499. doi: 10.1074/jbc.M400900200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bakheet T, Frevel M, Williams BR, Greer W, Khabar KS. ARED: human AU-rich element-containing mRNA database reveals an unexpectedly diverse functional repertoire of encoded proteins. Nucleic Acids Res. 2001;29:246–254. doi: 10.1093/nar/29.1.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carballo E, Lai WS, Blackshear PJ. Feedback inhibition of macrophage tumor necrosis factor-α production by tristetraprolin. Science. 1998;281:1001–1005. doi: 10.1126/science.281.5379.1001. [DOI] [PubMed] [Google Scholar]
- 6.Carballo E, Lai WS, Blackshear PJ. Evidence that tristetraprolin is a physiological regulator of granulocyte-macrophage colony-stimulating factor messenger RNA deadenylation and stability. Blood. 2000;95:1891–1899. [PubMed] [Google Scholar]
- 7.Lai WS, Carballo E, Strum JR, Kennington EA, Phillips RS, Blackshear PJ. Evidence that tristetraprolin binds to AU-rich elements and promotes the deadenylation and destabilization of tumor necrosis factor α mRNA. Mol. Cell. Biol. 1999;19:4311–4323. doi: 10.1128/mcb.19.6.4311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lai WS, Blackshear PJ. Interactions of CCCH zinc finger proteins with mRNA: tristetraprolin-mediated AU-rich element-dependent mRNA degradation can occur in the absence of a poly(A) tail. J. Biol. Chem. 2001;276:23144–23154. doi: 10.1074/jbc.M100680200. [DOI] [PubMed] [Google Scholar]
- 9.Cao H, Dzineku F, Blackshear PJ. Expression and purification of recombinant tristetraprolin that can bind to tumor necrosis factor-α mRNA and serve as a substrate for mitogen-activated protein kinases. Arch. Biochem. Biophys. 2003;412:106–120. doi: 10.1016/s0003-9861(03)00012-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blackshear PJ, Lai WS, Kennington EA, Brewer G, Wilson GM, Guan X, Zhou P. Characteristics of the interaction of a synthetic human tristetraprolin tandem zinc finger peptide with AU-rich element-containing RNA substrates. J. Biol. Chem. 2003;278:19947–19955. doi: 10.1074/jbc.M301290200. [DOI] [PubMed] [Google Scholar]
- 11.Worthington MT, Pelo JW, Sachedina MA, Applegate JL, Arseneau KO, Pizarro TT. RNA binding properties of the AU-rich element-binding recombinant Nup475/TIS11/tristetraprolin protein. J. Biol. Chem. 2002;277:48558–48564. doi: 10.1074/jbc.M206505200. [DOI] [PubMed] [Google Scholar]
- 12.Sawaoka H, Dixon DA, Oates JA, Boutaud O. Tristetraprolin binds to the 3′-untranslated region of cyclooxygenase-2 mRNA. A polyadenylation variant in a cancer cell line lacks the binding site. J. Biol. Chem. 2003;278:13928–13935. doi: 10.1074/jbc.M300016200. [DOI] [PubMed] [Google Scholar]
- 13.Dixon DA. Dysregulated post-transcriptional control of COX-2 gene expression in cancer. Curr. Pharm. Des. 2004;10:635–646. doi: 10.2174/1381612043453171. [DOI] [PubMed] [Google Scholar]
- 14.Yu H, Stasinopoulos S, Leedman P, Medcalf RL. Inherent instability of plasminogen activator inhibitor type 2 mRNA is regulated by tristetraprolin. J. Biol. Chem. 2003;278:13912–13918. doi: 10.1074/jbc.M213027200. [DOI] [PubMed] [Google Scholar]
- 15.Taylor GA, Carballo E, Lee DM, Lai WS, Thompson MJ, Patel DD, Schenkman DI, Gilkeson GS, Broxmeyer HE, Haynes BF, Blackshear PJ. A pathogenetic role for TNF α in the syndrome of cachexia, arthritis, and autoimmunity resulting from tristetraprolin (TTP) deficiency. Immunity. 1996;4:445–454. doi: 10.1016/s1074-7613(00)80411-2. [DOI] [PubMed] [Google Scholar]
- 16.Phillips K, Kedersha N, Shen L, Blackshear PJ, Anderson P. Arthritis suppressor genes TIA-1 and TTP dampen the expression of tumor necrosis factor α, cyclooxygenase 2, and inflammatory arthritis. Proc. Natl. Acad. Sci. U.S.A. 2004;101:2011–2016. doi: 10.1073/pnas.0400148101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DuBois RN, McLane MW, Ryder K, Lau LF, Nathans D. A growth factor-inducible nuclear protein with a novel cysteine/histidine repetitive sequence. J. Biol. Chem. 1990;265:19185–19191. [PubMed] [Google Scholar]
- 18.Worthington MT, Amann BT, Nathans D, Berg JM. Metal binding properties and secondary structure of the zinc-binding domain of Nup475. Proc. Natl. Acad. Sci. U.S.A. 1996;93:13754–13759. doi: 10.1073/pnas.93.24.13754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Amann BT, Worthington MT, Berg JM. A Cys3His zinc-binding domain from Nup475/tristetraprolin: a novel fold with a disklike structure. Biochemistry. 2003;42:217–221. doi: 10.1021/bi026988m. [DOI] [PubMed] [Google Scholar]
- 20.Carballo E, Cao H, Lai WS, Kennington EA, Campbell D, Blackshear PJ. Decreased sensitivity of tristetraprolin-deficient cells to p38 inhibitors suggests the involvement of tristetraprolin in the p38 signaling pathway. J. Biol. Chem. 2001;276:42580–42587. doi: 10.1074/jbc.M104953200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Taylor GA, Thompson MJ, Lai WS, Blackshear PJ. Phosphorylation of tristetraprolin, a potential zinc finger transcription factor, by mitogen stimulation in intact cells and by mitogen-activated protein kinase in vitro. J. Biol. Chem. 1995;270:13341–13347. doi: 10.1074/jbc.270.22.13341. [DOI] [PubMed] [Google Scholar]
- 22.Zhu W, Brauchle MA, Di Padova F, Gram H, New L, Ono K, Downey JS, Han J. Gene suppression by tristetraprolin and release by the p38 pathway. Am. J. Physiol. 2001;281:L499–L508. doi: 10.1152/ajplung.2001.281.2.L499. [DOI] [PubMed] [Google Scholar]
- 23.Chrestensen CA, Schroeder MJ, Shabanowitz J, Hunt DF, Pelo JW, Worthington MT, Sturgill TW. MAPKAP kinase 2 phosphorylates tristetraprolin on in vivo sites including Ser178, a site required for 14-3-3 binding. J. Biol. Chem. 2004;279:10176–10184. doi: 10.1074/jbc.M310486200. [DOI] [PubMed] [Google Scholar]
- 24.Mahtani KR, Brook M, Dean JL, Sully G, Saklatvala J, Clark AR. Mitogen-activated protein kinase p38 controls the expression and posttranslational modification of tristetraprolin, a regulator of tumor necrosis factor α mRNA stability. Mol. Cell. Biol. 2001;21:6461–6469. doi: 10.1128/MCB.21.9.6461-6469.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Johnson BA, Stehn JR, Yaffe MB, Blackwell TK. Cytoplasmic localization of tristetraprolin involves 14-3-3-dependent and -independent mechanisms. J. Biol. Chem. 2002;277:18029–18036. doi: 10.1074/jbc.M110465200. [DOI] [PubMed] [Google Scholar]
- 26.Adams A, Thorn JM, Yamabhai M, Kay BK, O'Bryan JP. Intersectin, an adaptor protein involved in clathrin-mediated endocytosis, activates mitogenic signaling pathways. J. Biol. Chem. 2000;275:27414–27420. doi: 10.1074/jbc.M004810200. [DOI] [PubMed] [Google Scholar]
- 27.Lai WS, Thompson MJ, Taylor GA, Liu Y, Blackshear PJ. Promoter analysis of Zfp-36, the mitogeninducible gene encoding the zinc finger protein tristetraprolin. J. Biol. Chem. 1995;270:25266–25272. doi: 10.1074/jbc.270.42.25266. [DOI] [PubMed] [Google Scholar]
- 28.Cao H, Sullivan TD, Boyer CD, Shannon JC. Bt1, a structural gene for the major 39–44 kDa amyloplast membrane polypeptides. Physiol. Plant. 1995;95:176–186. [Google Scholar]
- 29.Stoscheck CM. Quantitation of protein. Methods Enzymol. 1990;182:50–68. doi: 10.1016/0076-6879(90)82008-p. [DOI] [PubMed] [Google Scholar]
- 30.Sambrook J, Fritsch EF, Naniatis T. Molecular Cloning: A Laboratory Manual. 2nd Cold Spring Harbor Laboratory Press; Plainview, NY: 1989. [Google Scholar]
- 31.Shevchenko A, Wilm M, Vorm O, Mann M. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal. Chem. 1996;68:850–858. doi: 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- 32.Deterding LJ, Prasad R, Mullen GP, Wilson SH, Tomer KB. Mapping of the 5′-2-deoxyribose-5-phosphate lyase active site in DNA polymerase β by mass spectrometry. J. Biol. Chem. 2000;275:10463–10471. doi: 10.1074/jbc.275.14.10463. [DOI] [PubMed] [Google Scholar]
- 33.Cao H, James MG, Myers AM. Purification and characterization of soluble starch synthases from maize endosperm. Arch. Biochem. Biophys. 2000;373:135–146. doi: 10.1006/abbi.1999.1547. [DOI] [PubMed] [Google Scholar]
- 34.Fransen L, Muller R, Marmenout A, Tavernier J, Van der Heyden J, Kawashima E, Chollet A, Tizard R, Van Heuverswyn H, Van Vliet A, Ruysschaert M, Fiers W. Molecular cloning of mouse tumour necrosis factor cDNA and its eukaryotic expression. Nucleic Acids Res. 1985;13:4417–4429. doi: 10.1093/nar/13.12.4417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Setzer DR. Measuring equilibrium and kinetic constants using gel retardation assays. Methods Mol. Biol. 1999;118:115–128. doi: 10.1385/1-59259-676-2:115. [DOI] [PubMed] [Google Scholar]
- 36.Cao H, Imparl-Radosevich J, Guan H, Keeling PL, James MG, Myers AM. Identification of the soluble starch synthase activities of maize endosperm. Plant Physiol. 1999;120:205–216. doi: 10.1104/pp.120.1.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Johnson BA, Geha M, Blackwell TK. Similar but distinct effects of the tristetraprolin/TIS11 immediate-early proteins on cell survival. Oncogene. 2000;19:1657–1664. doi: 10.1038/sj.onc.1203474. [DOI] [PubMed] [Google Scholar]
- 38.Brooks SA, Connolly JE, Diegel RJ, Fava RA, Rigby WF. Analysis of the function, expression, and subcellular distribution of human tristetraprolin. Arthritis Rheum. 2002;46:1362–1370. doi: 10.1002/art.10235. [DOI] [PubMed] [Google Scholar]
- 39.Taylor GA, Thompson MJ, Lai WS, Blackshear PJ. Mitogens stimulate the rapid nuclear to cytosolic trans-location of tristetraprolin, a potential zinc-finger transcription factor. Mol. Endocrinol. 1996;10:140–146. doi: 10.1210/mend.10.2.8825554. [DOI] [PubMed] [Google Scholar]
- 40.Phillips RS, Ramos SB, Blackshear PJ. Members of the tristetraprolin family of tandem CCCH zinc finger proteins exhibit CRM1-dependent nucleocytoplasmic shuttling. J. Biol. Chem. 2002;277:11606–11613. doi: 10.1074/jbc.M111457200. [DOI] [PubMed] [Google Scholar]
- 41.Murata T, Yoshino Y, Morita N, Kaneda N. Identification of nuclear import and export signals within the structure of the zinc finger protein TIS11. Biochem. Biophys. Res. Commun. 2002;293:1242–1247. doi: 10.1016/S0006-291X(02)00363-7. [DOI] [PubMed] [Google Scholar]
- 42.Lai WS, Kennington EA, Blackshear PJ. Tristetraprolin and its family members can promote the cell-free deadenylation of AU-rich element-containing mRNAs by poly(A) ribonuclease. Mol. Cell. Biol. 2003;23:3798–3812. doi: 10.1128/MCB.23.11.3798-3812.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Manley DM, McComb ME, Perreault H, Donald LJ, Duckworth HW, O'Neil JD. Secondary structure and oligomerization of the E. coli glycerol facilitator. Biochemistry. 2000;39:12303–12311. doi: 10.1021/bi000703t. [DOI] [PubMed] [Google Scholar]
- 44.Hudson BP, Martinez-Yamout MA, Dyson HJ, Wright PE. Recognition of the mRNA AU-rich element by the zinc finger domain of TIS11d. Nat. Struct. Mol. Biol. 2004;11:257–264. doi: 10.1038/nsmb738. [DOI] [PubMed] [Google Scholar]
- 45.Brewer BY, Malicka J, Blackshear PJ, Wilson GM. RNA sequence elements required for high affinity binding by the zinc finger domain of tristetraprolin: Conformational changes coupled to the bipartite nature of AU-rich mRNAdestabilizing motifs. J. Biol. Chem. 2004;279:27870–27877. doi: 10.1074/jbc.M402551200. [DOI] [PubMed] [Google Scholar]
- 46.Cao H, Blackshear PJ. Characterization of tristetraprolin as a zinc-dependent mRNA ARE-binding protein. Mol. Cell. Proteomics. 2003;2:704. [Google Scholar]
- 47.Michel SL, Guerrerio AL, Berg JM. Selective RNA binding by a single CCCH zinc-binding domain from Nup475 (tristetraprolin) Biochemistry. 2003;42:4626–4630. doi: 10.1021/bi034073h. [DOI] [PubMed] [Google Scholar]
- 48.DeMaria CT, Brewer G. AUF1 binding affinity to A+U-rich elements correlates with rapid mRNA degradation. J. Biol. Chem. 1996;271:12179–12184. doi: 10.1074/jbc.271.21.12179. [DOI] [PubMed] [Google Scholar]
- 49.Blaxall BC, Pellett AC, Wu SC, Pende A, Port JD. Purification and characterization of β-adrenergic receptor mRNA-binding proteins. J. Biol. Chem. 2000;275:4290–4297. doi: 10.1074/jbc.275.6.4290. [DOI] [PubMed] [Google Scholar]
- 50.Blattner FR, Plunkett G, III, Bloch CA, Perna NT, Burland V, Riley M, Collado-Vides J, Glasner JD, Rode CK, Mayhew GF, Gregor J, Davis NW, Kirkpatrick HA, Goeden MA, Rose DJ, Mau B, Shao Y. The complete genome sequence of Escherichia coli K-12. Science. 1997;277:1453–1474. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- 51.Ivanov KI, Puustinen P, Merits A, Saarma M, Makinen K. Phosphorylation down-regulates the RNA binding function of the coat protein of potato virus A. J. Biol. Chem. 2001;276:13530–13540. doi: 10.1074/jbc.M009551200. [DOI] [PubMed] [Google Scholar]
- 52.Jammi NV, Beal PA. Phosphorylation of the RNA-dependent protein kinase regulates its RNA-binding activity. Nucleic Acids Res. 2001;29:3020–3029. doi: 10.1093/nar/29.14.3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen CY, Gherzi R, Ong SE, Chan EL, Raijmakers R, Pruijn GJ, Stoecklin G, Moroni C, Mann M, Karin M. AU binding proteins recruit the exosome to degrade ARE-containing mRNAs. Cell. 2001;107:451–464. doi: 10.1016/s0092-8674(01)00578-5. [DOI] [PubMed] [Google Scholar]
- 54.Stoecklin G, Stubbs T, Kedersha N, Wax S, Rigby WF, Blackwell TK, Anderson P. MK2-induced tristetraprolin: 14-3-3 complexes prevent stress granule association and ARE-mRNA decay. EMBO J. 2004;23:1313–1324. doi: 10.1038/sj.emboj.7600163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Carman JA, Nadler SG. Direct association of tristetraprolin with the nucleoporin CAN/Nup214. Biochem. Biophys. Res. Commun. 2004;315:445–449. doi: 10.1016/j.bbrc.2004.01.080. [DOI] [PubMed] [Google Scholar]
- 56.Twizere JC, Kruys V, Lefebvre L, Vanderplasschen A, Collete D, Debacq C, Lai WS, Jauniaux JC, Bernstein LR, Semmes OJ, Burny A, Blackshear PJ, Kettmann R, Willems L. Interaction of retroviral Tax oncoproteins with tristetraprolin and regulation of tumor necrosis factor-α expression. J. Natl. Cancer Inst. 2003;95:1846–1859. doi: 10.1093/jnci/djg118. [DOI] [PubMed] [Google Scholar]
- 57.Esclatine A, Taddeo B, Roizman B. Herpes Simplex Virus 1 Induces Cytoplasmic Accumulation of TIA-1/TIAR and both Synthesis and Cytoplasmic Accumulation of Tristetraprolin, Two Cellular Proteins That Bind and Destabilize AU-Rich RNAs. J. Virol. 2004;78:8582–8592. doi: 10.1128/JVI.78.16.8582-8592.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Brooks SA, Connolly JE, Rigby WFC. The role of mRNA turnover in the regulation of tristetraprolin expression: evidence for an extracellular signal-regulated kinase-specific, AU-rich element-dependent, autoregulatory pathway. J. Immunol. 2004;172:7263–7271. doi: 10.4049/jimmunol.172.12.7263. [DOI] [PubMed] [Google Scholar]
- 59.Tchen CR, Brook M, Saklatvala J, Clark AR. The stability of tristetraprolin mRNA is regulated by mitogen-activated protein kinase p38 and by tristetraprolin itself. J. Biol. Chem. 2004;279:32393–32400. doi: 10.1074/jbc.M402059200. [DOI] [PubMed] [Google Scholar]